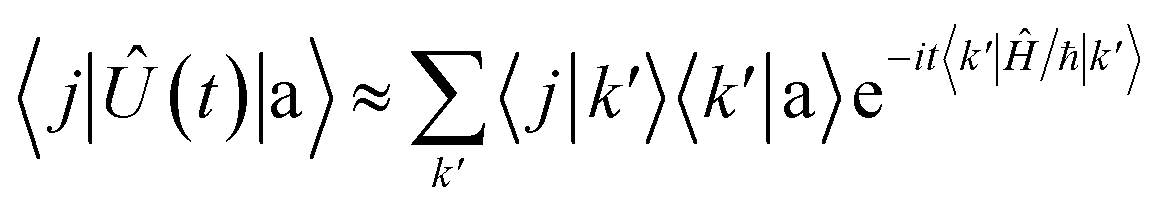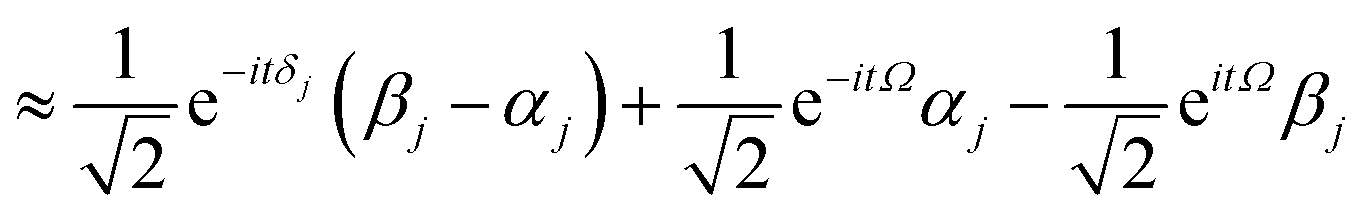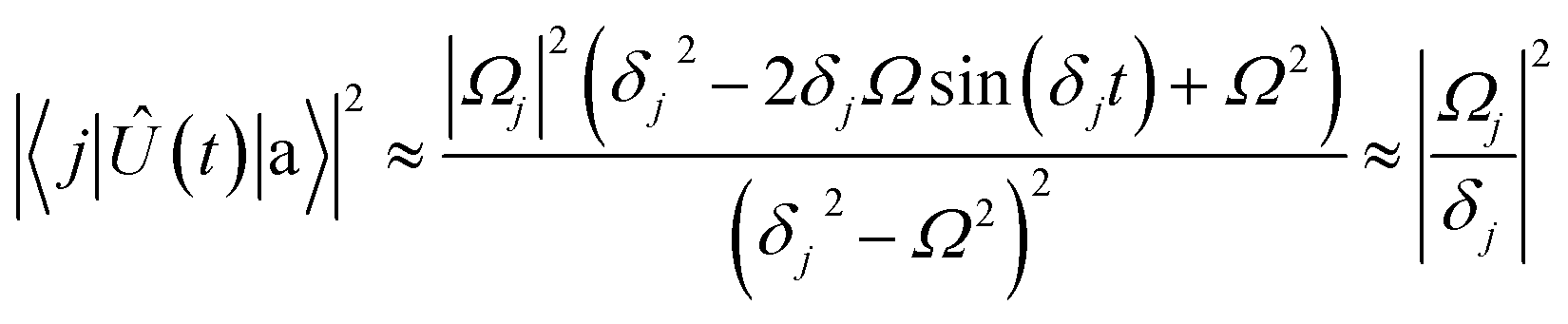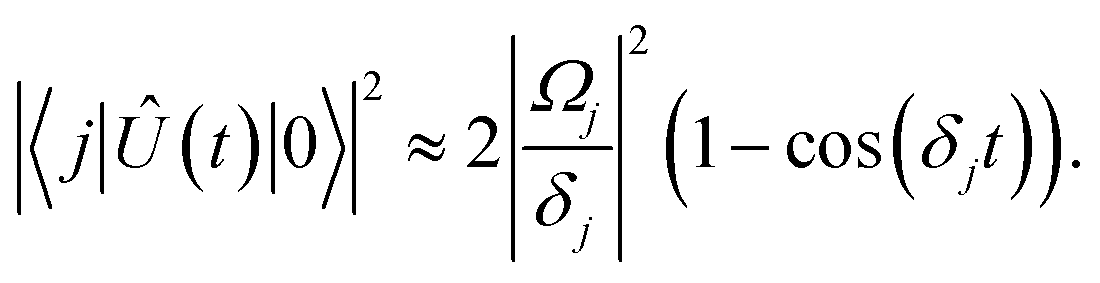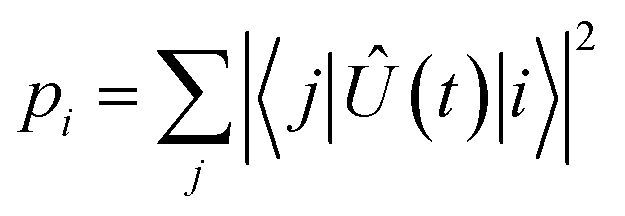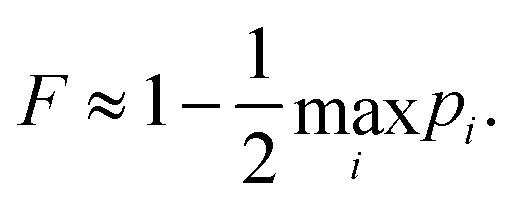DOI:
10.1039/C8SC02355G
(Edge Article)
Chem. Sci., 2018,
9, 6830-6838
Dipolar exchange quantum logic gate with polar molecules
Received
30th May 2018
, Accepted 12th July 2018
First published on 13th July 2018
Abstract
We propose a two-qubit gate based on dipolar exchange interactions between individually addressable ultracold polar molecules in an array of optical dipole traps. Our proposal treats the full Hamiltonian of the 1Σ+ molecule NaCs, utilizing a pair of nuclear spin states as storage qubits. A third rotationally excited state with rotation-hyperfine coupling enables switchable electric dipolar exchange interactions between two molecules to generate an iSWAP gate. All three states are insensitive to external magnetic and electric fields. Impacts on gate fidelity due to coupling to other molecular states, imperfect ground-state cooling, blackbody radiation and vacuum spontaneous emission are small, leading to potential fidelity above 99.99% in a coherent quantum system that can be scaled by purely optical means.
1 Introduction
Important progress has been made towards a laboratory quantum computer with state-of-the-art demonstrations reaching a combination of 5 qubits and 98.3% CNOT gate fidelity.1,2 The criteria for quantum computation3 have been identified as (1) a scalable system of qubits (2) initialization (3) coherence (4) universal set of qubit gates (5) measurement. Items 2 through 5 have been demonstrated at sufficient fidelities,4–8 showing that computation with many qubits9–11 may be possible. But the route toward scalability remains challenging. Here, we focus on the problem of producing a high-fidelity two-qubit gate using optically trapped dipolar molecules, with the hope that this physical system allows for easier scalability. Recent demonstrations of flexible optical tweezer arrays12,13 show a method by which many qubits could be rearranged to implement quantum algorithms.
Optically trapped, electrically dipolar neutral molecules have long been recognized as potential qubits14–18 where the dipole–dipole interaction between two molecules mediates a two-qubit gate. However, most proposals rely on static or oscillating dressing electric fields to polarize the molecules, where the molecular Stark energy is much larger than the dipolar interaction. This imposes stringent constraints on field stability.
Here we describe concretely how the natural dipolar interaction between two molecules can produce an iSWAP gate, without the need for additional polarizing fields, thereby removing an important source of implementation complexity and qubit decoherence. This iSWAP gate, together with single qubit rotations, forms a universal set of qubit gates.19,20 We exploit the rich molecular internal structure and use NaCs as an example to find molecular qubits that are expected to have long coherence (item 3). The gate relies on two-qubit interactions that are switched on by driving one qubit state to a third state via a microwave transition. We find parameters that allow gates with high fidelity (F > 1 − 10−4) in 10 ms at a magnetic field of 1 Gauss when light shifts due to the optical trap are neglected. For an optical trap depth of 600 kHz, the same fidelity and duration can be reached for a 35 Gauss magnetic field. The gate duration could be reduced by applying shaped pulses rather than the square pulses considered here.
2 Exchange and the iSWAP gate
It is a well known phenomenon that if two identical systems interact weakly, where one system has an energy excitation and the other does not, the excitation eventually transfers. This effect can form the basis for a two-qubit gate17,20–24 and has been discussed in the context of molecule-based quantum simulations of spin models.25,26 The transfer of excitation via the dipole–dipole interaction has been demonstrated for ultracold KRb molecules27,28 and atoms with large magnetic dipoles29 in optical lattices.
We rely on the natural dipole–dipole interaction between molecules to enable evolution of the type |0;e〉 ↔ |e;0〉 where |0〉 is a sub-level of the rotational ground state, and |e〉 is a sub-level of the first rotational excited state (see Fig. 1A and B). In the two-particle state, the first position refers to the first molecule, and the second position refers to the second molecule. This exchange interaction leaves the states |0;0〉 and |e;e〉 unchanged, and the overall unitary evolution in the basis |0;0〉, |0;e〉, |e;0〉, |e;e〉 is28
| | 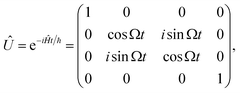 | (1) |
where
Ω =
D/
r3 is the interaction Rabi rate,
r is the molecule–molecule distance, and the duration
t = π/(2
Ω) produces the iSWAP gate. The factor
D (see
eqn (3)) depends on the choice of molecule, separation direction, choice of states, magnetic field, and light shift. In the 1 Gauss example of Section 5, two NaCs molecules have
t = 4 ms for
r = 2.9 μm separation along
![[x with combining circumflex]](https://www.rsc.org/images/entities/i_char_0078_0302.gif)
, which is also the magnetic field direction. For the 35 Gauss example, which includes the effects of a 600 kHz deep optical trap, we use an interaction duration of
t = 2 ms with
r = 2.5 μm along
and the magnetic field direction is
ẑ. While the resolution of optical tweezers with beam waist below 1 μm supports smaller separation and gates as fast as
t = 50 μs, two effects place additional constraints. (1) Off-resonant population leakage degrades the gate fidelity for short durations (Section 6) and (2) smaller molecule separation (
r) makes the gate more sensitive to motional excitation (Section 7).
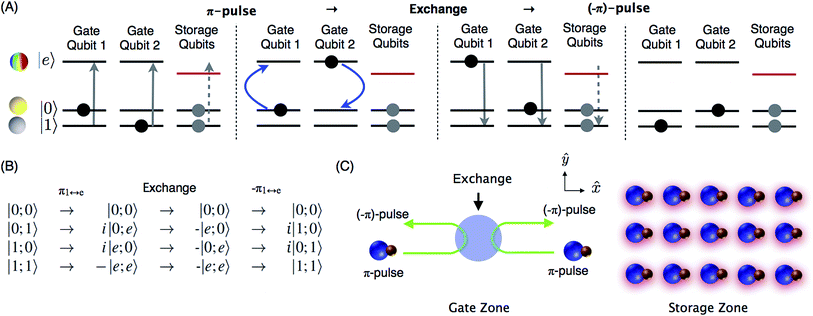 |
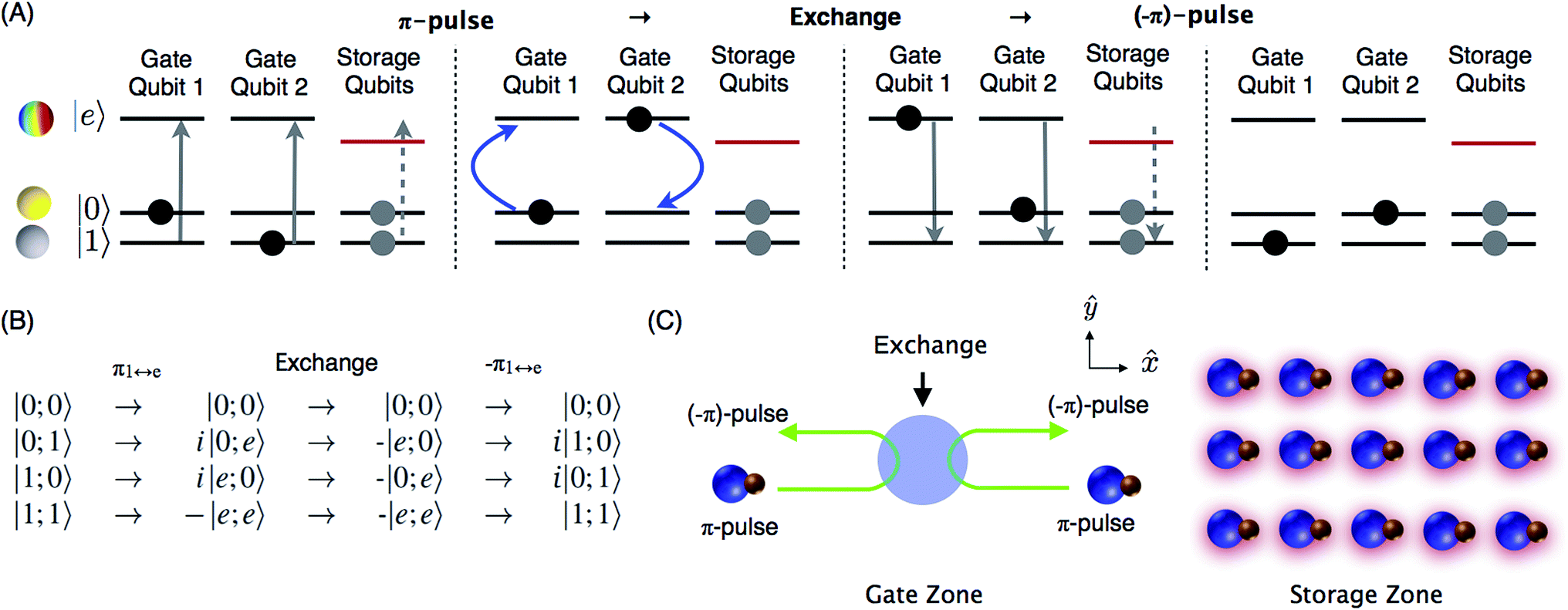
| | Fig. 1 (A) iSWAP gate based on dipolar exchange between a pair of molecular states with opposite parity. The colored sphere of the |e〉 state represents the wavefunction amplitude of the dipole direction for an N = 1, mN = 0 state, where the quantization axis is horizontal. The states |0〉 and |1〉 are hyperfine sublevels of the rotational ground state N = 0. Superpositions of |e〉 and |0〉 or |1〉 produce an electric dipole moment that oscillates at a frequency corresponding to approximately twice the rotational constant of the molecule and couples to a nearby qubit. The four panels in (A) show the initial state |0;1〉 evolving through the gate to i|1;0〉, where horizontal arrows indicate the flow of time. State |e〉 of the storage qubits is light shifted out of resonance, for individual addressability. (B) Includes other basis states and important details of quantum phases. (C) Qubit array based on molecular hyperfine states. Any pair of qubits can be moved from the storage zone to the gate zone in a flexible array of optical tweezers. During the gate operation, a spatially uniform microwave pulse transfers population from state |1〉 to |e〉 in the gate qubits, so that the amplitudes of the |0;e〉 and |e;0〉 states are exchanged. To achieve individual addressability with high spatial resolution, the light that shifts the storage qubits out of resonance (indicated by red shadows) can be produced in a similar way as the tweezer array. | |
Quantum computing requires two qubit states |0〉 and |1〉 that are coherent and couple minimally to the environment and other qubits. In this proposal, we utilize two hyperfine sublevels of the rotational ground state of a molecule as states |0〉 and |1〉. Long-lasting coherence of such states has recently been demonstrated in a gas of ultracold NaK molecules.30 While the hyperfine levels offer coherence, they do not produce strong dipole–dipole interactions between nearby molecules. To enable this interaction and produce an iSWAP gate, the |1〉 state in two molecules is temporarily transferred to the rotationally excited state |e〉 via a microwave π-pulse. Then, after energy exchange, the |e〉 population is transferred back to |1〉. The propagator in eqn (1) still applies, but now in the computational basis |0;0〉, |0;1〉, |1;0〉, |1;1〉 as shown in Fig. 1(B).
The above sequence requires an excited state |e〉 that couples to two different hyperfine levels (|0〉 and |1〉) of the ground state via electric fields. We rely on the hyperfine interaction term of the internal molecular Hamiltonian that couples molecular rotation to nuclear spin via the nuclear electric quadrupole moment31,32 to produce eigenstates that contain superpositions of different nuclear spin states. This interaction requires a nuclear spin greater than 1/2. Even though external electric fields do not change the nuclear spin directly, they can change the nuclear spins by driving transitions between states with different superpositions.33,34 In this manuscript, we use 23Na133Cs as an example because it has a large permanent electric dipole moment (4.6 Debye), and full quantum control of individually trapped molecules is being developed.35,36 A similar gate scheme that makes use of internal molecular couplings could also be applied to other ultracold polar molecules, including other bialkalis where a single internal quantum state can already be prepared34,37–40 and molecules of 2Σ electronic structure with spin-rotational coupling.41–44
3 Qubit array
For a quantum computer, a large number of molecular qubits could be held in an array of optical tweezers for storage (see Fig. 1C). Two arbitrary qubits can be selected for gate operations by means of a configurable tweezer array12,13 and moved so they are separated from the other qubits and initially far from each other. Off-resonant light is applied to the array of stored qubits to shift their |e〉 energy level so that the π-pulses have no effect. Individual addressability of qubits requires a spatial light pattern with high contrast ratio between stored and gate qubits, which can be generated by similar optics as the array of optical tweezers. The light causes only very small differential shifts for the storage states |0〉 and |1〉. A spatially uniform microwave π-pulse of well-defined polarization then transfers |1〉 to |e〉 for the two gate qubits. Then the qubits are moved near each other and back apart to allow for the interaction. This movement naturally produces a temporally shaped interaction with gradually rising and falling strength to reduce off-resonant population leakage. The final π-pulse transfers the |e〉 population to |1〉. An enhanced gate sequence can include a central spin-echo pulse to mitigate slowly drifting energies (Section 6). More advanced dynamical-decoupling sequences can be applied to further reduce decoherence and the sensitivity to qubit motion (Section 7).
4 Gate calculation
Because the molecule is in a 1Σ+ state, the electronic spin and orbital angular momentum are zero, and do not enter the calculation. We identify quantum states in the uncoupled basis by the quantum numbers N, mN, m1, and m2, where N is the angular momentum associated with molecular rotation and mN, m1, m2 are projections of this angular momentum and the two nuclear spins onto the magnetic field axis. While the uncoupled basis is convenient for calculations, the basis states generally do not coincide with eigenstates of the molecular Hamiltonian. Nevertheless, we label Hamiltonian eigenstates as |N, mN, m1, m2〉, where we use the quantum numbers of the uncoupled basis state with maximum overlap. Although this labeling scheme could in principle assign the same quantum numbers to two different eigenstates, we have verified that this does not occur for the specific states discussed here. When two-molecule states are described, they are written as |a;b〉 where a and b are the states of molecules 1 and 2 respectively.
We solve for the eigenstates associated with the molecular hyperfine Hamiltonian for NaCs32 at various magnetic fields and optical trap depths. The energy levels are shown as a function of magnetic field in Fig. 2 and trap depth in Fig. 4. The dipole–dipole interaction and electric-field-driven π-pulses both depend on the electric dipole moments of the molecules. The Hamiltonian associated with an externally applied electric field to molecule j is
| | ĤE = −![[d with combining circumflex]](https://www.rsc.org/images/entities/b_char_0064_0302.gif) j·E j·E | (2) |
where we use the interaction picture and rotating-wave approximation to remove the time-dependence of oscillating fields. The dipole–dipole interaction is
| | 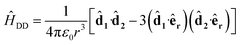 | (3) |
where
j is the dipole moment operator of molecule
j. Here,
êr is the unit vector along the separation direction for the two molecules and the dot products are evaluated as sums over the three spatial directions. The dipole moment operators are determined by the rotational part of the energy eigenstates and for a basis state
N, m
N, this axis has an orientation whose quantum wave function is the spherical harmonic
YmN. From the eigenstates of the molecular Hamiltonian we calculate the matrices associated with the dipole operators
![[d with combining circumflex]](https://www.rsc.org/images/entities/i_char_0064_0302.gif) x
x,
y,
z for the laboratory coordinate system.
28 These matrix elements are diagonal in the quantum numbers m
1 and m
2 of the uncoupled basis and can be reduced to Wigner-3j symbols.
31
 |
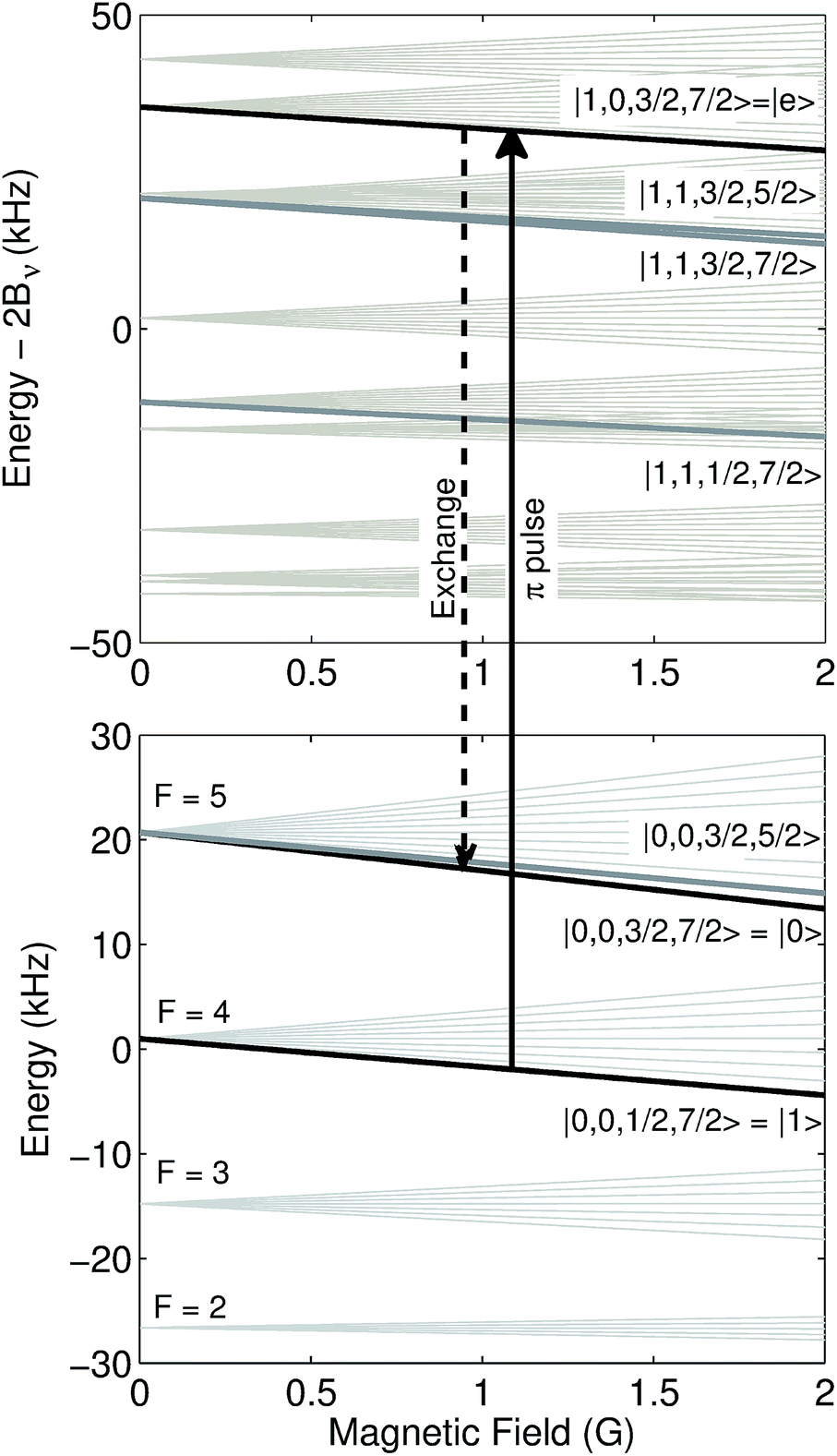
| | Fig. 2 Hyperfine and Zeeman energy levels as a function of magnetic field for the N = 1 (top) and N = 0 (bottom) states of 23Na133Cs (v = 0) in zero electric field. While a number of states are nearly degenerate with the states of interest |0〉, |1〉, and |e〉, selection rules prevent them from participating in the interactions. Bv = 1.7396 GHz is the molecular rotation constant. | |
 |
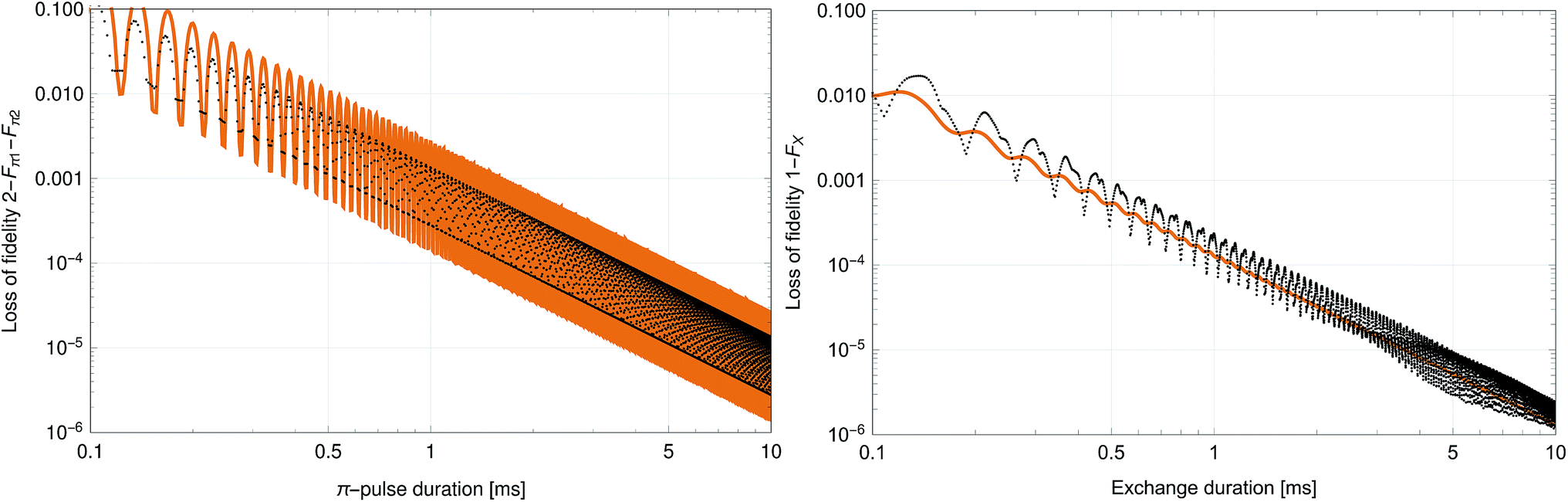
| | Fig. 3 Loss of fidelity as a function of duration for the two π-pulses (2 − Fπ1 − Fπ2) and exchange (1 − Fx). In both cases, the interaction strength (drive strength or molecule–molecule separation) is adjusted so the operation completes in the nominal duration. The ripples are caused by the square pulse shape whose sinc-function power-spectrum-minima move across off-resonant transitions as the duration is varied. The scaling behavior for the maximum fidelity points is 2 − Fπ1 − Fπ2 = 2.8 × 10−4t−2 and 1 − Fx = 8.5 × 10−5t−2 where t is given in ms. Details of the fidelity calculation are given in Section 4. The solid lines represent the perturbation theory results while the dotted lines correspond to the full Hamiltonian. | |
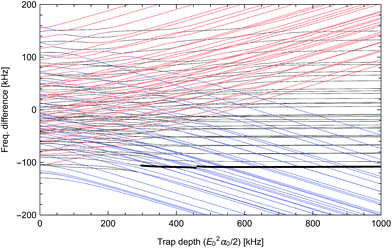 |
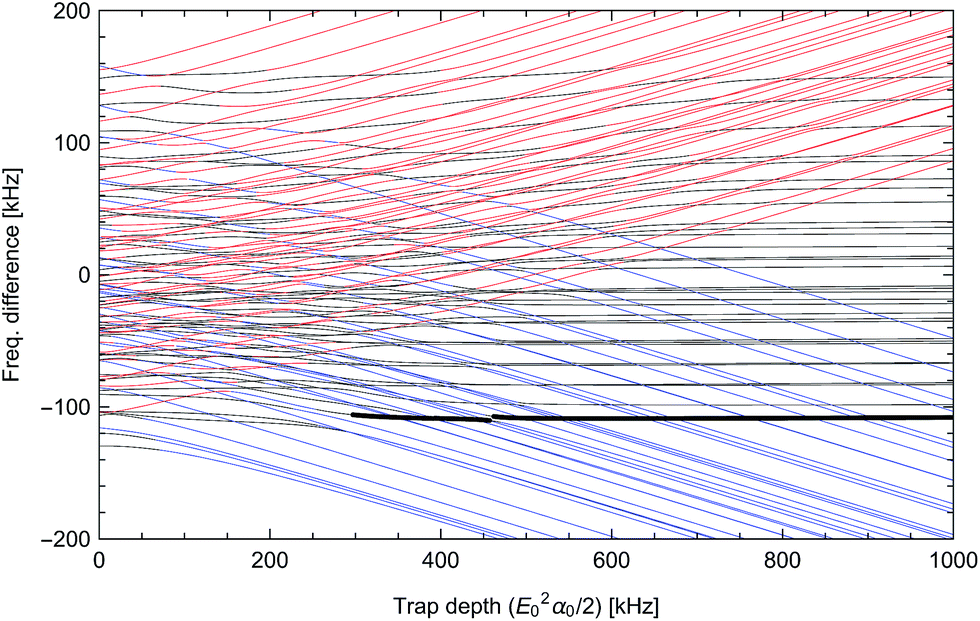
| | Fig. 4 Differential light shifts of excited rotational states (N = 1) with respect to the ground state, as a function of trap depth. The large offset of about 3.48 GHz has been subtracted. States with predominantly 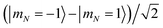 character with dipole moment along are shown in black. States with predominantly character with dipole moment along are shown in black. States with predominantly 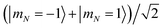 character with dipole moment along ŷ are blue. States with predominantly |mN = 0〉 character with dipole moment along ẑ are red. The thick line shows the state |e〉 = |1,−1,3/2,7/2〉. The polarization vector is character with dipole moment along ŷ are blue. States with predominantly |mN = 0〉 character with dipole moment along ẑ are red. The thick line shows the state |e〉 = |1,−1,3/2,7/2〉. The polarization vector is ![[small epsilon, Greek, circumflex]](https://www.rsc.org/images/entities/i_char_e107.gif) = = ![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) cosγ + iŷsinγ with ellipticity γ = 35.6091°, adjusted to null the slope of |e〉 at 600 kHz. A magnetic field of 35 Gauss lies along ẑ. cosγ + iŷsinγ with ellipticity γ = 35.6091°, adjusted to null the slope of |e〉 at 600 kHz. A magnetic field of 35 Gauss lies along ẑ. | |
For each step (π-pulse and exchange), we calculate unitary evolution according to the time-independent Hamiltonians in eqn (2) and (3). The resonant couplings drive the desired gate behavior, while off-resonant couplings to other states cause population leakage and Stark shifts. For a low magnetic field without light, the most important Hamiltonian terms are listed in Tables 1 and 2. The unitary transformation is applied to many test states in the computational basis to determine the minimum gate fidelity.45 The approximate population leakage can also be calculated more simply via perturbation theory (Appendix A).
Table 1 Non-zero coupling terms of the |1〉 ↔ |e〉 π-pulse Hamiltonian (interaction picture) in the rotating-wave approximation when σ+-polarized radiation at 3.48 GHz is applied to a molecule in 1 Gauss magnetic field. The electric field amplitude is 0.03 V m−1, such that the π-pulse duration is 3.01 ms (adjusted for maximum fidelity). The radiation frequency has been adjusted by 6.3 radians per second to compensate for dynamic Stark shifts. All terms connecting to |0〉, |1〉, or |e〉 are shown
| From (i) |
To (j) |
H
ij
/ℏ [s−1] |
H
jj
/ℏ [s−1] |
| |e〉 |
|1〉 |
521.9 |
0 |
| |1〉 |
|e〉 |
521.9 |
6.3 |
| |0〉 |
|0〉 |
— |
117851.0 |
| |1〉 |
|1,1,1/2,7/2〉 |
1135.6 |
−285144.0 |
| |1〉 |
|1,1,3/2,5/2〉 |
2.3 |
−82709.5 |
| |e〉 |
|0,0,3/2,5/2〉 |
470.8 |
122454.0 |
| |0〉 |
|1,1,3/2, 7/2〉 |
1249.8 |
−86537.2 |
Table 2 Non-zero coupling terms for |e;0〉 of the exchange Hamiltonian when two molecules are separated by 2.9 μm along the 1 Gauss magnetic field. Propagation of the Hamiltonian approximates the exchange evolution (eqn (1)) when applied for a duration 4.02 ms (adjusted for maximum fidelity). The terms |0;0〉 and |e;e〉 do not couple to other states
| From (i) |
To (j) |
H
ij
/ℏ [s−1] |
H
jj
/ℏ [s−1] |
| |e;0〉 |
|0;e〉 |
−390.7 |
0 |
| |e;0〉 |
|0;1,1,3/2,5/2〉 |
−195.6 |
−82715.8 |
| |e;0〉 |
|0,0,3/2,5/2;1,1,3/2,7/2〉 |
107.6 |
−81940.9 |
| |e;0〉 |
|0;1,1,1/2,7/2〉 |
180.0 |
−285151.0 |
| |e;0〉 |
|1;1,1,3/2,7/2〉 |
119.3 |
−204395.0 |
5 Gate speed and fidelity
In addition to the desired evolution in eqn (1), the iSWAP gate sequence causes off-resonant coupling to other molecular states. This causes a trade-off between gate speed and fidelity. Although two NaCs molecules could be brought to a separation below 1 μm, with an exchange duration of 50 μs, such an interaction would limit the gate fidelity to 99.6%. At low magnetic field with NaCs, we find that the interactions couple off-resonantly to other states with δ > 10 kHz detuning both for the π-pulses and the exchange. For the square pulses considered here, time-energy uncertainty causes off-resonant population transfer out of the computational basis states and a gate error of order (τδ)−2. Off-resonant coupling to other states also causes dynamic Stark shifts which result in small reproducible phase and frequency shifts. We expect that these can be corrected for without loss of fidelity.
We have calculated the unitary time evolution for all states that could couple via microwave pulses (ĤE) and dipole–dipole interactions (ĤDD) to the states in Fig. 1B. For low magnetic field (1 Gauss), details of the Hamiltonian terms are given is Tables 1 and 2, where we have chosen the states |0〉 = |0,0,3/2,7/2〉, |1〉 = |0,0,1/2,7/2〉 and |e〉 = |1,0,3/2,7/2〉.
We find that a gate with 10 ms duration has a fidelity of F = 1 − 3.6 × 10−5. The exchange part of the example gate holds the two molecules at a distance of 2.9 μm for a duration τx = 4.02 ms. The π-pulses of duration 3.01 ms utilize a σ+ polarized microwave electric field at a frequency of 3.48 GHz and amplitude 0.03 V m−1. The π-pulse and exchange fidelities due to population leakage are shown separately in Fig. 3. Population leakage out of the computational basis must be corrected for to support long gate sequences.46 Off-resonant coupling can likely be reduced by use of shaped pulses rather than square ones, to reach the same fidelity in a shorter duration.47 At a higher magnetic field of 35 Gauss with 600 kHz trap depth, the best states (F = 1 − 6 × 10−5 at 9.4 ms) are |1〉 = |0,0,3/2,5/2〉 and |e〉 = |1,−1,3/2,7/2〉, with |0〉 as above.
6 Intensity fluctuations and light shifts
If the molecule is optically trapped, light shifts significantly perturb the energy eigenstates and transition frequencies. To reach the wavefunction spread described in Section 7, we assume a 600 kHz trap depth (12.9 kW cm−2 intensity) with elliptical polarization.48 We also assume a trapping laser wavelength of 1030 nm. The resulting energy levels are shown in Fig. 4 for a magnetic field of 35 Gauss. The higher magnetic field was chosen, because the Zeeman splitting reduces off-resonant couplings during the |1〉 ↔ |e〉 π-pulses that are induced by the light shift Hamiltonian. Fig. 5 shows the magnitude of off-resonant coupling terms to other molecular states. For interactions that turn on and off sharply, the loss of fidelity due to off-resonant population leakage is given by a sum of terms involving off-resonant coupling strengths and detunings (see Appendix A). Overall, we find that the exchange interaction has reduced leakage compared to the 1 Gauss case, even for a faster interaction. The |1〉 ↔ |e〉 π-pulses cause slightly higher leakage, which may be reduced by shaped or DRAG pulse techniques.47
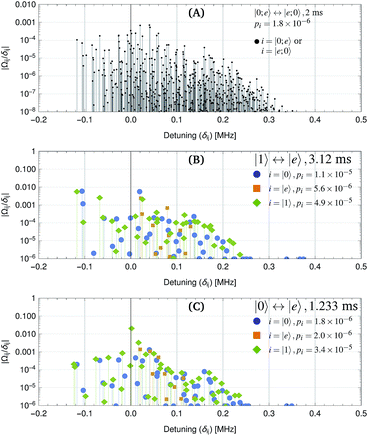 |
| | Fig. 5 Off-resonant coupling terms |Ωij/δij| that cause population leakage during the gate steps. Here, Ωij is the coupling Rabi rate between source state i and leakage state j, and δij is the frequency difference in radians per s. For each step, the probability of population leakage pi is shown (see Appendix A), where the summation is over all coupled states j. pi can be used to estimate off-resonant population leakage without calculating the full unitary time evolution. (A) Exchange interaction with separation of 2.5 μm along for a 2 ms exchange duration. The fidelity, calculated from unitary time evolution is F = 1 − 2.0 × 10−6. (B) |1〉 ↔ |e〉 π-pulse with electric field amplitude 0.0544 V m−1 along ẑ for a 3.12 ms pulse duration. The fidelity for a pair of π-pulses, calculated from the unitary time evolution is F = 1 − 5.4 × 10−5. (C) |0〉 ↔ |e〉 π-pulse with electric field amplitude 0.0157 V m−1 along for a 1.233 ms pulse duration. The state |1〉 can be neglected if its population has already been transferred to |e〉. | |
For an optical trap with elliptical polarization, the quadratic sensitivity to intensity is calculated to be Δf = (ΔI/I)2 × 12.6 kHz, leading to gate errors of 5 × 10−6 for relative intensity fluctuations of ΔI/I = 2 × 10−3. A more significant effect is polarization drift. For quartz waveplates with a temperature sensitivity of retardance of 10−4 K−1, one may expect polarization ellipticity fluctuations of 10−5 and light shift fluctuations of 4 × 10−6 (2.5 Hz). Because these fluctuations are slow thermal effects, spin echo pulses can reduce their impact. If the quadratic light shift changes slowly, e.g. due to recoil heating, the effect is also mitigated. One possible implementation is shown in Table 3 where a |0〉 ↔ |e〉 π-pulse is inserted in the middle of the gate sequence. This leads to first-order insensitivity of the gate error with respect to constant light shift errors. The result is equivalent to an iSWAP gate between the qubits, followed by inversion of the individual qubits. Note that this spin-echo example was chosen for simplicity, and other dynamical decoupling techniques to compensate slow drifts could also be applied. The |0〉 ↔ |e〉 π-pulses, which are used for the spin-echo have little off-resonant coupling, even for a 1 ms duration (see Fig. 5).
Table 3 Evolution of the computational basis states through the gate, which includes a central spin-echo pulse to cancel the phase evolution from slowly varying energy shifts between |0〉 and |e〉, such as light shifts. The exchange interaction is split into two parts, where each has one half the duration required for full exchange
|
|
π1↔e |
|
1/2 Ex. |
|
π0↔e |
|
1/2 Ex. |
|
(−π1↔e) |
|
| |0;0〉 |
→ |
|0;0〉 |
→ |
|0;0〉 |
→ |
−|e;e〉 |
→ |
−|e;e〉 |
→ |
|1;1〉 |
| |0;1〉 |
→ |
i|0;e〉 |
→ |

|
→ |
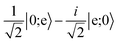
|
→ |
|0;e〉 |
→ |
−i|0;1〉 |
| |1;0〉 |
→ |
i|e;0〉 |
→ |
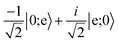
|
→ |
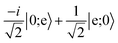
|
→ |
|e;0〉 |
→ |
−i|1;0〉 |
| |1;1〉 |
→ |
−|e;e〉 |
→ |
−|e;e〉 |
→ |
|0;0〉 |
→ |
|0;0〉 |
→ |
|0; 0〉 |
The differential light shift between two hyperfine ground states for NaK was measured30 as 5 × 10−5 Hz (W/cm−2)−1. With the assumption that the states |0〉 and |1〉 in NaCs have similar differential shifts, a beam with 10 kW cm−2 intensity and 1064 nm wavelength can Stark shift the |e〉 states of the stored qubits by 200 kHz while shifting their |0〉 and |1〉 states by only 0.6 Hz differentially. In this case, the polarization is adjusted for maximum differential Stark shift of |e〉 with respect to |0〉 and a spatially patterned beam can be generated in the same way as the tweezer array. If the Stark shifting beam has a relative intensity stability of 2 × 10−3, the loss of fidelity is 1 − F ≈ 7 × 10−10 for a single stored qubit. The large ratio between |0〉 − |e〉 and |0〉 − |1〉 sensitivities makes it possible to individually address certain molecules by light-shifting the |e〉 state of the other molecules.
7 Effects of molecule motion
In the ideal gate, both molecules are in the motional ground state of their optical tweezer, and the exchange interaction strength is always the same. However, ground-state cooling is imperfect and the molecules gain kinetic energy due to photon recoil. Given the imaginary polarizability of NaCs Im[α] ≈ 10−4 (atomic units),49 the total scattering rate for a 600 kHz deep trap is approximately 0.8 Hz with a heating rate of 0.44 quanta per second axially and 0.023 quanta per second for each radial direction. Therefore, it is desirable for the gate fidelity to exceed the ground-state occupation fidelity. Here we examine the effect of motional excitations where the spatial coupling constant of eqn (3) is modified. For the proposed situation where the molecular dipoles lie along , this coupling scales as (1 − 3cos2θ)/r3 where θ is the angle between the separation axis and , and r is the separation distance (2.5 μm here). For θ ≈ 0, a series expansion in terms of spatial coordinates yields a change in the interaction Rabi rate Ω per motional quantum of| | | Δ〈m|Ω|m〉/Δnj,k ≈ fkΩ0(sk/r)2 | (4) |
where |m〉 = |n1,x,n1,y,n1,z;n2,x,n2,y,n2,z〉 is the state of two-molecule motion, Ω0 = D/r3 is the interaction Rabi rate without motion of eqn (1), nj,k is the motional excitation number for molecule j in direction 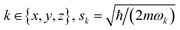 are the zero-point wave function spreads, fx = 12, fy,z = −6, and the approximation includes terms up to second order in position. The above trap depth corresponds to motional frequencies of ωx,y = 2π·12.4 kHz and ωz = 2π·2.8 kHz for NaCs, where a Gaussian beam radius of 1 μm is assumed. Because the loss of fidelity is proportional to the square of the Rabi rate deviation from the mean, 1 − F can calculated from the variance of the Rabi rate, which can be expressed in terms of mean motional quantum numbers
are the zero-point wave function spreads, fx = 12, fy,z = −6, and the approximation includes terms up to second order in position. The above trap depth corresponds to motional frequencies of ωx,y = 2π·12.4 kHz and ωz = 2π·2.8 kHz for NaCs, where a Gaussian beam radius of 1 μm is assumed. Because the loss of fidelity is proportional to the square of the Rabi rate deviation from the mean, 1 − F can calculated from the variance of the Rabi rate, which can be expressed in terms of mean motional quantum numbers ![[n with combining macron]](https://www.rsc.org/images/entities/i_char_006e_0304.gif) j,k for a thermal distribution of motional states:
j,k for a thermal distribution of motional states:| | 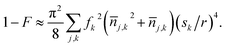 | (5) |
If the molecules are near the motional ground state with excitations dominated by imperfect cooling of Na,50 the mean excitation numbers of each molecule along x, y, and z are 0.019, 0.024, and 0.024 respectively. Then motional effects cause a fidelity loss of 9 × 10−6 for the above trap strength. The effect of motion on the two-qubit gate can be reduced by implementing a BB1 sequence.51,52 Because the sign of fz is opposite fx, it is possible to choose a separation direction that makes the interaction Rabi rate first-order insensitive to nj,z. This occurs for ![[r with combining circumflex]](https://www.rsc.org/images/entities/i_char_0072_0302.gif) = cosθ + ẑsinθ with θ = (1/4)cos−1(3/35), and the interaction strength is reduced by 20%. An optical lattice in the weakly confining direction would also reduce the effects of axial motion. Note that the term j,k2sk4 in eqn (5), which limits fidelity for j,k > 1, is independent of trap intensity when j,k is dominated by recoil heating.
= cosθ + ẑsinθ with θ = (1/4)cos−1(3/35), and the interaction strength is reduced by 20%. An optical lattice in the weakly confining direction would also reduce the effects of axial motion. Note that the term j,k2sk4 in eqn (5), which limits fidelity for j,k > 1, is independent of trap intensity when j,k is dominated by recoil heating.
The motional state also affects the average light shift, because the tweezer intensity drops away from the trap center. For a Gaussian beam trap with thermally excited molecules whose mean quantum numbers are k, we find the relative intensity variance 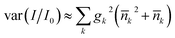 where gk = −4(sk/w)2 for the radial directions, gz = −2(szλ/(πw2))2 and w is the beam radius. The associated standard deviation in relative intensity is 2 × 10−3 for the residual motion after ground-state cooling described above. This corresponds to the intensity stability assumed in Section 6.
where gk = −4(sk/w)2 for the radial directions, gz = −2(szλ/(πw2))2 and w is the beam radius. The associated standard deviation in relative intensity is 2 × 10−3 for the residual motion after ground-state cooling described above. This corresponds to the intensity stability assumed in Section 6.
8 Other effects
Effects from blackbody radiation and spontaneous emission are small for the system we have outlined above. The dominant effect at room temperature is due to blackbody radiation and for NaCs the vibrational transition absorption rate is 1.7 × 10−3 s−1 while the rotational transition rate is negligible.53 The spontaneous emission rate from the N = 1 rotationally excited state is of order 10−8 s−1.
The rate of decoherence due to scattering of optical tweezer photons has not been calculated. However, where long coherence between hyperfine ground states of NaK was observed, the decoherence was attributed to spatial intensity variation in the optical trap rather than scattering.30 A calculation of scattering rates should distinguish between internal-state-preserving (Rayleigh) and internal-state-changing (Raman) scattering, as the rate of Raman scattering can be several orders of magnitude lower than Rayleigh scattering.54,55
Magnetic field fluctuations cause dephasing of quantum states if their energies have an unequal slope with respect to field changes. For both the 1 Gauss and 35 Gauss examples, we find that the relative sensitivities of the |0〉, |1〉, and |e〉 states are below 1 kHz Gauss−1. While this field sensitivity is small, the associated loss of fidelity grows with a factor N2 if maximally entangled states such as 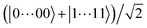 with N qubits are stored in the qubit array. The same scaling applies to light that differentially shifts the phase of |0〉 and |1〉 (see Section 6). Such common-mode dephasing errors can be reduced by use of dynamical decoupling or decoherence-free subspaces.56
with N qubits are stored in the qubit array. The same scaling applies to light that differentially shifts the phase of |0〉 and |1〉 (see Section 6). Such common-mode dephasing errors can be reduced by use of dynamical decoupling or decoherence-free subspaces.56
9 Single qubit rotations
Single qubit X and Y rotations are less complex than two-qubit gates and can be accomplished by individually addressing only one molecule and using a simplified sequence without dipole–dipole exchange. It may be advantageous to first perform a π-pulse from |0〉 to |e〉, then a rotation between |1〉 and |e〉 and finally a π-pulse from |e〉 to |0〉 as shown in Table 4. The |0〉 ↔ |e〉 π-pulses can have less off-resonant coupling than the previously discussed |1〉 ↔ |e〉 pulses (see Fig. 5), and high fidelity can be achieved for a shorter gate duration than in the two-qubit case. Rotation about the Z axis can be accomplished by a |0〉 ↔ |e〉 π-pulse with one oscillator phase, followed by a second |0〉↔ |e〉 π-pulse with a different oscillator phase.
Table 4 Single qubit rotation sequence where the π-pulses are performed as Y-rotations, and the “Rotation” step acts on the states |1〉 and |e〉
|
|
π0↔e |
|
Rot. |
|
−π0↔e |
|
| |1〉 |
→ |
|1〉 |
→ |
α|1〉 − β|e〉 |
→ |
α|1〉 + β|0〉 |
| |0〉 |
→ |
−|e〉 |
→ |
γ|1〉 − δ|e〉 |
→ |
γ|1〉 + δ|0〉 |
10 Conclusion
We have described a room-temperature scheme for quantum computing based on iSWAP gates performed by dipolar molecules, individually trapped in optical tweezers. Calculations indicate a potential gate fidelity above 0.9999 with low decoherence, although the rate of Raman scattering of optical tweezer photons remains to be determined. A modest magnetic field is used, and electric fields or field gradients are not needed. Scaling to many qubits would require an equal number of optical dipole traps in a movable pattern. For this gate to be realized, individual neutral ground-state molecules must still be produced, and improved state-measurement is needed. We expect that long gate sequences will require the mitigation of population leakage and recoil heating, e.g. by periodically teleporting the state of used molecules onto new ones.46 While this proposal utilizes two hyperfine ground levels of NaCs, thirty other ground levels exist, which might allow each molecule to contain several qubits.
Although the proposed iSWAP gate is slower than quantum gates in other systems, decoherence effects can be small due to the fact that the qubit states are isolated from the environment by the symmetry of 1Σ+ states. The longer gate duration also reduces the noise bandwidth of actively stabilized parameters. For the foreseeable future, experimental quantum computing will aim to increase the number of available qubits and gate fidelity, and molecules have the potential to advance both goals.
A. Off-resonant coupling
We wish to calculate the population leakage during gate interactions. In this case, there are two “main” states (here |a〉 and |b〉) and leakage states |j〉 to which the main states connect with large detuning and/or weak coupling. For simplicity, we assume that the Hamiltonian associated with the interaction turns on and off instantaneously to produce time-independent coupling. In a real implementation, it will be important to ramp the interactions up and down smoothly to minimize off-resonant coupling.
One approach for calculating leakage is to generate the full Hamiltonian matrix and diagonalize it to compute the unitary time evolution. While straightforward, this is computationally expensive when there are many leakage states. Complementary to the full calculation of unitary time evolution, we use first-order perturbation theory below to calculate the population leakage as a simple sum. A comparison of the techniques can be seen in Fig. 3. Note that this does not account for coherent population buildup, which may develop if several interactions are combined without phase randomization.
During interactions between degenerate primary states |a〉 and |b〉 with coupled leakage states |j〉, the Hamiltonian can be written in terms of “desired” coupling Ĥ0 and “leakage” couplings Ĥ′ as
| | 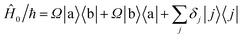 | (7) |
| | 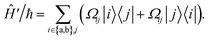 | (8) |
here, the basis state phases have been chosen to make the coefficients
Ω and
Ωij real and positive. The base Hamiltonian
Ĥ0 has eigenvectors |+〉, |−〉, |
j〉 with eigenvalues
ℏΩ, −
ℏΩ,
ℏδj and
| | 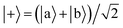 | (9) |
| | 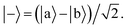 | (10) |
According to first-order perturbation theory, the perturbed eigenstates (assuming that different |j〉 states don't couple to one another) are
| | 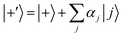 | (11) |
| | 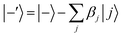 | (12) |
| |  | (13) |
where
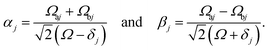
These states approximately diagonalize the time-evolution operator and can be used to estimate the transitions from the initial state |a〉 into states |
j〉 due to unitary time evolution
Û(
t) = e
−iĤt/ℏ:
| |  | (14) |
| | 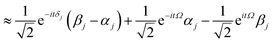 | (15) |
where the
eqn (14) sum is over all perturbed states. Simplification from
eqn (14) to
(15) utilizes unperturbed energies in the exponential terms. In our case for any
j, due to selection rules, only one of
Ωaj and
Ωbj is ever non-zero. We call the non-zero value
Ωj. If one makes the assumption
t = π/(2
Ω) (π-pulse), the population leakage is
| | 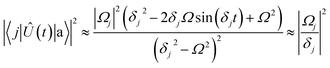 | (16) |
where the first approximation is due to the use of perturbation theory and the second approximation is valid when
Ω ≪
δj. Identical expressions are found for |〈
j|
Û(
t)|b〉|
2.
Eqn (16) describes the leakage out of states |1〉 and |e〉 during the |1〉 ↔ |e〉 π-pulse, states |0〉 and |e〉 during the |0〉 ↔ |e〉 π-pulse, and states |0;e〉 and |e;0〉 during the |0;e〉 ↔ |e;0〉 exchange. To treat leakage from |0〉 during the |1〉 ↔ |e〉 π-pulse, let
Ω = 0. Then
eqn (15) simplifies to
| | 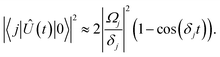 | (17) |
A careful choice of π-time reduces this leakage term, equivalent to minimizing leakage via alignment of power-spectrum zeros.
To evaluate the fidelity of each interaction step, we calculate the total population leakage probability from each nominally-populated state |i〉 as
| | 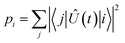 | (18) |
using
eqn (16) or (
17), as appropriate. The minimum fidelity is then
| | 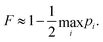 | (19) |
Conflicts of interest
There are no conflicts to declare.
References
- N. M. Linke, D. Maslov, M. Roetteler, S. Debnath, C. Figgatt, K. A. Landsman, K. Wright and C. Monroe, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 3305–3310 CrossRef PubMed.
- P. H. Leung, K. A. Landsman, C. Figgatt, N. M. Linke, C. Monroe and K. R. Brown, Phys. Rev. Lett., 2018, 120, 020501 CrossRef PubMed.
- D. P. DiVincenzo, Fortschr. Phys., 2000, 48, 771–783 CrossRef.
- K. R. Brown, A. C. Wilson, Y. Colombe, C. Ospelkaus, A. M. Meier, E. Knill, D. Leibfried and D. J. Wineland, Phys. Rev. A: At., Mol., Opt. Phys., 2011, 84, 030303 CrossRef.
- C. J. Ballance, T. P. Harty, N. M. Linke, M. A. Sepiol and D. M. Lucas, Phys. Rev. Lett., 2016, 117, 060504 CrossRef PubMed.
- J. P. Gaebler, T. R. Tan, Y. Lin, Y. Wan, R. Bowler, A. C. Keith, S. Glancy, K. Coakley, E. Knill, D. Leibfried and D. J. Wineland, Phys. Rev. Lett., 2016, 117, 060505 CrossRef PubMed.
- S. Sheldon, L. S. Bishop, E. Magesan, S. Filipp, J. M. Chow and J. M. Gambetta, Phys. Rev. A, 2016, 93, 012301 CrossRef.
- R. Barends, J. Kelly, A. Megrant, A. Veitia, D. Sank, E. Jeffrey, T. C. White, J. Mutus, A. G. Fowler, B. Campbell, Y. Chen, Z. Chen, B. Chiaro, A. Dunsworth, C. Neill, P. O'Malley, P. Roushan, A. Vainsencher, J. Wenner, A. N. Korotkov, A. N. Cleland and J. M. Martinis, Nature, 2014, 508, 500 CrossRef PubMed.
- A. R. Calderbank and P. W. Shor, Phys. Rev. A: At., Mol., Opt. Phys., 1996, 54, 1098–1105 CrossRef.
- A. Steane, Proc. R. Soc. London, Ser. A, 1996, 452, 2551–2577 CrossRef.
- J. Preskill, Proc. R. Soc. London, Ser. A, 1998, 454, 385–410 CrossRef.
- D. Barredo, S. de Léséleuc, V. Lienhard, T. Lahaye and A. Browaeys, Science, 2016, 354, 1021–1023 CrossRef PubMed.
- M. Endres, H. Bernien, A. Keesling, H. Levine, E. R. Anschuetz, A. Krajenbrink, C. Senko, V. Vuletic, M. Greiner and M. D. Lukin, Science, 2016, 354, 1024–1027 CrossRef PubMed.
- D. DeMille, Phys. Rev. Lett., 2002, 88, 067901 CrossRef PubMed.
- S. F. Yelin, K. Kirby and R. Côté, Phys. Rev. A: At., Mol., Opt. Phys., 2006, 74, 050301 CrossRef.
- J. Zhu, S. Kais, Q. Wei, D. Herschbach and B. Friedrich, J. Chem. Phys., 2013, 138, 024104 CrossRef PubMed.
- F. Herrera, Y. Cao, S. Kais and K. B. Whaley, New J. Phys., 2014, 16, 075001 CrossRef.
- M. Karra, K. Sharma, B. Friedrich, S. Kais and D. Herschbach, J. Chem. Phys., 2016, 144, 094301 CrossRef PubMed.
- D. P. DiVincenzo, Phys. Rev. A: At., Mol., Opt. Phys., 1995, 51, 1015–1022 CrossRef.
- N. Schuch and J. Siewert, Phys. Rev. A: At., Mol., Opt. Phys., 2003, 67, 032301 CrossRef.
- D. Loss and D. P. DiVincenzo, Phys. Rev. A: At., Mol., Opt. Phys., 1998, 57, 120–126 CrossRef.
- D. Mozyrsky, V. Privman and M. L. Glasser, Phys. Rev. Lett., 2001, 86, 5112–5115 CrossRef PubMed.
- A. Blais, R.-S. Huang, A. Wallraff, S. M. Girvin and R. J. Schoelkopf, Phys. Rev. A: At., Mol., Opt. Phys., 2004, 69, 062320 CrossRef.
- A. Andre, D. DeMille, J. M. Doyle, M. D. Lukin, S. E. Maxwell, P. Rabl, R. J. Schoelkopf and P. Zoller, Nat. Phys., 2006, 2, 636–642 Search PubMed.
- R. Barnett, D. Petrov, M. Lukin and E. Demler, Phys. Rev. Lett., 2006, 96, 190401 CrossRef PubMed.
- A. V. Gorshkov, S. R. Manmana, G. Chen, J. Ye, E. Demler, M. D. Lukin and A. M. Rey, Phys. Rev. Lett., 2011, 107, 115301 CrossRef PubMed.
- B. Yan, S. A. Moses, B. Gadway, J. P. Covey, K. R. A. Hazzard, A. M. Rey, D. S. Jin and J. Ye, Nature, 2013, 501, 521–525 CrossRef PubMed.
-
M. L. Wall, K. R. A. Hazzard and A. M. Rey, Quantum magnetism with ultracold molecules, in From atomic to mesoscale: The Role of Quantum Coherence in Systems of Various Complexities, ed. S. Malinovskaya and I. Novikova, World Scientific, 2015 Search PubMed.
- A. de Paz, A. Sharma, A. Chotia, E. Maréchal, J. H. Huckans, P. Pedri, L. Santos, O. Gorceix, L. Vernac and B. Laburthe-Tolra, Phys. Rev. Lett., 2013, 111, 185305 CrossRef PubMed.
- J. W. Park, Z. Z. Yan, H. Loh, S. A. Will and M. W. Zwierlein, Science, 2017, 357, 372–375 CrossRef PubMed.
-
R. Zare, Angular momentum: understanding spatial aspects in chemistry and physics, Wiley, 1988 Search PubMed.
- J. Aldegunde and J. M. Hutson, Phys. Rev. A, 2017, 96, 042506 CrossRef.
- J. Aldegunde, H. Ran and J. Hutson, Phys. Rev. A: At., Mol., Opt. Phys., 2009, 80, 043410 CrossRef.
- S. Ospelkaus, K.-K. Ni, G. Quéméner, B. Neyenhuis, D. Wang, M. H. G. de Miranda, J. L. Bohn, J. Ye and D. S. Jin, Phys. Rev. Lett., 2010, 104, 030402 CrossRef PubMed.
-
L. R. Liu, J. T. Zhang, Y. Yu, N. R. Hutzler, Y. Liu, T. Rosenband and K.-K. Ni, ArXiv: 1701.03121, 2017.
- L. R. Liu, J. D. Hood, Y. Yu, J. T. Zhang, N. R. Hutzler, T. Rosenband and K.-K. Ni, Science, 2018, 360, 900–903 CrossRef PubMed.
- T. Takekoshi, L. Reichsöllner, A. Schindewolf, J. M. Hutson, C. R. Le Sueur, O. Dulieu, F. Ferlaino, R. Grimm and H.-C. Nägerl, Phys. Rev. Lett., 2014, 113, 205301 CrossRef PubMed.
- S. A. Will, J. W. Park, Z. Z. Yan, H. Loh and M. W. Zwierlein, Phys. Rev. Lett., 2016, 116, 225306 CrossRef PubMed.
- P. D. Gregory, J. A. Blackmore, J. Aldegunde, J. M. Hutson and S. L. Cornish, Phys. Rev. A, 2017, 96, 021402 CrossRef.
- M. Guo, X. Ye, J. He, G. Quéméner and D. Wang, Phys. Rev. A, 2018, 97, 020501 CrossRef.
- J. F. Barry, D. J. McCarron, E. B. Norrgard, M. H. Steinecker and D. DeMille, Nature, 2014, 512, 286–289 CrossRef PubMed.
- S. Truppe, H. J. Williams, M. Hambach, L. Caldwell, N. J. Fitch, E. A. Hinds, B. E. Sauer and M. R. Tarbutt, Nat. Phys., 2017, 13, 1173 Search PubMed.
- L. Anderegg, B. L. Augenbraun, E. Chae, B. Hemmerling, N. R. Hutzler, A. Ravi, A. Collopy, J. Ye, W. Ketterle and J. M. Doyle, Phys. Rev. Lett., 2017, 119, 103201 CrossRef PubMed.
-
L. Anderegg, B. L. Augenbraun, Y. Bao, S. Burchesky, L. W. Cheuk, W. Ketterle and J. M. Doyle, ArXiv: 1803.04571, 2018.
-
M. Nielsen and I. Chuang, Quantum Computation and Quantum Information, Cambridge University Press, 2000 Search PubMed.
- A. G. Fowler, Phys. Rev. A: At., Mol., Opt. Phys., 2013, 88, 042308 CrossRef.
- F. Motzoi, J. M. Gambetta, P. Rebentrost and F. K. Wilhelm, Phys. Rev. Lett., 2009, 103, 110501 CrossRef PubMed.
-
T. Rosenband, D. D. Grimes and K.-K. Ni, Optics Express, ArXiv: 1804.01030, 2018, in press Search PubMed.
- R. Vexiau, D. Borsalino, M. Lepers, A. Orban, M. Aymar, O. Dulieu and N. Bouloufa-Maafa, Int. Rev. Phys. Chem., 2017, 36, 709–750 Search PubMed.
- Y. Yu, N. R. Hutzler, J. T. Zhang, L. R. Liu, J. D. Hood, T. Rosenband and K.-K. Ni, Phys. Rev. A, 2018, 97, 063423 CrossRef.
- S. Wimperis, J. Magn. Reson., Ser. A, 1994, 109, 221–231 CrossRef.
- J. A. Jones, Proc. R. Soc. London, Ser. A, 2003, 361, 1429–1440 Search PubMed.
- N. Vanhaecke and O. Dulieu, Mol. Phys., 2007, 105, 1723–1731 CrossRef.
- R. A. Cline, J. D. Miller, M. R. Matthews and D. J. Heinzen, Opt. Lett., 1994, 19, 207–209 CrossRef PubMed.
- H. Uys, M. J. Biercuk, A. P. VanDevender, C. Ospelkaus, D. Meiser, R. Ozeri and J. J. Bollinger, Phys. Rev. Lett., 2010, 105, 200401 CrossRef PubMed.
- P. Zanardi and M. Rasetti, Phys. Rev. Lett., 1997, 79, 3306–3309 CrossRef.
|
| This journal is © The Royal Society of Chemistry 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *abc,
Till
Rosenband
*abc,
Till
Rosenband
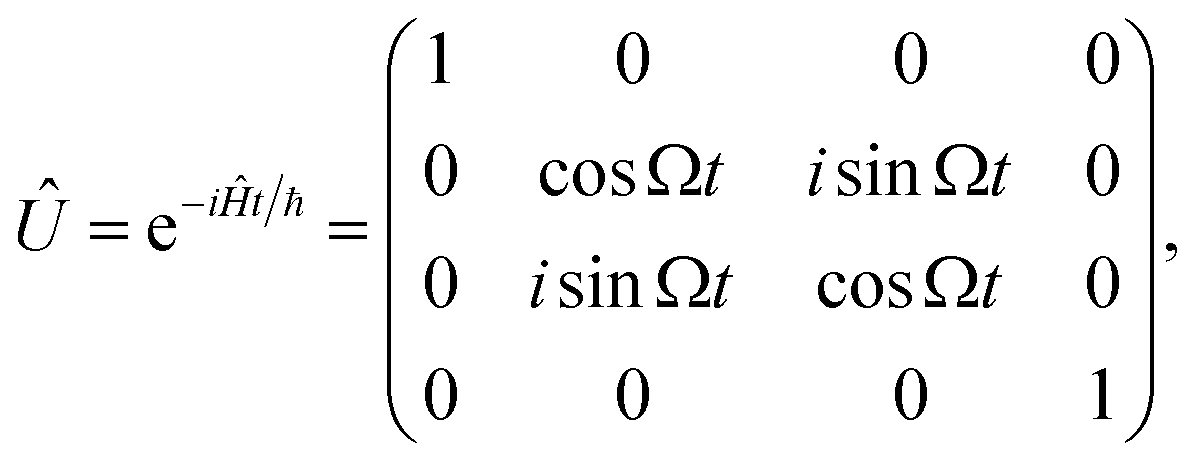
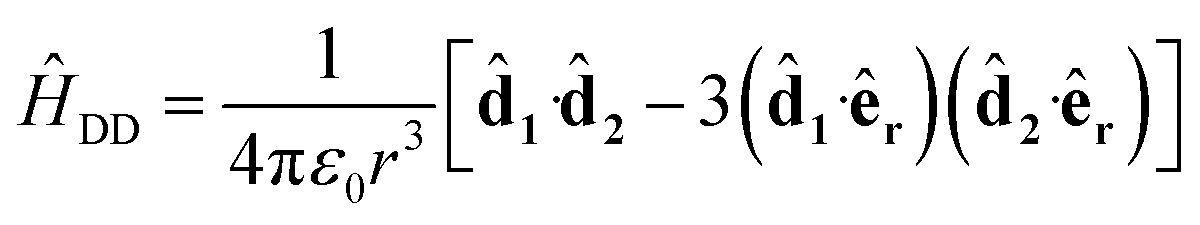
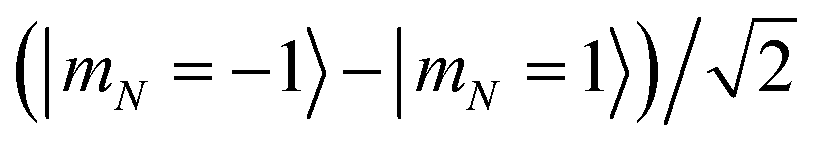 character with dipole moment along
character with dipole moment along 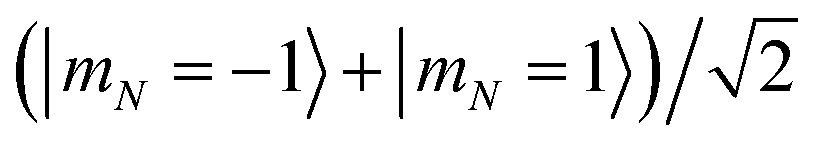 character with dipole moment along ŷ are blue. States with predominantly |mN = 0〉 character with dipole moment along ẑ are red. The thick line shows the state |e〉 = |1,−1,3/2,7/2〉. The polarization vector is
character with dipole moment along ŷ are blue. States with predominantly |mN = 0〉 character with dipole moment along ẑ are red. The thick line shows the state |e〉 = |1,−1,3/2,7/2〉. The polarization vector is 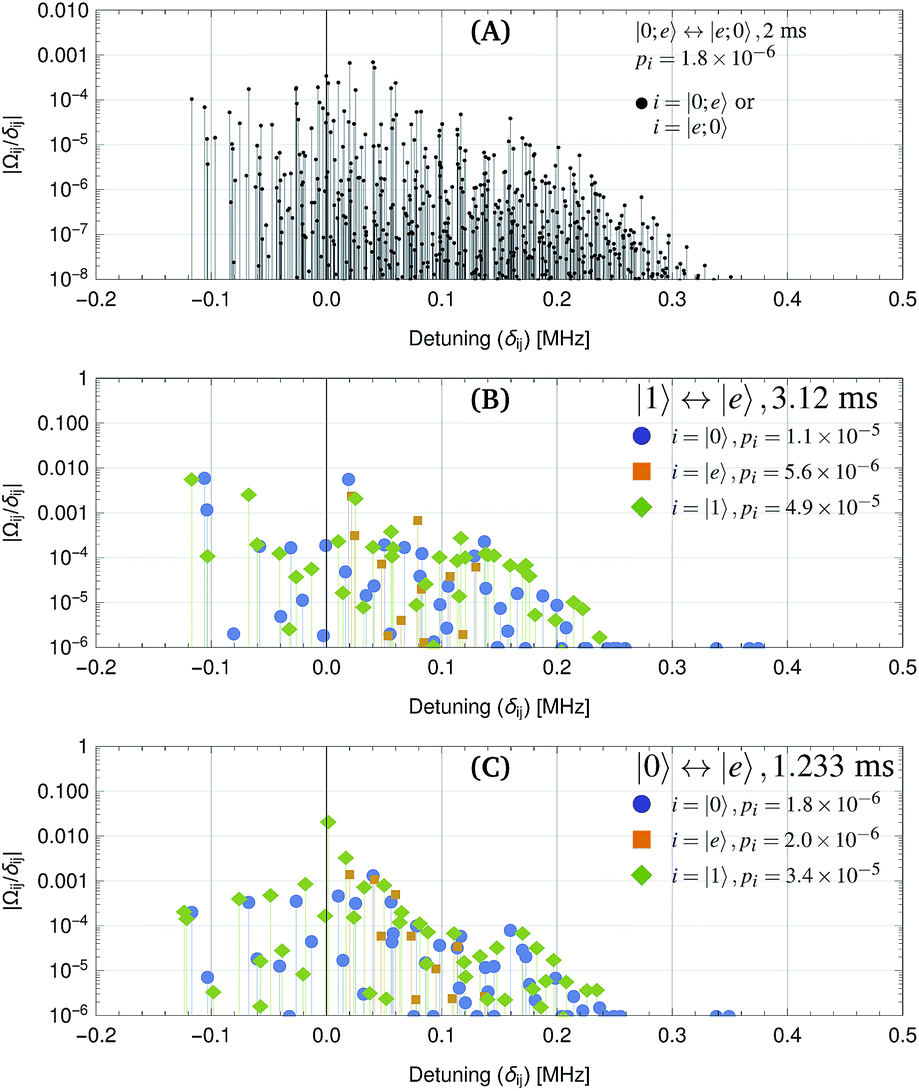
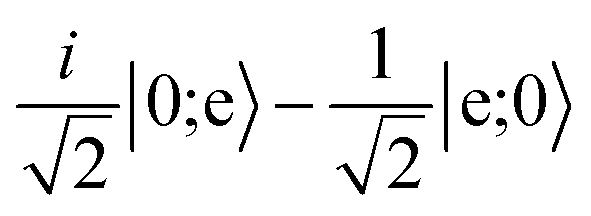
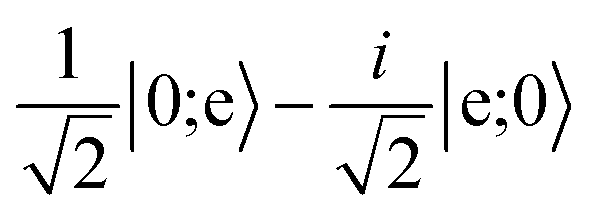

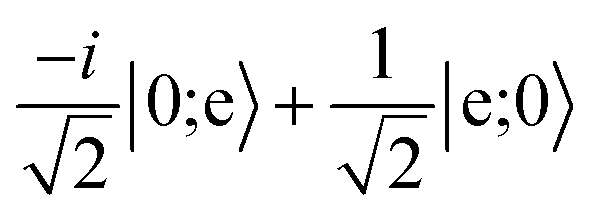
 are the zero-point wave function spreads, fx = 12, fy,z = −6, and the approximation includes terms up to second order in position. The above trap depth corresponds to motional frequencies of ωx,y = 2π·12.4 kHz and ωz = 2π·2.8 kHz for NaCs, where a Gaussian beam radius of 1 μm is assumed. Because the loss of fidelity is proportional to the square of the Rabi rate deviation from the mean, 1 − F can calculated from the variance of the Rabi rate, which can be expressed in terms of mean motional quantum numbers
are the zero-point wave function spreads, fx = 12, fy,z = −6, and the approximation includes terms up to second order in position. The above trap depth corresponds to motional frequencies of ωx,y = 2π·12.4 kHz and ωz = 2π·2.8 kHz for NaCs, where a Gaussian beam radius of 1 μm is assumed. Because the loss of fidelity is proportional to the square of the Rabi rate deviation from the mean, 1 − F can calculated from the variance of the Rabi rate, which can be expressed in terms of mean motional quantum numbers 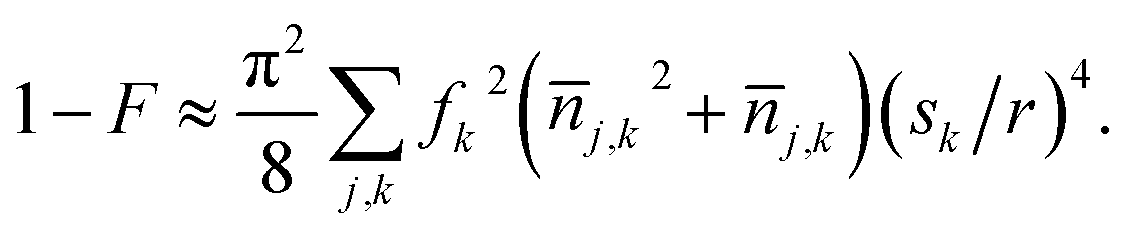
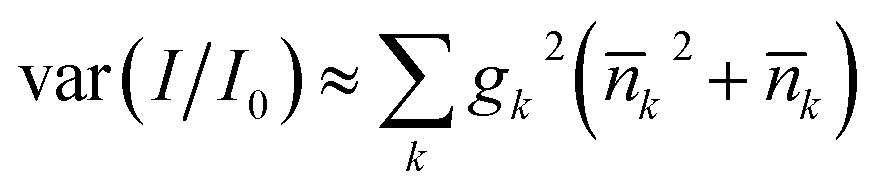 where gk = −4(sk/w)2 for the radial directions, gz = −2(szλ/(πw2))2 and w is the beam radius. The associated standard deviation in relative intensity is 2 × 10−3 for the residual motion after ground-state cooling described above. This corresponds to the intensity stability assumed in Section 6.
where gk = −4(sk/w)2 for the radial directions, gz = −2(szλ/(πw2))2 and w is the beam radius. The associated standard deviation in relative intensity is 2 × 10−3 for the residual motion after ground-state cooling described above. This corresponds to the intensity stability assumed in Section 6. with N qubits are stored in the qubit array. The same scaling applies to light that differentially shifts the phase of |0〉 and |1〉 (see Section 6). Such common-mode dephasing errors can be reduced by use of dynamical decoupling or decoherence-free subspaces.56
with N qubits are stored in the qubit array. The same scaling applies to light that differentially shifts the phase of |0〉 and |1〉 (see Section 6). Such common-mode dephasing errors can be reduced by use of dynamical decoupling or decoherence-free subspaces.56
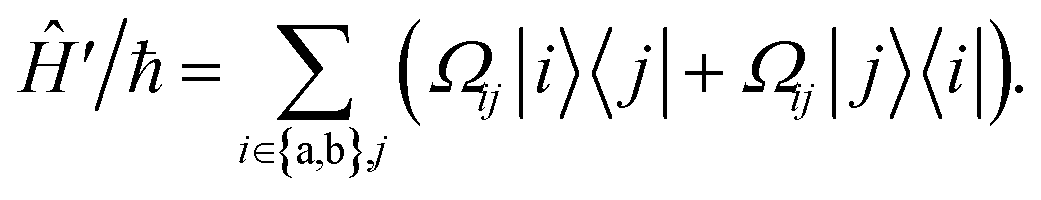
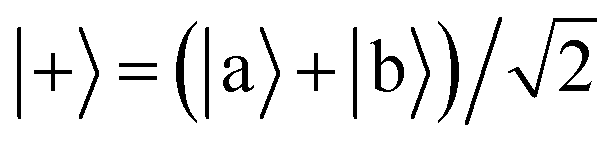
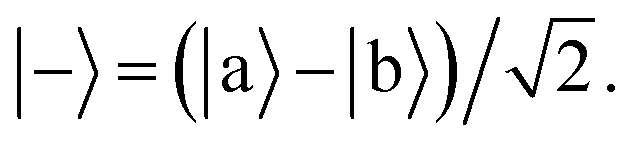
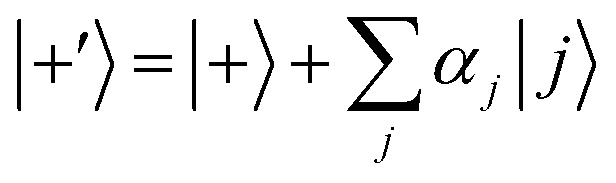
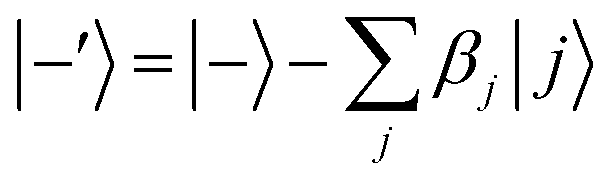
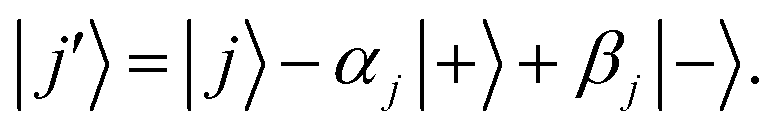
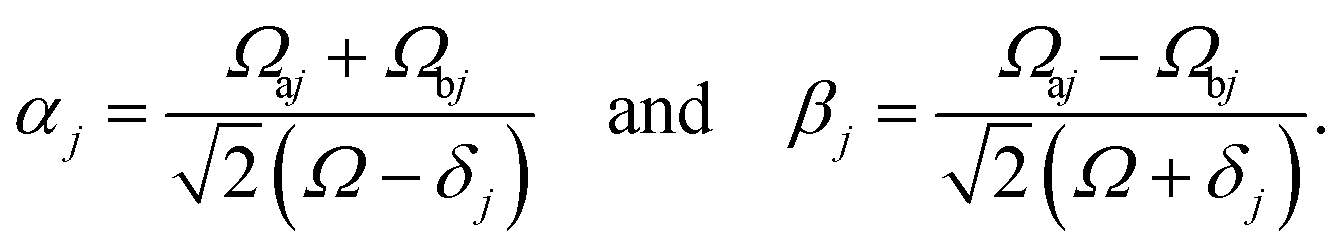 These states approximately diagonalize the time-evolution operator and can be used to estimate the transitions from the initial state |a〉 into states |j〉 due to unitary time evolution Û(t) = e−iĤt/ℏ:
These states approximately diagonalize the time-evolution operator and can be used to estimate the transitions from the initial state |a〉 into states |j〉 due to unitary time evolution Û(t) = e−iĤt/ℏ: