
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHighly diastereoselective synthesis of tricyclic fused-pyrans by sequential hydride shift mediated double C(sp3)–H bond functionalization†
Keiji
Mori
 ab,
Nobuaki
Umehara
a and
Takahiko
Akiyama
*a
ab,
Nobuaki
Umehara
a and
Takahiko
Akiyama
*a
aDepartment of Chemistry, Faculty of Science, Gakushuin University, 1-5-1 Mejiro, Toshima-ku, Tokyo 171-8588, Japan. E-mail: takahiko.akiyama@gakushuin.ac.jp; Fax: +81-3-3986-1029; Tel: +81-3-3986-0221
bDepartment of Applied Chemistry, Graduate School of Engineering, Tokyo University of Agriculture and Technology, 2-24-16 Nakacho, Koganei, Tokyo 184-8588, Japan
First published on 25th June 2018
Abstract
Described herein is the Brønsted acid-catalyzed double C(sp3)–H bond functionalization of alkyl phenethyl ether derivatives. In this process, a [1,5]-[1,5]-hydride shift occurred successively to afford tricyclic fused pyran derivatives in excellent chemical yields with excellent diastereoselectivities (up to >20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). The key to achieve this reaction was the introduction of two methyl groups at the benzylic position, which was effective in both hydride shift processes: (1) the Thorpe–Ingold effect for the first hydride shift and (2) conformational control in the second hydride shift.
:1). The key to achieve this reaction was the introduction of two methyl groups at the benzylic position, which was effective in both hydride shift processes: (1) the Thorpe–Ingold effect for the first hydride shift and (2) conformational control in the second hydride shift.
Introduction
Development of an efficient synthetic method is a major topic of research interest in modern synthetic organic chemistry. Direct transformation of inert C–H bonds, in particular, has attracted much attention because of its high potential for the development of new trends in the synthesis of complex organic molecules.1 Hydride shift triggered C(sp3)–H bond functionalization has recently emerged as a powerful tool for the rapid construction of various useful organic molecules.2Fig. 1 illustrates the details of this strategy. The key feature of this transformation is the [1,5]-hydride shift of the C(sp3)–H bond α to a heteroatom. Subsequent 6-endo cyclization to a cationic species A affords fused heterocycle 2.3–6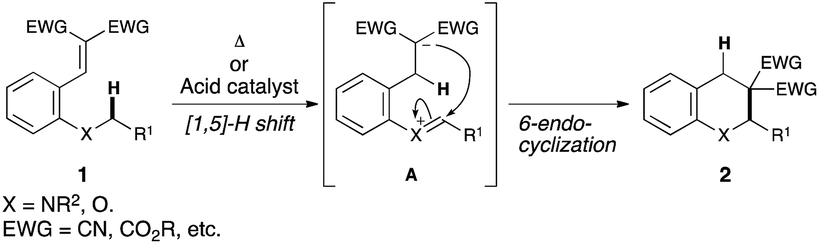 | ||
| Fig. 1 C(sp3)–H functionalization by the internal redox process. | ||
As part of our ongoing effort to develop a new synthetic transformation method involving hydride shift triggered C(sp3)–H bond functionalization,7 we have recently successfully applied this strategy to a sequential hydride shift system (double C(sp3)–H bond functionalization).8,9 The key to achieving this reaction was the use of the reactive α,β-unsaturated trifluoromethyl ketone as the electrophilic moiety. This setting effectively promoted an unprecedented [1,4]-[1,5]-hydride shift sequence, affording linear tricyclic piperidines in excellent chemical yields and with excellent diastereoselectivities (eqn (1), Fig. 2).8 The application of this sequential system to an oxygen analogue is also highly important because it would be a promising tool for rapid construction of multisubstituted tricyclic pyrans, which are found in some biologically active compounds. The two challenges to achieve this reaction are as follows: (1) the lower electron-donating ability of an oxygen atom than that of a nitrogen atom, which is disadvantageous for the hydride shift process, and (2) the propensity to eliminate the alkoxy group.10
 | ||
| Fig. 2 Double C(sp3)–H bond functionalization in alkyl phenethyl ether derivatives. | ||
We wish to report herein a solution to the issues related to the realization of the double C(sp3)–H bond functionalization in alkyl phenethyl ether derivatives (eqn (2), Fig. 2). The key to achieving this functionalization was to introduce two methyl groups at the benzylic position, thereby promoting the desired [1,5]-[1,5]-hydride shift/cyclization sequence smoothly to furnish the corresponding tricyclic pyrans in excellent chemical yields with excellent diastereoselectivities (up to >20:1 d.r.).
Results and discussion
An initial trial was conducted with cinnamylidene barbiturate 3 bearing a 2-benzyloxyethyl moiety at the ortho position (Scheme 1). Treatment of 3 with 10 mol% Yb(OTf)3, which exhibited excellent catalytic performance in the sequential internal redox reactions we previously developed,8,9 in refluxing ClCH2CH2Cl resulted in the recovery of the starting material 3. Gd(OTf)3 was also ineffective and the reaction did not proceed at all. | ||
| Scheme 1 Initial trial with simple cinnamylidene barbiturate 3. | ||
We assumed that these disappointing results were ascribed to the low electron-donating ability of the oxygen atom compared with the nitrogen atom (vide supra). To improve the hydride shift ability, a substrate 5 with a PMP group (electron-donating group)7c adjacent to the oxygen atom was employed (Scheme 2). Although the starting material 5 was mostly consumed, the resulting product was not the desired adduct 6 but the tetracyclic compound 7, which was produced by BnOH elimination, E/Z isomerization, followed by the inverse electron demand Diels–Alder reaction (IEDDA).
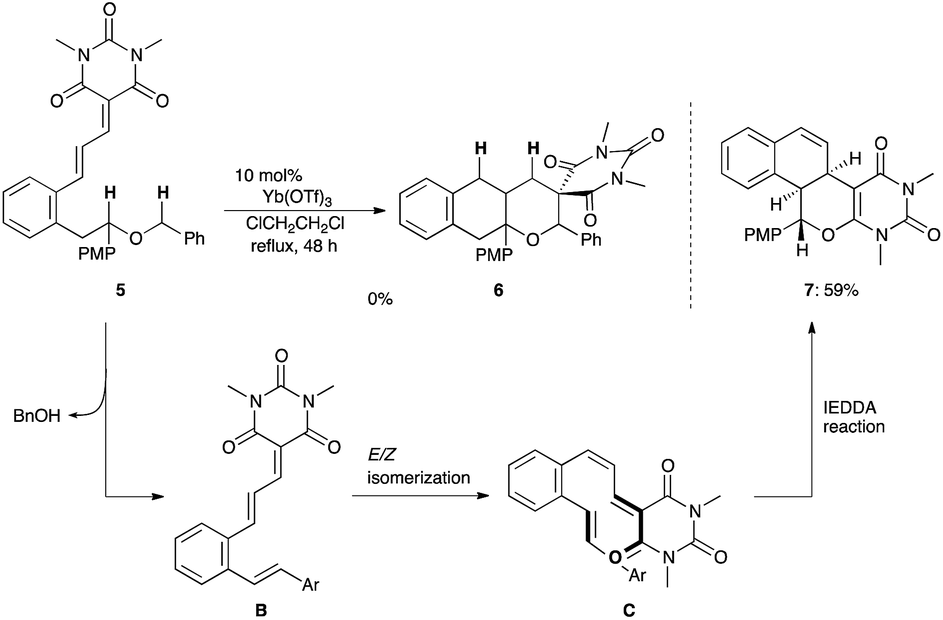 | ||
| Scheme 2 Reaction of 5 with a PMP group adjacent to the oxygen atom. | ||
The above results suggest that there are two obstacles to realize the sequential hydride shift reaction: (1) the low reactivity of the substrate and (2) the ease of eliminating the alkoxy group. We envisioned that 8 with two alkyl groups at the benzylic position would be a suitable substrate for overcoming these issues. This structural modification would be effective in both hydride shift processes (Fig. 3): (1) the Thorpe–Ingold effect for the first hydride shift and (2) the conformational control in the second hydride shift. The cooperative effect of these two factors would lead to a dramatic improvement of the reactivity. Another advantage of using this substrate is the suppression of elimination of the alkoxy group by the absence of benzylic hydrogens.
 | ||
| Fig. 3 Design of substrate 8 with a malonate moiety. | ||
As expected, the planned reaction proceeded smoothly with 8a. When 8a was treated with 10 mol% Yb(OTf)3 in refluxing ClCH2CH2Cl, the sequential [1,5]-hydride shift process proceeded to afford the tricyclic pyran derivative 9a in excellent chemical yield (90%). Only two diastereomers were observed in this reaction even though 9a has three stereogenic centers, and one diastereomer was slightly majored (d.r. = 3.3:1). Other metal triflates such as Sc(OTf)3, Gd(OTf)3, and Dy(OTf)3 also promoted the reaction albeit with lower diastereoselectivities (d.r. = <4.5:1, entries 2–4). The use of Mg(OTf)2 or TiCl4 resulted in the complete recovery of the starting material (entries 5 and 6). FeCl3 promoted the reaction effectively to furnish 9a in 87% yield with 5.6:1 diastereoselectivity (entry 7). TfOH exhibited high reactivity as well as satisfactory diastereoselectivity (86%, d.r. = 13.0:1, entry 9), although the reactivity of Tf2NH, also a strong Brønsted acid catalyst, was moderate (72%, d.r. = 5.0:1, entry 8). 2 mol% TfOH sufficed to complete the reaction with moderate diastereoselectivity (89%, d.r. = 6.7:1, entry 10). Excellent diastereoselectivity was realized even with a 2 mol% catalyst when the reaction was conducted in toluene (99%, d.r. = >20:1, entry 11). The structure of the major diastereomer was determined as described for 9a by X-ray crystallographic analysis, and those of others shown in Fig. 4 were surmised by analogy.11,12
 | ||
| Fig. 4 Substrate scope of diastereoselective synthesis of tricyclic pyrans. | ||
With the highly diastereoselective tricyclic pyran synthesis in our hand, the substrate scope of this reaction was examined (Fig. 4).13 Various substituents such as electron-donating groups (Me and OMe) and an electron-withdrawing group (F) were tolerated in this reaction, and the corresponding tricyclic pyrans 9b–f were obtained in excellent chemical yields with excellent diastereoselectivities (>94%, d.r. = >12.5:1). Although the diastereoselectivity was sufficient (d.r. = 11.2:1), the chemical yield was modest (30%) in the case of the naphthyl substrate 9g owing to the detachment of the benzyl group.14 Both allyl ether 8h and ethyl ether 8i were also suitable substrates, and excellent diastereoselectivities were achieved for both substrates (d.r. = >20:1). The construction of three contiguous stereogenic centers was also attainable, that is, the reaction of the substrate 8j with a monomethyl group at the benzylic position afforded the adduct 9j in 93% with 3.1:1 diastereoselectivity.11
To elucidate the reaction mechanism, additional experiments were conducted (Scheme 3). No primary kinetic isotope effect was observed in both hydride shift processes: The KIE (kH/D) values were determined to be 1.63 (for the first hydride shift) and 0.96 (for the second hydride shift). These results imply the reversibility of both hydride shift processes. To check their possible reversibility, the substrates 14 and 15 having the D atom in vinylic positions were subjected to the optimized reaction conditions. No incorporation of the D atom into the position adjacent to the oxygen atom was observed, which completely ruled out the reversibility of the hydride shift steps. Furthermore, the diastereomer ratio did not change even when the adduct 9a with low diastereoselectivity (d.r. = 6.7:1, entry 10 in Table 1) was subjected to slightly modified optimized reaction conditions (2 mol% TfOH, toluene, reflux, 48 h). Importantly, the relative stereochemistries of the two diastereomers of 9a were determined to be cis and trans in the ring junction [JH–H = 6.8 Hz in cis-9a and JH–H = 11.6 Hz in trans-9a (major diastereomer)], which indicates that the diastereoselectivity was determined in the first hydride shift/cyclization process. The above results suggest that the excellent diastereoselectivities were achieved under kinetic control, and the rate-determining step would be the first cyclization step, and not the hydride shift processes as in the case of the sequential hydride shift system we have recently reported.8
 | ||
| Scheme 3 Additional experiments for clarification of the reaction mechanism. | ||
|
|
||||
|---|---|---|---|---|
| Entry | Catalyst | Solvent | Yieldb [%] | d.r.c |
| a Unless otherwise noted, all reactions were conducted with 0.10 mmol of 8a in the presence of an acid catalyst (10 mol%) in ClCH2CH2Cl (1.0 mL) at refluxing temperature. b Isolated yield. c Determined by the comparison of integral values of the C3 (methine) proton of each diastereomer. d MS5Å was added. e 2 mol% catalyst. f The reaction was completed within 1 h. | ||||
| 1 | Yb(OTf)3 | ClCH2CH2Cl | 90 | 3.3:1 |
| 2 | Sc(OTf)3 | ClCH2CH2Cl | 88 | 4.2:1 |
| 3 | Gd(OTf)3 | ClCH2CH2Cl | 22 (68) | 2.5:1 |
| 4 | Dy(OTf)3 | ClCH2CH2Cl | 60 | 4.5:1 |
| 5 | Mg(OTf)2 | ClCH2CH2Cl | 0 (98) | — |
| 6 | TiCl4 | ClCH2CH2Cl | 0 (90) | — |
| 7 | FeCl3 | ClCH2CH2Cl | 87 | 5.6:1 |
| 8 | Tf2NH | ClCH2CH2Cl | 72 | 5.0:1 |
| 9 | TfOH | ClCH2CH2Cl | 86 | 13.0:1 |
| 10d | TfOH | ClCH2CH2Cl | 89 | 6.7:1 |
| 11d,e,f | TfOH | Toluene | 99 | >20:1 |

Conclusions
In summary, we have developed a highly diastereoselective synthesis of fused tricyclic pyrans via double C(sp3)–H bond functionalization triggered by sequential hydride shift processes. The key to achieving this goal was the careful design of the substrate with two methyl groups at the benzylic position, and the desired sequential reaction proceeded smoothly with low catalyst loading of TfOH (only 2 mol%). Various tricyclic pyrans with electron-donating and -withdrawing substituents on the aromatic ring were obtained in excellent chemical yields with excellent diastereoselectivities (up to d.r. = >20:1). Additional experiments suggested that the rate-determining step was not the hydride shift step but the first cyclization step. Further investigation on the hydride shift/cyclization sequence, particularly the catalytic asymmetric version, is underway in our laboratory.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was partially supported by a JSPS KAKENHI Grant No. JP26288053 and grants from the Uehara Memorial foundation, Naito foundation, and Inoue Foundation of Science.Notes and references
- For recent reviews on C–H activation, see: (a) K. Godula and D. Sames, Science, 2006, 312, 67 CrossRef PubMed; (b) R. G. Bergman, Nature, 2007, 446, 391 CrossRef PubMed; (c) D. Alberico, M. E. Scott and M. Lautens, Chem. Rev., 2007, 107, 174 CrossRef PubMed; (d) H. M. L. Davies and J. R. Manning, Nature, 2008, 451, 417 CrossRef PubMed; (e) X. Chen, K. M. Engle, D.-H. Wang and J.-Q. Yu, Angew. Chem., Int. Ed., 2009, 48, 5094 CrossRef PubMed; (f) R. Jazzar, J. Hitce, A. Renaudat, J. Sofack-Kreutzer and O. Baudoin, Chem.–Eur. J., 2010, 16, 2654 CrossRef PubMed; (g) T. W. Lyons and M. S. Sanford, Chem. Rev., 2010, 110, 1147 CrossRef PubMed; (h) H. M. L. Davies, J. Du Bois and J.-Q. Yu, Chem. Soc. Rev., 2011, 40, 1855 RSC; (i) T. Brückl, R. D. Baxter, Y. Ishihara and P. S. Baran, Acc. Chem. Res., 2012, 45, 826 CrossRef PubMed; (j) Y. Qin, J. Lv and S. Luo, Tetrahedron Lett., 2014, 55, 551 CrossRef; (k) H. M. L. Davies and D. Morton, J. Org. Chem., 2016, 81, 343 CrossRef PubMed See also, highlight on visible-light photocatalysis: X.-Q. Hu, J.-R. Chen and W.-J. Xiao, Angew. Chem., Int. Ed., 2017, 56, 1960 CrossRef PubMed.
- For recent reviews on internal redox process, see: (a) M. Tobisu and N. Chatani, Angew. Chem., Int. Ed., 2006, 45, 1683 CrossRef PubMed; (b) S. C. Pan, Beilstein J. Org. Chem., 2012, 8, 1374 CrossRef PubMed; (c) M. Wang, ChemCatChem, 2013, 5, 1291 CrossRef; (d) B. Peng and N. Maulide, Chem.–Eur. J., 2013, 19, 13274 CrossRef PubMed; (e) L. Wang and J. Xiao, Adv. Synth. Catal., 2014, 356, 1137 CrossRef; (f) M. C. Haibach and D. Seidel, Angew. Chem., Int. Ed., 2014, 53, 5010 CrossRef PubMed; (g) S. J. Kwon and D. Y. Kim, Chem. Rec., 2016, 16, 1191 CrossRef PubMed.
- These types of reactions are classified under the term “tert-amino effect.” For reviews, see: (a) O. Meth-Cohn and H. Suschitzky, Adv. Heterocycl. Chem., 1972, 14, 211 CrossRef; (b) W. Verboom and D. N. Reinhoudt, Recl. Trav. Chim. Pays-Bas, 1990, 109, 311 CrossRef; (c) O. Meth-Cohn, Adv. Heterocycl. Chem., 1996, 65, 1 CrossRef; (d) J. M. Quintela, Recent Res. Dev. Org. Chem., 2003, 7, 259 Search PubMed; (e) P. Mátyus, O. Elias, P. Tapolcsányi, A. Polonka-Bálint and B. Halász-Dajka, Synthesis, 2006, 2625 CrossRef.
- For selected examples of internal redox reactions, see: (a) A. Polonka-Bálint, C. Saraceno, K. Ludányi, A. Bényei and P. Mátyus, Synlett, 2008, 2846 Search PubMed; (b) C. Zhang, S. Murarka and D. Seidel, J. Org. Chem., 2009, 74, 419 CrossRef PubMed; (c) J. C. Ruble, A. R. Hurd, T. A. Johnson, D. A. Sherry, M. R. Barbachyn, P. L. Toogood, G. L. Bundy, D. R. Graber and G. M. Kamilar, J. Am. Chem. Soc., 2009, 131, 3991 CrossRef PubMed; (d) M. J. Mahoney, D. T. Moon, J. Hollinger and E. Fillion, Tetrahedron Lett., 2009, 50, 4706 CrossRef; (e) G. Zhou and J. Zhang, Chem. Commun., 2010, 46, 6593 RSC; (f) J. Cao, X. Yang, X. Hua, Y. Deng and G. Lai, Org. Lett., 2011, 13, 478 CrossRef PubMed; (g) I. D. Jurberg, B. Peng, E. Wöstefeld, M. Wasserloos and N. Maulide, Angew. Chem., Int. Ed., 2012, 51, 1950 CrossRef PubMed; (h) T. Sugiishi and H. Nakamura, J. Am. Chem. Soc., 2012, 134, 2504 CrossRef PubMed; (i) Y.-Y. Han, W.-Y. Han, X.-M. Zheng and W.-C. Yuan, Org. Lett., 2012, 14, 4054 CrossRef PubMed; (j) P. A. Vadpla, I. Carrea and D. Sames, J. Org. Chem., 2012, 77, 6689 CrossRef PubMed; (k) X. Gao, V. Gaddam, E. Altenhofer, R. R. Tata, Z. Cai, N. Yongpruksa, A. K. Garimallaprabhakaran and M. Harmata, Angew. Chem., Int. Ed., 2012, 51, 7016 CrossRef PubMed; (l) D.-F. Chen, Z.-Y. Han, Y.-P. He, J. Yu and L.-Z. Gong, Angew. Chem., Int. Ed., 2012, 51, 12307 CrossRef PubMed; (m) S. Shaaban and N. Maulide, Synlett, 2013, 1722 Search PubMed; (n) G. Cera, M. Chiarucci, F. Dosi and M. Bandini, Adv. Synth. Catal., 2013, 335, 2227 CrossRef; (o) A. Dieckmann, M. T. Richers, A. Y. Platonoova, C. Zhang, D. Seidel and N. K. Houk, J. Org. Chem., 2013, 78, 4132 CrossRef PubMed; (p) M. Alajarin, B. Bonillo, M. Marin-Luna, P. Sanchez-Andrada and A. Vidal, Chem.–Eur. J., 2013, 19, 16093 CrossRef PubMed; (q) A. Vidal, M. Martin-Luna and M. Alajarin, Eur. J. Org. Chem., 2014, 878 CrossRef; (r) L. Tóth, Y. Fu, H. Y. Zhang, A. Mándi, K. E. Köver, T.-Z. Iiiyés, A. Kiss-Szikszai, B. Balogh, T. Kurtán, S. Antus and P. Mátyus, Beilstein J. Org. Chem., 2014, 10, 2594 CrossRef PubMed; (s) Y.-Z. Chang, M.-L. Li, W.-F. Zhao, X. Wen, H. Sun and Q.-L. Xu, J. Org. Chem., 2015, 80, 9620 CrossRef PubMed; (t) C. W. Suh, S. J. Kwon and D. Y. Kim, Org. Lett., 2017, 19, 1334 CrossRef PubMed; (u) S.-S. Li, L. Zhou, L. Wang, H. Zhao, L. Yu and J. Xiao, Org. Lett., 2018, 20, 138 CrossRef PubMed.
- For other types of internal redox reactions, see: (a) N. K. Pahadi, M. Paley, R. Jana, S. R. Waetzig and J. A. Tunge, J. Am. Chem. Soc., 2009, 131, 16626 CrossRef PubMed; (b) C. Zhang and D. Seidel, J. Am. Chem. Soc., 2010, 132, 1798 CrossRef PubMed; (c) I. Deb and D. Seidel, Tetrahedron Lett., 2010, 51, 2945 CrossRef; (d) C. Zhang, D. Das and D. Seidel, Chem. Sci., 2011, 2, 233 RSC; (e) H. Mao, R. Xu, J. Wan, Z. Jiang, C. Sun and Y. Pan, Chem.–Eur. J., 2010, 16, 13352 CrossRef PubMed; (f) I. Deb, D. Das and D. Seidel, Org. Lett., 2011, 13, 812 CrossRef PubMed; (g) H. Mao, S. Wang, P. Yu, H. Lv, R. Xu and Y. Pan, J. Org. Chem., 2011, 76, 1167 CrossRef PubMed; (h) D. Das, M. T. Richers, L. Ma and D. Seidel, Org. Lett., 2011, 13, 6584 CrossRef PubMed; (i) L. Ma, W. Chen and D. Seidel, J. Am. Chem. Soc., 2012, 134, 15305 CrossRef PubMed; (j) D. Das, A. X. Sun and D. Seidel, Angew. Chem., Int. Ed., 2013, 52, 3765 CrossRef PubMed; (k) D. Das and D. Seidel, Org. Lett., 2013, 15, 4358 CrossRef PubMed; (l) Q.-H. Zheng, W. Meng, G.-J. Jiang and Z.-X. Yu, Org. Lett., 2013, 15, 5298 CrossRef PubMed; (m) W. Chen, Y. K. Kang, R. G. Wilde and D. Seidel, Angew. Chem., Int. Ed., 2014, 53, 5179 Search PubMed; (n) M. T. Richers, M. Breugst, A. Yu, A. Ullrich, A. Dieckmann, K. N. Houk and D. Seidel, J. Am. Chem. Soc., 2014, 136, 6123 CrossRef PubMed; (o) Y. Kang, W. Chen, M. Breugst and D. Seidel, J. Org. Chem., 2015, 80, 9628 CrossRef PubMed; (p) L. Ma and D. Seidel, Chem.–Eur. J., 2015, 21, 12908 CrossRef PubMed.
- For examples of enantioselective internal redox reactions, see: (a) S. Murarka, I. Deb, C. Zhang and D. Seidel, J. Am. Chem. Soc., 2009, 131, 13226 CrossRef PubMed; (b) Y. K. Kang, S. M. Kim and D. Y. Kim, J. Am. Chem. Soc., 2010, 132, 11847 CrossRef PubMed; (c) W. Cao, X. Liu, W. Wang, L. Lin and X. Feng, Org. Lett., 2011, 13, 600 CrossRef PubMed; (d) G. Zhou, F. Liu and J. Zhang, Chem.–Eur. J., 2011, 17, 3101 CrossRef PubMed; (e) Y.-P. He, Y.-L. Du, S.-W. Luo and L. Z. Gong, Tetrahedron Lett., 2011, 52, 7064 CrossRef; (f) L. Chen, L. Zhang, Z. Lv, J.-P. Cheng and S. Luo, Chem.–Eur. J., 2012, 18, 8891 CrossRef PubMed; (g) L. Zhang, L. Chen, Z. Lv, J.-P. Cheng and S. Luo, Chem.–Asian J., 2012, 7, 2569 CrossRef PubMed; (h) Z.-W. Jiao, S.-Y. Zhang, C. He, Y.-Q. Tu, S.-H. Wang, F.-M. Zhang, Y.-Q. Zhang and H. Li, Angew. Chem., Int. Ed., 2012, 51, 8811 CrossRef PubMed; (i) Y. K. Kang and D. Y. Kim, Adv. Synth. Catal., 2013, 355, 3131 CrossRef; (j) C. W. Suh, S. B. Woo and D. Y. Kim, Asian J. Org. Chem., 2014, 3, 399 CrossRef; (k) Y. K. Kang and D. Y. Kim, Chem. Commun., 2014, 50, 222 RSC; (l) C. W. Suh and D. Y. Kim, Org. Lett., 2014, 16, 5374 CrossRef PubMed; (m) J. Yu, N. Li, D.-F. Chen and S.-W. Luo, Tetrahedron Lett., 2014, 55, 2859 CrossRef; (n) W. Cao, X. Liu, J. Guo, L. Lin and X. Feng, Chem.–Eur. J., 2015, 21, 1632 CrossRef PubMed . See also, ref. 7h.
- For the internal redox reaction developed by our group, see: (a) K. Mori, Y. Ohshima, K. Ehara and T. Akiyama, Chem. Lett., 2009, 38, 524 CrossRef; (b) K. Mori, T. Kawasaki, S. Sueoka and T. Akiyama, Org. Lett., 2010, 12, 1732 CrossRef PubMed; (c) K. Mori, S. Sueoka and T. Akiyama, J. Am. Chem. Soc., 2011, 133, 2424 CrossRef PubMed; (d) K. Mori, S. Sueoka and T. Akiyama, Chem. Lett., 2011, 40, 1386 CrossRef; (e) K. Mori, T. Kawasaki and T. Akiyama, Org. Lett., 2012, 14, 1436 CrossRef PubMed; (f) K. Mori, K. Kurihara and T. Akiyama, Chem. Commun., 2014, 50, 3729 RSC; (g) K. Mori, N. Umehara and T. Akiyama, Adv. Synth. Catal., 2015, 357, 901 CrossRef; (h) T. Yoshida and K. Mori, Chem. Commun., 2017, 53, 4319 RSC; (i) M. Machida and K. Mori, Chem. Lett., 2018, 47, 868 CrossRef; (j) K. Yokoo and K. Mori, Chem. Commun., 2018, 54, 6927 RSC. For an asymmetric version of internal redox reaction catalyzed by chiral phosphoric acid, see: (k) K. Mori, K. Ehara, K. Kurihara and T. Akiyama, J. Am. Chem. Soc., 2011, 133, 6166 CrossRef PubMed.
- K. Mori, K. Kurihara, S. Yabe, M. Yamanaka and T. Akiyama, J. Am. Chem. Soc., 2014, 136, 3744 CrossRef PubMed . See, also: L. Wang, J. Xiao, Org. Chem. Front. 2016, 3, 635 Search PubMed.
- Asymmetric version of double C(sp3)–H bond functionalization: K. Mori, R. Isogai, Y. Kamei, M. Yamanaka and T. Akiyama, J. Am. Chem. Soc., 2018, 140, 6203 CrossRef PubMed.
- Internal redox reaction of oxygen analogue, see: (a) S. J. Pastine, K. M. McQuaid and D. Sames, J. Am. Chem. Soc., 2005, 127, 12180 CrossRef PubMed; (b) S. J. Pastine and D. Sames, Org. Lett., 2005, 7, 5429 CrossRef PubMed; (c) K. M. McQuaid and D. Sames, J. Am. Chem. Soc., 2009, 131, 402 CrossRef PubMed; (d) D. Shinkai, H. Murase, T. Hata and H. Urabe, J. Am. Chem. Soc., 2009, 131, 3166 CrossRef PubMed; (e) P. A. Vadola and D. Sames, J. Am. Chem. Soc., 2009, 131, 16525 CrossRef PubMed . See also, ref. 6h and 7b.
- The structures of 9a, 9c, and 9j were unambiguously established by single-crystal X-ray analysis. CCDC-1838261, 1838259, and 1838262 contains the supplementary crystallographic data of 9a, 9c, and 9j.
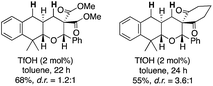
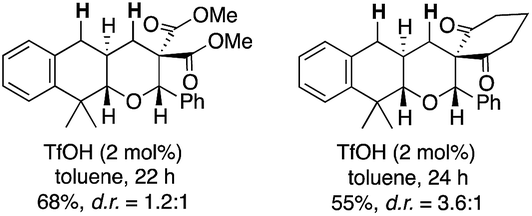
- The employment of barbituric acid moiety as an electrophilic portion was critical for the excellent results. The substrates with dimethyl malonate and 1,3-cyclohexanedione underwent the desired reactions smoothly to afford the tricyclic compounds, but their diastereomer ratio remained low to moderate (less than d.r. = <3.6:1)
 .
. - All of cinnamylidene barbiturates 8 were used as a mixture of E/Z isomer (E/Z = >4.3/1).
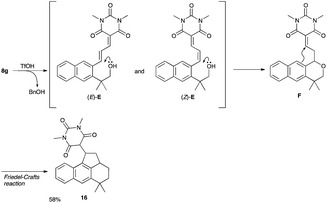
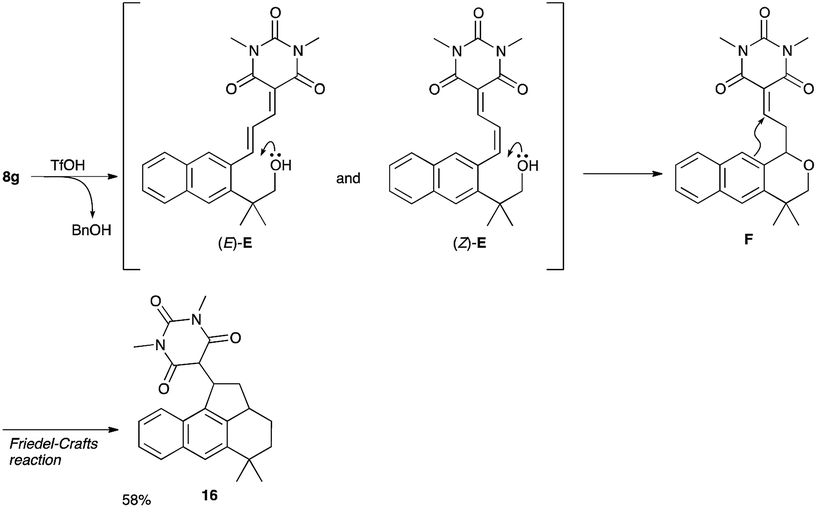
- The low chemical yield of 9g was ascribed to the formation of 16 (58%), which was produced via cleavage of benzyl group, intramolecular Michael addition of oxygen atom followed by intramolecular Friedel–Crafts reaction
 .
.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1838259, 1838261 and 1838262. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8sc02103a |
| This journal is © The Royal Society of Chemistry 2018 |