
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMembrane glycomics reveal heterogeneity and quantitative distribution of cell surface sialylation†
Diane Dayoung
Park
 *ab,
Gege
Xu
a,
Maurice
Wong
a,
Chatchai
Phoomak
c,
Mingqi
Liu
d,
Nathan E.
Haigh
e,
Sopit
Wongkham
c,
Pengyuan
Yang
d,
Emanual
Maverakis
e and
Carlito B.
Lebrilla
a
*ab,
Gege
Xu
a,
Maurice
Wong
a,
Chatchai
Phoomak
c,
Mingqi
Liu
d,
Nathan E.
Haigh
e,
Sopit
Wongkham
c,
Pengyuan
Yang
d,
Emanual
Maverakis
e and
Carlito B.
Lebrilla
a
aDepartment of Chemistry, University of California, Davis, CA 95616, USA
bDepartment of Surgery, Beth Israel Deaconess Medical Center, Harvard Medical School, Boston, MA 02115, USA. E-mail: dpark4@bidmc.harvard.edu
cDepartment of Biochemistry, Faculty of Medicine, Khon Kaen University, Khon Kaen 40002, Thailand
dDepartment of Chemistry, Institutes of Biomedical Sciences, Fudan University, Shanghai 200032, China
eDepartment of Dermatology, University of California, Davis School of Medicine, Sacramento, CA 95817, USA
First published on 27th June 2018
Abstract
Given that unnatural sugar expression is metabolically achieved, the kinetics and disposition of incorporation can lend insight into the temporal and localization preferences of sialylation across the cell surface. However, common detection schemes lack the ability to detail the molecular diversity and distribution of target moieties. Here we employed a mass spectrometric approach to trace the placement of azido sialic acids on membrane glycoconjugates, which revealed substantial variations in incorporation efficiencies between N-/O-glycans, glycosites, and glycosphingolipids. To further explore the propensity for sialylation, we subsequently mapped the native glycome of model epithelial cell surfaces and illustrate that while glycosylation sites span broadly across the extracellular region, a higher number of heterogeneous glycoforms occur on sialylated sites closest to the transmembrane domain. Beyond imaging techniques, this integrative approach provides unprecedented details about the frequency and structure-specific distribution of cell surface sialylation, a critical feature that regulates cellular interactions and homeostatic pathways.
Introduction
Sialic acids comprise a nine-carbon, α-keto family of monosaccharides that exist as diverse forms resulting from functional group variations and differences in connecting linkages. The prime location of sialic acids at the ends of membrane and secreted glycoproteins contributes in large part to their exploitation by viruses, bacteria, and toxins that recognize sialylated ligands with high specificity.1–4 Contributing to the pathogenesis of gastrointestinal and systemic infections, sialic acids can be taken up and utilized by bacteria as an energy source or incorporated into capsular or lipopolysaccharide components, protecting them from host immune recognition. Similarly, tumor cells have been shown to express aberrantly high levels of sialylation, which permits immune escape by blocking complement activation, cytotoxic granule release, and physical interaction.5 Following this complexity, ongoing studies actively seek efficient synthetic schemes in complement with detailed structural analysis of sialylated glycoconjugates toward improved understanding of their functions.Metabolic labeling has become the foremost strategy to install and selectively scan sialic acids on cell surfaces.6–9 This approach utilizes the cell's endogenous biosynthetic machinery to convert and activate exogenously administered variants of N-acetylmannosamine (ManNAc) which then get incorporated into native glycoconjugates as the corresponding N-acetylneuraminic acid (Neu5Ac). However, due to the non-templated fashion of glycan assembly, a vast complexity of structures is generated that are difficult to sequence compared to other macromolecules such as proteins and DNA. Consequently, information about the levels, efficiency, and products of incorporation remain unresolved, which limits its use for probing functional relevance. Structural characterization is particularly valuable given that certain sialylated moieties, such as sialyl Lewis X, sialyl Tn antigen, and sialylated poly-N-acetyllactosamine (poly-LacNAc), may have specialized functions.10–14 Although fluorescent tags are commonly used for visualizing labeled cells,15–17 this technique cannot locate the residue-specific sites of glycosylation that have converted to unnatural forms and may actually result in false positive measurements.18 Moreover, under the premise that sialic acids reside in high density on the surface of many types of cancer cells, identifying the sites where sialylation occurs on a glycoprotein is a central goal toward regulating their molecular interactions with therapeutics. Therefore, global mapping of the occurrence of sialylated sites and glycans, principally on specified domains of membrane proteins, would help elucidate how sialylated glycans amplify or demote biological activity.
In parallel with metabolic labeling, advanced analytical tools are emerging to enable full characterization of unnaturally glycosylated proteins using isotopic labeling or chemical ligation followed by affinity purification.19–21 Here, we present a platform to comprehensively survey the structures of modified glycans on the cell membrane in addition to their associated glycoproteins and glycosphingolipids with minimal sample manipulation. In our approach, the protein-, site-, glycan-, and lipid-specific locations of azido sialic acids were probed by using the monosaccharide as a mass label, removing the need for additional conjugation and capture steps. The wide range of observed incorporation rates of the unnatural sugar subsequently prompted in-depth identification of native cell surface glycoproteins. This detail was used to map the occurrence of sialic acids on the extracellular domain and explore the factors that dictate or accompany sialylation.
Results
Locating unnatural sialic acids on cell membrane glycans
To comprehensively survey cell surface sialic acid expression, azide functional groups (azido N-acetylneuraminic acid, SiaNAz) were metabolically installed across sialo-glycoconjugates for utility as unique mass reporters (Fig. 1). Within the mixture of released and purified N- and O-glycans sourced from various epithelial cell lines, SiaNAzylated glycans were distinguished from other structures by diagnostic ions originating from the unnatural monosaccharide. The SiaNAz residue has a signature mass of 332.10 Da, distinct from the mass of other monosaccharides. Using CID-MS/MS, fragment ions m/z 315.09 [SiaNAz − H2O + H]+ and m/z 333.10 [SiaNAz + H]+ unambiguously indicated the presence of the unnatural sialic acid in the selected glycan precursor. The acquired mass spectra of SiaNAz-incorporated structures that are representative of typical N-glycan classes and mucin type O-glycans are shown in Fig. S1.† In addition to compounds that incorporated SiaNAz residues on all possible sites of sialylation (Fig. 2A, top panel), we observed evidence of singly or partially incorporated structures (Fig. 2A, bottom panel). During the chemical release of O-glycans, which is performed in the presence of a reducing agent, we observed the conversion of the azide to an amine (Fig. S2†). Thus, glycans containing the reduced SiaNAz residue (SiaNAm, 306.11 Da) were also included as products of metabolic labeling. Amino sialic acids were otherwise absent in untreated controls. This analysis provided retention time stamps and chromatographic profiles of individual SiaNAz-incorporated N- and O-glycans, enabling consideration for quantitative measures.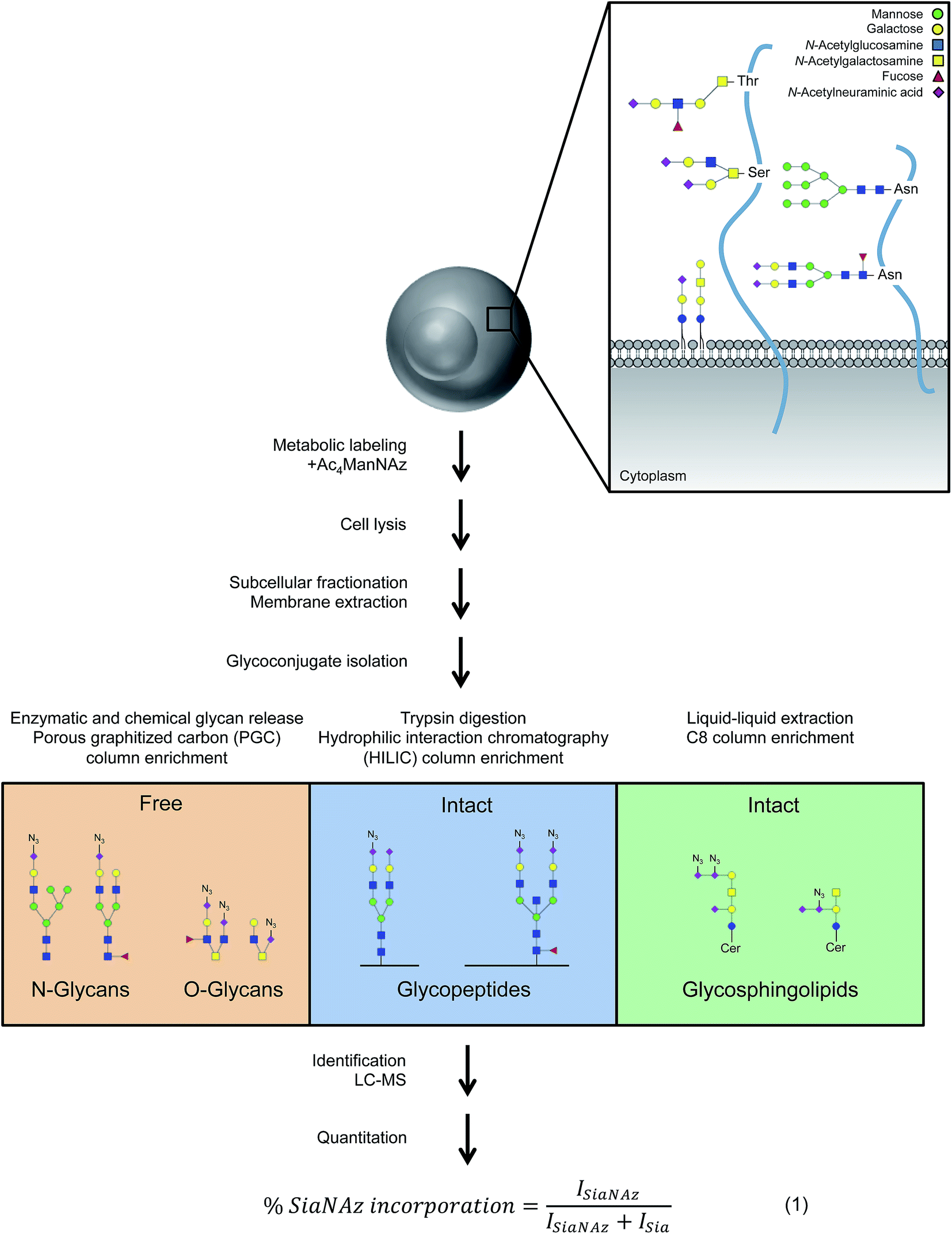 | ||
| Fig. 1 Isolation, identification, and quantitation of glycoconjugates associated with the plasma membrane. Each cell is uniquely decorated by signature glycoproteins and glycolipids that are of various compositions and lengths. Based on structural features, isolation techniques were optimized for free and intact glycoconjugate analysis. Upon identification, unnatural sialic acid incorporation was calculated according to abundances (I, intensity). | ||
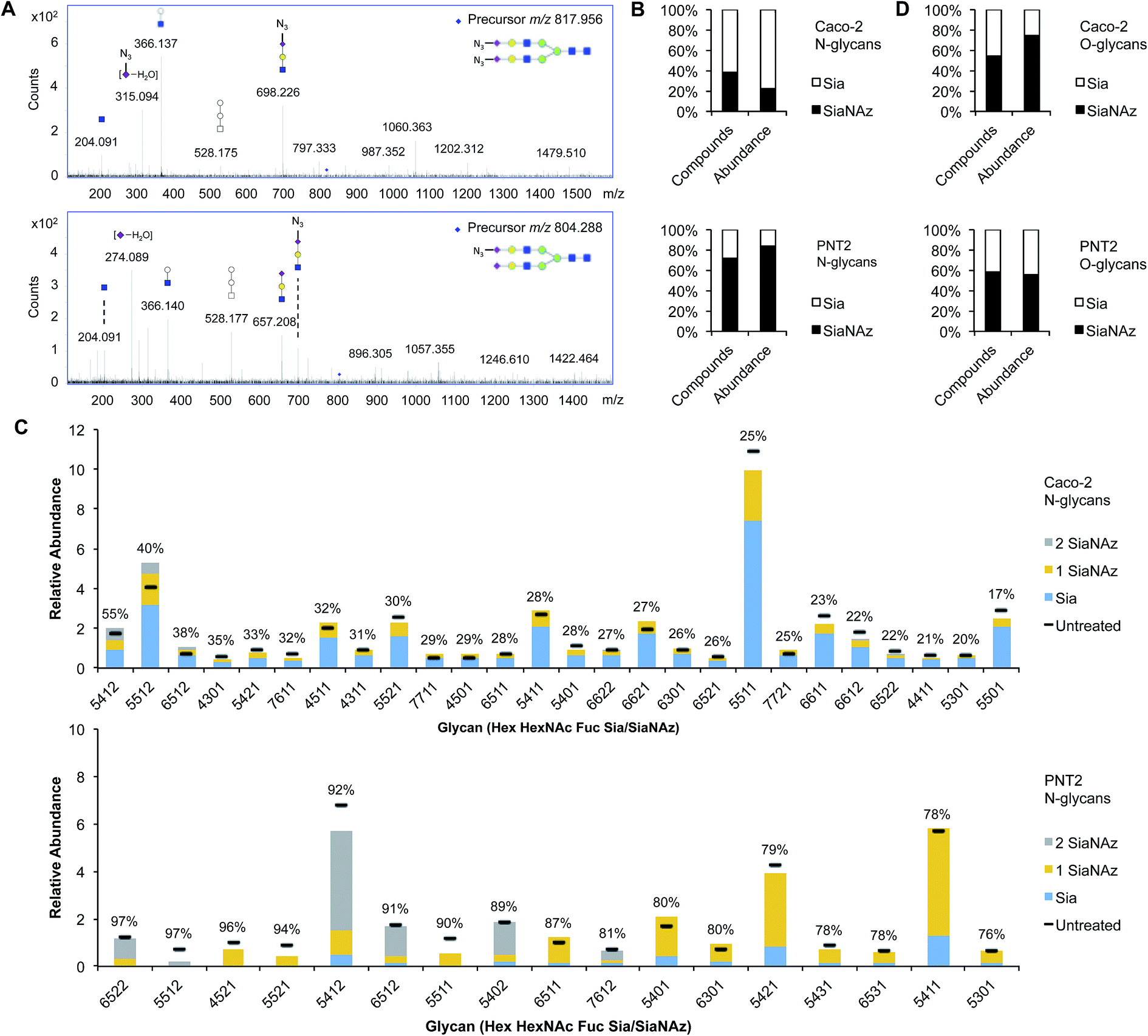 | ||
| Fig. 2 Profiling SiaNAzylated N- and O-glycans of the cell surface. (A) MS/MS spectra of N-glycan structures that have doubly (top panel) or singly (bottom panel) incorporated SiaNAz. Fragment ions m/z 292.10 (dehydrogenated m/z 274.08) and m/z 333.11 (dehydrogenated m/z 315.08) indicate the presence of Sia and SiaNAz, respectively, in the selected glycan precursor. (B) Net percent incorporation of SiaNAz onto N-glycans. (C) Relative abundances of sialylated N-glycans on ManNAz-treated Caco-2 and PNT2 (threshold >0.5%). Bar graphs are arranged by order of percent incorporation, as indicated in the inset (high to low, left to right). (D) Net percent incorporation of SiaNAz onto O-glycans. | ||
Quantitation of unnatural sialic acid incorporation
To accurately enumerate the efficiency and extent of incorporation across all sialylated species, addition of azido N-acetylmannosamine (ManNAz), the metabolic precursor of SiaNAz, was optimized by dose- and time-response measurements (Fig. S3†). It is evident that every cell bears its own N-glycan signature that is uniquely decorated by features such as fucosylation, sialylation, and mannosylation.22–26 Accordingly, among the epithelial cells we examined, the extent of SiaNAz incorporation was related to the tissue of origin (Fig. S4†). Of note, however, the starting amounts of sialylation did not heavily influence the incorporation efficiency. Q-RT-PCR measurements further showed that expression levels of sialyltransferases were not the primary basis for determining the incorporation efficiency (Fig. S5†). PNT2, derived from non-tumorigenic prostate epithelial cells, showed the highest level of incorporation. Approximately 72% of its sialylated N-glycans possessed at least one SiaNAz residue (Fig. 2B). Collectively, SiaNAzylated structures comprised 84% of the summed abundances of all its sialylated N-glycans. Differentiated enterocytic Caco-2 cells required the highest amount of ManNAz (100 μM) but showed the lowest level of incorporation. On average, of the 252 sialylated N-glycan structures identified on Caco-2, 105 (42%) possessed one or more SiaNAz residues. Based on abundances, SiaNAz-containing N-glycans accounted for 29% of the total cell surface sialylation. The reproducibility of incorporation was validated by replicate experiments (Fig. S6†). In subsequent analyses, therefore, we centered on PNT2 and Caco-2 glycosylation.For deeper comparison of the N-glycome between treated and untreated cells, we evaluated the incorporation efficiencies of individual sialylated structures with relative abundances >0.5%. In the presence of ManNAz, the identity of cell surface glycans generally remained similar to those native to the control (Fig. 3C), suggesting that the glycan processing sialyltransferases permit the addition of modified sialic acids during synthesis of nascent glycans. Significantly, our analysis indicated that incorporation varies with structure. Based on eqn (1) (Fig. 1), incorporation levels per N-glycan ranged from 17% to 55% in Caco-2 and 76% to 97% in PNT2. Although the introduction of a monosaccharide variant to cells did not cause major deviations in glycan biosynthesis, certain azido glycans were generated at higher levels than others. SiaNAzylated structures with the highest abundances corresponded to the most abundant non-SiaNAzylated structures (Hex5HexNAc5Fuc1Sia1/Hex5HexNAc5Fuc1SiaNAz1, Caco-2; Hex5HexNAc4Fuc1Sia1/Hex5HexNAc4Fuc1SiaNAz1, PNT2). However, the abundances of structures with the highest incorporation rate were low or only moderate (Hex5HexNAc4Fuc1Sia2, Caco-2; Hex6HexNAc5Fuc2Sia2 and Hex5HexNAc5Fuc1Sia2, PNT2). The efficiency of incorporation may be dictated by features of the originating proteins.
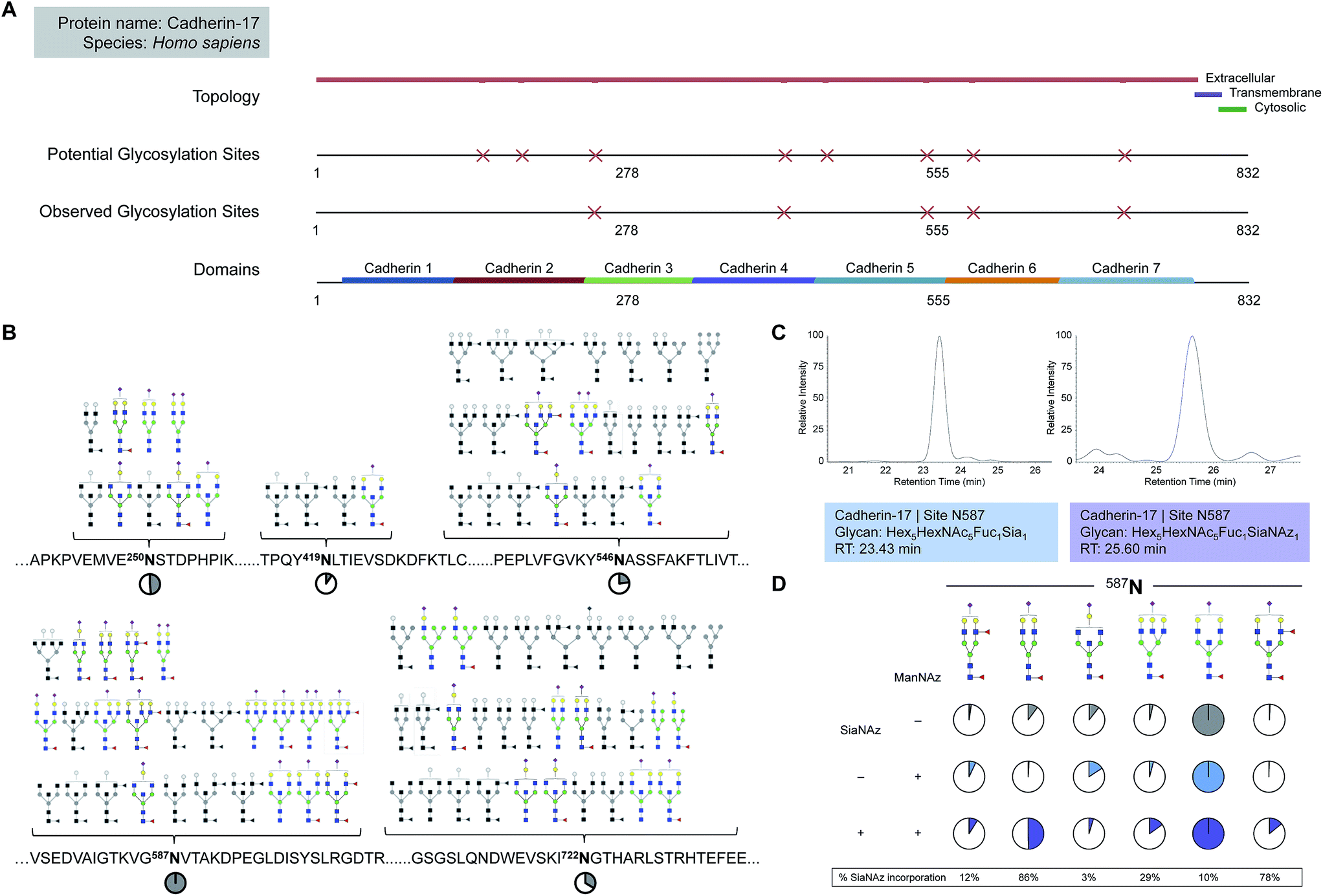 | ||
| Fig. 3 Quantitation of site-specific incorporation of SiaNAz. (A) Features of the glycosylated membrane protein, cadherin-17, shown in sequence view from position 1 to 832. (B) Site-specific glycan structures observed on cadherin-17. Non-sialylated glycoforms are drawn in grayscale. Pie charts show the intensity of each site relative to the most abundant site, N587 (denoted as 100%). (C) Chromatograms of the most abundant sialylated and SiaNAzylated glycoforms expressed on cadherin-17 at N587. (D) Site-specific abundances of sialylated glycoforms identified on cadherin-17 at N587. Pie charts indicate the contribution of each glycoform relative to the most abundant glycoform observed at each site per group. Colors differentiate between untreated and treated groups (+/− ManNAz) in addition to unmodified and modified sialic acid (+/− SiaNAz). | ||
Incorporation of SiaNAz onto O-linked glycans was in turn monitored by alkaline beta-elimination immediately following de-N-glycosylation. Interestingly, Caco-2 showed a low N-glycan incorporation rate overall but expressed SiaNAz-O-glycans with high efficiency. Of all sialylated O-glycan structures on Caco-2, about 55% incorporated SiaNAz, comprising 74% of the total sialylated O-glycan abundance (Fig. 2D). In contrast, PNT2 displayed a lower percentage of incorporated structures in its O- than its N-glycome. According to abundance, SiaNAzylated O-glycans accounted for 56% of all sialylated O-glycans. Unlike Caco-2, a major proportion of O-glycans exemplified natively on PNT2 were sialylated (84% by abundance) (Fig. S7†). In particular, a disialylated core 2 glycan (Hex2HexNAc2Sia2) was the predominant species, indicative of its association with mucin-type glycoproteins. Correspondingly, compounds that incorporated the highest amount of SiaNAz on PNT2 consisted of sialyl core 2 (Hex2HexNAc2Sia1SiaNAz1 and Hex2HexNAc2SiaNAz2) and core 1 (Hex2HexNAc2SiaNAz1) structures. In this regard, similar to N-glycan analysis, the most abundant SiaNAzylated O-glycans paralleled the most abundant sialylated O-glycans. However, percent incorporation differed by individual O-glycan structure, regardless of the abundance order (Table S1†). The variability in incorporation rates across glycan structures prompted investigation of where specific glycans are attached on membrane proteins.
Intra-protein incorporation differences
While free glycan analysis yields information about the diversity and composition of the glycan structures present in a sample, the parent protein and originating site of attachment are left unknown. Therefore, to compare between SiaNAzylated sites, we performed the study in the absence of PNGase F, preserving the site of glycan attachment, digested the proteins with trypsin, and searched specifically for glycopeptides with the SiaNAz mass label by C18-LC-MS/MS. Among the most abundant glycoproteins expressed on the Caco-2 cell membrane, cadherin-17 (CDH17) was distinctive in that all occupied sites were sialylated: N250, N419, N546, N587, and N722. Each site of glycosylation belonged to a distinctive extracellular cadherin domain (cadherin 3–7) (Fig. 3A) and uniquely expressed varying numbers of glycoforms (Fig. 3B). To calculate site-specific incorporation, we employed a label-free approach using the area under the peak (Fig. 3C) and normalized to the species with the highest abundance, according to eqn (1) (Fig. 1 and S8†).Among occupied sites, N587 was highest expressed and encompassed the highest diversity of sialylated structures (Fig. 3B). Within this site, six abundant singly SiaNAzylated glycans were identified: Hex5HexNAc4Fuc2SiaNAz1, Hex5HexNAc4Fuc1SiaNAz1, Hex4HexNAc5Fuc1SiaNAz1, Hex6HexNAc5Fuc1SiaNAz1, Hex5HexNAc5Fuc1SiaNAz1, and Hex5HexNAc5Fuc2SiaNAz1. Similar to glycomic analysis, the most abundant glycoform in untreated and treated cells was modified with a bisecting N-acetylglucosamine (GlcNAc) structure, Hex5HexNAc5Fuc1Sia1 and Hex5HexNAc5Fuc1SiaNAz1, respectively (Fig. 3B and D). A range of incorporation levels was observed for each glycoform at N587. However, the extent of SiaNAz incorporation at each site did not correlate with the order of abundance of glycans observed at a given site. For example, we observed only 10% incorporation on Hex5HexNAc5Fuc1SiaNAz1, despite its high abundance. In particular, the glycoform bearing Hex5HexNAc4Fuc1SiaNAz1 exhibited the highest level of incorporation (86%) while the glycoform bearing Hex4HexNAc5Fuc1SiaNAz1 showed the lowest (3%). Among all glycosylated sites of CDH17, the maximum level of incorporation was observed in the glycoform containing Hex5HexNAc4Fuc1SiaNAz1 on a remote site, N722. Of note, N722 is the last occupied glycosylation site of CDH17 nearest to its transmembrane domain (Fig. 3A). According to site-specific analysis, different sites on the same protein displayed a range of glycan compositions and each glycoform incorporated SiaNAz with varying efficiencies. As observed with glycan analysis, incorporation efficiencies within and between sites were not contingent on abundance order.
Site-specific map of membrane glycoproteins
Upon observing differential incorporation of SiaNAz residues on membrane glycans and between protein sites, we proposed that the general propensity for sialylation across sites may be influenced by the composition of surrounding amino acids and the chemical environment. Following the growing need to systematically define all glycosylated sites, we used a large-scale approach, which involves membrane extraction, protease digestion, glycopeptide enrichment, and informative fragmentation. Due to the low ionization efficiency of glycopeptides and suppression of ions during MS analysis, we performed high recovery pre-enrichment using iSPE®-HILIC, a newly designed hydroxyethyl amide-based matrix, which substantially outperformed other methods of enrichment (Fig. S9†). Using this technique, we observed multifold increases in the number and abundance of glycopeptides identified and more than 90% enrichment of glycopeptides over non-glycosylated peptides in the cell membrane fraction (Fig. S10†). In addition, iSPE®-HILIC resolved glycosites previously unidentified using conventional zwitterionic (ZIC)-HILIC.Often both glycan and protein information cannot be obtained in a single experiment because different energies are required to fragment the oligosaccharide and the peptide backbone. Here, we employed stepped collision energy generating ions by a combination of low, medium, and high energies. In this way, multiple component information arising from peptide, glycan, and glycopeptide fragmentation was simultaneously acquired to increase sequence coverage, identify saccharide structural motifs, and localize sites, respectively (Fig. S11†). To aid glycopeptide identifications, the compositions we elucidated with global glycan release were used to develop a focused library and bracket the search. Data were filtered strictly by protein p-value (|log prob| >2), delta mod (>10), and byonic score (>200) to eliminate false positives. Independent replicate analyses validated the identifications (Fig. S12†). As a result, we successfully characterized spectra matched to 444 N-glycan sites on 165 Caco-2 cell surface glycoproteins, giving rise to 2553 nonredundant glycoforms (Fig. 4A, and Tables S2 and S3†). Although uncommon, 47 additional, distinct glycoforms from short tryptic peptides with four or less amino acids were identified but excluded from subsequent analysis due to the ambiguity in assigning their originating protein. About 62% of all glycoproteins were classified as single-pass transmembrane proteins, of which 52% expose the N-terminus to the extracellular domain while 10% present the C-terminus (Fig. 4B). Additionally, 20% multi-pass transmembrane proteins contained polytopic loops with more than one region of the protein extending into and out of the extracellular region. The remaining 18% of identified glycoproteins were lipid-linked, peripheral, or secretory. On PNT2, 186 N-glycosites and 553 nonredundant glycoforms were identified on 112 cell surface glycoproteins in total (Tables S4 and S5†). The types of glycosylated transmembrane proteins were similar to those on Caco-2, where the majority of extracellular regions were N-terminal ends of proteins.
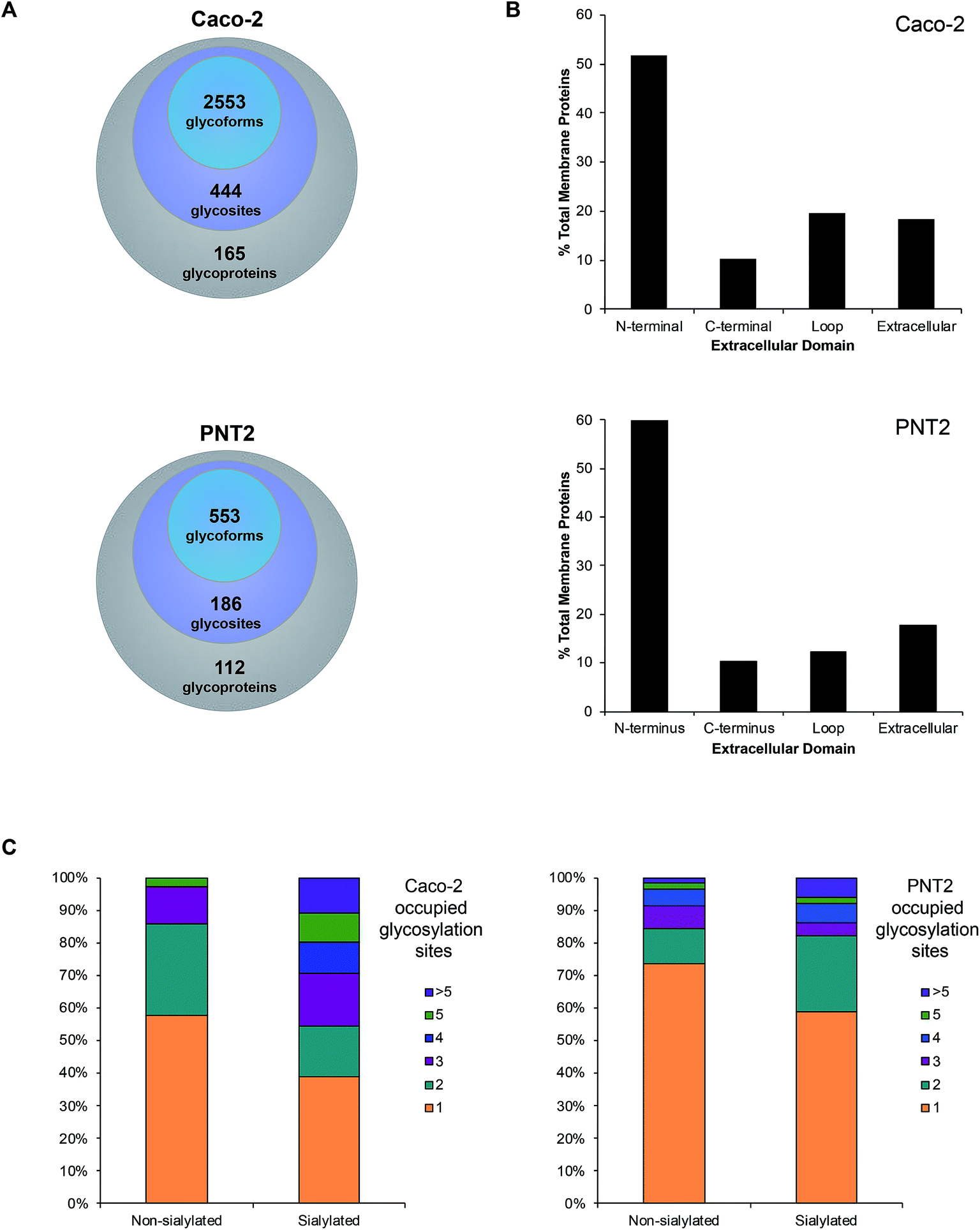 | ||
| Fig. 4 Characteristics of the total cell surface glycoproteome of Caco-2 and PNT2. (A) The number of membrane-associated proteins, sites, and glycoforms identified on Caco-2 and PNT2. (B) Classification of membrane-associated glycoproteins by the specific region(s) of the protein that is extended into the extracellular domain. (C) Comparison of the number of occupied glycosylated sites found on sialylated versus non-sialylated glycoproteins. | ||
With knowledge of the compositions of glycans associated at each site, we effectively distinguished sialylated glycoproteins from those lacking sialic acid residues. Based broadly on the respective polypeptide backbone of proteins, the presence of sialylation did not directly correlate with the properties of the naked protein alone, such as length, net charge, grand average of hydrophobicity (GRAND), or aliphatic index (Fig. S13†). With respect to molecular function, the highest percentage of proteins was classified as having binding, catalytic, or receptor activities regardless of the occurrence of sialylation. As a result of structural constraints, it has been predicted that processed complex type N-glycans occur at sites distant from disulfide bonds.27 On average, non-sialylated proteins possessed slightly more disulfide bonds than sialylated proteins. However, the number of disulfide bonds alone did not distinguish sialylated proteins from non-sialylated proteins. Notably, while the chemical and molecular features of their underlying proteins were similar, we observed that sialylated glycoproteins had higher numbers of occupied N-glycan sites than their non-sialylated counterparts. Approximately 61% of sialylated Caco-2 proteins were glycosylated at more than one site whereas 42% of non-sialylated Caco-2 proteins possessed multiple occupied sites of glycosylation (Fig. 4C). Similarly, more than one glycosylation site was occupied in about 41% of sialylated PNT2 proteins versus 26% of non-sialylated proteins. Unique to Caco-2, sialylated and non-sialylated proteins were differentiated (P < 0.05, Student's t-test) based solely on the number of occupied sites.
Sialylation dynamics of extracellular domains
Following the observation that glycosylation is limited to the extracellular space, the sequence of single pass transmembrane proteins was assigned a value ranging from 0 to 1, representing the region from the end of the transmembrane helix to the protein terminus (Fig. 5A). From this normalized pictorial representation, we observed that glycosites span broadly across the extracellular region. When we surveyed the lateral arrangement of sites with respect to the presence or absence of sialylation, sites with sialylated glycans were dispersed broadly as opposed to being polarized on any one region of the protein. By prediction modeling, the majority of glycosylated sites were located on coiled regions of the protein (Fig. S14†). We further screened each glycosylated site based on the occurrence of sialylation. Compared to non-sialylated sites, sialylated sites tended to exhibit greater heterogeneity. About 82% of sialylated Caco-2 and 72% of sialylated PNT2 sites showed more than one glycoform on each site (Fig. 5B). The maximum number of glycoforms observed on any one site was as high as 51 on sialylated Caco-2 and 25 on sialylated PNT2 membrane proteins. In comparison, at non-sialylated Caco-2 and PNT2 membrane sites, a maximum of eight and six glycoforms were observed, respectively. In general, a higher proportion of glycoproteins were identified that possessed the tri-amino acid motif Asn-Xaa-Thr than Asn-Xaa-Ser (about 1.3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, where Xaa is any amino acid except Pro) (Fig. S15†). When considering residue frequencies at surrounding positions, neither Xaa within the N-glycosylation motif sequence nor residues ±9 from the consensus motif distinguished sialylated sites from non-sialylated sites. Repeated leucine residues were more frequently located on the N-terminal side of glycosylated sites.
:1, where Xaa is any amino acid except Pro) (Fig. S15†). When considering residue frequencies at surrounding positions, neither Xaa within the N-glycosylation motif sequence nor residues ±9 from the consensus motif distinguished sialylated sites from non-sialylated sites. Repeated leucine residues were more frequently located on the N-terminal side of glycosylated sites.
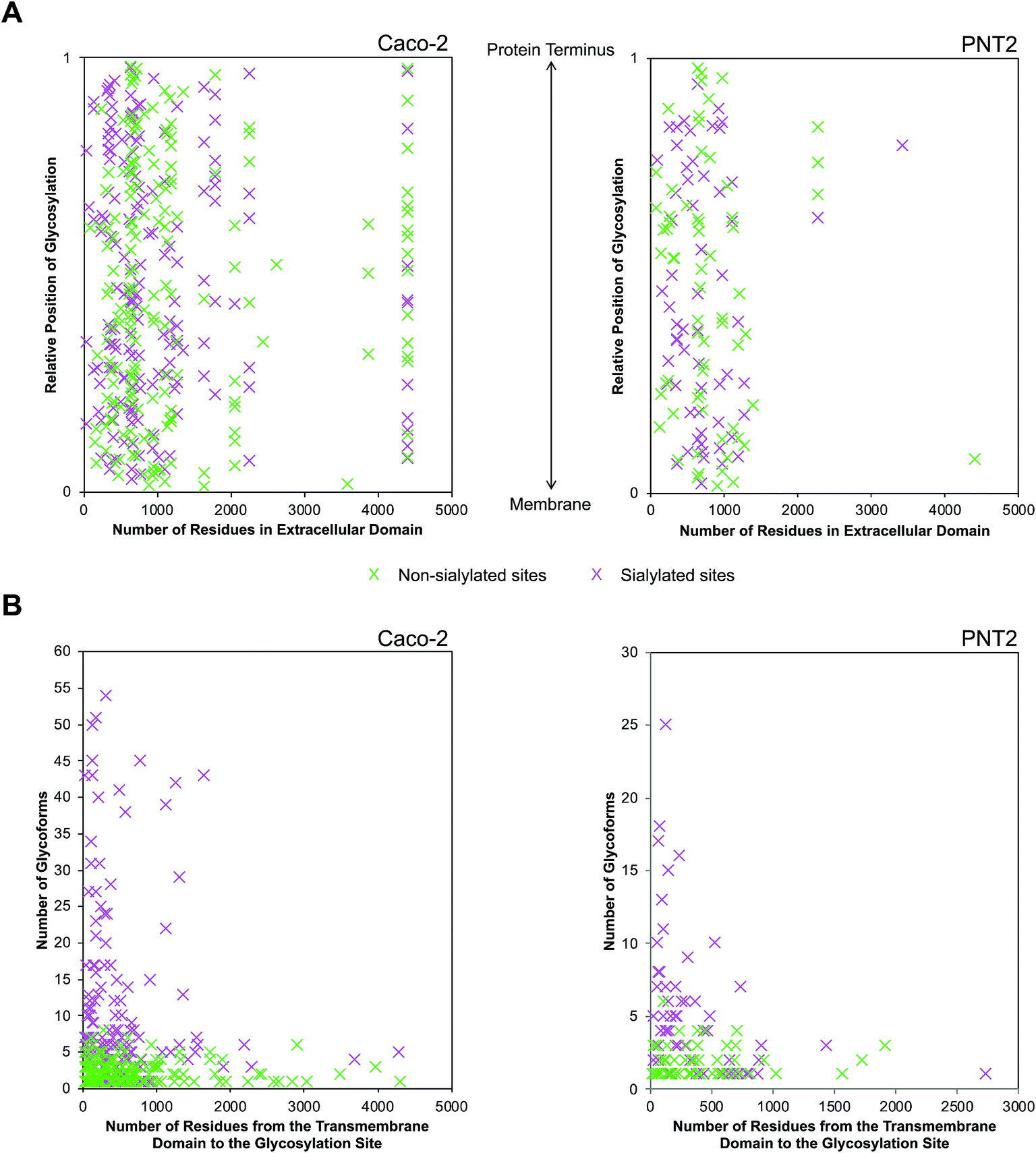 | ||
| Fig. 5 Distinctions between sialylated and non-sialylated sites of glycosylation on the extracellular domain. (A) Graphical representation of occupied sites of glycosylation specific to the extracellular domain. The relative position of glycosylation was given a value between 0 and 1, representing the transmembrane domain and the protein terminus, respectively. Sialylated and non-sialylated sites are indicated as green and pink markers, respectively. (B) Graphical representation of the number of glycoforms with respect to the position of their glycosylation site along the extracellular domain. | ||
Provided that sialylated sites are dispersed throughout the extracellular domain, we assessed whether the abundances of glycoforms were spatially related. Strikingly, for single pass transmembrane proteins, sites bearing the most number of glycoforms were found nearer to the transmembrane domain (Fig. 5B). Sites that were more than 1700 residues away from the transmembrane domain possessed less than ten glycoforms. From this analysis, we observed that the number of distinct glycoforms produced per site was influenced by the location of the extracellular glycosylation site relative to the transmembrane domain as well as the presence of sialylation. The same trend existed on high and low abundance glycoproteins and was independent of protein family. Graphical representations of glycoforms expressed along the extracellular domain on key membrane proteins are depicted in Fig. S16.†
Profiling sialylated glycosphingolipid diversity
Sialylation is a major component of glycosphingolipids (GSLs) interspersed across the cell membrane. These distinctive structures are composed of a polar head group (glycan), presented on the outer face of the membrane, and a hydrophobic tail (ceramide), which inserts into the phospholipid bilayer. The head group is assembled by a subset of specific glycosyltransferases and contains multiple potential sites of sialylation via glycosidic linkages to itself and/or galactose (terminal or sandwiched). We isolated intact GSLs from Caco-2 and PNT2 cell membranes to determine the extent of SiaNAz incorporation at the lipid level as compared to the observed rates at the N-/O-glycan and site levels. All identifications were verified by MS/MS (Fig. S17 and S18†). Untreated PNT2 exhibited high amounts of mono- and di-sialylated GSLs (Fig. 6A and Table S6†). Strikingly, unlike the patterns of membrane protein glycosylation, fucosylated GSLs were not observed. The most common head group was composed of Hex2HexNAc1Sia1 (GM2) while the most abundant ceramide tail was 42 carbons in length (Fig. 6B). The ceramide tail of all PNT2 GSLs was unsaturated and di-hydroxylated. Most of these species (80%) possessed one degree of unsaturation. In contrast, with a total of 53 unique sialylated GSL compositions, Caco-2 showed higher diversity than PNT2 in the types of GSLs embedded in the membrane (Fig. 6B and Table S7†). With regard to glycosylation, only monosialylated GSLs were observed on Caco-2. Comprising 78% of all glycosylated head groups, the most abundant sugar moiety was composed of the GM1 structure Hex3HexNAc3Sia1. The number of carbon atoms in the hydrophobic tail of any given GSL on Caco-2 varied from 32 to 44. Among the most abundant GSLs, nearly 40% possessed ceramides of 42-carbon units and 33% possessed ceramides of 34-carbon units in length. Intriguingly, hydroxylation was a key feature of all Caco-2 GSLs. Approximately 40% were mono-hydroxylated, 55% were tri-hydroxylated, and 0.1% possessed four hydroxyl groups. Moreover, the bulk of the ceramide tails were unsaturated (74%). Of these, 53% were mono-unsaturated and 21% were di-unsaturated. Di-unsaturation occurred more frequently in ceramides with 42 or more carbon units.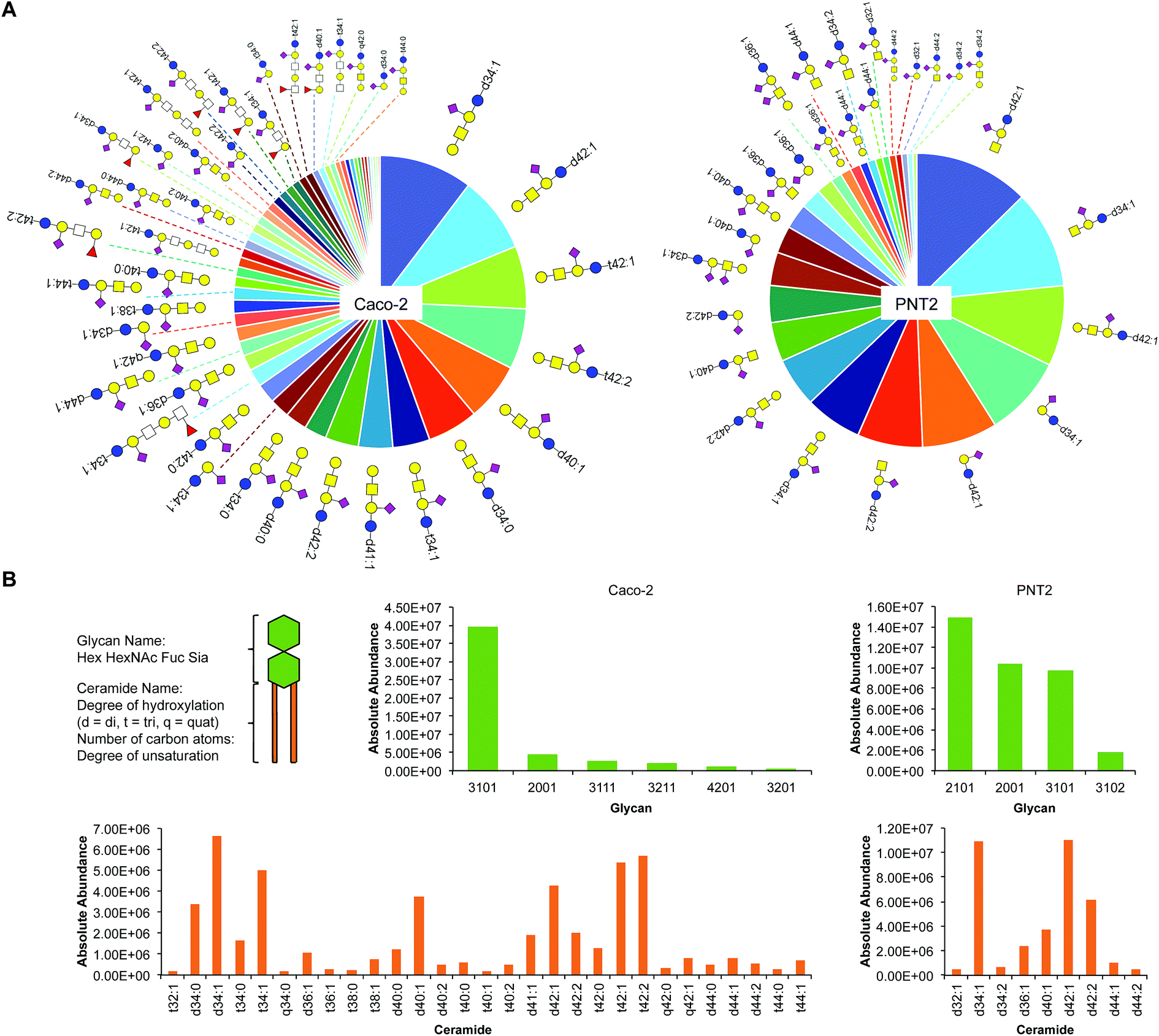 | ||
| Fig. 6 Glycosphingolipid (GSL) identification, quantitation, and classification. (A) Glycosphingolipid wheel depicting elucidated sialylated structures on Caco-2 and PNT2 by order of abundance (Tables S6 and S7†). Open squares represent positions where either N-acetylglucosamine (GlcNAc) or N-acetylgalactosamine (GalNAc) can be attached. (B) Comparison of sialylated GSLs (sGSLs) based separately on the polar head group and the ceramide tail. | ||
After ManNAz treatment, we observed complete conversion of sGSLs (sialylated GSLs or gangliosides) into SiaNAzylated GSLs on PNT2. Incorporation was highly efficient, where all identified sGSLs possessed one or more SiaNAz residues. The extent of incorporation varied with individual structure. In particular, the highest level of incorporation (92%) was observed in a GD1-based structure, Hex3HexNAc1NeuAc2/d34:1 (Fig. 7A), whereas a GM1-based structure, Hex3HexNAc1NeuAc1/d44:2, showed the lowest level of incorporation (42%). In all disialylated GSLs, we observed both singly and doubly incorporated structures. Similar to what was observed in protein glycosylation, the most abundant GSLs tended to express higher abundances of SiaNAz. In total, SiaNAzylated species accounted for 75% of the abundances of sGSLs (Fig. 7B). In Caco-2, 30 of the 53 (57%) sGSL compositions incorporated SiaNAz (Fig. 7B). Based on the summed abundances, an average incorporation rate of 8% was observed across all SiaNAzylated species after 72 h. The highest abundant SiaNAzylated GSL compositions were likewise the highest abundant non-SiaNAzylated GSL compositions (Fig. 7A).
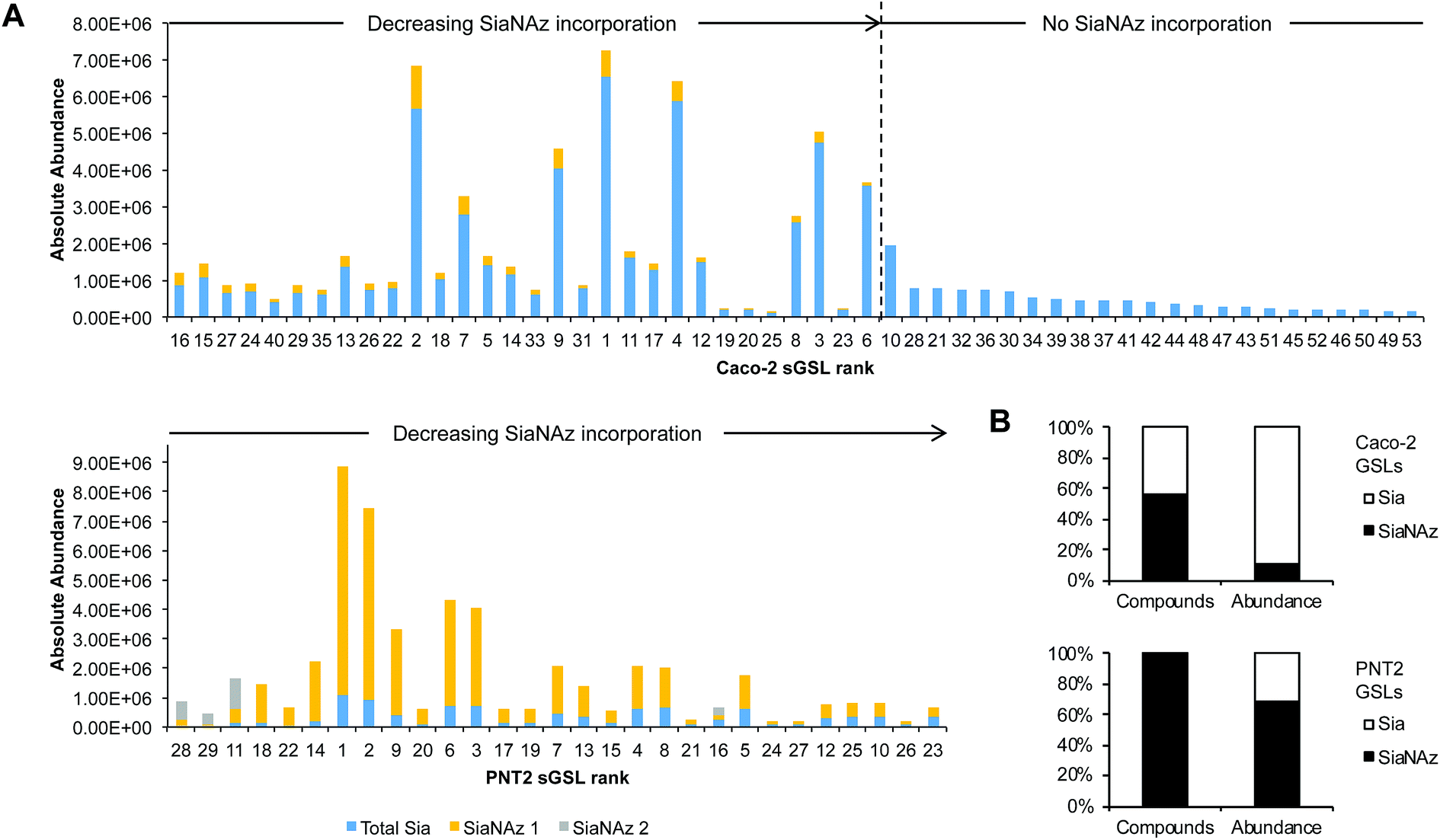 | ||
| Fig. 7 Quantitation of SiaNAz incorporation onto glycosphingolipids. (A) Variations in percent incorporation of SiaNAz onto individual sGSLs. Bar graphs are arranged by order of incorporation (high to low, left to right). Rank refers to GSLs by order of abundance in untreated controls. (B) Net incorporation of SiaNAz onto GSLs according to the number of compounds and relative abundance. | ||
Discussion
Sialylation occurs through the coordinated action of enzymes that transfer free sialic acids onto the terminal position of nascent glycans. The heterogeneity of potentially modified structures is vast, derived from differences in linkage positions, connectivity, and multimerity. An important aspect of our initial global analysis approach was porous graphitized carbon (PGC)-LC to separate the assortment of released N- and O-glycans, which encompasses sialylated as well as non-sialylated structures, according to parameters such as size, polarity, and linkage. We demonstrate that SiaNAzylated species are uniquely separated and use their retention differences as time stamps for ease of subsequent identification and quantitation. Furthermore, the application of a microfluidic nanoflow chip-based system permitted minimal injection volumes (μL), enabling avenues for high throughput screening.The overall efficiency of azido sialic acid incorporation differed by cell origin and glycan structure, predictably under the control of cell-specific factors such as glycosyl enzyme activity and monosaccharide metabolism. However, further analyses of protein structure and dynamics are needed to understand the causes underlying the notable differences in incorporation efficiencies between N- and O-glycans in addition to the differences observed between protein glycosylation sites. For example, turnover kinetics and residue accessibility could substantially affect the rate of monosaccharide incorporation. In particular, incorporation of metabolized sugars onto mucin-type glycoproteins, which are densely packed with repeating units of O-glycans, may require a different time scale than is necessary for glycoproteins with less density. Exogenous carbon sources have indeed been shown to affect mucin biosynthesis.28,29 Relatedly, disparities in the absolute amounts of N- and O-glycans present on cell surfaces may account for differences in incorporation. Future studies involve distinguishing sialyl linkages and introducing other unnatural modifications while quantifying incorporation rates. At large, it is important for metabolic labeling experiments to control cell culture parameters, which have been shown to meaningfully affect glycan expression.30 Enhancing the understanding of single cell metabolism will provide additional insight into maximizing cell surface sugar incorporation.
We and others have shown that the parent protein plays a pivotal role in defining the biological significance of its glycovariants such that its function cannot be recapitulated by the glycan(s) alone.31–33 Hence, it is important to regard the glycan as a component of a specific protein. Based on a full-length comparison between sialylated and non-sialylated proteins, amino acid sequence composition alone did not play a significant role in determining the predisposition to sialylation. Similar to the full-length protein analysis, features of the surrounding amino acids (local features), including hydropathy and amino acid residue frequency, failed to distinguish sialylated sites from non-sialylated sites. Laterally along the extracellular domain, occupied glycosylated sites on membrane-spanning proteins were broadly spaced as opposed to being polarized on the terminus. While the folded protein conformation may shape where glycosylation occurs on a protein, many structures remain unsolved. In addition, available crystal structures often exclude glycans and thus may not reflect the actual fold. In fact, comparative studies have shown that the conformation of a glycoprotein is significantly altered once glycosylation is removed.34,35 Bioinformatic tools predict that most of the glycosylation sites we elucidated in this study occur on coiled configurations of the protein. Three-dimensional structural characterization of glycoproteins in their native forms are needed to preserve the actual folded state and depict the spatial arrangement of sialic acids on the cell membrane with respect to nearby molecules. Site-specific maps obtained from our analysis provide details to assist in correlating sites of sialylation with the protein structure and conserved functional motifs.
Another interesting finding was garnered upon determining the heterogeneity of glycoforms associated with each site, which provided a more complete representation of native glycoproteins. Notably, sialylated sites possessed more glycoforms than their non-sialylated counterparts. Moreover, sites with greater heterogeneity were generally located closer to the transmembrane domain than to the protein terminus. While such site-specificity was not observed in all membrane glycoproteins, it was particularly prominent in those with an unusually high number of glycoforms (>10). Based on this intriguing trend, the glycosylation status of the transitional region between a glycoprotein's extracellular and transmembrane domains may contain information regarding its three-dimensional structure. Glycans mature after being transferred to their parent protein in the ER and Golgi. For initiation, the site of glycosylation must be present in the luminal side of the ER. Oligosaccharyltransferase (OST) then recognizes the consensus sequence and is responsible for the en bloc transfer of a glycan precursor onto the polypeptide substructure of the acceptor. It is known that the transmembrane domain of proteins lacks glycosylation as the region is inaccessible to OST. Given the nature of the biosynthetic pathway, glycosylated sites nearest to the transmembrane domain may be preferred sites for processing enzymes as they are exposed or energetically favorable for recognition. Although structural aspects in combination with enzymatic activity may play a role in promoting a site-specific increase in glycoforms, the implications of the generated diversity remain unclear.
Several caveats remain with the methods described here. Indeed, we have elucidated various new sites and glycoforms on cells but the identifications are not exhaustive. Additional identifications may be obtained, for example, by increasing yields in membrane protein extraction and by utilizing different cocktails of specific and non-specific proteases. To enable sufficient identifications without enrichment strategies, enhanced selection of glycopeptides during the LC-MS/MS experiment and data-independent tools need to be improved. The current study affords a detailed view of cell surface glycoproteins. A focused study of the glycosylation patterns that occur on intracellular high molecular weight transmembrane proteins is merited.
Apart from our efforts to characterize glycoproteins, we also tracked the incorporation of SiaNAz onto glycosphingolipids. Due to their amphipathic nature, their isolation and analysis have posed a significant challenge. We provide a comprehensive view of the types of sGSLs that exist on cells and therefore new insight into the total sialo-glycome of the cell surface. Glycosphingolipids participate in adhesion of adjoining cells via the carbohydrate moieties present on the outer layer of the plasma membrane.36,37 We identified the relevant surface molecules on model intestinal and prostate epithelial cells and demonstrate that incorporation of exogenous sialic acids can be exceptionally efficient. These results promote future studies harnessing the participating sialyltransferases. The localization of sGSLs along the membrane is difficult to define as they are not homogenously distributed.38 In fact, these molecules are predicted to be the main occupants of specialized regions named lipid rafts, which are dynamic in size and composition depending on the stimuli imposed on the cell.39,40 The clustering of glycans in these microdomains may augment opportunities for recognition by nearby biomolecules.
Sialylated GSLs not only effectively mediate cell adhesion but also activate cell signaling pathways via the ceramide portions.41 The GSL tail has two components, the sphingoid base and the fatty acid, linked together by an amide bond. Interestingly, the number of hydroxyl groups is variable in response to cell conditions and occurs at markedly higher frequencies in intestinal cells. Indeed, we observed GSLs with >2 hydroxyl group modifications on Caco-2 which were absent on PNT2. Furthermore, we observed rare structures with ceramide tails that seemingly possess an odd number of carbon atoms. It is postulated that these structures include odd-numbered fatty acids, some of which may have branched chains.42 Ceramides can thus be distinguished by the carbon length, hydroxyl group modifications, and the degree of unsaturation. The degree of unsaturation influences the ability of the tail to pack into the membrane,43 conferring rigidity, and may also govern the rates of glycolipid recycling or shedding from the membrane.44 Analyses are ongoing to probe the biological significance of the mixture of sGSLs in these contexts.
Analytical advancements, besides characterizing manufactured drug candidates, have the potential to guide the design of therapeutics with coordinated binding to specified regions of proteins. Hypersialylation, in particular, is a prevalent feature of disease progression, reported in cases of cancer, diabetes, and rheumatoid arthritis.45–48 Moreover, the level of sialic acid expression significantly influences the biological activity of biopharmaceutical proteins49,50 and potentiates binding of cell surface receptors.51,52 Controlling the frequency, efficiency, and site specificity of sialylated determinants may thus create advantageous binding opportunities for the targets and/or therapeutics.
Conclusions
In addition to the multiplicity of occupied sites on a given glycoprotein, the number of glycoforms that exist within a single site adds sufficient challenge to glycoconjugate characterization and manufacturing. Herein we used metabolic incorporation of unnatural sialic acids as a method for installing a unique mass label on cell surfaces to investigate the tendency of sialylation. This technique allowed for extensive characterization of membrane-associated glycoproteins and glycolipids, revealing cellular, structural, and spatial preferences for sialic acid expression. The methods presented here can probe structural diversity and be applied towards the design of therapeutics, including those to block or accentuate binding of key glycan–protein interactions that mediate cellular homeostasis, cancer, and immunity.Experimental
Cell culture
Caco-2 intestinal epithelial cells were obtained from the American Type Culture Collection (ATCC, VA) and grown 14 days post-confluence in Eagle's Minimum Essential Medium (EMEM) supplemented with non-essential amino acids, 1 g L−1D-glucose, 2 mM L-glutamine, 10% (v/v) fetal bovine serum, and 100 U mL−1 penicillin and streptomycin. KKU-213 and KKU-213L5 cholangiocarcinoma cells were obtained from the Japanese Collection of Research Bioresources Cell Bank (JCRB, Osaka, Japan) and grown in Ham's F12 medium supplemented with non-essential amino acids, 1.8 g L−1D-glucose, 1 mM L-glutamine, 10% (v/v) fetal bovine serum, and 100 U mL−1 penicillin and streptomycin. PNT2 prostate epithelial cells were grown in RPMI-1640 medium supplemented with non-essential amino acids, 2 g L−1D-glucose, 2 mM L-glutamine, 10% (v/v) fetal bovine serum, and 100 U mL−1 penicillin and streptomycin. HT-29 colorectal epithelial cells were obtained from ATCC and grown 7 days post-confluence in McCoy's 5A medium supplemented with non-essential amino acids, 3 g L−1D-glucose, 1.5 mM L-glutamine, 10% (v/v) fetal bovine serum, and 100 U mL−1 penicillin and streptomycin. All cells were maintained at 37 °C with 5% CO2 in a humidified incubator.Metabolic labeling of sialic acid-containing glycans
Metabolic labeling of cells has been described previously.15,53,54 In brief, per-O-acetylated azido N-acetylmannosamine (ManNAz) (Invitrogen, CA) was solubilized in dimethyl sulfoxide (DMSO) at 100X and added to the growing media (final concentration of DMSO <0.1%). Cells were incubated at 37 °C with 5% CO2 for an additional 72 h, washed three times with PBS, and harvested by scraping.Cell membrane extraction
Details of the isolation of the cell membrane fraction have been described previously.55 Briefly, harvested cell pellets were resuspended in homogenization buffer containing 0.25 M sucrose, 20 mM HEPES-KOH (pH 7.4), and a 1:100 protease inhibitor cocktail (EMD Millipore, CA). Cells were lysed on ice using a probe sonicator (Qsonica, CT) operated with alternating on and off pulses of 5 and 10 s, respectively. Lysates were pelleted by centrifugation at 2000 × g for 10 min to remove the nuclear fraction and unlysed cells. The supernatant was transferred to high speed tubes (Beckman Coulter, CA), loaded onto a Beckman Optima TLX Ultracentrifuge at 4 °C, and centrifuged at 200000 × g for 45 min in series to remove other non-membrane subcellular fractions. The resulting membrane pellet was stored at −20 °C until further processing.
Real-time PCR analysis
Quantitative real-time PCR was performed as described previously.55,56 Cells were harvested by scraping, washed twice with PBS, and resuspended in RNAlater (Life Technologies, CA). Total RNA was extracted using an RNeasy plus mini kit (Qiagen, CA) and the quantity and quality of RNA were determined by using a Qubit Fluorometer (Life Technologies) and TapeStation 2200 (Agilent Technologies, CA) following the manufacturer's protocol. Total RNA was reverse transcribed to cDNA using the iScript Reverse Transcription Supermix (Bio-Rad Laboratories, CA) following the manufacturer's instructions. Predesigned human glycosylation PrimePCR plates (Bio-Rad Laboratories) were used for real-time PCR using the CFX96 Touch Real-Time PCR detection system (Bio-Rad Laboratories). Analysis was performed using CFX Manager 3.1 software (Bio-Rad Laboratories). Gene expression was normalized to reference genes GAPDH, TBP, and HPRT1.Preparation of N-glycans
Proteins were suspended with 100 μL of 100 mM NH4HCO3 in 5 mM dithiothreitol and heated at 100 °C for 10 s to thermally denature the proteins. To release the glycans, 2 μL of peptide N-glycosidase F (New England Biolabs, MA) were added to the samples, which were then incubated in a microwave reactor (CEM Corporation, NC) at 20 watts for 10 min. After addition of 400 μL of cold ethanol, samples were incubated at −80 °C for 1.5 h to precipitate deglycosylated proteins and centrifuged at 21000 × g for 20 min. The precipitated proteins were dried and stored at −20 °C for O-glycan preparation and analysis. The supernatant containing N-glycans was collected and dried. The released N-glycans were purified by solid-phase extraction using porous graphitized carbon (PGC) packed cartridges (Grace, IL). The cartridges were first equilibrated with nanopure water and a solution of 80% (v/v) acetonitrile and 0.05% (v/v) trifluoroacetic acid in water. The dried samples were solubilized, loaded onto the cartridge, and washed with nanopure water at a flow rate of 1 mL min−1 to remove salts and buffer. N-Glycans were eluted with a solution of 40% (v/v) acetonitrile and 0.05% (v/v) trifluoroacetic acid in water and dried prior to mass spectrometric analysis.
Preparation of O-glycans
Dried de-N-glycosylated proteins were suspended in 90 μL of nanopure water and sonicated for 20 min. Subsequently, 10 μL of 2 M NaOH and 100 μL of 2 M NaBH4 were added and mixed. The mixture was incubated at 45 °C for 18 h before cold 10% acetic acid was added slowly on ice to adjust the pH to 5–7. After centrifugation at 21000 × g for 20 min, the supernatant containing free O-glycans was desalted using PGC cartridges as described above. The eluted O-glycans were dried and further purified using iSPE®-HILIC cartridges (Nest Group, MA). The cartridges were conditioned with acetonitrile, 0.1% (v/v) trifluoroacetic acid in water, and 89% (v/v) acetonitrile with 1% (v/v) trifluoroacetic acid in water. The dried samples were solubilized and loaded onto the column. The flow-through was collected and reloaded two times. The column was then washed with 89% (v/v) acetonitrile with 1% (v/v) trifluoroacetic acid in water. Purified O-glycans were eluted with a solution of 0.1% (v/v) trifluoroacetic acid in water and dried prior to mass spectrometric analysis.
Glycomic LC-MS/MS analysis
Glycan samples were reconstituted in nanopure water and analyzed using an Agilent nano-LC/ESI QTOF MS system (Agilent Technologies, CA). The samples were introduced into the MS with a microfluidic chip, which consists of a 40 nL enrichment and 43 × 0.075 mm ID analytical columns packed with porous graphitized carbon and a nanoelectrospray tip. Separation was carried out at a constant flow rate of 0.4 μL min−1: (A) 3% (v/v) acetonitrile and 0.1% (v/v) formic acid in water and (B) 90% (v/v) acetonitrile in 1% (v/v) formic acid in water. MS spectra were acquired at 1.5 s per spectrum over a mass range of m/z 500–2000 in positive ionization mode. Mass inaccuracies were corrected with reference m/z 1221.991.Collision-induced fragmentation was performed with nitrogen gas using a series of collision energies (Vcollision) dependent on the m/z values of the N-glycans, based on the equation Vcollision = m/z (1.8/100 Da) V–2.4 V.
Glycomic data analysis
N-Glycan compositions were identified according to accurate mass using an in-house retrosynthetic library constructed based on the knowledge of the mammalian N-glycan biosynthetic pathway.57 SiaNAz-labeled glycans were identified by the presence of fragment ions m/z 315.09 [SiaNAz − H2O + H]+ and m/z 333.10 [SiaNAz + H]+. Amino sialic acid (SiaNAm)-containing O-glycans resulting from the reduction of SiaNAz were identified by the presence of fragment ions m/z 289.10 [SiaNAm − H2O + H]+ and m/z 307.11 [SiaNAm + H]+. Signals above a signal-to-noise ratio of 5.0 were filtered and deconvoluted using MassHunter Qualitative Analysis B.06.01 (Agilent, CA). Deconvoluted masses were compared to theoretical masses using a mass tolerance of 20 ppm and a false discovery rate of 0.6%. Relative abundances were determined by integrating peak areas for observed glycan masses and normalizing to the summed peak areas of all glycans detected.Preparation of glycopeptides
Proteins were denatured in 8 M urea at 55 °C, reduced with 18 mM dithiothreitol, alkylated with 27 mM iodoacetamide, diluted to 1 M urea with 50 mM ammonium bicarbonate, and incubated with 1 μg trypsin at 37 °C overnight. The resulting peptides were concentrated in vacuo. Glycopeptides were enriched by solid-phase extraction using ZIC-HILIC or iSPE®-HILIC cartridges (Nest Group, MA). Cartridges were conditioned with acetonitrile, 0.1% (v/v) trifluoroacetic acid in water, followed by 1% (v/v) trifluoroacetic acid and 80% (v/v) acetonitrile in water. Peptides were loaded onto the column (×3) and washed with 1% (v/v) trifluoroacetic acid and 80% (v/v) acetonitrile in water. The enriched products were eluted with a solution of 0.1% (v/v) trifluoroacetic acid in water and dried prior to mass spectrometric analysis.Glycoproteomic LC-MS/MS analysis
Samples were loaded using 2% (v/v) acetonitrile and 0.1% (v/v) trifluoroacetic acid in water and separated using a reverse-phase Michrom Magic C18AQ column (200 μm, 150 mm) coupled with a Q Exactive Plus Orbitrap mass spectrometer through a Proxeon nano-spray source (Thermo Scientific, CA). A binary gradient was applied using 0.1% (v/v) formic acid in (A) water and (B) acetonitrile: 0–75 min, 5–35% (B); 75–82 min, 35–80% (B); 82–84 min, 80% (B); 84–85 min, 80–5% (B). Per acquisition, the instrument was run in data-dependent mode as follows: spray voltage, 2.2 kV; ion transfer capillary temperature, 200 °C; full scan mass range, m/z 700–2000; MS automatic gain control, 1 × 106; MS maximum injection time, 30 ms; MS/MS automatic gain control, 5 × 104; MS/MS maximum injection time, 50 ms; dynamic exclusion, 10 s; precursor resolution, 70000; product ion resolution, 17500; precursor ion isolation width, 1.6 m/z; stepped normalized collision energy, 17, 27, 37.
Glycoproteomic data analysis
Raw data were exported using xCalibur version 2.0 (Thermo Scientific, CA). Proteins were identified using Byonic version 2.7.4 (Protein Metrics, CA) against the reviewed Swiss-Prot human protein database with sample-specific parameters as determined from Preview (Protein Metrics, CA): mass tolerances of 5–10 ppm for the precursor and 10–20 ppm for fragment ions; carbamidomethylation of cysteine as a fixed modification; oxidation of methionine and proline, deamidation of asparagine and glutamine, methylation of lysine and arginine, and acetylation of the protein N-terminus as variable modifications; N-glycosylation of asparagine (in-house human database or custom list derived from released glycans); two missed cleavage sites. Identifications were filtered with a 1% false discovery rate (FDR) and were accepted if the following conditions were met: |log prob| >2; delta mod >10; byonic score >200. SiaNAz-labeled peptides were identified using a SiaNAzylated N-glycan database (383 entries, multiple Sia and SiaNAz combinations allowed) and validated by the presence of fragment ions m/z 315.09 [SiaNAz − H2O + H]+ and m/z 333.10 [SiaNAz + H]+. The secondary structure of glycosylated sites was predicted by NetSurfP version 1.1.58 Functional annotation analysis was performed using the PANTHER database.59 Grand average of hydropathicity (GRAVY) values were calculated from the hydropathy values of each amino acid residue in the sequence.60 Amino acid frequency plots were generated using WebLogo version 2.8.2.61 Glycopeptide fragmentation patterns were annotated using Byonic or pGlyco.62 Individual protein abundances were normalized to the total intensity of membrane proteins using Byonic. Statistical analysis was performed using a two-tailed Student's t-test.Site-specific quantitation was performed using the area under the curve. All abundances were normalized to the maximal of each component as follows: the abundance of each site within a protein was normalized to the most abundant occupied site on the protein; the abundance of each glycoform within a site was normalized to the abundances of the most abundant glycoform observed on the given site.
Glycosphingolipid extraction
Glycosphingolipid extraction was performed with modifications from previous reports.63 To each prepared membrane fraction of untreated and ManNAz-treated Caco-2 and PNT2 cells, a 500 μL solution of water:methanol:chloroform (3:8:4) was added. Samples were vortexed and centrifuged at 21000 × g for 1 min. The supernatant was collected into a separate vial. The extraction solution was added to the remaining precipitate and the procedure repeated. Supernatants from the first and second extractions were combined. To facilitate phase separation, 100 μL of 1 M KCl was added to the solution. The upper layer was collected and dried in vacuo. GSLs were then purified by solid-phase extraction using C8 cartridges (Supelco, PA) and concentrated in vacuo prior to mass spectrometric analysis.
Glycolipidomic LC-MS/MS analysis
Purified glycosphingolipid samples were reconstituted in methanol:water (1:1, v/v) and analyzed using an Agilent nano-LC/ESI QTOF MS system (Agilent, CA). Samples were loaded onto the enrichment column packed with ZORBAX C18 (5 μm) at 3 μL min−1. Separation was carried out on the analytical column at a constant flow rate of 0.3 μL min−1 using a binary gradient as follows: (A) 20 mM ammonium acetate and 0.1% (v/v) acetic acid in water; (B) 20 mM ammonium acetate and 0.1% (v/v) acetic acid in methanol:isopropanol (85:15, v/v); 70–75% (B), 0–1 min; 75–85% (B), 1–4 min; 85–100% (B), 4–40 min; 100% (B), 40–55 min; 100–70% (B), 55–58 min; 70% (B), 58–60 min. Per acquisition, one MS scan was followed by five MS/MS scans in data-dependent mode. Collision-induced dissociation was performed using a series of collision energies based on optimized conditions according to the formula Vcollision = m/z (1.2/100 Da) + 12.
Glycolipidomic data analysis
Data analysis was done with MassHunter Qualitative Analysis B.07 and Profinder B.08 (Agilent, CA). GSL compounds were identified with the find by molecular feature function, and filtered through an in-house library of GSL compositions. The glycan and lipid composition of the GSLs was further confirmed through tandem MS fragmentation spectra. A library that includes the chemical formulas and retention times of identified GSLs was composed from the data exported from MassHunter. This library was then used in Profinder to perform batch targeted feature extraction and to quantitate the peak areas of the identified GSL compounds. The program included both proton and ammonium adducts in its integration of compound signals. A mass tolerance of 10 ppm was applied for signal extraction. Peak smoothing using a Gaussian function was applied prior to integration.Data accessibility
Raw MS data and annotated MS/MS spectra have been uploaded to the MASSIVE repository under access ID MSV000082036.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Ling-Yu Wang and Hsing-Jien Kung of the University of California, Davis School of Medicine for providing PNT2 cells and Anthony Herren of the University of California, Davis for assistance with Orbitrap performance. This work was supported by the National Institutes of Health under award numbers R01GM049077 (to C. B. L.) and DP2OD008752 (to E. M.).References
- E. R. Vimr, ISRN Microbiol., 2013, 2013, 816713 CrossRef PubMed.
- S. Kelm and R. Schauer, Int. Rev. Cytol., 1997, 175, 137–240 CrossRef PubMed.
- N. M. Varki and A. Varki, Lab. Invest., 2007, 87, 851–857 CrossRef PubMed.
- C. Traving and R. Schauer, Cell. Mol. Life Sci., 1998, 54, 1330–1349 CrossRef PubMed.
- C. Bull, M. H. den Brok and G. J. Adema, Biochim. Biophys. Acta, 2014, 1846, 238–246 Search PubMed.
- P. V. Chang, X. Chen, C. Smyrniotis, A. Xenakis, T. Hu, C. R. Bertozzi and P. Wu, Angew. Chem., 2009, 48, 4030–4033 CrossRef PubMed.
- S. T. Laughlin, N. J. Agard, J. M. Baskin, I. S. Carrico, P. V. Chang, A. S. Ganguli, M. J. Hangauer, A. Lo, J. A. Prescher and C. R. Bertozzi, Meth. Enzymol., 2006, 415, 230–250 CrossRef PubMed.
- M. R. Bond, H. Zhang, P. D. Vu and J. J. Kohler, Nat. Protoc., 2009, 4, 1044–1063 CrossRef PubMed.
- N. W. Charter, L. K. Mahal, D. E. Koshland Jr and C. R. Bertozzi, Glycobiology, 2000, 10, 1049–1056 CrossRef PubMed.
- S. Hakomori, Curr. Opin. Immunol., 1991, 3, 646–653 CrossRef PubMed.
- S. Nakamori, M. Kameyama, S. Imaoka, H. Furukawa, O. Ishikawa, Y. Sasaki, T. Kabuto, T. Iwanaga, Y. Matsushita and T. Irimura, Cancer Res., 1993, 53, 3632–3637 Search PubMed.
- C. M. Nycholat, R. McBride, D. C. Ekiert, R. Xu, J. Rangarajan, W. Peng, N. Razi, M. Gilbert, W. Wakarchuk, I. A. Wilson and J. C. Paulson, Angew. Chem., 2012, 51, 4860–4863 CrossRef PubMed.
- C. Ohyama, S. Tsuboi and M. Fukuda, EMBO J., 1999, 18, 1516–1525 CrossRef PubMed.
- A. Takada, K. Ohmori, T. Yoneda, K. Tsuyuoka, A. Hasegawa, M. Kiso and R. Kannagi, Cancer Res., 1993, 53, 354–361 Search PubMed.
- S. T. Laughlin and C. R. Bertozzi, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 12–17 CrossRef PubMed.
- K. W. Dehnert, B. J. Beahm, T. T. Huynh, J. M. Baskin, S. T. Laughlin, W. Wang, P. Wu, S. L. Amacher and C. R. Bertozzi, ACS Chem. Biol., 2011, 6, 547–552 CrossRef PubMed.
- M. Sawa, T. L. Hsu, T. Itoh, M. Sugiyama, S. R. Hanson, P. K. Vogt and C. H. Wong, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 12371–12376 CrossRef PubMed.
- W. Qin, K. Qin, X. Fan, L. Peng, W. Hong, Y. Zhu, P. Lv, Y. Du, R. Huang, M. Han, B. Cheng, Y. Liu, W. Zhou, C. Wang and X. Chen, Angew. Chem., 2017, 57, 1817–1820 CrossRef PubMed.
- C. M. Woo, A. T. Iavarone, D. R. Spiciarich, K. K. Palaniappan and C. R. Bertozzi, Nat. Methods, 2015, 12, 561–567 CrossRef PubMed.
- J. M. Smeekens, W. Chen and R. Wu, J. Am. Soc. Mass Spectrom., 2015, 26, 604–614 CrossRef PubMed.
- D. R. Spiciarich, R. Nolley, S. L. Maund, S. C. Purcell, J. Herschel, A. T. Iavarone, D. M. Peehl and C. R. Bertozzi, Angew. Chem., 2017, 56, 8992–8997 CrossRef PubMed.
- N. Fujitani, J. Furukawa, K. Araki, T. Fujioka, Y. Takegawa, J. Piao, T. Nishioka, T. Tamura, T. Nikaido, M. Ito, Y. Nakamura and Y. Shinohara, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 2105–2110 CrossRef PubMed.
- H. Hemmoranta, T. Satomaa, M. Blomqvist, A. Heiskanen, O. Aitio, J. Saarinen, J. Natunen, J. Partanen, J. Laine and T. Jaatinen, Exp. Hematol., 2007, 35, 1279–1292 CrossRef PubMed.
- M. Anugraham, F. Jacob, S. Nixdorf, A. V. Everest-Dass, V. Heinzelmann-Schwarz and N. H. Packer, Mol. Cell. Proteomics, 2014, 13, 2213–2232 CrossRef PubMed.
- Y. Wang, D. Park, A. G. Galermo, D. Gao, H. Liu and C. B. Lebrilla, Proteomics, 2016, 16, 2977–2988 CrossRef PubMed.
- S. Chen, T. Zheng, M. R. Shortreed, C. Alexander and L. M. Smith, Anal. Chem., 2007, 79, 5698–5702 CrossRef PubMed.
- E. P. Go, A. Cupo, R. Ringe, P. Pugach, J. P. Moore and H. Desaire, J. Virol., 2015, 90, 2884–2894 CrossRef PubMed.
- L. Montagne, C. Piel and J. P. Lalles, Nutr. Rev., 2004, 62, 105–114 CrossRef PubMed.
- M. Faure, D. Moennoz, F. Montigon, C. Mettraux, D. Breuille and O. Ballevre, J. Nutr., 2005, 135, 486–491 CrossRef PubMed.
- D. Park, G. Xu, M. Barboza, I. M. Shah, M. Wong, H. Raybould, D. A. Mills and C. B. Lebrilla, Glycobiology, 2017, 27, 847–860 CrossRef PubMed.
- K. Higai, Y. Aoki, Y. Azuma and K. Matsumoto, Biochim. Biophys. Acta, 2005, 1725, 128–135 CrossRef PubMed.
- S. M. Alam, B. Aussedat, Y. Vohra, R. Ryan Meyerhoff, E. M. Cale, W. E. Walkowicz, N. A. Radakovich, K. Anasti, L. Armand, R. Parks, L. Sutherland, R. Scearce, M. G. Joyce, M. Pancera, A. Druz, I. S. Georgiev, T. Von Holle, A. Eaton, C. Fox, S. G. Reed, M. Louder, R. T. Bailer, L. Morris, S. S. Abdool-Karim, M. Cohen, H. X. Liao, D. C. Montefiori, P. K. Park, A. Fernandez-Tejada, K. Wiehe, S. Santra, T. B. Kepler, K. O. Saunders, J. Sodroski, P. D. Kwong, J. R. Mascola, M. Bonsignori, M. A. Moody, S. Danishefsky and B. F. Haynes, Sci. Transl. Med., 2017, 9, eaai7521 CrossRef PubMed.
- S. Krishnan, M. Shimoda, R. Sacchi, M. J. Kailemia, G. Luxardi, G. A. Kaysen, A. N. Parikh, V. N. Ngassam, K. Johansen, G. M. Chertow, B. Grimes, J. T. Smilowitz, E. Maverakis, C. B. Lebrilla and A. M. Zivkovic, Sci. Rep., 2017, 7, 43728 CrossRef PubMed.
- X. Huang, J. J. Barchi Jr, F. D. Lung, P. P. Roller, P. L. Nara, J. Muschik and R. R. Garrity, Biochemistry, 1997, 36, 10846–10856 CrossRef PubMed.
- M. J. Feige, S. Nath, S. R. Catharino, D. Weinfurtner, S. Steinbacher and J. Buchner, J. Mol. Biol., 2009, 391, 599–608 CrossRef PubMed.
- C. C. Blackburn and R. L. Schnaar, J. Biol. Chem., 1983, 258, 1180–1188 Search PubMed.
- R. T. Huang, Nature, 1978, 276, 624–626 CrossRef PubMed.
- A. Kusumi and Y. Sako, Curr. Opin. Cell Biol., 1996, 8, 566–574 CrossRef PubMed.
- K. Kasahara and Y. Sanai, Glycoconj. J., 2000, 17, 153–162 CrossRef PubMed.
- M. Sorice, I. Parolini, T. Sansolini, T. Garofalo, V. Dolo, M. Sargiacomo, T. Tai, C. Peschle, M. R. Torrisi and A. Pavan, J. Lipid Res., 1997, 38, 969–980 Search PubMed.
- S. Hakomori, K. Handa, K. Iwabuchi, S. Yamamura and A. Prinetti, Glycobiology, 1998, 8, xi–xix CrossRef PubMed.
- A. Poulos, Lipids, 1995, 30, 1–14 CrossRef PubMed.
- S. N. Pinto, L. C. Silva, A. H. Futerman and M. Prieto, Biochim. Biophys. Acta Biomembr., 2011, 1808, 2753–2760 CrossRef PubMed.
- F. Chang, R. Li and S. Ladisch, Exp. Cell Res., 1997, 234, 341–346 CrossRef PubMed.
- E. C. Seales, G. A. Jurado, B. A. Brunson, J. K. Wakefield, A. R. Frost and S. L. Bellis, Cancer Res., 2005, 65, 4645–4652 CrossRef PubMed.
- L. Vazquez-Moreno, M. C. Candia-Plata and M. R. Robles-Burgueno, Clin. Biochem., 2001, 34, 35–41 CrossRef PubMed.
- S. Nagamine, T. Yamazaki, K. Makioka, Y. Fujita, M. Ikeda, M. Takatama, K. Okamoto, H. Yokoo and Y. Ikeda, Neuropathology, 2016, 36, 333–345 CrossRef PubMed.
- M. A. Elliott, H. G. Jorgensen and K. D. Smith, Pharm. Pharmacol. Commun., 1998, 4, 545–547 Search PubMed.
- B. Byrne, G. G. Donohoe and R. O'Kennedy, Drug discovery today, 2007, 12, 319–326 CrossRef PubMed.
- J. C. Egrie, E. Dwyer, J. K. Browne, A. Hitz and M. A. Lykos, Exp. Hematol., 2003, 31, 290–299 CrossRef PubMed.
- S. Kelm, A. Pelz, R. Schauer, M. T. Filbin, S. Tang, M. E. de Bellard, R. L. Schnaar, J. A. Mahoney, A. Hartnell and P. Bradfield, et al. , Curr. Biol., 1994, 4, 965–972 CrossRef PubMed.
- N. Razi and A. Varki, Glycobiology, 1999, 9, 1225–1234 CrossRef PubMed.
- J. A. Prescher and C. R. Bertozzi, Cell, 2006, 126, 851–854 CrossRef PubMed.
- S. J. Luchansky, S. Argade, B. K. Hayes and C. R. Bertozzi, Biochemistry, 2004, 43, 12358–12366 CrossRef PubMed.
- D. Park, K. A. Brune, A. Mitra, A. I. Marusina, E. Maverakis and C. B. Lebrilla, Mol. Cell. Proteomics, 2015, 14, 2910–2921 CrossRef PubMed.
- D. Park, N. Arabyan, C. C. Williams, T. Song, A. Mitra, B. C. Weimer, E. Maverakis and C. B. Lebrilla, Mol. Cell. Proteomics, 2016, 15, 3653–3664 CrossRef PubMed.
- S. R. Kronewitter, H. J. An, M. L. de Leoz, C. B. Lebrilla, S. Miyamoto and G. S. Leiserowitz, Proteomics, 2009, 9, 2986–2994 CrossRef PubMed.
- B. Petersen, T. N. Petersen, P. Andersen, M. Nielsen and C. Lundegaard, BMC Struct. Biol., 2009, 9, 51 CrossRef PubMed.
- P. D. Thomas, A. Kejariwal, M. J. Campbell, H. Mi, K. Diemer, N. Guo, I. Ladunga, B. Ulitsky-Lazareva, A. Muruganujan, S. Rabkin, J. A. Vandergriff and O. Doremieux, Nucleic Acids Res., 2003, 31, 334–341 CrossRef PubMed.
- J. Kyte and R. F. Doolittle, J. Mol. Biol., 1982, 157, 105–132 CrossRef PubMed.
- G. E. Crooks, G. Hon, J. M. Chandonia and S. E. Brenner, Genome Res., 2004, 14, 1188–1190 CrossRef PubMed.
- W. F. Zeng, M. Q. Liu, Y. Zhang, J. Q. Wu, P. Fang, C. Peng, A. Nie, G. Yan, W. Cao, C. Liu, H. Chi, R. X. Sun, C. C. Wong, S. M. He and P. Yang, Sci. Rep., 2016, 6, 25102 CrossRef PubMed.
- H. Lee, J. B. German, R. Kjelden, C. B. Lebrilla and D. Barile, J. Agric. Food Chem., 2013, 61, 9689–9696 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8sc01875h |
| This journal is © The Royal Society of Chemistry 2018 |