
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCarbene derived diradicaloids – building blocks for singlet fission?†
Julian
Messelberger
a,
Annette
Grünwald
a,
Piermaria
Pinter
b,
Max M.
Hansmann
 c and
Dominik
Munz
*a
c and
Dominik
Munz
*a
aFriedrich-Alexander Universität Erlangen-Nürnberg, Anorganische und Allgemeine Chemie, Egerlandstr. 1, 91058 Erlangen, Germany. E-mail: dominik.munz@fau.de
bTechnische Universität Dresden, Physikalische Organische Chemie, Bergstr. 66, 01069 Dresden, Germany
cGeorg-August Universität Göttingen, Institut für Organische und Biomolekulare Chemie, Tammannstraße 2, 37073 Göttingen, Germany
First published on 2nd July 2018
Abstract
Organic singlet diradicaloids promise application in non-linear optics, electronic devices and singlet fission. The stabilization of carbon allotropes/cumulenes (C1, C2, C4) by carbenes has been equally an area of high activity. Combining these fields, we showed recently that carbene scaffolds allow as well for the design of diradicaloids. Herein, we report a comprehensive computational investigation (CASSCF/NEVPT2; fractional occupation DFT) on the electronic properties of carbene–bridge–carbene type diradicaloids. We delineate how to adjust the properties of these ensembles through the choice of carbene and bridge and show that already a short C2 bridge results in remarkable diradicaloid character. The choice of the carbene separately tunes the energies of the S1 and T1 excited states, whereas the bridge adjusts the overall energy level of the excited states. Accordingly, we develop guidelines on how to tailor the electronic properties of these molecules. Of particular note, fractional occupation DFT is an excellent tool to predict singlet–triplet gaps.
Introduction
The synthesis and spectroscopic scrutiny of organic diradicals is a vibrant area of research.1–5 These molecules are suitable for organic field-effect transistors (OFETs),6,7 non-linear optics,8 two-photon absorption (TPA),8 energy storage9 and organic spintronics10 due to their unique physico-chemical properties. Of particular importance, organic diradicals allow for the construction of solar cells based on singlet fission.11–14 Singlet fission, i.e. the conversion of one excited singlet state to two triplet states, promises a breakthrough for a new generation of photovoltaics. While current materials are typically limited to a maximum of 33% (Shockley–Queisser limit),15 singlet fission permits in principle quantum efficiencies of 200%.16,17 One of the major limitations for singlet fission based light harvesting is the limited number and structural similarity of chromophores currently known to undergo this process.It has consequently been emphasized that it is “essential that additional classes of efficient singlet fission chromophores be discovered”.18 One very well-known example for a Kekulé diradicaloid is Tschitschibabin's hydrocarbon (Fig. 1).19 This molecule can be understood by two resonance structures, which correspond to the cumulenic closed-shell singlet state and the diradical open-shell state, which could be either an open-shell singlet or a triplet (Fig. 1).20,21 The relative weights of these closed-shell and open-shell resonance structures are associated with the diradicaloid character of the molecule. Spectroscopic investigations of organic diradicals are unfortunately challenging due to their typically high reactivity. Therefore, much work has been devoted on taming these reactive compounds through the extension of the π-system,22,23 introduction of stabilizing heteroatoms,24–28 and/or the kinetic stabilization by steric bulk. We introduced carbenes as suitable building blocks for the isolation of carbene–bridge–carbene ensembles with very high diradicaloid character (Fig. 2).29 The synthetic approach followed a straightforward and highly modular route, which allows for the combination of any stable free carbene with a large variety of different connecting bridges. Our synthetic efforts were inspired and guided by the fact that cyclic (alkyl)(amino) carbenes (CAACs)30 stabilize organic radicals very well,31–35 whereas N-heterocyclic carbene (NHC)36 derived radicals37,38 appear to be more reactive.
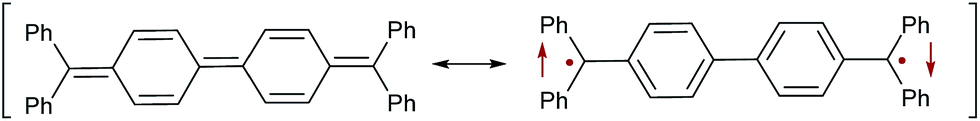 | ||
| Fig. 1 Closed-shell singlet (left) as well as open-shell singlet (right) resonance structures of Tschitschibabin's diradicaloid. | ||
 | ||
| Fig. 2 Modular synthesis of carbene derived diradicaloids and cumulenes. | ||
Further examples of cumulenes connected by two NHCs with saturated39,40 and unsaturated41 backbones were very recently reported. Equally, heterocyclic carbenes can stabilize cumulenes and carbon allotropes,42,43 which show unique optoelectronic properties.44–48 We conclude therefore that carbenes are very likely to find many more applications in these areas. Key for the design of singlet or triplet diradicaloids is their electronic structure, which is most prominently reflected by their degree of diradicaloid character (i.e., population of the lowest unoccupied molecular orbital LUMO).49 Computational approaches offer here a convenient method to design such molecules.50–55
Herein, we report a detailed and comprehensive investigation using high-level CASSCF/NEVPT2 calculations on the (di)radical properties of carbene functionalized extended π-systems. We elucidate the influence of the carbene end groups and put them into perspective to other well-known diradicaloids and polyaromatic hydrocarbons (PAHs). We will quantify the effects of the cumulenic bridge and of the aromaticity. In particular, we describe which carbene leads to high diradical character. Eventually, we derive guidelines for tailoring the electronic properties of these diradicaloids for singlet fission in solar cells or two-photon absorption.
Results and discussion
Seven well-studied singlet diradicaloids were chosen as references (Fig. 3). Tschitschibabin's diradicaloid hydrocarbon (1) is related to Thiele's quinoid hydrocarbon (2), which was reported to be considerably more stable.56 Tetracene (3) and pentacence (4), which have been extensively studied in the context of singlet fission, were picked as representative for fused π-systems.57 Other prominent examples of this class include e.g. zethrenes,58–60 phenalenyles,61,62 quinodimethanes,63–67 or diindeno fused π-systems.68,69 The bisnitroxide (5),70–74 bisoxoverdazyl (6),75 and bisquinone (7)76–79 were chosen as examples for heteroatom incorporation and supposedly very high diradical character.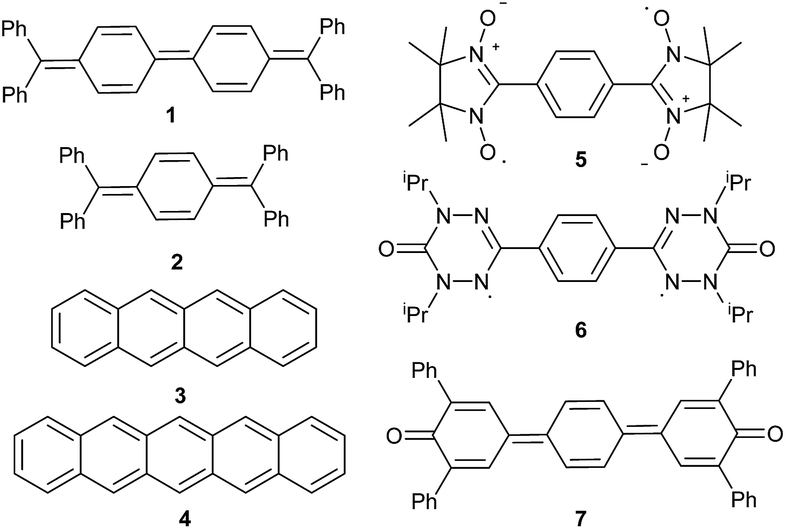 | ||
| Fig. 3 Isolated and herein studied molecular scaffolds with singlet diradicaloid character. | ||
The electronic and steric properties of singlet carbenes can be tuned in a straightforward fashion by adjacent π-donors, ring size, as well as aromaticity.80–82 Cyclic (alkyl)(amino) carbenes (CAACs) with one nitrogen heteroatom adjacent to the carbene (Fig. 4; 8, 10–14) allow for comparably efficient electron delocalization from the π-system of a bridge through their π-acceptor capabilities.
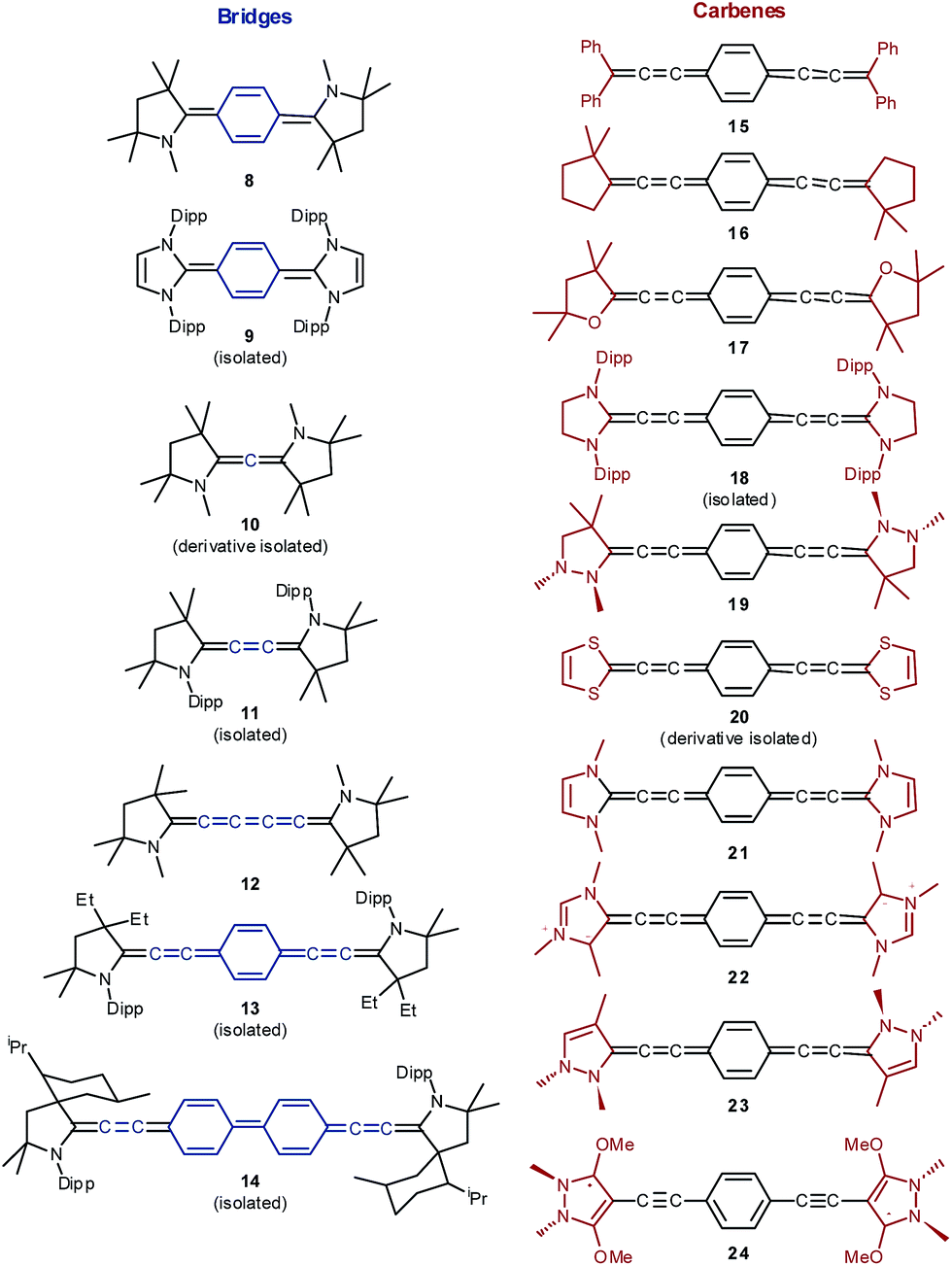 | ||
| Fig. 4 Isolated and proposed carbene stabilized Kekulé diradicaloids studied herein. Structures 19, 23 and 24 show pyramidalized amino groups. | ||
These derivatives were chosen in order to study the influence of the conjugated bridge, because most (10,8311,42,4313,2914![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 29) are isolable. Additionally, we included the very recently synthesized NHC congener 9.41 Olefin substituents as modeled in 15 and 16 (i.e., alkylidenes or dialkylcarbenes) and cyclic vinyl ether substituents (i.e., Fischer carbenes) 17 should lead to even stronger electron delocalization. On the contrary, bisheteroatom substitution should give rise to electron richer derivatives (18, 19). The compounds 20–23 feature moderate to comparably strong aromatic character of the respective free carbenes. The attachment of mesoionic carbenes (22) and carbenes with significant carbodicarbene character (respectively bent allenes, 24)84–88 with significant population of the carbene's π-orbital gives access to very electron rich ensembles. Note that the bentallene 24 shows only very weak aromatic character as evidenced by the experimentally observed pyramidalization of the nitrogen atoms.89
29) are isolable. Additionally, we included the very recently synthesized NHC congener 9.41 Olefin substituents as modeled in 15 and 16 (i.e., alkylidenes or dialkylcarbenes) and cyclic vinyl ether substituents (i.e., Fischer carbenes) 17 should lead to even stronger electron delocalization. On the contrary, bisheteroatom substitution should give rise to electron richer derivatives (18, 19). The compounds 20–23 feature moderate to comparably strong aromatic character of the respective free carbenes. The attachment of mesoionic carbenes (22) and carbenes with significant carbodicarbene character (respectively bent allenes, 24)84–88 with significant population of the carbene's π-orbital gives access to very electron rich ensembles. Note that the bentallene 24 shows only very weak aromatic character as evidenced by the experimentally observed pyramidalization of the nitrogen atoms.89
Computational description of singlet diradicaloids90
The computational modeling of compounds with singlet-diradicaloid character is not a straightforward task. Although unrestricted DFT is in principle capable of describing such systems using the broken-symmetry formalism,91–94 the multi-reference singlet state is not well modeled for small singlet–triplet gaps. Spin decontamination procedures have been proposed to address the shortcomings – however, they also do not always lead to results in agreement with the experiment.95–98 Indeed, we observed for the bisCAAC cumulenes that the description using DFT strongly underestimated the stability of the closed-shell singlet state and hence was not an appropriate method to predict the absorption spectra.29 Multireference methods like the complete active space – self consistent field method (CASSCF)99 are suitable for properly treating such systems.100–105 Unfortunately, CASSCF is not a “black-box” procedure like DFT and the selection, i.e. quality, of the active orbitals typically determines the outcome of the calculation. The application of computational investigations using this and related methods remain therefore even for organic singlet diradicaloids comparably scarce,50–52,55,103,106–117 whereas unrestricted DFT calculations are still much more commonly applied.118–127A common descriptor of diradicaloid character are the diradical indices y0 and y1. They correspond to the natural orbital occupation number (NOON) of the lowest unoccupied natural orbital (LUNO) and LUNO+1, respectively, and are of course connected with the overall bond order.128 A population of y0 = 1 and y1 = 0 signifies a diradical, whereas y0 = 1 and y1 = 1 describes a tetraradical. Non-linear optical properties like two-photon absorption are related with the second-order hyperpolarizability.129–131 This property has been theoretically as well as experimentally shown to be enhanced for systems with a moderate singlet diradical contribution of about 36% (i.e., y0 = 0.36). It has also been suggested that molecules with a diradicaloid character above y0 = 0.1 are in principle candidates for singlet fission as long as y1 is considerably smaller than y0.132–134 Molecules with comparably low to intermediate y0 values are also here expected to show higher energy efficiency.135 The values of y0 and y1 are of course good indicators for the excitation energies related with these orbitals. They are therefore directly connected with the energy matching conditions for singlet fission processes.127 A related approach, which relies on the relative weight of the closed-shell (“20”) and double-excited configurations (“02”) in a “two electrons in two orbitals” CI calculation has been proposed by Neese.92,136
Singlet fission is believed to be only feasible if the energy of the first excited singlet state E(S1) exceeds twice the energy of the triplet state E(T1) (eqn (1)).
| 2E(T1) ≈ E(S1) and/or 2E(T1) < E(S1) | (1) |
For practical applications, the energy of the S1 and twice the T1 states should be in the same order of magnitude and close or moderately higher than 2.0 eV. Furthermore, twice the energy of the excited triplet state should be smaller than the energy of the second excited triplet state in order to avoid recombination processes of triplet excitons (eqn (2)).
| 2E(T1) < E(T2) | (2) |
However, note that these two requirements are obviously not necessarily sufficient for the observation of singlet fission. Additionally, intermolecular interactions for this bimolecular process (electronic coupling) as well as molecular vibrations, which are associated with relaxation processes (vibronic coupling) are important.54 Evidently, the stability of the excited states and the extinction coefficient are as well significant for light harvesting purposes. Another (computational) interpretive tool for the estimation of diradicaloid character, which relies on the fractional occupation number weighted electron density (NFOD), was recently introduced by Grimme.137,138 Of particular interest, this method comes at an extremely low computational cost. It is based on smearing the molecule's electrons over the molecular orbitals using finite temperature DFT and is a measure of static electron correlation. Molecules with a delocalized FOD and a large NFOD have multireference character.
Computational methods
All complete active-space self-consistent-field (CASSCF)99 calculations were performed with ORCA 4.0.1 (ref. 139) using the def2-TZVPP140 basis set. The resolution of identity approximation and the related basis sets for both Coulomb and HF exchange integrals were used (RI-JK).141 Tighter than default convergence criteria were chosen (tightscf). The second order perturbation theory NEVPT2 was applied to account for the effects of dynamic electron correlation.142 The reported diradical indices as well as molecular orbital plots relate to the singlet ground states for all molecules. Five roots each were calculated for the state averaged modeling of the absorption spectra for the singlet states and triplet states. For the state-averaged CASSCF(14,14) calculations, three roots were calculated for the triplet- and four roots for the singlet states. Calculations using different number of roots (e.g., 5 or 10) showed only very small deviation for the transition to the S1 state and reordering of the states after the NEVPT2 correction is unproblematic when calculating at least 5 roots. Reported energies relate accordingly to vertical excitation from the S0 states. Note that the geometry optimization of the excited states at the CAS level of theory is computationally extremely demanding. The structural parameters (“reorganization energy”) of the molecules change moderately upon excitation. Geometry optimizations of 13 in the triplet state, closed-shell singlet state as well as broken-symmetry open-shell singlet state (B3LYP/def2-SVP) afforded similar structural parameters. E.g., the distortion of the two acetylene units differs between the calculations by only about 10°. See the ESI† for CASSCF energies of the DFT optimized open-shell singlet, closed-shell singlet as well as triplet state of 13. Nevertheless, the calculated adiabatic singlet–triplet gap is 0.73 eV, while the vertical excitations 1S → 3T for the solid state structure and the B3LYP closed-shell optimized structure are 0.80 eV and 0.98 eV, respectively. The choice of the active space for each molecule is delineated in detail in the ESI.† For the effect of enlarging the active space from 2 electrons in 2 orbitals, see as well the ESI.† An evaluation of basis set effects (def2-SVP, def2-TZVP, def2-TZVPP, def2-TZVPD, ma-def2-TZVPP, def2-QZVPP, cc-pVTZ, aug-cc-pVTZ) indicates smooth convergence toward the complete basis limit and that def2-TZVPP is of sufficient quality for predicting the energies of the S1 and T1 states with a deviation of 0.04 eV from the largest basis set (ESI†). Typically, all the π-orbitals of the conjugated system were included up to 14 electrons in 14 orbitals [CASSCF(14,14)]. The NEVPT2 calculation for pentacene (4) evolved to be very time consuming for CASSCF(14,14) – therefore, the given energies relate to CASSCF(12,12). The predicted absorption wavelengths (energies of S1 states, respectively) show a root mean square deviation RMSD from the experimentally determined values of 0.19 eV (mean deviation: 0.15 eV), which we consider an excellent fit (ESI†). Time dependent DFT using the B3LYP/def2-TZVPP level of theory underestimates the energy level of the S1 state of e.g.14 even without truncation by more than 0.5 eV. The deviation for the T1 states, which is mainly due to the constrained geometric parameters (vide supra), appears to be larger and the calculations seem to systematically overestimate the singlet–triplet gap by up to 0.3 eV. However, note that experimental energies have only been reported for tetracene and pentacene, whereas the expected error for molecules with smaller π-system like 13 is expected to be smaller (the RMSD value for these two T1 states and the calculated adiabatic singlet–triplet gap of 13 amount to 0.28 eV). The SMD implicit solvation model was applied for benchmark studies with experimental reported absorption spectra and led to moderately improved results (RMSD for S1 states: 0.18 eV).143,144 Increasing solvent polarity leads for 13 to a moderately increased level of the T1 state, whereas the effects on the S1 state appear to not follow a straightforward trend (ESI†). The absorption wavelengths reported in the manuscript are not corrected for solvation effects for comparability.The structural parameters of 1–7, 11, 13, 14, 18 were obtained by optimization of the hydrogen atom positions from their solid state structures (B3LYP/def2-SVP). The D3 dispersion correction145 with Becke–Johnson damping146 was applied. All diisopropylphenyl (Dipp) groups and the menthyl substituent of structure 14 were truncated by methyl substituents. The iso-propyl groups of 6 and the phenyl substituents of structure 7 were modeled by methyl groups. The structures of all the other molecules were optimized in the singlet state without symmetry or internal coordinate constrains and were verified as true minima by the absence of negative eigenvalues in the harmonic vibrational frequency analysis. The restricted formalism, which showed very good agreement with the solid state structures for the CAAC derived molecules,29 was used. The fractional occupation number weighted electron density (FOD) analysis was carried out with the default values as implemented in ORCA (TPSS/def2-TZVP; 5000 K). Calculated structures and molecular orbitals were visualized with Chemcraft, Avogadro 1.1.1147 and IBOView.148
Comparison carbene vs. non-carbene derived diradicaloids
The analysis of the electronic structure of 13 suggests strong cumulenic character and reveals that the localization of the two radicals as shown in Fig. 1 is an oversimplification (Fig. 5).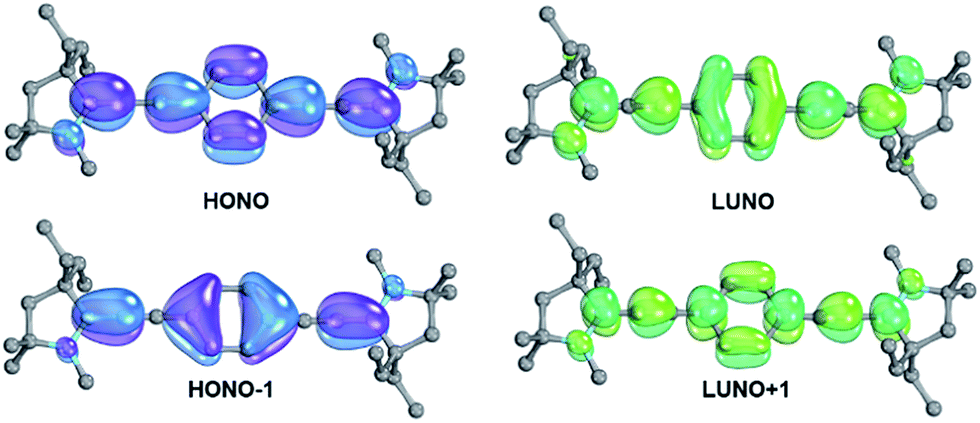 | ||
| Fig. 5 Frontier orbitals of 13 as obtained from CASSCF(12,12) calculation. | ||
The diradicaloid character is mainly associated with the two frontier orbitals, which extend over the whole π-system and show an occupation of 1.71 (highest occupied natural orbital, HONO) and 0.29 (lowest unoccupied natural orbital, LUNO), respectively. Note that the CAAC moieties show a strong contribution of their pz orbitals as well as of the adjacent amine groups. The occupancies of the HONO−1 (1.89) as well as the LUNO+1 (0.12) point at moderate tetraradical contributions.
In sight of an ideal diradical index of y0 = 0.36 for a large two-photon absorption cross section (vide supra), we conclude that 13 should be a very good candidate for two-photon absorption.8,131 The vertical excitation to the S1 state can be approximated with a double excitation (29%) from the HONO to the LUNO and smaller single excitations from the HONO and HONO−1 to the LUNO (18%) and LUNO+1 (16%), respectively. This transition is expected to have very low intensity (fosc ≈ 0.0). The excitation to the S2 state, which is only slightly higher in energy, involves mainly excitation of one electron to the LUNO (44%) with strong intensity (fosc ≈ 0.9). Hence, 13 qualifies as a class III chromophore (vide infra).11
The energy level of the S1 state shows an energy gap of 2.49 eV in relation to the S0 state, which is reasonable for applications associated with singlet fission. The vertical energy gap to the triplet state T1 is a bit too low (0.80), whereas the level of the T2 state is sufficiently high in energy (2.90 eV). To put the carbene derived singlet diradicaloid into perspective with well-studied congeners, we compare the CAAC derived diradicaloid 13 with Tschitschibabin's (1) as well as Thiele's (2) hydrocarbons and tetracene (3) as well as pentacene (4). Table 1 shows the most significant parameters for an evaluation of the electronic character of these molecules. The calculated absorption bands agree very well with the experimental values (1: expλ = 576 nm;1492: “orange”;563: expλ = 475 nm,1504: expλ = 578 nm,11,15113: expλ = 551 nm29). The calculated energies of the excited T1 states appear to be a bit too high in comparison to the experimental values (e.g., 3: expE(S1): 2.61 eV; expE(T1): 1.34 eV; 4: expE(S1): 2.13 eV; expE(T1): 0.95 eV),11,151,152 which is mainly due to conformational changes associated with the transition to the T1 states (ESI†). As evidenced by y0 and NFOD, the CAAC derived molecule shows significant diradicaloid character, which lies between Thiele's and Tschitschibabin's hydrocarbons and exceeds the one of tetracene. Note that the NFOD values as well as plots (ESI†) indicate significant multireference character for all molecules. Experimentally, tetracene is well known to undergo singlet fission, although the process is slightly endergonic, i.e. 2E(T1) > E(S1) (“thermally activated singlet fission”).11 Tetracene (note fosc = 0.1) shows typically a very slow singlet-fission rate, which underlines the need for the development of novel materials.153,154 Oxygen sensitive pentacene on the contrary shows experimentally slightly exoergic singlet fission.
| a The biradical indices relate to CASSCF(14,14), whereas the energies were only calculated with CASSCF(12,12) due to prohibitive computational demand for the NEVPT2 correction. | |||||
|---|---|---|---|---|---|
| Compound | 1 | 2 | 3 | 4 | 13 |
| y 0 | 0.47 | 0.19 | 0.18 | 0.19a | 0.29 |
| y 1 | 0.12 | 0.08 | 0.08 | 0.09a | 0.12 |
| N FOD | 1.36 | 0.60 | 0.52 | 0.89 | 0.85 |
| E(S1) in [eV] | 2.13 | 2.41 | 2.46 | 2.07 | 2.49 |
| E(T1) in [eV] | 0.41 | 1.59 | 1.65 | 1.23 | 0.80 |
| E(T2) in [eV] | 2.79 | 3.90 | 2.90 | 2.50 | 2.90 |
| Absorption S0 → S1 (S2) in [nm] | 581 (339 S2) | 514 | 503 | 599 | 498 (444 S2) |
| f osc S0 → S1 (S2) in [nm] | 0.0 (0.01) | 0.6 | 0.1 | 0.13 | 0.0 (0.9) |
Comparison of carbene derived diradicaloids
The CAAC (8) and NHC (9) derivatives of Thiele's hydrocarbon 2 (Table 2) are predicted to show a quite similar diradicaloid character (2: y0 = 0.19; 8: y0 = 0.16; 9: y0 = 0.13). The energy levels of the excited T1 states are moderately elevated [2: E(T1) = 1.59 eV; 8: E(T1) = 1.70 eV; 9: E(T1) = 1.78 eV]. Interestingly, the diradical character of 2, 8 and 9 is much smaller than the one of the nitroxide 5 or oxoverdazyl 6. Both these compounds are almost perfect diradicals with energetically degenerate open-shell singlet and triplet ground states. The unpaired electrons are essentially localized on the heteroatoms with only very small contribution of the phenylene linkers (ESI†). The quinone congener 7 is predicted to show a diradical index, which is in between (y0 = 0.32).| Compound | 5 | 6 | 7 | 8 | 9 |
| Substituent | Nitroxide | Oxo-verdazyl | Quinone | CAAC | NHC |
| y 0 | 1.0 | 1.0 | 0.32 | 0.16 | 0.13 |
| y 1 | 0.16 | 0.14 | 0.09 | 0.09 | 0.09 |
| N FOD | 2.14 | 2.10 | 1.34 | 0.47 | 0.69 |
| E(S1) in [eV] | 2.30 | 3.09 | 2.19 | 2.74 | 2.46 |
| E(T1) in [eV] | 0.0 | 0.0 | 0.75 | 1.70 | 1.78 |
| E(T2) in [eV] | 2.2 | 2.97 | 2.90 | 3.51 | 2.79 |
| Absorption S0 → S1 (S2) in [nm] | 538 | 400 | 566 | 461 | 503 |
| f osc S0 → S1 | 0.02 | 0.2 | 1.3 | 0.6 | 0.9 |
Table 3 collects all data for the carbene analogues of 13. Olefin substitution by a “diphenylalkylidene” substituent (15) leads to quite low lying S1 and T1 states [E(S1) = 1.59 eV; E(T1) = 0.54 eV] and a high diradical index y0 of 0.41 (Table 3). It is interesting to note that the “dialkylalkylidene” substituted diradicaloid 16 shows comparably elevated S1 and T1 states and reduced diradical character [E(S1) = 2.56 eV, E(T1) = 0.86 eV, y0 = 0.30]. The very π-electron deficient “dialkylmethylidene” group stabilizes accordingly the closed-shell cumulene. The high diradical character of 15 in comparison to 16 is – as also evidenced by electron delocalization of the relevant orbitals onto the phenyl rings – due to the radical-stabilizing influence of the aromatic phenyl substituents.
| Compound | 15 | 16 | 17 | 13 | 18 | 19 | 20 | 21 | 22 | 23 | 24 |
| Substituent | Diphenyl-carbene | Cyclopentylidene | Cyclic Fischer carbene | CAAC | saNHC | Pyrazolidin-ylidene | TTF | NHC | MIC | Pyrazolin-ylidene | Bent allene |
| y 0 | 0.41 | 0.30 | 0.29 | 0.29 | 0.26 | 0.30 | 0.38 | 0.28 | 0.27 | 0.39 | 1 |
| y 1 | 0.11 | 0.13 | 0.12 | 0.12 | 0.11 | 0.13 | 0.11 | 0.11 | 0.14 | 0.12 | 0.11 |
| N FOD | 1.20 | 0.73 | 0.76 | 0.85 | 0.83 | 0.91 | 1.14 | 1.28 | 1.58 | 1.16 | 2.41 |
| E(S1) in [eV] | 1.59 | 2.56 | 2.55 | 2.49 | 2.35 | 2.46 | 2.07 | 1.67 | 0.96 | 1.89 | 2.37 |
| E(T1) in [eV] | 0.54 | 0.86 | 0.87 | 0.80 | 0.91 | 0.80 | 0.62 | 0.75 | 0.72 | 0.61 | 0 |
| E(T2) in [eV] | 2.64 | 2.90 | 2.90 | 2.90 | 2.97 | 2.83 | 2.55 | 2.19 | 0.84 | 2.31 | 2.30 |
| Absorption S0 → S1 (S2, S3) in [nm] | 680 | 483 (276, S3) | 484 (288, S3) | 498 (444, S2) | 527 | 504 (433, S2) | 598 | 742 | 1297 | 625 (422, S2) | 522 |
| f osc S0 → S1 (S2, S3) | 1.0 | 0.0 (0.1, S3) | 0.0 (0.2, S3) | 0.0 (0.9, S2) | 1.1 | 0.0 (1.1, S2) | 1.1 | 1.2 | 0.2 | 0.0 (0.4, S2) | 0.01 |
Dialkylcarbenes in their (excited) singlet state are of course more π-acidic than a Fischer carbene or CAAC with one π-electron donating oxo substituent or amine group, respectively. Decreasing the π-acceptor properties through introduction of an oxygen atom (17) reduces the diradical character as well as energy of the S1 state very slightly [y0 = 0.29, E(S1) = 2.55 eV]. This trend holds when going to the amino derivative 13 [y0 = 0.29, E(S1) = 2.49 eV],80,155,156 and the bisamino derivative 18 [y0 = 0.26, E(S1) = 2.35 eV]. The pyrazolidinylidene 19 [E(S1) = 2.46 eV] shows similar properties. The experimental UV-Vis spectra reported for 13 and 18 agree well with the computational predictions with bands around 551 nm (13) and 595 nm (18). Both molecules appear to be electronically quite similar. We suggest therefore that the capability of saturated NHCs to stabilize organic radicals appears to be much stronger than commonly believed in the literature. Of particular interest is the comparison with the compounds 20–23, where the free carbenes are considerably aromatic. These molecules show much lower energies for their S1 states, which do not feature double excitation from the HONO. Especially the mesoionic carbene derivative 22 was predicted to be very reactive [E(S1) = 0.96 eV; E(T1) = 0.72 eV]. It does therefore not come as a surprise that it was recently experimentally found that saturated NHCs stabilize radicals much better than aromatic NHCs.38 Likewise, 21 is predicted to be much more reactive [E(S1) = 1.64 eV] than 18. The vertical singlet–triplet gap is also calculated to be comparably small [E(T1) = 0.75 eV]. Eventually, we evaluated the electronic properties of 24, which features two cyclic bent-allene molecules.86,157–160 Bent allenes can be described as aliphatic carbene derivatives with a filled pz orbital, i.e. they behave as π-donors.
Strikingly, perfect diradical character (y0 = 1) and a very high NFOD of 2.41 was calculated. The S1 energy level of 24 [E(S1) = 2.37 eV] is in the same order of magnitude as found for the other non-aromatic carbene derivatives 13–19.
Fig. 6 illustrates the effect of all carbene substituents on the levels of the S1 and T1. Our calculations suggest accordingly that the population of the pz orbital of the free aliphatic carbenes allows mainly for a tuning of the energy level of the T1 state. On the contrary, aromaticity of the carbene has a very strong influence on the energy levels of mainly the S1 states.
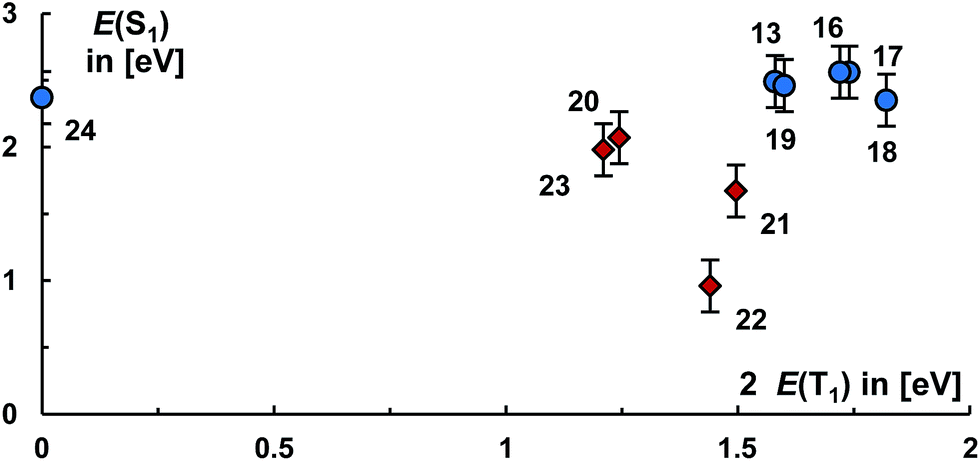 | ||
| Fig. 6 Energy levels of S1 and twice the T1 states for 13 and 16–24. Derivatives of aliphatic carbenes are labeled with blue cycles, carbenes with aromatic character with red squares. Error bars relate to the root mean square deviation for the S1 states from the experimentally available values. | ||
Comparison of bridges
Increasing the length of the linker leads to an increase of the diradicaloid character through insulation of the two formal radical centers. Especially the biphenylene bridged bisCAAC compound 14 shows a very high diradical index of y0 = 0.78 (Table 4), which is well reflected in the reduced electron density on the linker in the HONO and LUNO (Fig. 7). The singlet–triplet gap of this molecule becomes consequently very small [E(T1) = 0.21 eV]. The calculated absorption spectrum is well in line with the experiment, where a band of small intensity was observed at 767 nm and a band of strong intensity at 653 nm.161 The transition to S2 (fosc = 1.0) is mainly associated with a promotion of one electron from the HONO to the LUNO, whereas the S1 state (fosc = 0.001) corresponds to the double excitation as was obtained for 13 (vide supra). This chromophore qualifies accordingly also as a class III chromophore featuring a “doubly excited singlet state”.11 It is interesting to note that polyenes are typical class III chromophores, whereas aromatics usually belong to either class I or class II.11| Compound | 10 | 11 | 12 | 8 | 13 | 14 |
| Bridge | C1 | C2 | C4 | C6H4 | CC–C6H4–CC | CC–C6H4–C6H4–CC |
| y 0 | 0.07 | 0.22 | 0.16 | 0.16 | 0.29 | 0.78 |
| y 1 | 0 | 0.04 | 0.07 | 0.09 | 0.12 | 0.11 |
| N FOD | 0.06 | 0.50 | 0.47 | 0.47 | 0.85 | 1.43 |
| E(S1) in [eV] | 5.38 | 2.88 | 2.82 | 2.74 | 2.49 | 1.58 |
| E(T1) in [eV] | 4.27 | 1.41 | 1.68 | 1.70 | 0.80 | 0.21 |
| E(T2) in [eV] | 4.54 | 5.62 | 4.4 | 3.51 | 2.90 | 2.10 |
| Absorption S0 → S1 (S2) in [nm] | 231 | 430 | 442 | 461 | 498 (444) | 784 (610) |
| f osc S0 → S1 (S2) | 0.0 | 0.6 | 0.0 | 0.6 | 0.0 (0.9) | 0.0 (1.0) |
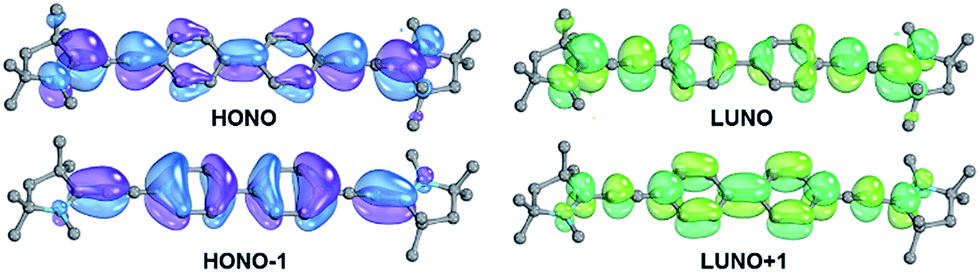 | ||
| Fig. 7 Frontier orbitals of 14 as obtained from CASSCF(14,14) calculation. | ||
The other compounds 8–12 on the contrary are class I chromophores, where the transition to the S1 state is due to a single HONO–LUNO excitation. The CAAC stabilized carbon(0) (allene, respectively) 10 shows of course only negligible diradicaloid character. Surprisingly, already the C2 derivative 11 has an energetically low-lying triplet state [E(T1) = 1.4 eV] and is significantly diradicaloid (y0 = 0.22, NFOD = 0.5). Its diradical character is comparable to Thiele's hydrocarbon and tetracene (vide supra).
This unexpected diradical character has not been noticed in previous calculations,43,162 but is equally reflected by the overall trend when going from 8 (y0 = 0.16, NFOD = 0.5) to “C2-extended” 13 (y0 = 0.29, NFOD = 0.8). Equally, it explains perfectly why the NHC congener 21, which is according to Tables 3 and 4 expected to show even larger diradicaloid character, could not be isolated in a previous synthetic study.163 Note furthermore that 11, which has been reported to be even stable at temperatures as high as 240 °C,43 appears to be a good candidate for singlet fission [E(S1) = 2.88 eV, E(T1) = 1.41 eV] with a reasonable fosc (0.63) for the relevant transition from the S0 to the S1 state.
Moving to carbene stabilized tetracarbon (12) or a phenylene (8) bridge reduces the energies of the S1 states and elevates the energies of the T1 states slightly [8: E(S1) = 2.74 eV, E(T1) = 1.70 eV; 12: E(S1) = 2.82 eV; E(T1) = 1.68 eV]. The T1 energy levels of 8, 11, 12, are in an excellent range for practical applications as is found in tetracene.153 Overall, we find therefore that enlarging the bridge affects both the energy levels of the S1 and T1 states. Fig. 8 puts the S1 energy levels for the CAAC compounds 8, 10–14 into relation with the doubled value of the T1 states. Importantly, the obtained fit [E(S1) = 0.89E(T1) + 1.48 eV] reveals that the S1 as well as T1 energy levels appear to be fairly linear dependent on the nature of the bridge. The choice of the bridge is accordingly an excellent tool for tailoring the overall level of the S1 state.
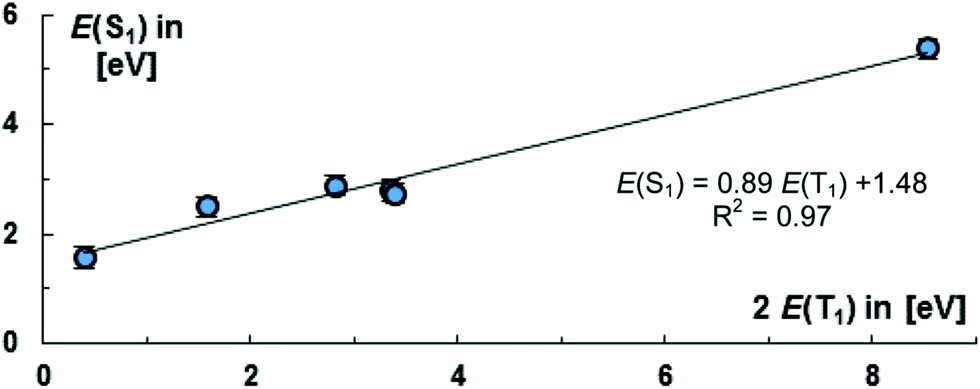 | ||
| Fig. 8 Suitability of 8, 10–14 for singlet fission according to energy matching condition of the S1 and T1 states. Error bars relate to the root mean square deviation for the S1 states from the experimentally available values. | ||
Eventually, we would like to put all the molecules studied herein into perspective to each other. Fig. 9, left, relates the energies of the S1 states with twice the energy of the T1 states.
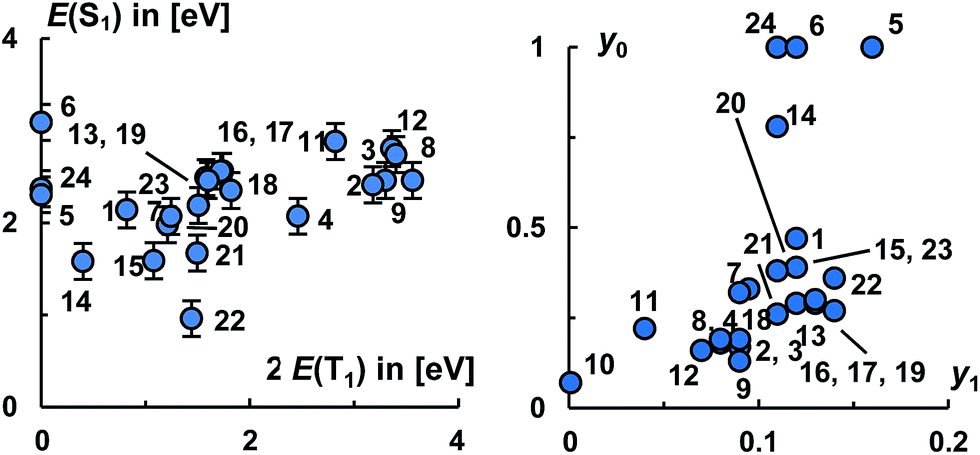 | ||
| Fig. 9 Suitability of investigated diradicaloids for singlet fission according to energy matching condition of S1 and T1 states (left; 10 has been omitted for clarity) and the value of the diradical indices y0 and y1 (right). Error bars relate to the root mean square deviation for the S1 states from the experimentally available values. | ||
Evidently, carbenes allow for the synthesis of diradicaloids with a large electronic diversity, where most investigated structures satisfy the energy criteria for singlet fission. Overall, the calculated levels of the S1 states range from 1.7 eV to 3.1 eV and the levels of the T1 states from 0 eV to 1.8 eV. Plotting the occupancies of the LUNO (y0) as well as LUNO+1 (y1) suggests likewise (Fig. 9, right) that many molecules studied herein qualify as very good candidates for singlet fission (0.1 < y0 < 0.5; y1 ≪ y0) and for two-photon absorption (y0 ≈ 0.36).
The molecules 8, 11, 12, 13, 16–21, 23 appear to be overall most promising for singlet fission, whereas 13, 15–23 should be good targets for two-photon absorption. Equally note that many of these molecules (16–21, 23) show comparably (in relation to their S1 states and the values obtained for pentacene and especially tetracene) low lying T1 states, which allows for considerably exoergic singlet fission. Of particular interest, we obtained an exponential relation [ln(NFOD) = −0.81E(T1) + 0.68] between the NFOD and the energies of the excited T1 states with a very good R2 value of 96% (Fig. 10). The correlation of the NFOD with the energies of the S1 states, the population of the LUNO (y0), or the correlation between y0 and the energies of the T1 states appears to be weaker (ESI†). We conclude therefore that the NFOD is a reliable and time-efficient method for the estimation of the singlet–triplet gaps of these molecules.
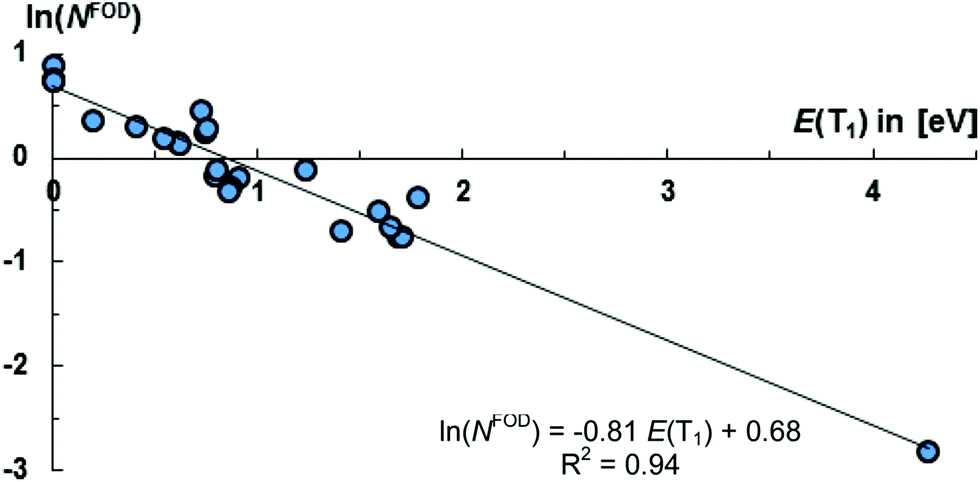 | ||
| Fig. 10 The NFOD is a good descriptor for the singlet–triplet gap.164 | ||
Conclusions
Detailed CASSCF/NEVPT2 computations outline how to design carbene derived diradicaloids with tailored electronic properties for optoelectronic applications. Non-aromatic carbenes allow mainly for a tuning of the energies of the T1 states. Aromaticity of the carbene groups has a very strong influence and reduces especially the energy level of the S1 states. Overall, π-electron poor carbenes are predicted to lead to comparably low diradicaloid character, whereas π-electron rich C-donor ligands like bent allenes lead to “pristine” diradicals. The design of the bridge (“insulator”) permits for an adjustment of the energy levels of both the first excited singlet and triplet states S1 and T1. Surprisingly, even very short bridges like carbene stabilized dicarbon (C2) is predicted to show quite strong diradicaloid character. We observe that the capability of saturated NHCs to stabilize organic radicals appears to be stronger than commonly believed in the literature.The fractional occupation number weighted electron density (NFOD) is correlated with the energy levels of the T1 state. Hence, it is an excellent predictive tools for the singlet–triplet gap.
Overall, the computations suggest that a considerable number of molecules studied herein are good candidates for application in singlet fission considering the energy matching criteria whereas some are suitable candidates for two-photon absorption. Our findings delineate how to obtain diradicaloids with desired electronic properties and give guidelines for merging carbene chemistry with singlet diradicaloid synthesis. We are convinced that diradicaloids derived from CAACs and NHCs with a saturated backbone are excellent candidates for optoelectronic applications and are therefore currently investigating their experimental behavior.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the RRZE Erlangen for computational resources. D. M. and M. M. H. thank the Fonds der Chemischen Industrie FCI for Liebig fellowships. Support by K. Meyer and M. Alcarazo is gratefully acknowledged. We also thank D. Guldi for his support.Notes and references
- M. Abe, Chem. Rev., 2013, 113, 7011–7088 CrossRef PubMed.
- T. Y. Gopalakrishna, W. Zeng, X. Lu and J. Wu, Chem. Commun., 2018, 54, 2186–2199 RSC.
- W. T. Borden, H. Iwamura and J. A. Berson, Acc. Chem. Res., 1994, 27, 109–116 CrossRef.
- Organic Redox Systems, ed. T. Nishinaga, John Wiley & Sons, Inc, Hoboken, New Jersey, 2016 Search PubMed.
- Z. Zeng, X. Shi, C. Chi, J. T. Lopez Navarrete, J. Casado and J. Wu, Chem. Soc. Rev., 2015, 44, 6578–6596 RSC.
- M. Chikamatsu, T. Mikami, J. Chisaka, Y. Yoshida, R. Azumi, K. Yase, A. Shimizu, T. Kubo, Y. Morita and K. Nakasuji, Appl. Phys. Lett., 2007, 91, 043506 CrossRef.
- H. Koike, M. Chikamatsu, R. Azumi, J. y. Tsutsumi, K. Ogawa, W. Yamane, T. Nishiuchi, T. Kubo, T. Hasegawa and K. Kanai, Adv. Funct. Mater., 2016, 26, 277–283 CrossRef.
- K. Kamada, K. Ohta, T. Kubo, A. Shimizu, Y. Morita, K. Nakasuji, R. Kishi, S. Ohta, S.-i. Furukawa, H. Takahashi and M. Nakano, Angew. Chem., Int. Ed., 2007, 46, 3544–3546 CrossRef PubMed.
- Y. Morita, S. Nishida, T. Murata, M. Moriguchi, A. Ueda, M. Satoh, K. Arifuku, K. Sato and T. Takui, Nat. Mater., 2011, 10, 947–951 CrossRef PubMed.
- V. A. Dediu, L. E. Hueso, I. Bergenti and C. Taliani, Nat. Mater., 2009, 8, 707–716 CrossRef PubMed.
- M. B. Smith and J. Michl, Chem. Rev., 2010, 110, 6891–6936 CrossRef PubMed.
- B. J. Walker, A. J. Musser, D. Beljonne and R. H. Friend, Nat. Chem., 2013, 5, 1019–1024 CrossRef PubMed.
- J. Zirzlmeier, D. Lehnherr, P. B. Coto, E. T. Chernick, R. Casillas, B. S. Basel, M. Thoss, R. R. Tykwinski and D. M. Guldi, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 5325–5330 CrossRef PubMed.
- N. J. Thompson, M. W. B. Wilson, D. N. Congreve, P. R. Brown, J. M. Scherer, T. S. Bischof, M. Wu, N. Geva, M. Welborn, T. V. Voorhis, V. Bulović, M. G. Bawendi and M. A. Baldo, Nat. Mater., 2014, 13, 1039–1043 CrossRef PubMed.
- W. Shockley and H. J. Queisser, J. Appl. Phys., 1961, 32, 510–519 CrossRef.
- D. N. Congreve, J. Lee, N. J. Thompson, E. Hontz, S. R. Yost, P. D. Reusswig, M. E. Bahlke, S. Reineke, T. Van Voorhis and M. A. Baldo, Science, 2013, 340, 334–337 CrossRef PubMed.
- O. E. Semonin, J. M. Luther, S. Choi, H.-Y. Chen, J. Gao, A. J. Nozik and M. C. Beard, Science, 2011, 334, 1530–1533 CrossRef PubMed.
- M. B. Smith and J. Michl, Annu. Rev. Phys. Chem., 2013, 64, 361–386 CrossRef PubMed.
- A. E. Tschitschibabin, Ber. Dtsch. Chem. Ges., 1907, 40, 1810–1819 CrossRef.
- L. K. Montgomery, J. C. Huffman, E. A. Jurczak and M. P. Grendze, J. Am. Chem. Soc., 1986, 108, 6004–6011 CrossRef PubMed.
- P. Ravat and M. Baumgarten, Phys. Chem. Chem. Phys., 2015, 17, 983–991 RSC.
- Z. Zeng, Y. M. Sung, N. Bao, D. Tan, R. Lee, J. L. Zafra, B. S. Lee, M. Ishida, J. Ding, J. T. López Navarrete, Y. Li, W. Zeng, D. Kim, K.-W. Huang, R. D. Webster, J. Casado and J. Wu, J. Am. Chem. Soc., 2012, 134, 14513–14525 CrossRef PubMed.
- C. Jiang, Y. Bang, X. Wang, X. Lu, Z. Lim, H. Wei, S. El-Hankari, J. Wu and Z. Zeng, Chem. Commun., 2018, 54, 2389–2392 RSC.
- Y. Su, X. Wang, X. Zheng, Z. Zhang, Y. Song, Y. Sui, Y. Li and X. Wang, Angew. Chem., Int. Ed., 2014, 53, 2857–2861 CrossRef PubMed.
- Y. Su, X. Wang, L. Wang, Z. Zhang, X. Wang, Y. Song and P. P. Power, Chem. Sci., 2016, 7, 6514–6518 RSC.
- S. Zheng, S. Barlow, C. Risko, T. L. Kinnibrugh, V. N. Khrustalev, S. C. Jones, M. Y. Antipin, N. M. Tucker, T. V. Timofeeva, V. Coropceanu, J.-L. Brédas and S. R. Marder, J. Am. Chem. Soc., 2006, 128, 1812–1817 CrossRef PubMed.
- T. Li, G. Tan, D. Shao, J. Li, Z. Zhang, Y. Song, Y. Sui, S. Chen, Y. Fang and X. Wang, J. Am. Chem. Soc., 2016, 138, 10092–10095 CrossRef PubMed.
- G. Tan and X. Wang, Acc. Chem. Res., 2017, 50, 1997–2006 CrossRef PubMed.
- M. M. Hansmann, M. Melaimi, D. Munz and G. Bertrand, J. Am. Chem. Soc., 2018, 140, 2546–2554 CrossRef PubMed.
- For the first report of a CAAC, see: (a) V. Lavallo, Y. Canac, C. Prasang, B. Donnadieu and G. Bertrand, Angew. Chem., Int. Ed., 2005, 44, 5705–5709 CrossRef PubMed. For reviews on CAACs, see: (b) M. Soleilhavoup and G. Bertrand, Acc. Chem. Res., 2015, 48, 256–266 CrossRef PubMed; (c) S. Roy, K. C. Mondal and H. W. Roesky, Acc. Chem. Res., 2016, 49, 357–369 CrossRef PubMed; (d) M. Melaimi, R. Jazzar, M. Soleilhavoup and G. Bertrand, Angew. Chem., Int. Ed., 2017, 56, 10046–10068 CrossRef PubMed; (e) U. S. D. Paul and U. Radius, Eur. J. Inorg. Chem., 2017, 3362–3375 CrossRef.
- J. K. Mahoney, D. Martin, F. Thomas, C. E. Moore, A. L. Rheingold and G. Bertrand, J. Am. Chem. Soc., 2015, 137, 7519–7525 CrossRef PubMed.
- J. K. Mahoney, D. Martin, C. E. Moore, A. L. Rheingold and G. Bertrand, J. Am. Chem. Soc., 2013, 135, 18766–18769 CrossRef PubMed.
- D. Munz, J. Chu, M. Melaimi and G. Bertrand, Angew. Chem., Int. Ed., 2016, 55, 12886–12890 CrossRef PubMed.
- D. Martin, M. Soleilhavoup and G. Bertrand, Chem. Sci., 2011, 2, 389–399 RSC.
- M. M. Hansmann, M. Melaimi and G. Bertrand, J. Am. Chem. Soc., 2018, 140, 2206–2213 CrossRef PubMed.
- For thematic issues and books on NHCs, see: (a) T. Rovis and S. P. Nolan, Synlett, 2013, 24, 1188–1189 CrossRef; (b) A. J. Arduengo and G. Bertrand, Chem. Rev., 2009, 109, 3209–3210 CrossRef PubMed; (c) S. Diez Gonzalez, N-Heterocyclic Carbenes: From Laboratory Curiosities to Efficient Synthetic Tools, Royal Society of Chemistry, Cambridge, 2010 RSC; (d) S. P. Nolan, N-Heterocyclic Carbenes: Effective Tools for Organometallic Synthesis, Wiley-VCH, Weinheim, 2014 Search PubMed.
- R. Ghadwal, D. Rottschäfer, B. Neumann, H.-G. Stammler, M. van Gastel and D. Andrada, Angew. Chem., Int. Ed., 2018, 130, 4855–4859 CrossRef.
- P. L. Arnold and S. T. Liddle, Organometallics, 2006, 25, 1485–1491 CrossRef.
- B. Barry, G. Soper, J. Hurmalainen, A. Mansikkamäki, K. N. Robertson, W. L. McClennan, A. J. Veinot, T. L. Roemmele, U. Werner-Zwanziger, R. T. Boeré, H. M. Tuononen, J. Clyburne and J. Masuda, Angew. Chem., Int. Ed., 2018, 57, 749–754 CrossRef PubMed.
- R. S. Ghadwal, D. Rottschafer, B. Neumann, G. Stammler and D. M. Andrada, Chem. Sci., 2018, 9, 4970–4976 RSC.
- D. Rottschäfer, N. K. T. Ho, B. Neumann, H. G. Stammler, M. v. Gastel, D. M. Andrada and R. S. Ghadwal, Angew. Chem., Int. Ed., 2018, 57, 5838–5842 CrossRef PubMed.
- L. Jin, M. Melaimi, L. Liu and G. Bertrand, Org. Chem. Front., 2014, 1, 351–354 RSC.
- Y. Li, K. C. Mondal, P. P. Samuel, H. Zhu, C. M. Orben, S. Panneerselvam, B. Dittrich, B. Schwederski, W. Kaim, T. Mondal, D. Koley and H. W. Roesky, Angew. Chem., Int. Ed., 2014, 53, 4168–4172 CrossRef PubMed.
- Acetylene Chemistry, ed. P. J. S. François Diederich, P. J. Stang and R. R. Tykwinski, Wiley-VCH Verlag GmbH & Co. KGaA, 2005 Search PubMed.
- M. Kivala and F. Diederich, Acc. Chem. Res., 2009, 42, 235–248 CrossRef PubMed.
- F. Diederich and M. Kivala, Adv. Mater., 2010, 22, 803–812 CrossRef PubMed.
- P. Rivera-Fuentes and F. Diederich, Angew. Chem., Int. Ed., 2012, 51, 2818–2828 CrossRef PubMed.
- D. Wendinger and R. R. Tykwinski, Acc. Chem. Res., 2017, 50, 1468–1479 CrossRef PubMed.
- T. Chen, L. Zheng, J. Yuan, Z. An, R. Chen, Y. Tao, H. Li, X. Xie and W. Huang, Sci. Rep., 2015, 5, 10923 CrossRef PubMed.
- T. Zeng, N. Ananth and R. Hoffmann, J. Am. Chem. Soc., 2014, 136, 12638–12647 CrossRef PubMed.
- J. Wen, Z. Havlas and J. Michl, J. Am. Chem. Soc., 2015, 137, 165–172 CrossRef PubMed.
- A. F. Schwerin, J. C. Johnson, M. B. Smith, P. Sreearunothai, D. Popović, J. Černý, Z. Havlas, I. Paci, A. Akdag, M. K. MacLeod, X. Chen, D. E. David, M. A. Ratner, J. R. Miller, A. J. Nozik and J. Michl, J. Phys. Chem. A, 2010, 114, 1457–1473 CrossRef PubMed.
- D. Lopez-Carballeira, D. Casanova and F. Ruiperez, Phys. Chem. Chem. Phys., 2017, 19, 30227–30238 RSC.
- S. Ito, T. Nagami and M. Nakano, J. Photochem. Photobiol., C, 2018, 34, 85–120 CrossRef.
- S. Ito and M. Nakano, J. Phys. Chem. C, 2015, 119, 148–157 CrossRef.
- J. Thiele and H. Balhorn, Ber. Dtsch. Chem. Ges., 1904, 37, 1463–1470 CrossRef.
- J. M. Robertson, V. C. Sinclair and J. Trotter, Acta Crystallogr., 1961, 14, 697–704 CrossRef.
- F. Hinkel, J. Freudenberg and U. H. Bunz, Angew. Chem., Int. Ed., 2016, 55, 9830–9832 CrossRef PubMed.
- W. Zeng, Z. Sun, T. S. Herng, T. P. Gonçalves, T. Y. Gopalakrishna, K.-W. Huang, J. Ding and J. Wu, Angew. Chem., Int. Ed., 2016, 55, 8615–8619 CrossRef PubMed.
- S. Lukman, J. M. Richter, L. Yang, P. Hu, J. Wu, N. C. Greenham and A. J. Musser, J. Am. Chem. Soc., 2017, 139, 18376–18385 CrossRef PubMed.
- W. Zeng, S. Lee, M. Son, M. Ishida, K. Furukawa, P. Hu, Z. Sun, D. Kim and J. Wu, Chem. Sci., 2015, 6, 2427–2433 RSC.
- T. Kubo, Chem. Rec., 2015, 15, 218–232 CrossRef PubMed.
- K. Ohashi, T. Kubo, T. Masui, K. Yamamoto, K. Nakasuji, T. Takui, Y. Kai and I. Murata, J. Am. Chem. Soc., 1998, 120, 2018–2027 CrossRef.
- D. Xia, A. Keerthi, C. An and M. Baumgarten, Org. Chem. Front., 2017, 4, 18–21 RSC.
- J. Casado, Top. Curr. Chem., 2017, 375, 73 CrossRef PubMed.
- H. Isobe, Y. Takano, Y. Kitagawa, T. Kawakami, S. Yamanaka, K. Yamaguchi and K. N. Houk, J. Phys. Chem. A, 2003, 107, 682–694 CrossRef.
- J. L. Segura and N. Martín, Chem. Rev., 1999, 99, 3199–3246 CrossRef PubMed.
- J. Ma, J. Liu, M. Baumgarten, Y. Fu, Y.-Z. Tan, K. S. Schellhammer, F. Ortmann, G. Cuniberti, H. Komber, R. Berger, K. Müllen and X. Feng, Angew. Chem., Int. Ed., 2017, 56, 3280–3284 CrossRef PubMed.
- G. E. Rudebusch, J. L. Zafra, K. Jorner, K. Fukuda, J. L. Marshall, I. Arrechea-Marcos, G. L. Espejo, R. Ponce Ortiz, C. J. Gómez-García, L. N. Zakharov, M. Nakano, H. Ottosson, J. Casado and M. M. Haley, Nat. Chem., 2016, 8, 753–759 CrossRef PubMed.
- A. Caneschi, P. Chiesi, L. David, F. Ferraro, D. Gatteschi and R. Sessoli, Inorg. Chem., 1993, 32, 1445–1453 CrossRef.
- P. Ravat, Y. Ito, E. Gorelik, V. Enkelmann and M. Baumgarten, Org. Lett., 2013, 15, 4280–4283 CrossRef PubMed.
- C. Train, L. Norel and M. Baumgarten, Coord. Chem. Rev., 2009, 253, 2342–2351 CrossRef.
- L. Catala, J. Le Moigne, N. Kyritsakas, P. Rey, J. J. Novoa and P. Turek, Chem.–Eur. J., 2001, 7, 2466–2480 CrossRef.
- S. Tolstikov, E. Tretyakov, S. Fokin, E. Suturina, G. Romanenko, A. Bogomyakov, D. Stass, A. Maryasov, M. Fedin, N. Gritsan and V. Ovcharenko, Chem.–Eur. J., 2014, 20, 2793–2803 CrossRef PubMed.
- J. B. Gilroy, S. D. J. McKinnon, P. Kennepohl, M. S. Zsombor, M. J. Ferguson, L. K. Thompson and R. G. Hicks, J. Org. Chem., 2007, 72, 8062–8069 CrossRef PubMed.
- J. Zhou and A. Rieker, J. Chem. Soc., Perkin Trans. 2, 1997, 931–938 RSC.
- R. West, J. A. Jorgenson, K. L. Stearley and J. C. Calabrese, J. Chem. Soc., Chem. Commun., 1991, 1234–1235 RSC.
- D. Schmidt, M. Son, J. M. Lim, M.-J. Lin, I. Krummenacher, H. Braunschweig, D. Kim and F. Würthner, Angew. Chem., Int. Ed., 2015, 54, 13980–13984 CrossRef PubMed.
- S. Lee, F. Miao, H. Phan, T. S. Herng, J. Ding, J. Wu and D. Kim, ChemPhysChem, 2017, 18, 591–595 CrossRef PubMed.
- O. Back, M. Henry-Ellinger, C. D. Martin, D. Martin and G. Bertrand, Angew. Chem., Int. Ed., 2013, 52, 2939–2943 CrossRef PubMed.
- K. Verlinden, H. Buhl, W. Frank and C. Ganter, Eur. J. Inorg. Chem., 2015, 2416–2425 CrossRef.
- D. Munz, Organometallics, 2018, 37, 275–289 CrossRef.
- For the report of the benzannulated congener, see: W. Grahn, H.-H. Johannes, J. Rheinheimer, B. Knieriem and E.-U. Würthwein, Liebigs Ann., 1995, 6, 1003–1009 CrossRef.
- W. C. Chen, J. S. Shen, T. Jurca, C. J. Peng, Y. H. Lin, Y. P. Wang, W. C. Shih, G. P. Yap and T. G. Ong, Angew. Chem., Int. Ed., 2015, 54, 15207–15212 CrossRef PubMed.
- W.-C. Chen, Y.-C. Hsu, C.-Y. Lee, G. P. A. Yap and T.-G. Ong, Organometallics, 2013, 32, 2435–2442 CrossRef.
- C. A. Dyker, V. Lavallo, B. Donnadieu and G. Bertrand, Angew. Chem., Int. Ed., 2008, 47, 3206–3209 CrossRef PubMed.
- H. Schmidbaur and A. Schier, Angew. Chem., Int. Ed., 2013, 52, 176–186 CrossRef PubMed.
- R. Tonner, F. Öxler, B. Neumüller, W. Petz and G. Frenking, Angew. Chem., Int. Ed., 2006, 45, 8038–8042 CrossRef PubMed.
- H. V. Huynh, in The Organometallic Chemistry of N-heterocyclic Carbenes, John Wiley & Sons, Ltd, 2017, pp. 293–329 Search PubMed.
- For a review on the computational modelling of singlet fission, see: D. Casanova, Chem. Rev., 2018 DOI:10.1021/acs.chemrev.7b00601.
- J. Grafenstein, E. Kraka, M. Filatov and D. Cremer, Int. J. Mol. Sci., 2002, 3, 360–394 CrossRef.
- F. Neese, J. Phys. Chem. Solids, 2004, 65, 781–785 CrossRef.
- R. Caballol, O. Castell, F. Illas, I. d. P. R. Moreira and J. P. Malrieu, J. Phys. Chem. A, 1997, 101, 7860–7866 CrossRef.
- E. R. Davidson and W. T. Borden, J. Phys. Chem., 1983, 87, 4783–4790 CrossRef.
- N. Ferre, N. Guihery and J. P. Malrieu, Phys. Chem. Chem. Phys., 2015, 17, 14375–14382 RSC.
- J. P. Malrieu and G. Trinquier, J. Phys. Chem. A, 2012, 116, 8226–8237 CrossRef PubMed.
- S. Yamanaka, T. Kawakami, H. Nagao and K. Yamaguchi, Chem. Phys. Lett., 1994, 231, 25–33 CrossRef.
- L. Noodleman, J. Chem. Phys., 1981, 74, 5737–5743 CrossRef.
- B. O. Roos, P. R. Taylor and P. E. M. Siegbahn, Chem. Phys., 1980, 48, 157–173 CrossRef.
- J. Pittner, P. Nachtigall, P. Čársky and I. Hubač, J. Phys. Chem. A, 2001, 105, 1354–1356 CrossRef.
- X. Li and J. Paldus, J. Chem. Phys., 2008, 129, 054104 CrossRef PubMed.
- E. R. Davidson and A. E. Clark, Int. J. Quantum Chem., 2005, 103, 1–9 CrossRef.
- J. P. Malrieu, R. Caballol, C. J. Calzado, C. de Graaf and N. Guihéry, Chem. Rev., 2014, 114, 429–492 CrossRef PubMed.
- D. Casanova and M. Head-Gordon, Phys. Chem. Chem. Phys., 2009, 11, 9779–9790 RSC.
- S. J. Stoneburner, J. Shen, A. O. Ajala, P. Piecuch, D. G. Truhlar and L. Gagliardi, J. Chem. Phys., 2017, 147, 164120 CrossRef PubMed.
- P. M. Zimmerman, F. Bell, D. Casanova and M. Head-Gordon, J. Am. Chem. Soc., 2011, 133, 19944–19952 CrossRef PubMed.
- A. Das, T. Muller, F. Plasser and H. Lischka, J. Phys. Chem. A, 2016, 120, 1625–1636 CrossRef PubMed.
- M. R. Momeni, J. Chem. Theory Comput., 2016, 12, 5067–5075 CrossRef PubMed.
- S. Radenković, S. Marković and V. Milenković, Chem. Phys., 2012, 545, 132–137 Search PubMed.
- H. K. Powell and W. T. Borden, J. Org. Chem., 1995, 60, 2654–2655 CrossRef.
- C. M. Isborn, E. R. Davidson and B. H. Robinson, J. Phys. Chem. A, 2006, 110, 7189–7196 CrossRef PubMed.
- O. Kwon and G. Chung, Bull. Korean Chem. Soc., 2008, 29, 2140–2144 CrossRef.
- V. Barone, I. Cacelli, P. Cimino, A. Ferretti, S. Monti and G. Prampolini, J. Phys. Chem. A, 2009, 113, 15150–15155 CrossRef PubMed.
- J. Hachmann, J. J. Dorando, M. Avilés and G. K.-L. Chan, J. Chem. Phys., 2007, 127, 134309 CrossRef PubMed.
- Z. Qu, D. Zhang, C. Liu and Y. Jiang, J. Phys. Chem. A, 2009, 113, 7909–7914 CrossRef PubMed.
- M. Melle-Franco, Chem. Commun., 2015, 51, 5387–5390 RSC.
- A. Das, T. Müller, F. Plasser, D. B. Krisiloff, E. A. Carter and H. Lischka, J. Chem. Theory Comput., 2017, 13, 2612–2622 CrossRef PubMed.
- D. Peng, X. Hu, D. Devarajan, D. H. Ess, E. R. Johnson and W. Yang, J. Chem. Phys., 2012, 137, 114112 CrossRef PubMed.
- D. H. Ess, E. R. Johnson, X. Hu and W. Yang, J. Phys. Chem. A, 2011, 115, 76–83 CrossRef PubMed.
- C. U. Ibeji and D. Ghosh, Phys. Chem. Chem. Phys., 2015, 17, 9849–9856 RSC.
- M. Bendikov, H. M. Duong, K. Starkey, K. N. Houk, E. A. Carter and F. Wudl, J. Am. Chem. Soc., 2004, 126, 7416–7417 CrossRef PubMed.
- H. F. Bettinger, Pure Appl. Chem., 2010, 82, 905–915 Search PubMed.
- D.-e. Jiang and S. Dai, J. Phys. Chem. A, 2008, 112, 332–335 CrossRef PubMed.
- R. Rakhi and C. H. Suresh, Phys. Chem. Chem. Phys., 2016, 18, 24631–24641 RSC.
- Y. Yang, E. R. Davidson and W. Yang, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, E5098–E5107 CrossRef PubMed.
- D. H. Ess and T. C. Cook, J. Phys. Chem. A, 2012, 116, 4922–4929 CrossRef PubMed.
- I. Paci, J. C. Johnson, X. Chen, G. Rana, D. Popović, D. E. David, A. J. Nozik, M. A. Ratner and J. Michl, J. Am. Chem. Soc., 2006, 128, 16546–16553 CrossRef PubMed.
- D. Doehnert and J. Koutecky, J. Am. Chem. Soc., 1980, 102, 1789–1796 CrossRef.
- M. Nakano, H. Fukui, T. Minami, K. Yoneda, Y. Shigeta, R. Kishi, B. Champagne, E. Botek, T. Kubo, K. Ohta and K. Kamada, Theor. Chem. Acc., 2011, 130, 711–724 Search PubMed.
- K. Kamada, K. Ohta, A. Shimizu, T. Kubo, R. Kishi, H. Takahashi, E. Botek, B. Champagne and M. Nakano, J. Phys. Chem. Lett., 2010, 1, 937–940 Search PubMed.
- M. Nakano, R. Kishi, S. Ohta, H. Takahashi, T. Kubo, K. Kamada, K. Ohta, E. Botek and B. Champagne, Phys. Rev. Lett., 2007, 99, 033001 CrossRef PubMed.
- T. Minami, S. Ito and M. Nakano, J. Phys. Chem. Lett., 2013, 4, 2133–2137 CrossRef.
- S. Ito, T. Minami and M. Nakano, J. Phys. Chem. C, 2012, 116, 19729–19736 CrossRef.
- T. Minami and M. Nakano, J. Phys. Chem. Lett., 2012, 3, 145–150 CrossRef.
- Note that the morphology is of course very important in the solid state.
- D. Herebian, K. E. Wieghardt and F. Neese, J. Am. Chem. Soc., 2003, 125, 10997–11005 CrossRef PubMed.
- C. A. Bauer, A. Hansen and S. Grimme, Chem.–Eur. J., 2017, 23, 6150–6164 CrossRef PubMed.
- N. D. Mermin, Phys. Rev., 1965, 137, A1441–A1443 CrossRef.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 73–78 Search PubMed.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- F. Weigend, J. Comput. Chem., 2008, 29, 167–175 CrossRef PubMed.
- C. Angeli, R. Cimiraglia, S. Evangelisti, T. Leininger and J. P. Malrieu, J. Chem. Phys., 2001, 114, 10252–10264 CrossRef.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef PubMed.
- C. J. Cramer and D. G. Truhlar, Acc. Chem. Res., 2008, 41, 760–768 CrossRef PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef PubMed.
- M. D. Hanwell, D. E. Curtis, D. C. Lonie, T. Vandermeersch, E. Zurek and G. R. Hutchison, J. Cheminf., 2012, 4, 1–17 Search PubMed.
- G. Knizia, J. Chem. Theory Comput., 2013, 9, 4834–4843 CrossRef PubMed.
- J. Bourdon and M. Calvin, J. Org. Chem., 1957, 22, 101–116 CrossRef.
- T. Okamoto, T. Suzuki, H. Tanaka, D. Hashizume and Y. Matsuo, Chem.–Asian J., 2012, 7, 105–111 CrossRef PubMed.
- C. Hellner, L. Lindqvist and P. C. Roberge, J. Chem. Soc., Faraday Trans. 2, 1972, 68, 1928–1937 RSC.
- J. J. Burdett, A. M. Müller, D. Gosztola and C. J. Bardeen, J. Chem. Phys., 2010, 133, 144506 CrossRef PubMed.
- A. B. Pun, S. N. Sanders, E. Kumarasamy, M. Y. Sfeir, D. N. Congreve and L. M. Campos, Adv. Mater., 2017, 29, 1701416 CrossRef PubMed.
- H. L. Stern, A. Cheminal, S. R. Yost, K. Broch, S. L. Bayliss, K. Chen, M. Tabachnyk, K. Thorley, N. Greenham, J. M. Hodgkiss, J. Anthony, M. Head-Gordon, A. J. Musser, A. Rao and R. H. Friend, Nat. Chem., 2017, 9, 1205–1212 CrossRef PubMed.
- S. V. C. Vummaleti, D. J. Nelson, A. Poater, A. Gomez-Suarez, D. B. Cordes, A. M. Z. Slawin, S. P. Nolan and L. Cavallo, Chem. Sci., 2015, 6, 1895–1904 RSC.
- A. Liske, K. Verlinden, H. Buhl, K. Schaper and C. Ganter, Organometallics, 2013, 32, 5269–5272 CrossRef.
- V. Lavallo, C. A. Dyker, B. Donnadieu and G. Bertrand, Angew. Chem., Int. Ed., 2009, 48, 1540–1542 CrossRef.
- I. Fernández, C. A. Dyker, A. DeHope, B. Donnadieu, G. Frenking and G. Bertrand, J. Am. Chem. Soc., 2009, 131, 11875–11881 CrossRef PubMed.
- M. M. Hanninen, A. Peuronen and H. M. Tuononen, Chem.–Eur. J., 2009, 15, 7287–7291 CrossRef PubMed.
- M. Christl and B. Engels, Angew. Chem., Int. Ed., 2009, 48, 1538–1539 CrossRef PubMed.
- Note that the experimental UV-Vis spectrum relates to a mixture of E and Z isomers.
- J. L. Dutton and D. J. D. Wilson, Angew. Chem., Int. Ed., 2012, 51, 1477–1480 CrossRef PubMed.
- D. C. Georgiou, B. D. Stringer, C. F. Hogan, P. J. Barnard, D. J. D. Wilson, N. Holzmann, G. Frenking and J. L. Dutton, Chem.–Eur. J., 2015, 21, 3377–3386 CrossRef PubMed.
- Compounds 7, 9, 21, 22 (which have NOT been omitted from the fit) appear to be outliers. Exploratory calculations suggest that enlarging the active space beyond CASSCF(14,14) leads to a better fit for these compounds.
Footnote |
| † Electronic supplementary information (ESI) available: Atomic coordinates, energies, delineation of the choice of the active space, FOD-plots, benchmark results, correlation plots, weight of configurations, molecular orbitals. See DOI: 10.1039/c8sc01999a |
| This journal is © The Royal Society of Chemistry 2018 |