
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceQuasi-single-crystalline CoO hexagrams with abundant defects for highly efficient electrocatalytic water oxidation†
Zuozhong
Liang
a,
Zhehao
Huang
c,
Haitao
Yuan
a,
Zhiyuan
Yang
a,
Chaochao
Zhang
a,
Yang
Xu
a,
Wei
Zhang
 a,
Haoquan
Zheng
*a and
Rui
Cao
*ab
a,
Haoquan
Zheng
*a and
Rui
Cao
*ab
aKey Laboratory of Applied Surface and Colloid Chemistry, Ministry of Education, School of Chemistry and Chemical Engineering, Shaanxi Normal University, Xi'an 710119, China. E-mail: zhenghaoquan@snnu.edu.cn
bDepartment of Chemistry, Renmin University of China, Beijing, 100872, China. E-mail: ruicao@ruc.edu.cn
cDepartment of Materials and Environmental Chemistry, Stockholm University, SE-10691 Stockholm, Sweden
First published on 20th July 2018
Abstract
Defects and structural long-range ordering have been recognized as two crucial characters for advanced electrocatalysts. However, these two features have rarely been achieved together. Herein, we report the preparation of single-crystalline CoO hexagrams and demonstrate their exceptional electrocatalytic properties for water oxidation. The quasi-single-crystalline CoO hexagrams, prepared at the critical phase transition point of β-Co(OH)2/Co(OH)F hexagrams, possess both abundant oxygen vacancies as defects and structural long-range ordering. The matching between the b-axis of Co(OH)F crystals and the a-axis of β-Co(OH)2 crystals is critical for the formation of the CoO hexagram single crystals. The quasi-single-crystalline CoO hexagrams with abundant defects are highly efficient for water oxidation by delivering a 10 mA cm−2 current density at a low overpotential of 269 mV in a 1 M KOH aqueous solution.
Introduction
Electrocatalysis has attracted tremendous interest in energy conversion and storage and also in organic synthesis.1–8 Electrocatalytic water oxidation is a key process in artificial photosynthesis.9–12 Recently, first-row transition metal oxides have been found to be active water oxidation catalysts (WOCs),13–38 but their efficiencies are required to be further improved. One strategy is to generate defects.39 Many types of defects in metal oxides have been reported, including anion and/or cation vacancies, intrinsic defects, dislocations, stepped surfaces, and boundaries.40–51 Defects can alter the electronic and surface properties of materials, usually leading to enhanced electrocatalytic performance.Single or quasi-single crystals with structural long-range ordering are essential to study the relationship between the structure and catalytic activity.52–58 However, only a few single crystals with abundant defects have been investigated as WOCs, and they are limited to simple low dimensional materials. For example, Qiao and co-workers reported that single-crystalline CoO nanorods with oxygen vacancies are highly active for water oxidation.52 Defect generation and structural long-range ordering are rarely achieved together in a hierarchical material. In consideration that a hierarchical material is typically a better electrocatalyst than its low dimensional building units,59,60 it is needed to prepare hierarchical single or quasi-single crystals with abundant defects for electrocatalytic studies.
Recently, we prepared hierarchical β-Co(OH)2/Co(OH)F hexagrams composed of plate-like β-Co(OH)2 hexagonal cores appended with six rod-like Co(OH)F branches (Fig. 1).61 By investigating the formation mechanism of this superstructure, we found that Co(OH)F nanorods epitaxially grow along β-Co(OH)2 hexagon edges due to the matching between the b-axis of Co(OH)F crystals and the a-axis of β-Co(OH)2 crystals. We therefore propose that this β-Co(OH)2/Co(OH)F hexagram is an ideal precursor to prepare defect-rich hierarchical CoO quasi-single crystals because (1) both Co(OH)2 and Co(OH)F can be converted to CoO upon phase transitions and (2) the matching between the β-Co(OH)2 and Co(OH)F crystals will lead to long-range ordering. Herein, we report the simple preparation of quasi-single-crystalline CoO hexagrams with abundant defects. The phase transfer of β-Co(OH)2/Co(OH)F hexagrams at the critical phase transition point gives CoO hexagrams with a single-crystal feature. This CoO superstructure shows exceptional activity for electrocatalytic water oxidation.
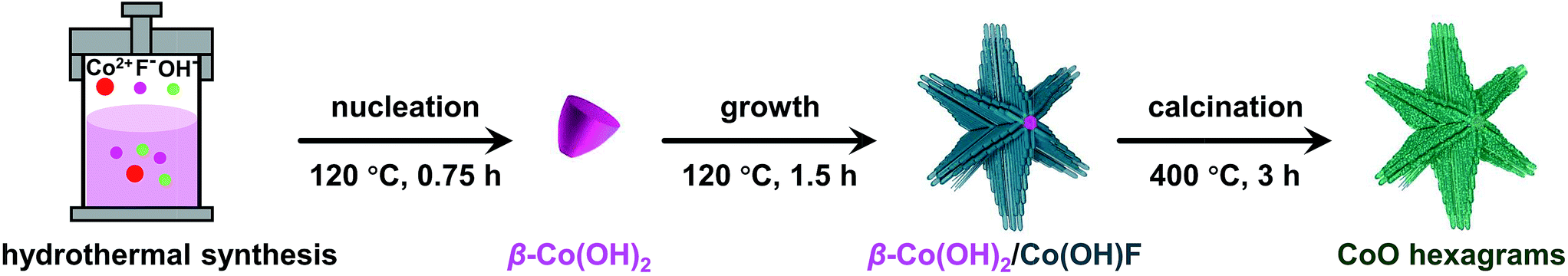 | ||
| Fig. 1 Schematic illustration of the preparation of quasi-single-crystalline CoO hexagrams. | ||
Experimental section
Methods and materials
All chemicals were commercially available and were used as received without further purification. CoCl2·6H2O (99%) was purchased from Shanghai Titanchem Co., Ltd. NH4F (98%) and hexamethylenetetramine (HMTA, 99.5%) were purchased from Energy Chemical. Nafion (5%) was purchased from DuPont. Ethanol (AR, 99.7%) was purchased from Tianjin Fuyu Fine Chemical Co., Ltd.Synthesis of quasi-single-crystalline CoO hexagrams
In a typical procedure, 1 mmol of CoCl2·6H2O, 2.5 mmol of NH4F and 0.6 mmol of HMTA were dissolved in 40 mL of deionized water with magnetic stirring to form a transparent solution. The solution was then transferred into a 100 mL Teflon-lined stainless-steel autoclave followed by heating the autoclave in an oven at 120 °C for different reaction times (0.75 h, 1 h, 1.5 h, and 12 h). After naturally cooling down to 25 °C, the resulting products were collected by centrifugation at 6000 rpm and were washed with deionized water and pure ethanol. The powders were dried in an oven at 60 °C for 24 h. Finally, the CoO was prepared by heating the β-Co(OH)2/Co(OH)F prepared at 1.5 h in a tube furnace under argon (flow rate: 60 mL min−1) for 3 h at a specific temperature between 200 and 600 °C with a heating rate of 2 °C min−1. The P-T (T = pyrolysis temperature, P = pyrolysis step) was used to define the as-prepared CoO products at different calcination temperatures, such as P-400.Synthesis of CoO nanoplates and CoO nanorods
The pure phase β-Co(OH)2 nanoplates were prepared by a hydrothermal method at 120 °C for 0.75 h with 1 mmol of CoCl2·6H2O, 2.5 mmol of NH4F and 0.6 mmol of HMTA dissolved in 40 mL of deionized water. The products were washed with deionized water and pure ethanol. The CoO nanoplates were then obtained by calcinating the pure phase β-Co(OH)2 at 400 °C for 3 h under argon.The pure phase Co(OH)F nanorods were obtained by a hydrothermal method at 120 °C for 12 h by dissolving 1 mmol of CoCl2·6H2O, 5.0 mmol of NH4F and 0.6 mmol of HMTA in 40 mL of deionized water. Then the CoO nanorods were obtained by the calcination process of Co(OH)F nanorods at 400 °C for 3 h under argon.
Characterization
Powder X-ray diffraction (PXRD) patterns of the as-prepared materials were obtained with an X-ray diffractometer (D8 Advance, Cu Kα, 40 kV/40 mA, Bruker). Fourier-transform infrared (FTIR) spectra were obtained using an IR spectrometer (Bruker, Tensor27) by making thin pellets with dried KBr powder. The morphology of the as-prepared material was observed with a scanning electron microscope (SEM, Hitachi, TM3000; SEM, Hitachi, SU8020). Transmission electron microscopy (TEM) images, high-resolution TEM (HRTEM) images and selected-area electron diffraction (SAED) patterns were obtained with a TEM (Tecnai G2 F20, FEI; JEM-2100, JEOL). The X-ray photoelectron spectroscopy (XPS) analysis of the materials was performed with a Kratos AXIS ULTRA XPS. The electron paramagnetic resonance (EPR) spectrum was obtained with a spectrometer at 9.43 GHz (Bruker, ELEXSYS E500 plus).Electrochemical studies
Electrochemical measurements were performed using a CH Instruments (Model CHI660E Electrochemical Analyzer) at 25 °C. Cyclic voltammetry (CV) of the as-prepared catalysts was carried out in 1.0 M KOH solution with a three-electrode system. We selected a glassy carbon (GC) electrode with a diameter of 3 mm (0.07 cm2) as the working electrode, Pt wire as the auxiliary electrode, and saturated Ag/AgCl as the reference electrode. The prepared catalysts were dropped onto the GC working electrode after polishing with Al2O3 powder of 50 nm particles and then ultrasonicating in ultrapure water. In a typical procedure, 4 mg of catalysts and 30 μL of Nafion solution (5 wt%, DuPont) were added into 1 mL of water/ethanol solution (Vwater![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Vethanol = 2:1) and then ultrasonicated for 1 h to prepare a homogeneous slurry. Finally, 5 μL of the catalyst was coated onto the GC working electrode using a pipette. Compensation for iR drop was carried out for all CV measurements (100%). The overpotential (η) was calculated versus the reversible hydrogen electrode (vs. RHE) based on the following equation: η = EAg/AgCl + (0.197 + 0.059 × pH) − 1.23.
:Vethanol = 2:1) and then ultrasonicated for 1 h to prepare a homogeneous slurry. Finally, 5 μL of the catalyst was coated onto the GC working electrode using a pipette. Compensation for iR drop was carried out for all CV measurements (100%). The overpotential (η) was calculated versus the reversible hydrogen electrode (vs. RHE) based on the following equation: η = EAg/AgCl + (0.197 + 0.059 × pH) − 1.23.
The Tafel plots were measured by carrying out linear sweep voltammetry (LSV) at a scan rate of 2 mV s−1. The electrochemical surface area (ECSA) was calculated by the following formula: ECSA = Cdl/Cs. The specific estimated capacitance Cs of 27 μF cm−2 for Co-based materials was used based on the value reported previously.62 Electrochemical impedance spectroscopy (EIS) was performed at a potential of 1.60 V (vs. RHE). The detection of O2 was carried out with a bipotentiostat (760E, CH Instruments) and rotator (Pine Research Instrumentation) by coating the catalyst on a rotating ring-disk electrode (RRDE) at a scan rate of 5 mV s−1 and a rotation rate of 1600 rpm in N2-saturated 1.0 M KOH solution. The RRDE has a GC disk with an area of 0.247 cm2 and a Pt ring with an area of 0.186 cm2. The O2 reduction process was performed by holding the potential of the Pt ring electrode at 0.40 V (vs. RHE). The faradaic efficiency was measured with O2 sensor analysis during controlled potential electrolysis at 1.60 V (vs. RHE) with P-400 coated on an indium tin oxide (ITO) electrode. The time dependence of the current density was measured at 1.60 V (vs. RHE) for P-400, commercial Ir/C, and a blank ITO electrode in 1.0 M KOH solution. The CV current (i)–potential (E) response was monitored (E: 1.51 → 0.91 → 1.71 → 1.51) for P-400 in 1.0 M KOH solution at different scan rates: 0.1, 0.2, 0.5, 1.0 and 2.0 V s−1. Consecutive CV with 10 cycles was performed using a freshly prepared GC electrode coated with P-400 in KOH solutions with different concentrations: 0.1, 0.2, 0.5, 1.0 and 2.0 M. The bubbles on the electrode surface were eliminated after each scan cycle. The iR compensation (100%) was conducted before each scan cycle.
Results and discussion
Characterization of quasi-single-crystalline CoO hexagrams with abundant defects
The β-Co(OH)2/Co(OH)F hexagrams were first synthesized according to the method we developed.61 Powder X-ray diffraction (PXRD) patterns show that the hexagrams are a mixture of β-Co(OH)2 and Co(OH)F (Fig. 2a). Scanning electron microscopy (SEM) and transmission electron microscopy (TEM) images of β-Co(OH)2/Co(OH)F hexagrams with different reaction times confirmed the formation of the hexagram superstructure, which is built by a β-Co(OH)2 nanoplate core and six Co(OH)F nanorod branches (Fig. 2b–f and S1–S3, ESI†). As shown in Fig. 2g and h, the electron diffraction (SAED) of the selected area in Fig. 2e indicates that the six Co(OH)F nanorods grow along the b-axis. Therefore, as shown in Fig. 2i, β-Co(OH)2/Co(OH)F hexagrams are epitaxial growth of Co(OH)F nanorods on the β-Co(OH)2 core due to the length matching between the b-axis of Co(OH)F (a = 10.275 Å, b = 3.118 Å, c = 4.684 Å, Pnma) and the a-axis of β-Co(OH)2 (a = 3.173 Å, c = 4.640 Å, P![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m1), which has been demonstrated in detail in a previous report (Fig. 2j).61
m1), which has been demonstrated in detail in a previous report (Fig. 2j).61
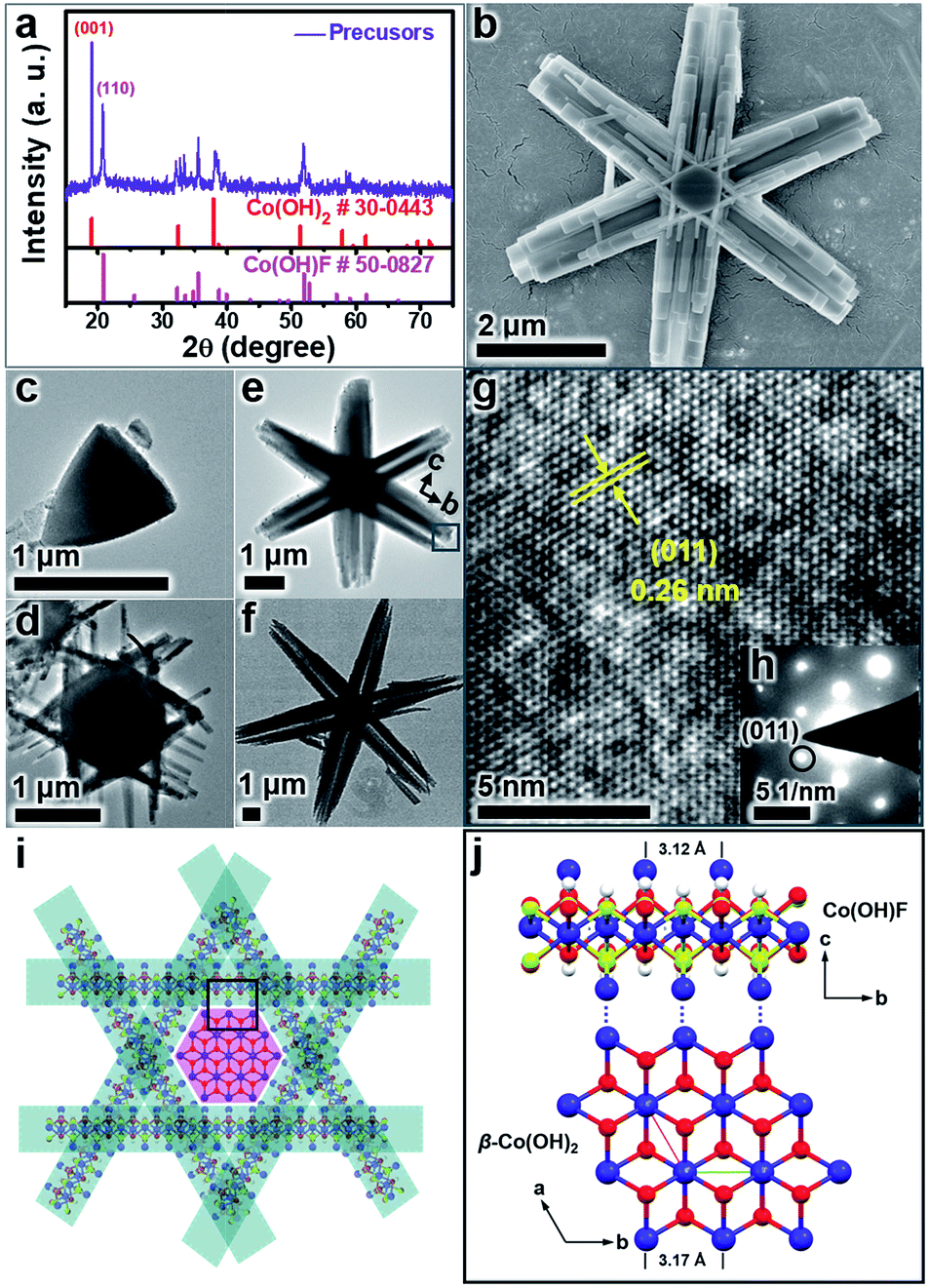 | ||
| Fig. 2 XRD pattern (a), SEM image (b), TEM image (e), HRTEM image (g), and SAED of the branch (h), and schematic illustration of the interface of Co(OH)F arms and the β-Co(OH)2 core (i and j) for the as-prepared six-branched β-Co(OH)2/Co(OH)F hexagram. TEM images of β-Co(OH)2/Co(OH)F hexagrams prepared at 0.75 h (c), 1 h (d), 1.5 h (e), and 12 h (f). | ||
The β-Co(OH)2/Co(OH)F was then phase transferred to CoO by calcination under argon. This phase transition at the critical point is expected to involve a symmetry breaking process and a decrease of structural ordering, which makes the resulting CoO defect-rich. In order to find out the critical phase transition point, we performed calcination at temperatures from 200 to 600 °C. The PXRD patterns of the samples calcined at different temperatures are shown in Fig. 3a. At 350 °C, β-Co(OH)2 started to transform. At 400 °C, the material was completely converted to CoO (JCPDS 43-1004), which was further confirmed by infrared spectroscopy (Fig. S4, ESI†). Therefore, 400 °C is critical for the phase transformation. P-600 is prepared as a control, since P-600 has the same structure as P-400 but has fewer defects.
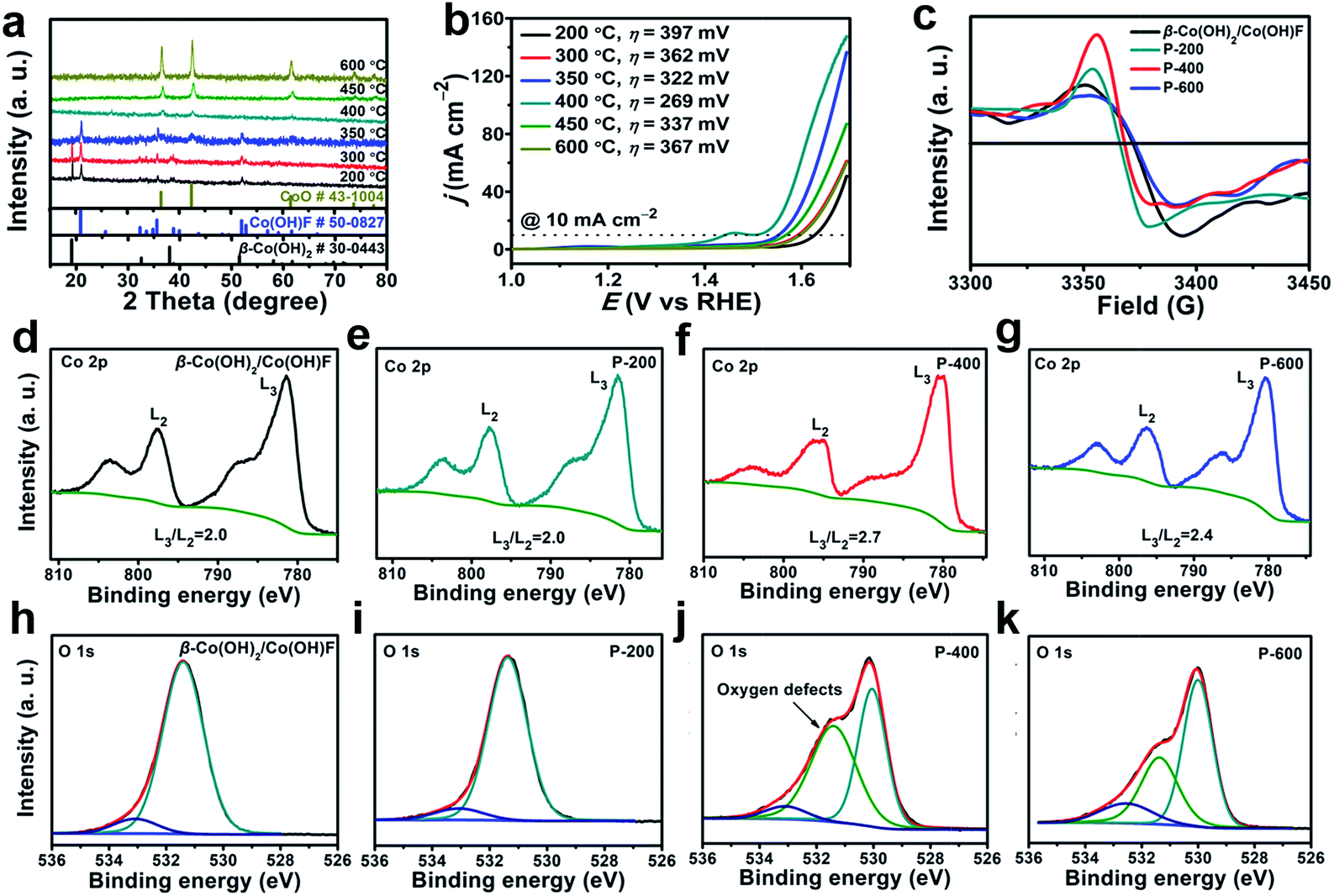 | ||
| Fig. 3 XRD patterns (a) and LSV data (b) of β-Co(OH)2/Co(OH)F hexagrams calcined at different temperatures. (c) EPR for the β-Co(OH)2/Co(OH)F, P-200, P-400, and P-600. High-resolution XPS Co 2p spectra (d–g), and O 1s spectra (h–k) for the β-Co(OH)2/Co(OH)F (d and h), P-200 (e and i), P-400 (f and j), and P-600 (g and k). | ||
The water oxidation performance of these materials was evaluated. Linear sweep voltammetry (LSV) data are shown in Fig. 3b. The iR compensation (100%) was conducted before each scan cycle. The overpotential (η) required to get current density j = 10 mA cm−2 decreases significantly from 397 mV for P-200 to 269 mV for P-400. Further increase of the calcination temperature will lead to activity loss. For P-600, η = 367 mV is required to get the same current. As a result, P-400 prepared at the critical phase transition point exhibits the highest activity for water oxidation.
To investigate the reason for the enhanced OER activity, we further performed electron paramagnetic resonance (EPR) and high-resolution X-ray photoelectron spectroscopy (XPS). The EPR signal at 3370 G is identified as electrons trapped in oxygen vacancies. The strong signal intensity infers that there are abundant oxygen vacancies in P-400.63 The high-resolution of XPS spectra were used to get more detailed information about the oxygen vacancies (Fig. 3d–i). The L3/L2 ratio in high-resolution XPS transition metal 2p spectra can be used to determine the oxygen vacancies.64 As shown in Fig. 3d–f, P-400 exhibits a higher L3/L2 ratio (2.7) compared to β-Co(OH)2/Co(OH)F (2.0), P-200 (2.0), and P-600 (2.4), indicating many more oxygen vacancies in P-400. The peak at 531.4 eV is assigned to the defects with low oxygen coordination.65 β-Co(OH)2/Co(OH)F shows similar XPS peaks of O 1s spectra to those of P-200, which may be attributed to the same crystalline structure of β-Co(OH)2/Co(OH)F and P-200. The area of the peak at 531.4 eV for P-400 is much larger than that for β-Co(OH)2/Co(OH)F, P-200, and P-600, which further indicates that a larger number of O-vacancies are present in P-400. The abundant oxygen vacancies as defects in P-400 are beneficial for the OER process.39
SEM, TEM, and high-resolution TEM (HRTEM) were used to investigate the detailed structure of P-400 (Fig. 4, S5 and S6, ESI†). P-400 remained the hexagram superstructure with a roughened surface (Fig. S5, ESI†). TEM, HRTEM images and N2 adsorption–desorption curves of P-400 further confirm the formation of mesoporous structures and abundant defects (Fig. S6 and S7, ESI†). Although the arms have a quiet rough surface and porous structure, their continuous crystal structures show that each arm is a single crystal. The electron diffraction (ED) patterns of three different arms and the centre part of an individual CoO hexagram are very similar, showing single-crystalline electron diffraction patterns which indicates a structural long-range ordering. As shown in Fig. 4b–d, by viewing from the same direction along [110], the identified lattice d-spacings of 0.21 and 0.25 nm can be assigned to the (200) and (111) facets of CoO, respectively. However, the three arms showed different crystal orientations, as the a axis (h00 reflections) is always along the short edge of the arms. This observation means that there is a rotation of 60° between arms, when they have the same exposed facets. This result fits well with the original orientations of the arms in Co(OH)F, where arms have intersection angles of 60°.61 Interestingly, the ED pattern selected in the central part of the hexagram remains as a single-crystalline pattern (Fig. 4a inset IV), which indicates that the crystal lattices of the arms and the centre nanoplate should be continuously connected. The HRTEM image of the interface between the nanorod and core has been taken, which demonstrates continuous connection of the crystal lattices at the interface (Fig. 4e). The length matching between the b-axis of Co(OH)F and the a-axis of β-Co(OH)2 at the interface ensures the continuous connection of the crystal lattices after calcination. Therefore, this matching is not only critical for the formation of this unique hexagram-like morphology but also for the quasi-single-crystallinity of the derived CoO hexagram. In addition, more severely diffused scattering (prolonged elliptical spots) can be observed in the central part. It has a similar d-spacing to that in the arms. The diffused scattering indicates that the central part is defect-rich. During the phase transformation, each edge of the center β-Co(OH)2 nanoplate was fitting the lattice of the connected Co(OH)F arms with almost the same interspacing distance, which led to the generation of a defect-rich quasi-single-crystalline CoO structure. The defects are generated where β-Co(OH)2 and Co(OH)F transferred to a denser phase CoO. The increase in the density of CoO (6.424 g cm−3) from Co(OH)F (4.214 g cm−3) and β-Co(OH)2 (3.732 g cm−3) generates the roughness and defects, and the larger the difference, the more defects were generated.
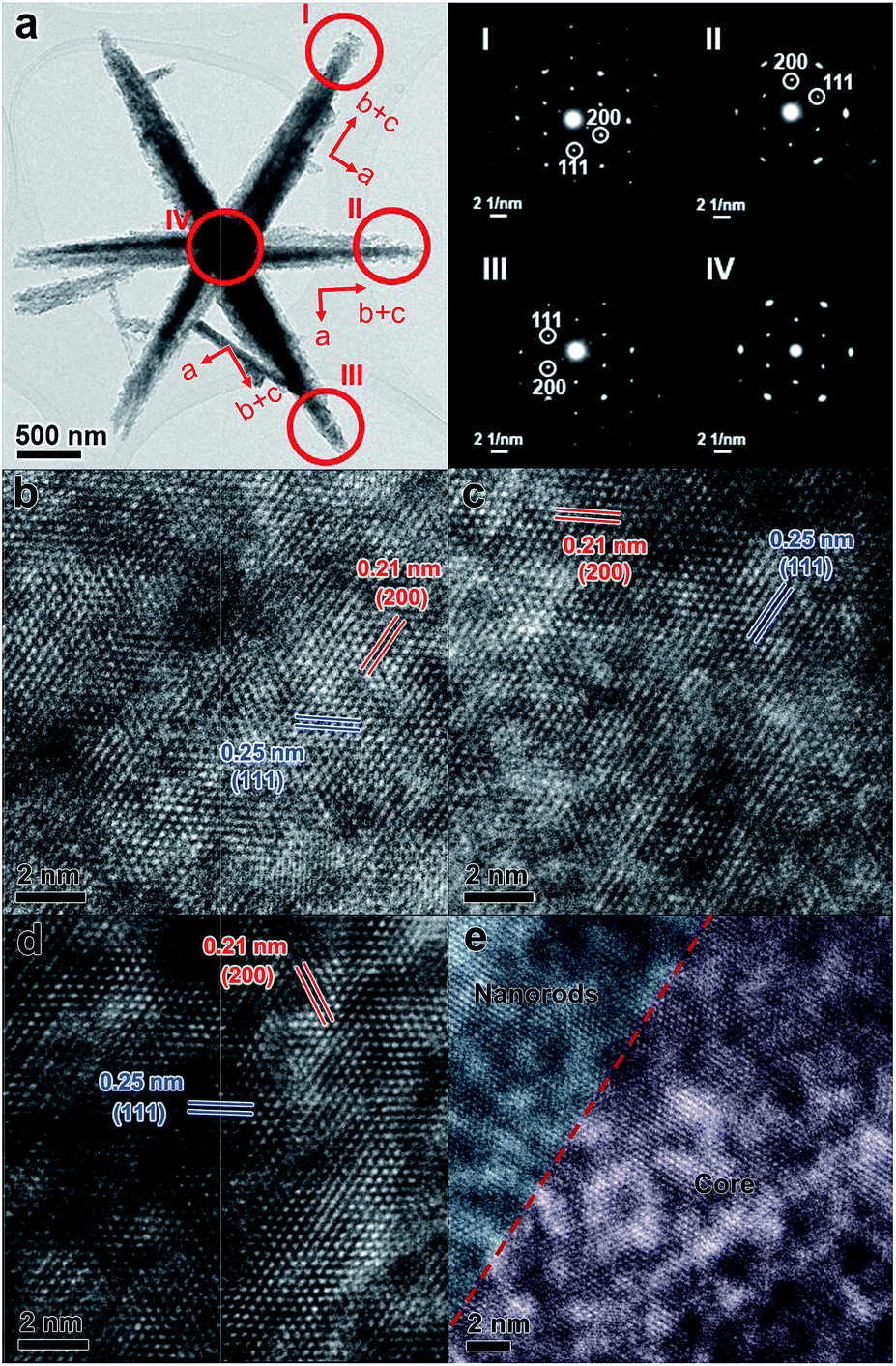 | ||
| Fig. 4 TEM image (a) and ED patterns obtained from different arms of P-400. HRTEM images (b–d) of P-400 viewing along [110]. They were obtained from arms I–III, respectively. HRTEM image (e) of the connection part between the nanorod and core in P-400. | ||
The EDS study shows that the Co/O ratio of P-400 CoO is c.a. 2 (Fig. S8, ESI†). It deviates a lot from CoO (the theoretical Co/O is 1), suggesting that there are a lot of oxygen-missing defects in the crystal. The diffused scattering in the ED pattern indicates that the central part is highly defect-rich (Fig. 4a). P-600 was found to be composed of CoO nanoparticles (Fig. S9, ESI†). The matching between the b-axis of Co(OH)F crystals and the a-axis of β-Co(OH)2 crystals is critical for the formation of single-crystal CoO hexagrams. On the basis of the abovementioned results, P-400 has abundant oxygen vacancies as defects and structural long-range ordering, which have rarely been observed in a hierarchical material.
Electrochemical studies of quasi-single-crystalline CoO hexagrams with abundant defects
We compared the electrocatalytic performance of P-400 with defect-rich quasi-single-crystalline CoO nanorods and nanoplates, which were prepared by calcining pure Co(OH)F nanorods and β-Co(OH)2 nanoplates at 400 °C, respectively. The η value at j = 10 mA cm−2 is 269 mV for P-400, 334 mV for CoO nanorods, and 339 mV for CoO nanoplates (Fig. 5a). Importantly, P-400 is more active than the commercial Ir/C catalyst (η = 295 mV), and represents the most efficient Co-based WOC in the literature (Table S1, ESI†). It is necessary to note that the exceptional activity of P-400 is assessed using a simple glassy carbon (GC) electrode rather than using carbon cloth, carbon paper, or metal foam electrodes with large supportive areas and high non-faradaic background currents.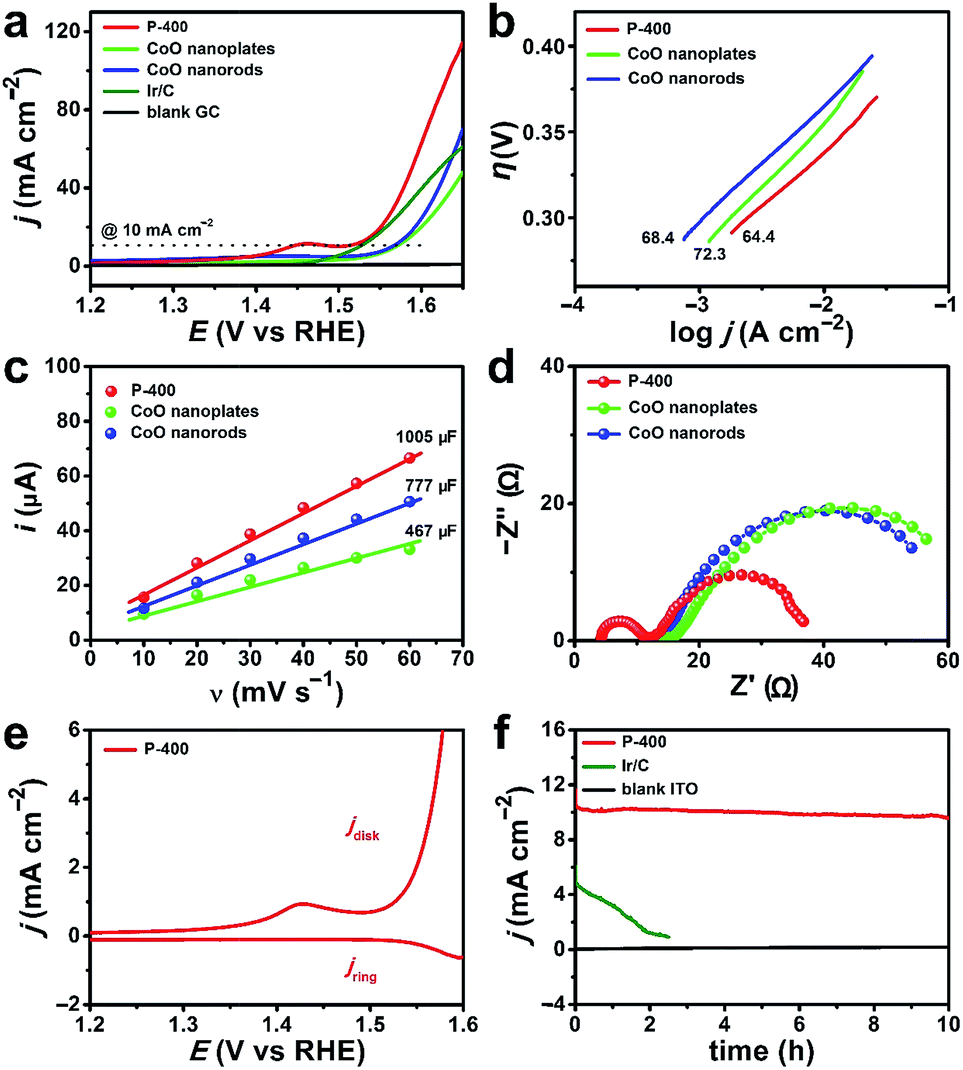 | ||
| Fig. 5 (a) LSV curves, (b) Tafel plots, (c) capacitance currents plotted versus scan rates, (d) EIS Nyquist plots at 1.60 V (vs. RHE), (e) RRDE measurement in N2-saturated 1.0 M KOH solution at 1600 rpm, and (f) current plots during 10 h controlled potential electrolysis in 1.0 M KOH solution for the catalysts. | ||
Electrochemical studies show that P-400 has a small Tafel slope of 64.4 mV dec−1, while these numbers are 68.4 and 72.3 mV dec−1 for CoO nanorods and nanoplates, respectively (Fig. 5b). The electrochemical surface area (ECSA) is determined by measuring the double-layer capacitance (Cdl) (Fig. 5c and S10, ESI†), giving 37.2 cm2 for P-400, 28.8 cm2 for CoO nanorods, and 17.3 cm2 for CoO nanoplates. This result further confirms the more exposed active sites in P-400. Normalized current density based on the calculated ECSA of catalysts shows that the enhanced electrocatalytic activity of P-400 is not only attributed to the increased ECSA (Fig. S11, ESI†). Electrochemical impedance spectroscopy (EIS) shows that the charge/mass transfer resistance of P-400 is much lower than that of CoO nanorods and nanoplates (Fig. 5d). The quasi-single-crystalline electrocatalysts can promote electronic conduction during electrocatalysis. All these factors may account for the significantly enhanced activity of P-400 for electrocatalytic water oxidation.
To prove the O2 evolution during water oxidation, rotating ring-disk electrode (RRDE) measurement was carried out in N2-saturated solutions (Fig. 5e). The formed O2 on the GC disk electrode was detected on the Pt ring electrode. The increase of the anodic catalytic current is synchronous with the increase of the cathodic Pt ring current, confirming that the catalytic current is due to the oxidation of water into O2. The faradaic efficiency was calculated to be 98% (Fig. S12, ESI†). During 10 h electrolysis, the current remained constant at 10 mA cm−2 for P-400 (Fig. 5f and S13, ESI†). After electrolysis, the morphology and the Co 2p XPS spectrum of P-400 remained almost unchanged (Fig. S14 and S15, ESI†). In the O 1s XPS spectrum, a new peak appeared at 529.7 eV, which agreed well with the peak of CoOOH in Co-based catalysts after electrolysis.60,66
Typical cyclic voltammetry (CV) current responses of i, i/ν, and i/ν1/2 with scan rate (ν) ranging from 0.1 to 2.0 V s−1 are shown in Fig. 6a–c. These data are used to describe the kinetic control by catalytic reaction, double layer charging capacitive current, and diffusion, respectively. The redox wave at 1.50 V versus the reversible hydrogen electrode (vs. RHE) can be assigned to the CoIII/CoIV oxidation (Fig. 6c).60,66 The OER process was activated well-behind the full occurrence of the CoIII/IV event. In addition, the CoIII/IV event is reversible when performing the cathodic return scan, which should not occur if there is no CoV species. Thus, the CoIV species must undergo further oxidation at higher potential to form the active species, which might be –CoV oxo/oxyl, to initiate water oxidation. This phenomenon was also observed and explained by other scientists.60,67 The i–ν1/2 response of the CoIV → CoIII reduction peak indicates that the kinetic control process is determined by the diffusion (Fig. 6c, inset). As shown in Fig. 6d and S16, ESI,† the catalytic currents increase with pH. The precatalytic redox peaks (E1 and E2) shift to the negative potential with increasing pH (Fig. 6e). The linear dependence of peak potentials versus pH values is plotted in Fig. 6f, confirming the proton-coupled electron transfer (PCET) process for the formation of CoIV species. The reaction order in pH (ρpH) can be determined from the following equation:68,69

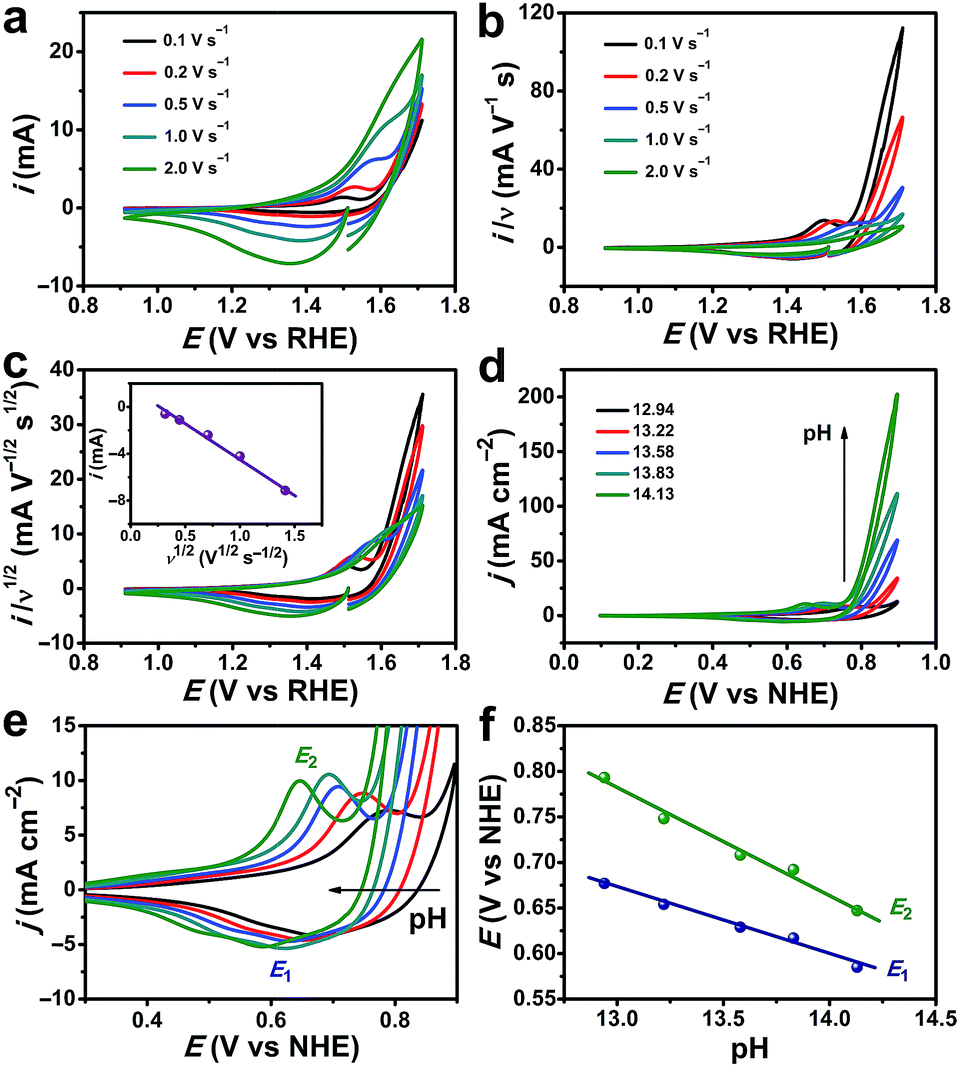 | ||
| Fig. 6 (a–c) CV current–potential responses of P-400 at different scan rates. Inset in (c): i vs. ν1/2. (d) CVs of P-400 in different pH KOH solutions. (e) The enlarged part showing the CVs in (d). (f) The dependence of the precatalytic redox peak potentials E1 and E2 on pH. | ||
Thus, the ρpH is equal to the negative of the slope of the potential vs. pH divided by the Tafel slope. At a fixed current density (j = 7 mA cm−2), the potential vs. pH produces an average slope of −125 mV per pH (Fig. S17, ESI†). At a specific pH, the Tafel slope is 64.4 mV dec−1. Therefore, the proton reaction order is calculated to be 1.94. This indicates that the oxygen evolution by the CoO hexagram exhibits an inverse second-order dependence on proton activity under alkaline conditions.
Conclusions
In conclusion, we reported the preparation of defect-rich quasi-single-crystalline CoO hexagrams and their exceptional electrocatalytic activity for water oxidation. The phase transfer of β-Co(OH)2/Co(OH)F at the critical point gave CoO hexagrams, which possess abundant oxygen vacancies as defects and structural long-range ordering. The matching between the b-axis of Co(OH)F crystals and the a-axis of β-Co(OH)2 crystals is critical for the formation of these rarely observed quasi-single-crystalline CoO hexagrams. The CoO hexagrams can catalyze water oxidation at η = 269 mV to get j = 10 mA cm−2. They are more active than CoO nanorods and nanoplates, and represent the most active Co-based material in the literature. This work proposes a simple strategy to prepare desired hierarchical materials, which have abundant defects and maintain the morphology and the long-range-ordered structure during defect generation.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful for support from the “Thousand Talents Program” of China, the National Natural Science Foundation of China (Grant No. 21101170, 21573139, 21601118, and 21773146), the Fundamental Research Funds for the Central Universities, and the Research Funds of Shaanxi Normal University. We thank Dr Cheuk-Wai Tai (Stockholm University) for valuable discussions and comments.Notes and references
- V. R. Stamenkovic, D. Strmcnik, P. P. Lopes and N. M. Markovic, Nat. Mater., 2017, 16, 57–69 CrossRef PubMed.
- Z. W. Seh, J. Kibsgaard, C. F. Dickens, I. Chorkendorff, J. K. Nørskov and T. F. Jaramillo, Science, 2017, 355, eaad4998 CrossRef PubMed.
- Y. Jiao, Y. Zheng, M. Jaroniec and S. Z. Qiao, Chem. Soc. Rev., 2015, 44, 2060–2086 RSC.
- A. Holewinski, J. C. Idrobo and S. Linic, Nat. Chem., 2014, 6, 828–834 CrossRef PubMed.
- A. Jutand, Chem. Rev., 2008, 108, 2300–2347 CrossRef PubMed.
- R. Francke and R. D. Little, Chem. Soc. Rev., 2014, 43, 2492–2521 RSC.
- D. Xu, X. Liu, H. Lv, Y. Liu, S. Zhao, M. Han, J. Bao, J. He and B. Liu, Chem. Sci., 2018, 9, 4451–4455 RSC.
- G.-R. Xu, J. Bai, J.-X. Jiang, J.-M. Lee and Y. Chen, Chem. Sci., 2017, 8, 8411–8418 RSC.
- W. Zhang, W. Z. Lai and R. Cao, Chem. Rev., 2017, 117, 3717–3797 CrossRef PubMed.
- D. G. Nocera, Acc. Chem. Res., 2012, 45, 767–776 CrossRef PubMed.
- Q. Lu, G. S. Hutchings, W. T. Yu, Y. Zhou, R. V. Forest, R. Z. Tao, J. Rosen, B. T. Yonemoto, Z. Y. Cao, H. M. Zheng, J. Q. Xiao, F. Jiao and J. G. Chen, Nat. Commun., 2015, 6, 6567 CrossRef PubMed.
- N. Danilovic, R. Subbaraman, K. C. Chang, S. H. Chang, Y. J. Kang, J. Snyder, A. P. Paulikas, D. Strmcnik, Y. T. Kim, D. Myers, V. R. Stamenkovic and N. M. Markovic, Angew. Chem., Int. Ed., 2014, 53, 14016–14021 CrossRef PubMed.
- L. Han, S. J. Dong and E. K. Wang, Adv. Mater., 2016, 28, 9266–9291 CrossRef PubMed.
- J. Qi, W. Zhang and R. Cao, Adv. Energy Mater., 2018, 8, 1701620 CrossRef.
- W. T. Hong, M. Risch, K. A. Stoerzinger, A. Grimaud, J. Suntivich and Y. Shao-Horn, Energy Environ. Sci., 2015, 8, 1404–1427 RSC.
- X. H. Gao, H. X. Zhang, Q. G. Li, X. G. Yu, Z. L. Hong, X. W. Zhang, C. D. Liang and Z. Lin, Angew. Chem., Int. Ed., 2016, 55, 6290–6294 CrossRef PubMed.
- Y. Liang, Y. F. Yu, Y. Huang, Y. M. Shi and B. Zhang, J. Mater. Chem. A, 2017, 5, 13336–13340 RSC.
- B. C. Weng, F. H. Xu, C. L. Wang, W. W. Meng, C. R. Grice and Y. F. Yan, Energy Environ. Sci., 2017, 10, 121–128 RSC.
- D. Y. Guo, F. F. Chen, W. Zhang and R. Cao, Sci. Bull., 2017, 62, 626–632 CrossRef.
- Y. Y. Liang, Y. G. Li, H. L. Wang, J. G. Zhou, J. Wang, T. Regier and H. J. Dai, Nat. Mater., 2011, 10, 780–786 CrossRef PubMed.
- L. Liardet and X. L. Hu, ACS Catal., 2018, 8, 644–650 CrossRef PubMed.
- M. B. Stevens, C. D. M. Trang, L. J. Enman, J. Deng and S. W. Boettcher, J. Am. Chem. Soc., 2017, 139, 11361–11364 CrossRef PubMed.
- W. Zhang, Y. Z. Wu, J. Qi, M. X. Chen and R. Cao, Adv. Energy Mater., 2017, 7, 1602547 CrossRef.
- F.-X. Ma, L. Yu, C.-Y. Xu and X. W. Lou, Energy Environ. Sci., 2016, 9, 862–866 RSC.
- Y. L. Deng, A. D. Handoko, Y. H. Du, S. B. Xi and B. S. Yeo, ACS Catal., 2016, 6, 2473–2481 CrossRef.
- B. S. Yeo and A. T. Bell, J. Am. Chem. Soc., 2011, 133, 5587–5593 CrossRef PubMed.
- C. L. Hu, L. Zhang, Z.-J. Zhao, J. Luo, J. Shi, Z. Q. Huang and J. L. Gong, Adv. Mater., 2017, 29, 1701820 CrossRef PubMed.
- I. Zaharieva, P. Chernev, M. Risch, K. Klingan, M. Kohlhoff, A. Fischer and H. Dau, Energy Environ. Sci., 2012, 5, 7081–7089 RSC.
- C.-H. Kuo, I. M. Mosa, A. S. Poyraz, S. Biswas, A. M. El-Sawy, W. Q. Song, Z. Luo, S.-Y. Chen, J. F. Rusling, J. He and S. L. Suib, ACS Catal., 2015, 5, 1693–1699 CrossRef.
- H. Y. Jin, J. Wang, D. F. Su, Z. Z. Wei, Z. F. Pang and Y. Wang, J. Am. Chem. Soc., 2015, 137, 2688–2694 CrossRef PubMed.
- Z. Y. Lu, H. T. Wang, D. S. Kong, K. Yan, P.-C. Hsu, G. Y. Zheng, H. B. Yao, Z. Liang, X. M. Sun and Y. Cui, Nat. Commun., 2014, 5, 4345 CrossRef PubMed.
- M. R. Gao, W. C. Sheng, Z. B. Zhuang, Q. R. Fang, S. Gu, J. Jiang and Y. S. Yan, J. Am. Chem. Soc., 2014, 136, 7077–7084 CrossRef PubMed.
- J. B. Gerken, S. E. Shaner, R. C. Massé, N. J. Porubsky and S. S. Stahl, Energy Environ. Sci., 2014, 7, 2376–2382 RSC.
- J. H. Wang, W. Cui, Q. Liu, Z. C. Xing, A. M. Asiri and X. P. Sun, Adv. Mater., 2016, 28, 215–230 CrossRef PubMed.
- R. D. L. Smith, M. S. Prévot, R. D. Fagan, S. Trudel and C. P. Berlinguette, J. Am. Chem. Soc., 2013, 135, 11580–11586 CrossRef PubMed.
- J. Landon, E. Demeter, N. İnoğlu, C. Keturakis, I. E. Wachs, R. Vasić, A. I. Frenkel and J. R. Kitchin, ACS Catal., 2012, 2, 1793–1801 CrossRef.
- M. Huynh, T. Ozel, C. Liu, E. C. Lau and D. G. Nocera, Chem. Sci., 2017, 8, 4779–4794 RSC.
- B. Liu, C. H. Kuo, J. Chen, Z. Luo, S. Thanneeru, W. Li, W. Song, S. Biswas, S. L. Suib and J. He, Angew. Chem., Int. Ed., 2015, 54, 9061–9065 CrossRef PubMed.
- D. F. Yan, Y. X. Li, J. Huo, R. Chen, L. M. Dai and S. Y. Wang, Adv. Mater., 2017, 29, 1606459 CrossRef PubMed.
- L. Xu, Q. Q. Jiang, Z. H. Xiao, X. Y. Li, J. Huo, S. Y. Wang and L. M. Dai, Angew. Chem., Int. Ed., 2016, 55, 5277–5281 CrossRef PubMed.
- M. Behrens, F. Studt, I. Kasatkin, S. Kühl, M. Hävecker, F. Abild-Pedersen, S. Zander, F. Girgsdies, P. Kurr, B.-L. Kniep, M. Tovar, R. W. Fischer, J. K. Nørskov and R. Schlögl, Science, 2012, 336, 893 CrossRef PubMed.
- W. J. Xu, F. L. Lyu, Y. C. Bai, A. Q. Gao, J. Feng, Z. X. Cai and Y. D. Yin, Nano Energy, 2018, 43, 110–116 CrossRef.
- J. Swaminathan, R. Subbiah and V. Singaram, ACS Catal., 2016, 6, 2222–2229 CrossRef.
- M.-H. Liu, Y.-W. Chen, X. Y. Liu, J.-L. Kuo, M.-W. Chu and C.-Y. Mou, ACS Catal., 2016, 6, 115–122 CrossRef.
- Y. X. Lin, L. Yang, Y. K. Zhang, H. L. Jiang, Z. J. Xiao, C. Q. Wu, G. B. Zhang, J. Jiang and L. Song, Adv. Energy Mater., 2018, 8, 1703623 CrossRef.
- J. W. Wan, W. X. Chen, C. Y. Jia, L. R. Zheng, J. C. Dong, X. S. Zheng, Y. Wang, W. S. Yan, C. Chen, Q. Peng, D. S. Wang and Y. D. Li, Adv. Mater., 2018, 1703623 Search PubMed.
- Y. W. Liu, X. M. Hua, C. Xiao, T. F. Zhou, P. C. Huang, Z. P. Guo, B. C. Pan and Y. Xie, J. Am. Chem. Soc., 2016, 138, 5087–5092 CrossRef PubMed.
- Y. C. Wang, T. Zhou, K. Jiang, P. M. Da, Z. Peng, J. Tang, B. Kong, W. B. Cai, Z. Q. Yang and G. F. Zheng, Adv. Energy Mater., 2014, 4, 1400696 CrossRef.
- S. L. Zhao, M. Li, M. Han, D. D. Xu, J. Yang, Y. Lin, N. E. Shi, Y. A. Lu, R. Yang, B. T. Liu, Z. H. Dai and J. C. Bao, Adv. Funct. Mater., 2018, 28, 1706018 CrossRef.
- J. R. Petrie, H. Jeen, S. C. Barron, T. L. Meyer and H. N. Lee, J. Am. Chem. Soc., 2016, 138, 7252–7255 CrossRef PubMed.
- J. Xie, S. Li, X. Zhang, J. Zhang, R. Wang, H. Zhang, B. Pan and Y. Xie, Chem. Sci., 2014, 5, 4615–4620 RSC.
- T. Ling, D. Y. Yan, Y. Jiao, H. Wang, Y. Zheng, X. L. Zheng, J. Mao, X. W. Du, Z. P. Hu, M. Jaroniec and S. Z. Qiao, Nat. Commun., 2016, 7, 12876 CrossRef PubMed.
- C. Gao, Q. Q. Meng, K. Zhao, H. J. Yin, D. W. Wang, J. Guo, S. L. Zhao, L. Chang, M. He, Q. X. Li, H. J. Zhao, X. J. Huang, Y. Gao and Z. Y. Tang, Adv. Mater., 2016, 28, 6485–6490 CrossRef PubMed.
- Y. G. Li, B. Tan and Y. Y. Wu, J. Am. Chem. Soc., 2006, 128, 14258–14259 CrossRef PubMed.
- L. Tian, H. L. Zou, J. X. Fu, X. F. Yang, Y. Wang, H. L. Guo, X. H. Fu, C. L. Liang, M. M. Wu, P. K. Shen and Q. M. Gao, Adv. Funct. Mater., 2010, 20, 617–623 CrossRef.
- C.-W. Tung, Y.-Y. Hsu, Y.-P. Shen, Y. X. Zheng, T.-S. Chan, H.-S. Sheu, Y.-C. Cheng and H. M. Chen, Nat. Commun., 2015, 6, 8106 CrossRef PubMed.
- H. Huang, W. J. Zhu, X. Y. Tao, Y. Xia, Z. Y. Yu, J. W. Fang, Y. P. Gan and W. K. Zhang, ACS Appl. Mater. Interfaces, 2012, 4, 5974–5980 CrossRef PubMed.
- W. Li, X. Gao, D. Xiong, F. Xia, J. Liu, W.-G. Song, J. Xu, S. M. Thalluri, M. F. Cerqueira, X. Fu and L. Liu, Chem. Sci., 2017, 8, 2952–2958 RSC.
- M. Rauber, I. Alber, S. Müller, R. Neumann, O. Picht, C. Roth, A. Schökel, M. E. Toimil-Molares and W. Ensinger, Nano Lett., 2011, 11, 2304–2310 CrossRef PubMed.
- S. H. Wan, J. Qi, W. Zhang, W. N. Wang, S. K. Zhang, K. Q. Liu, H. Q. Zheng, J. L. Sun, S. Y. Wang and R. Cao, Adv. Mater., 2017, 29, 1700286 CrossRef PubMed.
- Z. Z. Liang, Z. Y. Yang, Z. H. Huang, J. Qi, M. X. Chen, W. Zhang, H. Q. Zheng, J. L. Sun and R. Cao, Electrochim. Acta, 2018, 271, 526–536 CrossRef.
- C. C. L. McCrory, S. Jung, J. C. Peters and T. F. Jaramillo, J. Am. Chem. Soc., 2013, 135, 16977–16987 CrossRef PubMed.
- J. Ma, H. Wu, Y. Liu and H. He, J. Phys. Chem. C, 2014, 118, 7434–7441 CrossRef.
- Z. L. Wang, J. S. Yin and Y. D. Jiang, Micron, 2000, 31, 571–580 CrossRef PubMed.
- B. Jian, Z. Xiaodong, F. Bo, Z. Jiajia, Z. Min, Y. Wenlong, H. Xin, W. Hui, P. Bicai and X. Yi, Angew. Chem., Int. Ed., 2015, 54, 7399–7404 CrossRef PubMed.
- J. Qi, W. Zhang and R. Cao, Chem. Commun., 2017, 53, 9277–9280 RSC.
- C. Costentin, T. R. Porter and J.-M. Savéant, J. Am. Chem. Soc., 2016, 138, 5615–5622 CrossRef PubMed.
- Y. Surendranath, D. A. Lutterman, Y. Liu and D. G. Nocera, J. Am. Chem. Soc., 2012, 134, 6326–6336 CrossRef PubMed.
- M. Huynh, D. K. Bediako and D. G. Nocera, J. Am. Chem. Soc., 2014, 136, 6002–6010 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8sc02294a |
| This journal is © The Royal Society of Chemistry 2018 |