
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDynamic tungsten diselenide nanomaterials: supramolecular assembly-induced structural transition over exfoliated two-dimensional nanosheets†
Adem Ali
Muhabie
b,
Ching-Hwa
Ho
 a,
Belete Tewabe
Gebeyehu
a,
Shan-You
Huang
a,
Chih-Wei
Chiu
b,
Juin-Yih
Lai
ace,
Duu-Jong
Lee
cde and
Chih-Chia
Cheng
*a
a,
Belete Tewabe
Gebeyehu
a,
Shan-You
Huang
a,
Chih-Wei
Chiu
b,
Juin-Yih
Lai
ace,
Duu-Jong
Lee
cde and
Chih-Chia
Cheng
*a
aGraduate Institute of Applied Science and Technology, National Taiwan University of Science and Technology, Taipei 10607, Taiwan. E-mail: cccheng@mail.ntust.edu.tw
bDepartment of Materials Science and Engineering, National Taiwan University of Science and Technology, Taipei 10607, Taiwan
cDepartment of Chemical Engineering, National Taiwan University of Science and Technology, Taipei 10607, Taiwan
dDepartment of Chemical Engineering, National Taiwan University, Taipei 10617, Taiwan
eR&D Center for Membrane Technology, Chung Yuan Christian University, Chungli, Taoyuan 32043, Taiwan
First published on 31st May 2018
Abstract
A simple and effective method for direct exfoliation of tungsten diselenide (WSe2) into few-layered nanosheets has been successfully developed by employing a low molecular weight adenine-functionalized supramolecular polymer (A-PPG). In this study, we discover A-PPG can self-assemble into a long-range, ordered lamellar microstructure on the surface of WSe2 due to the efficient non-covalent interactions between A-PPG and WSe2. Morphological and light scattering studies confirmed the dynamic self-assembly behavior of A-PPG has the capacity to efficiently manipulate the transition between contractile and extended lamellar microstructures on the surface of metallic 1T-phase and semiconducting 2H-phase WSe2 nanosheets, respectively. The extent of WSe2 exfoliation can be easily controlled by systematically adjusting the amount of A-PPG in the composites, to obtain nanocomposites with the desired functional characteristics. In addition, the resulting composites possess unique liquid–solid phase transition behavior and excellent thermoreversible properties, revealing the self-assembled lamellar structure of A-PPG functions as a critical factor to manipulate and tailor the physical properties of exfoliated WSe2. This newly developed method of producing exfoliated WSe2 provides a useful conceptual and potential framework for developing WSe2-based multifunctional nanocomposites to extend their application in solution-processed semiconductor devices.
Introduction
Nanostructured layered tungsten diselenide (WSe2) materials, a class of newly emerging two dimensional (2D) transition-metal dichalcogenide (TMD) nanomaterials, have attracted extensive interest in the development of sensors,1 tunneling devices,2,3 transistors,4,5 optoelectronics,6 hydrogen evolution7–10 and flexible electronics11,12 due to their unique thermal, gas-sensitive, electrical, mechanical, photoelectrical and optical properties.13 WSe2 materials are also promising candidates for many optical and electrical applications due to their controllable band structure and intrinsic photoluminescence, features not found in graphene.14 The single layer of WSe2 is composed of a 6.7 Å-thick slab of a Se–W–Se sandwich layer. Atomically-thin WSe2 is unique as it is chemically homogeneous, but exhibits both a semiconducting 2H crystal structure and metallic 1T phase. The 2H phase, in which two layers per unit cell stack in hexagonal symmetry with a trigonal prismatic coordination, and the metastable 1T metallic phase, which possesses one-layer unit cell in tetragonal symmetry with octahedral coordination, are observed in 2D nanomaterials prepared by intercalation-assisted-exfoliation.15 Recently, the exfoliation of WSe2 nanosheets from pristine WSe2 has attracted a great deal of attention, as understanding the adsorption properties of functional materials dispersed on the surface of WSe2 is fundamental to enable rational control of surface functionalization, in order to generate high-efficiency optoelectronic devices for efficient hydrogen evolution, field-effect transistors and photodetectors/light emitting diodes.16,17 Hence, exfoliation techniques play a crucial role in the production of WSe2 for a variety of applications.A number of synthetic methods have been developed to fabricate WSe2 nanosheets, such as chemical vapor deposition, micromechanical cleavage, and chemical and sonication-assisted liquid phase exfoliation methods.2 In principle, the chemical vapor deposition technique enables fabrication of macroscopic areas of TMD films, though generation of single continuous layers on the macroscopic scale remains challenging. The commonly used micromechanical method has low repeatability, precision and limited ability to control the thickness and sizes of the sheets during production of exfoliated WSe2.18 Hence, many of the existing synthesis methods are not convenient; the scalability and practical applications of WSe2 nanosheets are limited by their high cost, low yield, restacking and complex synthetic procedures. Design of an environmentally friendly, low-cost, efficient and simple fabrication technique remains a significant challenge, but would be enormously valuable for both research and commercial applications.19
Compared to the aforementioned methods, the sonication-assisted liquid phase exfoliation method is more promising,20 as it can produce 2D nanosheets of high structural quality with stable, uniformly dispersed flakes.21–23 However, the major limitation of liquid-phase exfoliation is that the WSe2 nanosheets produced restack into a bulk structure via van der Waals interactions, which significantly limits extensive application of these nanomaterials.14 In order to resolve these problems and provide a realistic solution, it would be highly desirable to establish an efficient dispersal technique that effectively exfoliates WSe2 into an organic solution in which restacking of dispersed WSe2 is impeded, to enable the synthesis of high-quality dispersed WSe2 for a wide range of applications across many fields of engineering and science.15,16
Non-covalently functionalized supramolecular polymers have been acquired from functional macromers, and exhibit tailorable physical properties and viscoelastic functionality due to the dynamic bonds between monomeric units.24–26 Recently, our research group discovered an ureido-cytosine-functionalized supramolecular polymer, which we used to directly delaminate graphene from graphite into micrometer-sized few-layered sheets. Application of these supramolecular polymers for the production of 2D nanomaterials enabled well-controlled exfoliation, manipulation and successive dispersion of graphite to be easily achieved. The self-assembled lamellar microstructure of supramolecular polymers and electric conductivity of polymer-graphene composites have been characterized.27 Subsequently, we developed a simple, efficient method for exfoliation of hexagonal boron nitride (h-BN) into few-layered nanosheets using adenine end-capped polypropylene glycol (A-PPG) as a dispersing and stabilizing agent, and confirmed A-PPG self-assembled into either lamellar or micelle structures on the surface of h-BN nanosheets via π–π stacking. Furthermore, A-PPG/h-BN composites exhibited well-defined phase transition behavior and excellent thermal stability over a wide temperature range due to the presence of the adenine moieties and their reversible hydrogen-bond interactions.20 A-PPG is also a biocompatible stimuli-responsive polymer with potential for biomedical applications such as controlled drug delivery and bioimaging.28 Based on our previous findings, the combination of A-PPG with WSe2 represents a promising strategy for producing few-layer WSe2 nanosheets at high efficiency via a simple process, with potential for various applications.
In the present study, we demonstrate A-PPG can be used successfully to assist high-efficiency liquid phase exfoliation of WSe2 in tetrahydrofuran (THF) via ultrasonic treatment. Furthermore, morphological studies indicated formation of well-ordered lamellar nanostructures among the adsorbed A-PPG on the surface of WSe2 strongly manipulates the self-assembly behavior and physical properties of exfoliated WSe2 (Scheme 1). Moreover, adsorption of A-PPG on the surface of the 1T and 2H phases of WSe2 led to a spontaneous structural transformation between extended and contracted lamellar microstructures, resulting in excellent microstructural stability and stable thermoreversible behavior in the bulk state. To date, there has been no reported example of the production of exfoliated WSe2 nanosheets using a supramolecular polymer with the aim of efficiently manipulating contracted/expanded lamellar microstructures and achieving thermoreversible phase transition behavior on WSe2 surfaces. Hence, employing the adenine-based supramolecular A-PPG for exfoliation of WSe2 crystals provides a simple, efficient pathway towards the development of high-quality multifunctional exfoliated WSe2 for a wide variety of semiconductor applications.
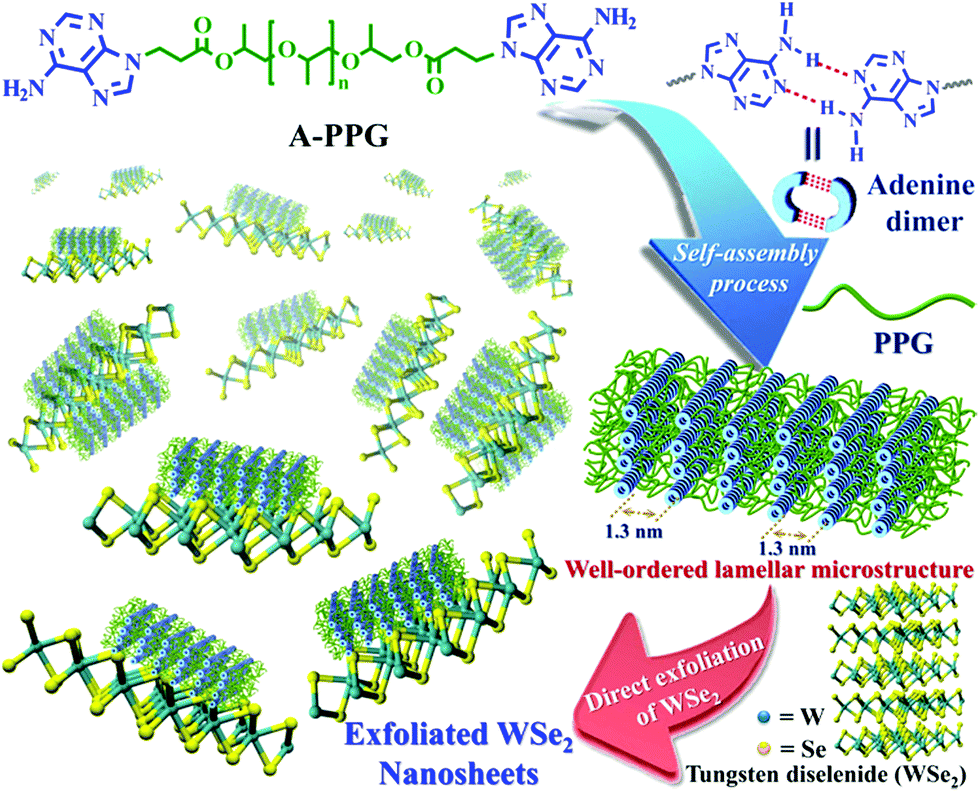 | ||
| Scheme 1 Chemical structures of the A-PPG macromer and schematic representation of the non-covalent liquid-phase exfoliation of WSe2 nanosheets in the presence of A-PPG dispersant. | ||
Results and discussion
A novel self-complementary adenine end-capped A-PPG was successfully prepared using a simple one-step synthesis method described in our previous work.20 The synthesized A-PPG spontaneously assembled into a well-ordered lamellar nanostructure (with an interlamellar distance of 1.33 nm, determined by wide angle X-ray diffraction) due to supramolecular polymerization of A-PPG via the doublet hydrogen bonding interactions between adenine moieties (Scheme 1).20 Subsequently, the WSe2 and A-PPG blend was sonicated in THF for 1 h at 25 °C. After ultrasonic treatment, exfoliated WSe2 was scattered throughout the solvent, resulting in a dark brown dispersion (inset photograph i in Fig. 1a). The solution remained stable for over one month at room temperature with no obvious signs of precipitation, implying attachment of A-PPG to the WSe2 sheets acts as a strong stabilizing agent against aggregation and re-stacking of single/few-layer WSe2 nanosheets. In addition, unlike pure A-PPG solution, the 50/50 WSe2/A-PPG composite solution demonstrated the Tyndall effect in THF (inset photograph ii in Fig. 1a); the path of the laser beam could be obviously seen within the solution due to scattering by the WSe2 nanosheets – suggesting uniform dispersion of the exfoliated WSe2 nanosheets – and exhibited red fluorescence due to the band transition after exfoliation.30 Photoluminescence (PL) spectroscopy analyses of these composites in THF were employed using an excitation wavelength of 420 nm. The A and B excitonic absorption peaks shown in Fig. 1b are the result of optical transitions involving a spin–orbit split valence band and degenerate conduction band at the K point of the Brillouin zone.31,32 The two splitting absorption peaks, A′ and B′, in the WSe2/A-PPG composites are the result of the A and B peaks splitting due to inter- and intralayer effects induced by the noncovalent interaction between A-PPG and WSe2. In addition, WSe2/A-PPG composites exhibited an increase in the emission intensity of the excitonic absorption peak B and split exciton peaks (A′ and B′), indicating the flakes thinned to a few layers as the concentration of A-PPG increased. This result also implies that the increased loading of A-PPG favors the active phase formation of WSe2. The WSe2 band structure underwent an indirect-to-direct optical gap transition when reduced in thickness to a few layers, which when exfoliated, yielded a 2D nanomaterial.33,34 Ultraviolet-visible (UV-vis) spectra of the 50/50 WSe2/A-PPG composite (Fig. 1a) contained an absorption peak at 773 nm, a feature of the A exciton of WSe2 (Fig. 1b) and attributed to the smallest direct excitonic transition in the WSe2 layers, suggesting the potential of these layers for photothermal materials.35 This observation also implies that the A-PPG macromer was stably adsorbed on the WSe2 surface due to the high-affinity interaction between WSe2 and A-PPG.36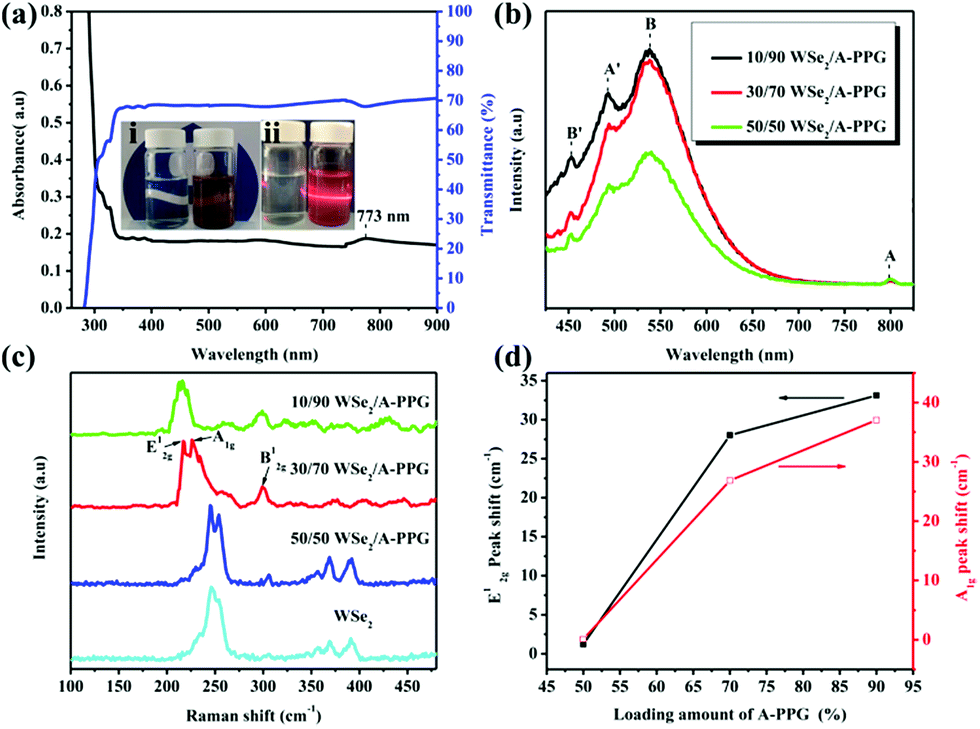 | ||
| Fig. 1 (a) UV-vis absorption spectrum for a THF dispersion of the 50/50 WSe2/A-PPG composite (concentration, 0.5 mg mL−1). The insets are photographs of (i) THF solutions of A-PPG (left) and WSe2/A-PPG (right) and (ii) the same solutions illuminated from the right using a 650 nm laser beam. (b) PL and (c) Raman spectra of WSe2 nanosheets with different contents of A-PPG. (d) Raman peak shift data extracted from peak frequency range (c). | ||
To further understand the interface structure relationships in the WSe2/A-PPG composites, Raman spectroscopy was used to detect the presence of the metallic 1T phase in as-exfoliated WSe2 nanosheets at high sensitivity, as presented in Fig. 1c. The characteristics of the peak at the respective in-plane E12g, out-of-plane A1g, B12g and small peaks in the high wavenumber region differed with the 2D WSe2 sheet thickness.12,16 Raman peak shifts are the major phenomenon indicative of exfoliation: as the amount of A-PPG loaded increased to 90%, the Raman peak of the WSe2 nanosheets underwent a significant red-shift compared to bulk WSe2, in agreement with previous reports.37 The frequency difference between the 10/90 WSe2/A-PPG and WSe2 peaks was 32.6 cm−1, which corresponds to three layers or fewer – as indicated by the extracted peak shift value (Fig. 1d).38 However, an increase in the intensity of the B12g peak provides a fingerprint of an increase in the proportion of 1T-phase Raman active modes, which are not allowed in the 2H phase,39 suggesting the pristine WSe2 layers stacked in 2H order (2H–WSe2) were successfully converted into 1T-phase WSe2 (1T-WSe2) nanosheets by incorporating A-PPG into the WSe2 layer. Notably, in the 1T-WSe2 Raman spectrum, the intensity of the E12g peak due to the remaining 2H–WSe2 was almost overwhelmed by the signal strength of the A1g peak. In contrast, the intact 2H–WSe2 spectrum contained a stronger E12g peak than A1g peak as the amount of A-PPG increased.40 In other words, as the amount of A-PPG increased, peak splitting and the intensity of the A1g mode increased significantly as the content of 1T-WSe2 increased. The absence of the small peaks (2M modes, related to second order and combinational Raman modes) in the higher frequency region (350–400 cm−1) of the Raman spectrum of exfoliated WSe2 further confirm formation of the high-quality, few-layer 1T-WSe2 phase.12,34,41 This result further indicates that the extent of exfoliation and proportion of the 1T-WSe2 phase can be controlled by systematically tuning the A-PPG content of the composites; this feature is highly desirable yet extremely rare within traditional nanocomposites and dispersing complex material systems.
In order to verify the presence of A-PPG macromers on the surface of WSe2, X-ray diffraction (XRD) was performed at 25 °C. As shown in Fig. 2a and b, the intensity of the diffraction peak at 13.70° assigned to the (002) crystallographic plane in the XRD pattern of bulk 2H–WSe2 dramatically reduced as the content of A-PPG increased. In addition, a shoulder peak emerged at about 13.40°, which originates from the 1T-WSe2 phase – as indicated by the enlarged (002) diffraction peak.9 After exfoliation, the intensity of the (002) reflection peak for each composite substantially reduced and slightly shifted from 13.45° (pristine WSe2) to 13.97° (10/90 WSe2/A-PPG) as the A-PPG content increased from 50 to 90%, leading to simultaneous disappearance of the (006) and (008) reflection peaks at 41.5° and 56.7°, possibly due to the effect of the non-covalent interactions between WSe2 and A-PPG. Therefore, three new small reflection peaks at 31.3°, 37.6° and 47.2° appeared in the XRD profiles, corresponding to the (100), (103) and (105) crystal planes (Fig. S1†), a consequence of assembly of A-PPG on the surface of WSe2, which thus tended to form highly exfoliated WSe2 nanosheets containing a high proportion of the metallic 1T phase.19,42 The weakening of the diffraction peaks revealed the exfoliated WSe2 has low crystallinity, much weaker stacking in the c direction, no defects and small crystallite size.43,44 This further suggests that the introduction of A-PPG into the WSe2 matrix significantly affects the phase behavior of WSe2 due to the self-complementary hydrogen-bonded adenine–adenine interactions involved in the self-assembly of A-PPG.20
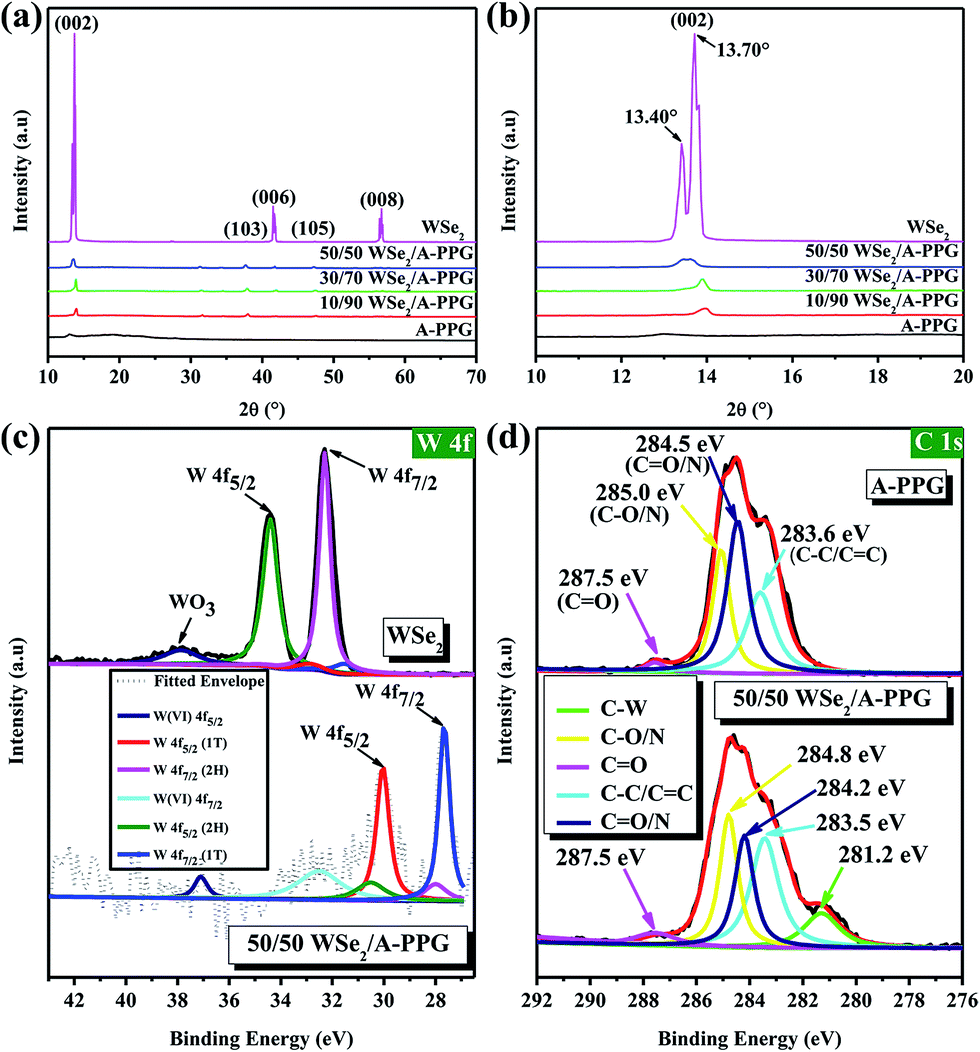 | ||
| Fig. 2 (a) XRD patterns for pure A-PPG, pure WSe2 and WSe2/A-PPG composites. (b) Enlarged view of the (002) diffraction peaks for all samples. High-resolution XPS spectra of (c) W 4f for bulk WSe2 and 50/50 WSe2/A-PPG, (d) C 1s for A-PPG and 50/50 WSe2/A-PPG. | ||
To further confirm how these specific interactions influence the WSe2 exfoliation process, X-ray photoelectron spectroscopy (XPS) was employed to confirm the bonding characteristics of the atomic interaction between WSe2 and A-PPG. As shown in Fig. 2c, the high-resolution W 4f photoelectron spectrum of pristine WSe2 displayed peaks at binding energies of 32.3 and 34.4 eV, corresponding to the W 4f7/2 and W 4f5/2 states, respectively,44,45 while lower binding energies of 27.8 for W 4f7/2 and 30.0 eV for W 4f5/2 were observed for the 50/50 WSe2/A-PPG composite. This confirms semiconducting 2H-phase WSe2 can be successfully converted into metallic 1T-phase WSe2 by incorporating A-PPG into the WSe2 layer, as the binding energies shifted negatively for the W 4f7/2 and W 4f5/2 peaks after exfoliation of the layers.46–48 In addition, peak deconvolution also showed that the percentage of 1T-WSe2 increased to 80% compared to pristine WSe2 (2.6%) in the bulk, consistent with the Raman analysis.45,49
Fig. 2d displays the high-resolution C 1s spectra of the A-PPG and 50/50 WSe2/A-PPG composite deconvoluted into several individual peaks. The appearance of the C–C/C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C, C–N/CN and CO XPS peaks in the composite at 283.5, 284.2, 284.8 and 287.5 eV, respectively, reflect the intermolecular interactions between the adenine/ester groups of A-PPG and exfoliated WSe2 nanosheets. The additional C–W peak (281.2 eV), which was absent in pure A-PPG, mainly reflects the π–π stacking of the adenine unit of A-PPG and electron-rich atoms on the WSe2 basal plane. Similar binding energy shifts also occurred for N 1s, O 1s and Se 3d in the WSe2/A-PPG composites compared to pristine A-PPG (Fig. S2†). The lower binding energy of the elements after exfoliation can be mainly attributed to the van der Waals interactions between the large electronegative atoms in A-PPG and electron-deficient atoms on the edge sites of WSe2.5,20 In other words, the nitrogen and electron-rich structure of adenine-based supramolecular A-PPG is indeed highly attractive for induction of noncovalent interactions with WSe2, including N–Se, N–W and W/Se–π–π coordination, as well as binding through charge transfer.4,26 Overall, the Raman, XRD and XPS results confirm that incorporation of A-PPG further induced a phase transition from the semiconductor 2H–WSe2 to metallic 1T-WSe2 and a microstructural transition from crystal layered structures to exfoliated nanosheets.
C, C–N/CN and CO XPS peaks in the composite at 283.5, 284.2, 284.8 and 287.5 eV, respectively, reflect the intermolecular interactions between the adenine/ester groups of A-PPG and exfoliated WSe2 nanosheets. The additional C–W peak (281.2 eV), which was absent in pure A-PPG, mainly reflects the π–π stacking of the adenine unit of A-PPG and electron-rich atoms on the WSe2 basal plane. Similar binding energy shifts also occurred for N 1s, O 1s and Se 3d in the WSe2/A-PPG composites compared to pristine A-PPG (Fig. S2†). The lower binding energy of the elements after exfoliation can be mainly attributed to the van der Waals interactions between the large electronegative atoms in A-PPG and electron-deficient atoms on the edge sites of WSe2.5,20 In other words, the nitrogen and electron-rich structure of adenine-based supramolecular A-PPG is indeed highly attractive for induction of noncovalent interactions with WSe2, including N–Se, N–W and W/Se–π–π coordination, as well as binding through charge transfer.4,26 Overall, the Raman, XRD and XPS results confirm that incorporation of A-PPG further induced a phase transition from the semiconductor 2H–WSe2 to metallic 1T-WSe2 and a microstructural transition from crystal layered structures to exfoliated nanosheets.
Further validation of the microstructures and morphologies of pristine WSe2 and the functionalized composites was performed by scanning electron microscopy (SEM) and high-resolution transmission electron microscopy (HRTEM). Fig. 3 shows the SEM micrographs of pristine A-PPG and WSe2 in comparison with their exfoliated counterparts. High-magnification SEM (Fig. 3e) revealed A-PPG can self-assemble into a micellar-like morphology with an average diameter of ca. 14 nm, in agreement with the dynamic light-scattering results (Fig. S3a†).20,50 This suggests the A-PPG chains are able to form large aggregates in polar THF solution, and form supramolecular micelles via the intermolecular hydrogen bonding interaction between adenine–adenine moieties. In the bulk state, the flat and smooth surface of stack-layered pristine WSe2 is clearly visible in Fig. 3a and c, along with the polygonal edge arrangements and thicknesses in the few micrometer-range.13,45 In contrast, the 50/50 WSe2/A-PPG composite exhibited a disordered morphology of folding WSe2 nanosheets, with a lateral size of 5–10 μm (Fig. 3b), indicating A-PPG acts as an effective dispersant to prevent reaggregation and restacking of the WSe2 nanosheets. Surprisingly, high-magnification SEM image (Fig. 3d) revealed the self-assembled micelles of A-PPG could self-assemble to form highly clustered structures and adsorbed on the surface of the exfoliated WSe2 sheets, which was diametrically opposite to the smooth surface of bare WSe2 (Fig. 3c). These observations indicate that A-PPG has a high binding affinity for WSe2 nanosheets due to the strong specific interactions and is stably adsorbed onto the WSe2 surface (Fig. 3f).51–53
 | ||
| Fig. 3 SEM images of (a, c) pristine WSe2, (b, d) exfoliated 50/50 WSe2/A-PPG composite and (e) pristine A-PPG. (f) Schematic diagram illustrating the interaction between spherical micelles and WSe2 nanosheets. | ||
Self-assembly of the adenine units in A-PPG played a significant role in the construction of the highly ordered hierarchical microstructure on the surface of WSe2 nanosheets. Thus, HRTEM was performed at 25 °C under high vacuum to further observe the self-assembled nanostructures of A-PPG in the 50/50 WSe2/A-PPG composite. As shown in Fig. 4, pristine WSe2 microstructures were distributed as irregular flakes with a high density of crystal surfaces. In addition, the two-dimensional fast Fourier transform (FFT) pattern in the inset of Fig. 4a illustrates the ordered crystalline structure of WSe2 with a 0.32 nm lattice distance.4,16 We carefully examined the structures from randomly selected areas of a WSe2 crystal sheet (in the green and red squares of Fig. 4b) and confirmed the coexistence of hexagonal structures, which are typically seen in the 1T phase, and also honeycomb lattices, characteristic of the 2H phase (Fig. 4c and d).29,54 These results were consistent with the XPS and Raman data. In the 50/50 WSe2/A-PPG composite, self-assembly of A-PPG into contracted and extended lamellar microstructures on the surface of exfoliated WSe2 nanosheets was clearly observed (Fig. 5c, labelled as ‘I’ and ‘II’ respectively), with a contracted lamellar d-spacing of 1.0 nm and an extended lamellar d-spacing of 1.8 nm. In addition, the two-dimensional FFT pattern in the right upper corner of Fig. 5a shows a disordered crystal structure with a 0.26 nm lattice distance, which could be attributed to adsorption of the lamellar microstructure of A-PPG over the surface of WSe2via a “specific interaction” between the polar moieties of A-PPG and the surface of the WSe2 nanosheets (Fig. 5b).
 | ||
| Fig. 4 (a) HRTEM image of pristine WSe2 and the corresponding electron diffraction pattern (insert). (b) Enlarged view of (a) showing the surface morphology of WSe2. The squares indicate 1T-WSe2 (green) and 2H–WSe2 (red) crystalline structures, which are shown in detail in (c) and (d), respectively. | ||
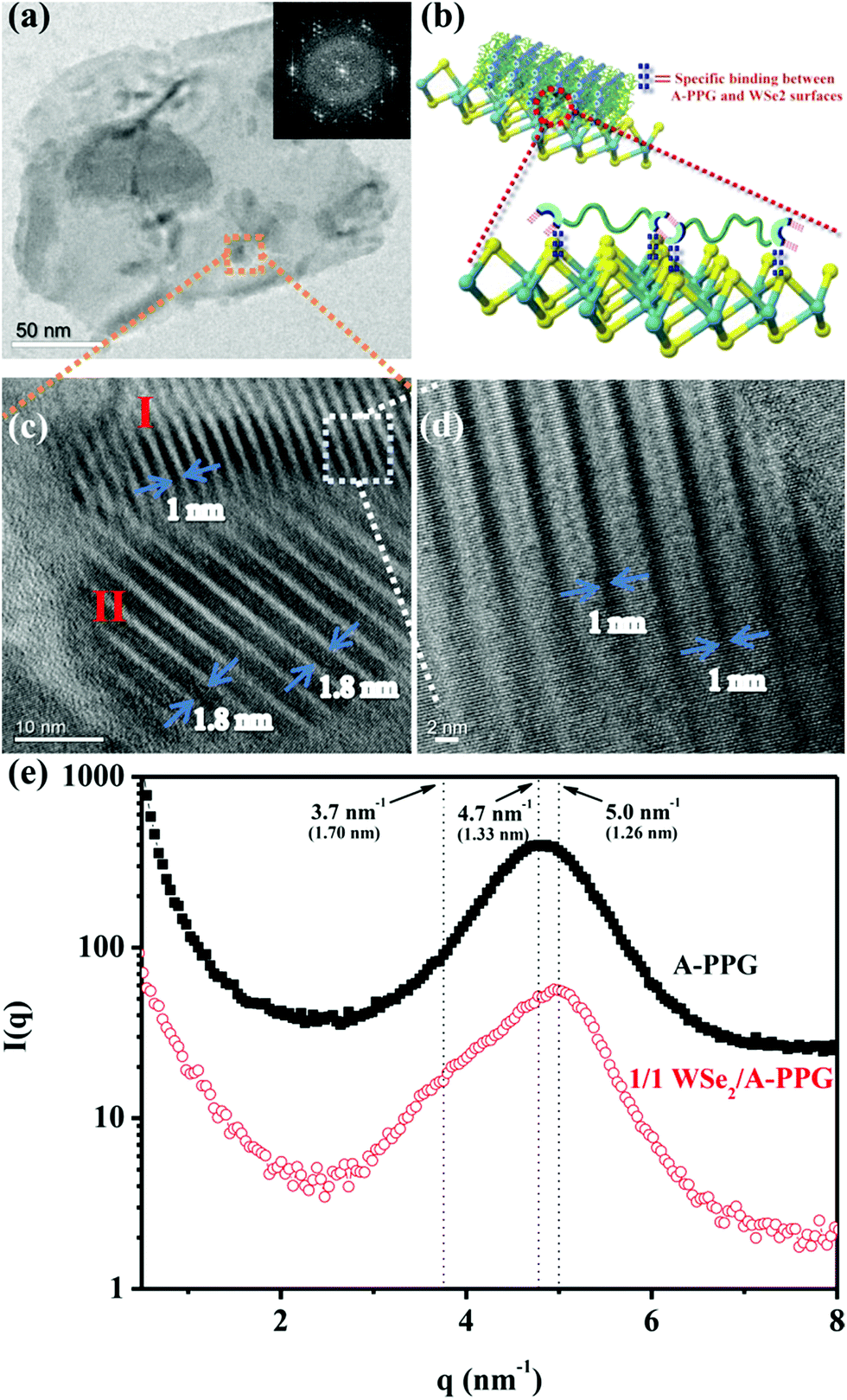 | ||
| Fig. 5 (a) HRTEM image of 50/50 WSe2/A-PPG composite and the corresponding electron diffraction pattern (given as insert). (b) Schematic illustrating the specific interaction between A-PPG and WSe2 nanosheets. (c) Enlarged view of (a) showing the contracted and extended lamellar microstructure of A-PPG on the surface of WSe2. (d) HRTEM image of the region enclosed by the white square in panel (c). (e) SAXS data for A-PPG and 50/50 WSe2/A-PPG recorded at 25 °C. | ||
Most notably, the d-spacing values of the contracted and extended lamellar microstructures, approximately 1.0 nm and 1.8 nm respectively, were significantly different to the theoretical PPG chain length (1.33 nm),20 possibly due to coexistence of the 1T-WSe2 and 2H–WSe2 phases that possess different physical properties and may contribute to dynamic contraction/extension-motion in the flexible PPG backbone during the self-assembly process. Thus, the interlamellar distance of the lamellar structure and transition mechanisms of A-PPG can be reasonably explained by a theory of chain contraction/extension transition within the flexible polymer.55 However, the 1T-structure is comprised of a negatively charged ion8,56 that possesses superior electrical conductivity and catalytic activity (compared to the 2H phase) and has shown enhanced performance in electrocatalytic hydrogen evolution51 and supercapacitors.22,23 Therefore, the electron-rich 1T phase in the surface of exfoliated WSe2 nanosheets (‘II’ region of Fig. 5c) may trigger formation of an extended PPG backbone and thus significantly increase lamellar d-spacing by electron transfer to A-PPG compared to the surface of 2H–WSe2 phase.29,57 The unique 1T-WSe2/A-PPG architecture maximizes the interfacial atomic contact between metallic 1T-WSe2 and A-PPG. 1T-WSe2 enhances the electron density of nitrogen donor atoms in the adenine units via a resonance effect to promote a stable interaction between A-PPG adenine moieties and WSe2, resulting in stretching of the A-PPG linear chain and an increase in interlamellar distance.54,56 The chemically stable 2H–WSe2 phase in the surface of exfoliated WSe2 nanosheets (‘I’ region of Fig. 5c) may have an insignificant effect on extension of the A-PPG polymer segment. However, the 2H–WSe2 phase may enable more contraction of the flexible PPG backbone compared to the theoretical d-spacing value (1.33 nm) of free A-PPG due to possible weakening of intermolecular interactions between WSe2 and A-PPG leading to a more thermodynamically stable A-PPG structure. Based on the above findings and description, we reasonably conjecture that coexistence of the 2H and 1T phases trigger the self-assembly of A-PPG into contracted and extended lamellar morphologies, respectively. Contraction and extension of the interlamellar distance in the PPG backbone could be easily tuned by regulating the controllable hydrogen-bonded structure between adenine units, which depends on the surface reactivity of the heterogeneous phase structure of WSe2.
In order further to confirm the d-spacing values of the adsorbed contracted and extended lamellar microstructures of A-PPG on the WSe2 surface, small-angle X-ray scattering (SAXS) measurements were used to identify the lamellar crystalline phases associated with A-PPG in pure A-PPG and the 50/50 WSe2/A-PPG composite. As shown in Fig. 5f, the SAXS profile of 50/50 WSe2/A-PPG composite at 25 °C exhibited two scattering peaks at q = 5.0 nm−1 (d = 1.26 nm) and q = 3.7 nm−1 (d = 1.70 nm). The peak at q = 5.0 nm−1 may be attributed to a decrease in lamellar d-spacing of the linear A-PPG polymer due to the inactive surface of the 2H–WSe2 phase, resulting in formation of a slightly contracted lamellar microstructure on the 2H–WSe2 surface. The other scattering peak located at q = 3.7 nm−1, with significantly longer d-spacing than that of pristine A-PPG (1.33 nm), is possibly attributed to the highly reactive surface of 1T-phase WSe2,55 which enhances chain extension of the flexible PPG backbone. In addition, the SAXS results showed similar trends as the HRTEM images (Fig. 5c). The HRTEM observations also revealed 1T-WSe2 and A-PPG interact via an effective noncovalent interaction, with each interacting junction contributing partial π bonds and charges to form a stable charge-transfer complex at the interface between WSe2 and A-PPG.4,26 On the other hand, the contractile and extended lamellar structures may help to stabilize the modified chemical structure and prevent degradation of WSe2 and increase charge transport across the interface.22,23
Generally, when WSe2 nanosheets were highly covalently functionalized with A-PPG, their surfaces were absorbed by randomly distributed domains of well-ordered lamellar or micelle A-PPG microstructures, which prevented restacking of the exfoliated WSe2 nanosheets and also improved their structural stability (Scheme 2). Mechanistically, we realize the adenine moieties of A-PPG are an essential unit that has the affinity to self-assemble spontaneously into desirable microstructures on the WSe2 surface, primarily due to the presence of the favorable hydrogen bonding interaction between self-complementary adenine moieties. When A-PPG is present in excess in the composites, at the beginning of the blending process, A-PPG tends to generate a well-defined lamellar structure with excellent long-range order due to the low concentration of A-PPG on the surface of WSe2 (Scheme 2a). Subsequently, excess A-PPG forms a large number of micellar aggregates over the flat surface of the lamellar hierarchical structures (Scheme 2c). Due to the presence of both the 1T and 2H phases in the WSe2 nanosheets, A-PPG not only drives the transition between contractile and extended lamellar structures, but also effectively manipulates the intermolecular d-spacing of the lamellar microstructure on the surfaces of the exfoliated 1T-WSe2 and 2H–WSe2 phases (Scheme 2b). Collectively, this study demonstrates that contraction and extension of the lamellar microstructure depend on the hydrogen bond motion of adenine dimers, interfacial affinity between WSe2 and A-PPG, and the different surface activity of the WSe2 phases. In addition, tuning the content of A-PPG in the composite can easily control the extent of WSe2 exfoliation and enables manipulation of the formation of lamellar structures or micellar aggregates over the surface of WSe2via noncovalent polymerization of A-PPG.55 Therefore, this new discovery suggests the self-assembly behavior of A-PPG can be employed to effectively tune the intrinsic physical properties of inorganic 2D nanomaterials and related heterostructures without additional chemical treatments.
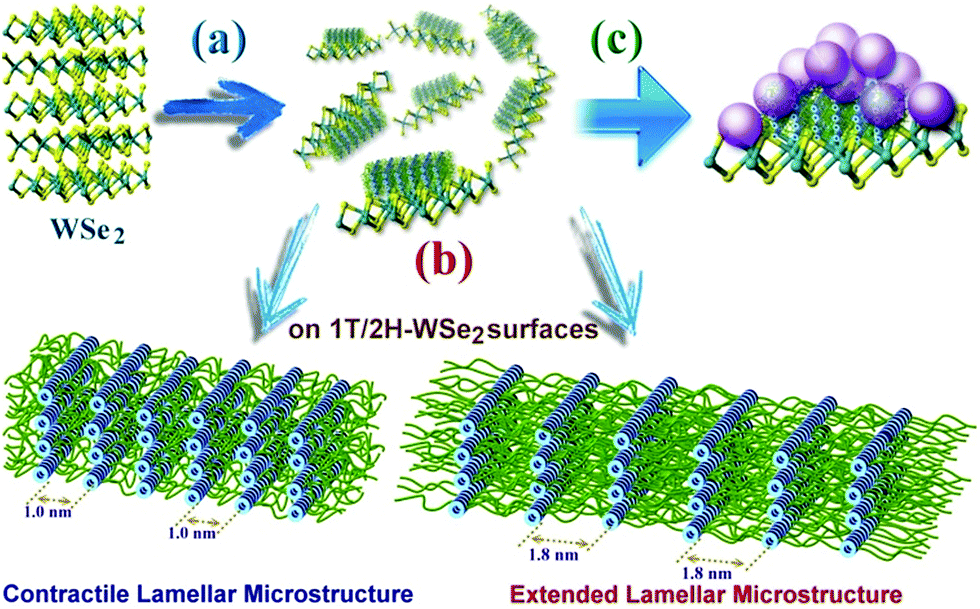 | ||
| Scheme 2 Proposed exfoliation processes for adsorption of (a) self-assembled structures, (b) contracted/extended lamellar microstructures and (c) micellar aggregates of A-PPG on the surface of exfoliated WSe2 nanosheets. | ||
In order to determine the number of WSe2 layers in the composites, atomic force microscopy (AFM) was performed to measure the step height of exfoliated WSe2 nanosheets on a silicon substrate. The theoretical WSe2 monolayer thickness is 0.64 nm.12 The measured step height of 50/50 WSe2/A-PPG layer was 3.0–3.5 nm (line scan in Fig. S4†), corresponding to fewer than five layers of WSe2 nanosheets, with a lateral dimension of 0.2–2 μm, consistent with the DLS results (Fig. S3b†). In addition, the edges of WSe2 had higher heights than the central structures; this may be due to the edges being either wrinkled or the formation of micelle-like or long-range ordered lamellar structures of A-PPG over the surface of WSe2 due to the noncovalent specific interaction between WSe2 and A-PPG.7,20,58,59 Differential scanning calorimetry (DSC) and variable-temperature Fourier transform infrared (VT-FTIR) spectroscopy were also employed to investigate thermoreversible stability and phase transition behavior within the WSe2/A-PPG system. The DSC and VT-FTIR (Fig. S5–S7†) showed the WSe2/A-PPG composites appeared to undergo a thermoreversible transition between ordered crystals and disordered fluids, and the hydrogen-bonded supramolecular interactions between adenine moieties reverted back perfectly to the original state as the samples were slowly cooled from 120 °C to 25 °C, as a result of the relationship between the thermoreversible hydrogen bond interactions and phase transition behavior. (The DSC and VT-FTIR are described in more detail in the ESI†). Furthermore, thermogravimetric analysis (TGA) (Fig. S8†) confirmed the WSe2/A-PPG composites possess excellent thermal stability due to the presence of the exfoliated WSe2 nanosheets in the matrix phase, which indicates the potential to achieve temperature-responsive functional composites with higher thermal stability.60 Based on results obtained in this study, the physical properties of WSe2/A-PPG composites – in particular their contracted/extended lamellar microstructures and thermoresponsive behavior (compared to pristine WSe2) – are highly suitable for potential applications in solution-processed photoluminescent sensors and semiconductor devices.61,62
Conclusions
We have designed a new, simple, effective exfoliation method using A-PPG macromer as a dispersant and stabilizer to produce exfoliated 2D WSe2 nanosheets in organic solution. Spectroscopic, compositional and morphological studies confirmed formation of micelle-like and lamellar structures of highly adsorbed A-PPG on the surface of WSe2 due to the specific noncovalent interaction between A-PPG and the WSe2 nanosheets. Moreover, the electron-rich metallic 1T-WSe2 phase may trigger the self-assembly of A-PPG resulting in formation of an extended lamellar microstructure by increasing the electron density of the nitrogen donor atoms in the adenine unit via a resonance effect, which enhanced the structural stability of exfoliated WSe2 through 1T-WSe2/A-PPG interactions. In contrast, the chemically stable semiconductor 2H–WSe2 phase assisted the formation of a contracted lamellar A-PPG microstructure due to weakening of the intermolecular interactions between 2H–WSe2 and A-PPG. Tuning the A-PPG content of the composites can be used to control the layer thickness of the exfoliated WSe2 nanosheets and physical and chemical properties of WSe2 in order to obtain nanocomposites with the desired functional characteristics. In addition, these newly-developed composites exhibit unique liquid–solid phase transition behavior and excellent thermoreversible properties due to the presence of the highly stable, reversible hydrogen-bonded adenine network in the composites. To the best of our knowledge, this WSe2/A-PPG system is the first method to exploit A-PPG macromers to produce highly exfoliated WSe2 and enable controlled, self-assembly of multifunctional supramolecular polymer structures on the surface of WSe2. This strategy may provide a useful conceptual framework for further developing and extending the applications of WSe2-based nanocomposites in solution-processed temperature-switching semiconductor devices.Experimental section
All chemical materials, characterizations and instrumentation used in this work are described in the ESI.†Synthesis of adenine end-capped PPG (A-PPG)
A-PPG was produced from low molecular weight PPG diacrylate (average molecular weight ∼800, ca. 14 repeat units) and adenine via Michael addition reaction using potassium tert-butoxide as an initiator. The synthesis procedure is described in detail in our previous work.20,28Preparation of bulk tungsten diselenide (WSe2)
WSe2 single crystals were grown by chemical vapor transport.29 This method contains two steps. First, prior to crystal growth, a powdered mixture of the pure starting material was prepared (W: 99.99% pure and Se: 99.999%) by reaction at 1050 °C for 10 days in evacuated quartz ampules. To improve the stoichiometry, 2 mol% excess selenium was added with respect to the stoichiometric mixture of the constituent elements. About 10 g of this mixture was placed into a quartz ampule (22 mm OD, 17 mm ID, 20 cm length), which was then evacuated to a pressure of 10−6 Torr and sealed. The mixture was slowly heated to 1050 °C. In the second step, chemical transport was achieved by mixing an appropriate amount of material and transport agent (I2 at 12 mg cm−3), which was placed in a quartz tube (22 mm OD, 17 mm ID, 20 cm length), cooled with liquid nitrogen, evacuated to 10−6 Torr and sealed. The growth temperature was set from 1050 to 960 °C with a gradient of −4.5 °C cm−1 (with crystal growth occurring in the cooler section). The reaction was maintained for 480 h, producing large single crystals with mirror-like surfaces.Preparation of WSe2/A-PPG composites
WSe2/A-PPG composites were produced by the sonication-assisted liquid exfoliation method. A-PPG was added into the THF and stirred at room temperature until PPG completely dissolved. Choosing a moderate polar THF for preparing composite materials can possibly be used to maintain the hydrogen-bonded structures of A-PPG in solution.20,27 Various contents of WSe2 were added to the same solution and sonicated for 1 h at 25 °C to produce stable, homogenous dispersions of WSe2/A-PPG. The synthesized impregnated WSe2/A-PPG composites were dark brown, and were dried completely by removing the solvent and volatile impurities in a vacuum oven at 60 °C. For comparison, pristine WSe2 and A-PPG samples were prepared under the same conditions.Conflicts of interest
The authors declare no conflicts of interest in this article.Acknowledgements
This study was supported financially by the Ministry of Science and Technology, Taiwan, under contracts MOST 104-2221-E-011-153 and MOST 105-2628-E-011-006-MY2.Notes and references
- M.-Y. Tsai, A. Tarasov, Z. R. Hesabi, H. Taghinejad, P. M. Campbell, C. A. Joiner, A. Adibi and E. M. Vogel, ACS Appl. Mater. Interfaces, 2015, 7, 12850–12855 Search PubMed.
- C. A. Joiner, P. M. Campbell, A. A. Tarasov, B. R. Beatty, C. J. Perini, M.-Y. Tsai, W. J. Ready and E. M. Vogel, ACS Appl. Mater. Interfaces, 2016, 8, 8702–8709 Search PubMed.
- A. Pant, Z. Mutlu, D. Wickramaratne, H. Cai, R. K. Lake, C. Ozkan and S. Tongay, Nanoscale, 2016, 8, 3870–3887 RSC.
- B. Liu, Y. Ma, A. Zhang, L. Chen, A. N. Abbas, Y. Liu, C. Shen, H. Wan and C. Zhou, ACS Nano, 2016, 10, 5153–5160 CrossRef PubMed.
- C. Lu, Y. Liu, Y. Ying and J. Liu, Langmuir, 2017, 33, 630–637 CrossRef PubMed.
- H. Taghinejad, M. Taghinejad, A. Tarasov, M.-Y. Tsai, A. H. Hosseinnia, H. Moradinejad, P. M. Campbell, A. A. Eftekhar, E. M. Vogel and A. Adibi, ACS Photonics, 2016, 3, 700–707 CrossRef.
- D. Jariwala, A. R. Davoyan, G. Tagliabue, M. C. Sherrott, J. Wong and H. A. Atwater, Nano Lett., 2016, 16, 5482–5487 CrossRef PubMed.
- B. Mahler, V. Hoepfner, K. Liao and G. A. Ozin, J. Am. Chem. Soc., 2014, 136, 14121–14127 CrossRef PubMed.
- Z. Liu, H. Zhao, N. Li, Y. Zhang, X. Zhang and Y. Du, Inorg. Chem. Front., 2016, 3, 313–319 RSC.
- M. Zou, J. Chen, L. Xiao, H. Zhu, T. Yang, M. Zhang and M. Du, J. Mater. Chem. A, 2015, 3, 18090–18097 Search PubMed.
- D. Akinwande, N. Petrone and J. Hone, Nat. Commun., 2014, 5, 5678 CrossRef PubMed.
- H. Zhou, C. Wang, J. C. Shaw, R. Cheng, Y. Chen, X. Huang, Y. Liu, N. O. Weiss, Z. Lin, Y. Huang and X. Duan, Nano Lett., 2015, 15, 709–713 CrossRef PubMed.
- H. Li, Z. Peng, J. Qian, M. Wang, C. Wang and X. Fu, ACS Appl. Mater. Interfaces, 2017, 9, 28704–28715 Search PubMed.
- L. Ci, L. Song, C. Jin, D. Jariwala, D. Wu, Y. Li, A. Srivastava, Z. F. Wang, K. Storr, L. Balicas, F. Liu and P. M. Ajayan, Nat. Mater., 2010, 9, 430–435 CrossRef PubMed.
- P. Cheng, K. Sun and Y. H. Hu, Nano Lett., 2016, 16, 572–576 CrossRef PubMed.
- M. Z. M. Nasir, C. C. Mayorga-Martinez, Z. Sofer and M. Pumera, ACS Nano, 2017, 11, 5774–5784 CrossRef PubMed.
- X. Yu and K. Sivula, Chem. Mater., 2017, 29, 6863–6875 CrossRef.
- C. Tan, X. Cao, X.-J. Wu, Q. He, J. Yang, X. Zhang, J. Chen, W. Zhao, S. Han, G.-H. Nam, M. Sindoro and H. Zhang, Chem. Rev., 2017, 117, 6225–6331 CrossRef PubMed.
- M.-Y. Tsai, S. Zhang, P. M. Campbell, R. R. Dasari, X. Ba, A. Tarasov, S. Graham, S. Barlow, S. R. Marder and E. M. Vogel, Chem. Mater., 2017, 29, 7296–7304 CrossRef.
- A. A. Muhabie, C. C. Cheng, J. J. Huang, Z. S. Liao, S. Y. Huang, C. W. Chiu and D. J. Lee, Chem. Mater., 2017, 29, 8513–8520 CrossRef.
- G. Z. Magda, J. Pető, G. Dobrik, C. Hwang, L. P. Biró and L. Tapasztó, Sci. Rep., 2015, 5, 14714 CrossRef PubMed.
- G. Eda, T. Fujita, H. Yamaguchi, D. Voiry, M. Chen and M. Chhowalla, ACS Nano, 2012, 6, 7311–7317 CrossRef PubMed.
- J. Brivio, D. T. L. Alexander and A. Kis, Nano Lett., 2011, 11, 5148–5153 CrossRef PubMed.
- F. Huang and O. A. Scherman, Chem. Soc. Rev., 2012, 41, 5879–5880 RSC.
- P. Wei, X. Yan and F. Huang, Chem. Soc. Rev., 2015, 44, 815–832 RSC.
- R. C. Selhorst, E. Puodziukynaite, J. A. Dewey, P. Wang, M. D. Barnes, A. Ramasubramaniam and T. Emrick, Chem. Sci., 2016, 7, 4698–4705 RSC.
- C. C. Cheng, F. C. Chang, J. H. Wang, J. K. Chen, Y. C. Yen and D. J. Lee, Nanoscale, 2016, 8, 723–728 RSC.
- C. C. Cheng, J. J. Huang, A. A. Muhable, Z. S. Liao, S. Y. Huang, S. C. Lee, C. W. Chiu and D. J. Lee, Polym. Chem., 2017, 8, 2292–2298 RSC.
- C. H. Ho, W. H. Chen, K. K. Tiong, K. Y. Lee, A. Gloter, A. Zobelli, O. Stephan and L. H. G. Tizei, ACS Nano, 2017, 11, 11162–11168 CrossRef PubMed.
- Y. Lin, T. V. Williams and J. W. Connell, J. Phys. Chem. Lett., 2010, 1, 277–283 CrossRef.
- R. Coehoorn, C. Haas and R. A. de Groot, Phys. Rev. B: Condens. Matter Mater. Phys., 1987, 35, 6203–6206 CrossRef.
- A. R. Beal, J. C. Knights and W. Y. Liang, J. Phys. C: Solid State Phys., 1972, 5, 3540–3551 CrossRef.
- W. Zhao, Z. Ghorannevis, L. Chu, M. Toh, C. Kloc, P.-H. Tan and G. Eda, ACS Nano, 2013, 7, 791–797 CrossRef PubMed.
- W. Zhao, Z. Ghorannevis, K. K. Amara, J. R. Pang, M. Toh, X. Zhang, C. Kloc, P. H. Tan and G. Eda, Nanoscale, 2013, 5, 9677–9683 RSC.
- G. S. Bang, S. Cho, N. Son, G. W. Shim, B.-K. Cho and S.-Y. Choi, ACS Appl. Mater. Interfaces, 2016, 8, 1943–1950 Search PubMed.
- J. Gu, X. Yang, Z. Lv, N. Li, C. Liang and Q. Zhang, Int. J. Heat Mass Transfer, 2016, 92, 15–22 CrossRef.
- X. Wang, X. Chen, Y. Zhou, C. Park, C. An, Y. Zhou, R. Zhang, C. Gu, W. Yang and Z. Yang, Sci. Rep., 2017, 7, 46694 CrossRef PubMed.
- D.-H. Kang, J. Shim, S. K. Jang, J. Jeon, M. H. Jeon, G. Y. Yeom, W.-S. Jung, Y. H. Jang, S. Lee and J.-H. Park, ACS Nano, 2015, 9, 1099–1107 CrossRef PubMed.
- X. Luo, Y. Zhao, J. Zhang, M. Toh, C. Kloc, Q. Xiong and S. Y. Quek, Phys. Rev. B: Condens. Matter Mater. Phys., 2013, 88, 195313 CrossRef.
- Y. Ma, B. Liu, A. Zhang, L. Chen, M. Fathi, C. Shen, A. N. Abbas, M. Ge, M. Mecklenburg and C. Zhou, ACS Nano, 2015, 9, 7383–7391 CrossRef PubMed.
- A. Berkdemir, H. R. Gutiérrez, A. R. Botello-Méndez, N. Perea-López, A. L. Elías, C.-I. Chia, B. Wang, V. H. Crespi, F. López-Urías, J.-C. Charlier, H. Terrones and M. Terrones, Sci. Rep., 2013, 3, 1755 CrossRef.
- W. Zhu, X. Gao, Q. Li, H. Li, Y. Chao, M. Li, S. M. Mahurin, H. Li, H. Zhu and S. Dai, Angew. Chem., Int. Ed., 2016, 55, 10766–10770 CrossRef PubMed.
- P. D. Antunez, D. H. Webber and R. L. Brutchey, Chem. Mater., 2013, 25, 2385–2387 CrossRef.
- M. Zou, J. Zhang, H. Zhu, M. Du, Q. Wang, M. Zhang and X. Zhang, J. Mater. Chem. A, 2015, 3, 12149–12153 Search PubMed.
- A. Y. S. Eng, A. Ambrosi, Z. Sofer, P. Šimek and M. Pumera, ACS Nano, 2014, 8, 12185–12198 CrossRef PubMed.
- D. Voiry, M. Salehi, R. Silva, T. Fujita, M. Chen, T. Asefa, V. B. Shenoy, G. Eda and M. Chhowalla, Nano Lett., 2013, 13, 6222–6227 CrossRef PubMed.
- M. A. Lukowski, A. S. Daniel, F. Meng, A. Forticaux, L. Li and S. Jin, J. Am. Chem. Soc., 2013, 135, 10274–10277 CrossRef PubMed.
- Y.-C. Lin, D. O. Dumcenco, Y.-S. Huang and K. Suenaga, Nat. Nanotechnol., 2014, 9, 391–396 CrossRef PubMed.
- A. Anto Jeffery, C. Nethravathi and M. Rajamathi, J. Phys. Chem. C, 2014, 118, 1386–1396 Search PubMed.
- B. T. Gebeyehu, S.-Y. Huang, A. W. Lee, J. K. Chen, J. Y. Lai, D. J. Lee and C. C. Cheng, Macromolecules, 2018, 51, 1189–1197 CrossRef.
- D. A. Henckel, O. Lenz and B. M. Cossairt, ACS Catal., 2017, 7, 2815–2820 CrossRef.
- X. Wang, Y. Chen, B. Zheng, F. Qi, J. He, P. Li and W. Zhang, Electrochim. Acta, 2016, 222, 1293–1299 CrossRef.
- T. Morishita and H. Okamoto, ACS Appl. Mater. Interfaces, 2016, 8, 27064–27073 Search PubMed.
- A. Ambrosi, Z. Sofer and M. Pumera, Chem. Commun., 2015, 51, 8450–8453 RSC.
- C. C. Cheng, J. H. Wang, W. T. Chuang, Z. S. Liao, J. J. Huang, S. Y. Huang, W. L. Fan and D. J. Lee, Polym. Chem., 2017, 8, 3294–3299 RSC.
- C. Zhao, J. M. Ang, Z. Liu and X. Lu, Chem. Eng. J., 2017, 330, 462–469 CrossRef.
- S. S. Chou, N. Sai, P. Lu, E. N. Coker, S. Liu, K. Artyushkova, T. S. Luk, B. Kaehr and C. J. Brinker, Nat. Commun., 2015, 6, 8311 CrossRef PubMed.
- Y. Liu, C. Tan, H. Chou, A. Nayak, D. Wu, R. Ghosh, H.-Y. Chang, Y. Hao, X. Wang, J.-S. Kim, R. Piner, R. S. Ruoff, D. Akinwande and K. Lai, Nano Lett., 2015, 15, 4979–4984 CrossRef PubMed.
- W. Zhang, Y. Cao, P. Tian, F. Guo, Y. Tian, W. Zheng, X. Ji and J. Liu, ACS Appl. Mater. Interfaces, 2016, 8, 32440–32449 Search PubMed.
- R. M. Silverstein, G. C. Bassler and T. C. Morrill, Spectrometric Identification of Organic Compounds, John Wiley & Sons, NewYork, 1991 Search PubMed.
- H. Li, G. Lu, Y. L. Wang, Z. Y. Yin, C. X. Cong, Q. Y. He, L. Wang, F. Ding, T. Yu and H. Zhang, Small, 2013, 9, 1974–1981 CrossRef PubMed.
- H. Li, J. Wu, Z. Yin and H. Zhang, Acc. Chem. Res., 2014, 47, 1067–1075 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: General methods, materials, and instrumental information are shown. See DOI: 10.1039/c8sc01778f |
| This journal is © The Royal Society of Chemistry 2018 |