
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCarboxylate-directed C–H allylation with allyl alcohols or ethers†
Xiao-Qiang
Hu
,
Zhiyong
Hu
,
A. Stefania
Trita
,
Guodong
Zhang
and
Lukas J.
Gooßen
 *
*
Evonik Chair of Organic Chemistry, Ruhr-Universität Bochum, ZEMOS, Universitätsstr. 150, 44801 Bochum, Germany. E-mail: lukas.goossen@rub.de
First published on 21st May 2018
Abstract
A [Ru(p-cymene)Cl2]2 catalyst activates allyl alcohols and ethers for the regioselective ortho-C–H allylation of aromatic and heteroaromatic carboxylates. The reaction is orthogonal to most C–H functionalisations with allyl alcohols in that allyl arenes rather than carbonyl compounds are obtained. A wide range of substrates are thus smoothly transformed to allylarenes at 50 °C in phosphate-buffered 2,2,2-trichloroethanol. The reaction concept combines the use of abundant reagents and directing groups in a sustainable, waste-minimised method for C–C bond formation.
Introduction
Allylarene motifs are widely found in natural products and biologically active molecules.1 The regiospecific introduction of allyl groups into functionalised arene substrates is generally achieved by coupling pre-formed or in situ-generated aryl-metal species with pre-activated allyl electrophiles,2 such as allyl acetates,3 carbonates,4 phosphates,5 halides,6 or allenes.7 The regiochemistry of C–H allylations is usually ensured by strong directing groups (Scheme 1a). The use in C–H functionalisations of non-activated allyl alcohols, with OH as the leaving group, would be highly desirable from the point of view of step- and atom economy. Allyl alcohols are widely available and would release only H2O as a by-product in dehydrogenative arylations.8 However, OH is such a poor leaving group that allyl alcohols usually react via a β-H elimination pathway leading to carbonyl compounds. The resulting Heck-type products are predominant not only in Pd-catalysed couplings of aryl halides, but also in Rh-catalysed oxidative ortho-C–H functionalisations (Scheme 1b).9 Examples for C–H allylations with alcohols as the allyl source are limited to Kanai's and Sundararaju's cobalt-catalysed allylation of nitrogen heterocycles,10 Matsunaga and Yoshino's allylation of 6-arylpurines and benzamides,11 and Kapur's ruthenium-catalysed C–H allylation of indoles bearing a pyridine directing group.12 All these reactions employ directing groups that are arduous to install and remove, and require high loadings of Ag or Cu additives. In our eyes, the ideal entry to allylarenes would consist of a regioselective C–H allylation directed by a simple, widely available substituent, and use non-derivatised alcohols as the allylating agent along with catalytic amounts of an inexpensive metal. In this respect, benzoic acids appeared to be particularly attractive starting materials, because carboxylate groups are abundant and can be tracelessly removed or act as anchor point for further transformations.13,14 Despite the low coordinating ability of carboxylates, efficient carboxylate-directed C–H functionalisations have been developed,14 such as arylations,15 acylations,16 alkylations,17 and alkenylations.18 Allylations are only possible starting from pre-formed allyl esters, and have a narrow substrate scope even at 135 °C.19 | ||
| Scheme 1 Strategies for C(sp2)–H allylation and reaction design. | ||
There is ample evidence that arenecarboxylates (1) can react with Ru-catalysts to give five-membered ruthenacycles (A).15c,17c,20 The first challenge was to tune the catalyst in a way that it would coordinate a simple allyl alcohol (2) and insert into its non-activated C–C double bond. The resulting ruthenacycle C had to be expected to undergo β-hydride elimination, leading to carbonyl compounds.9 However, if internal rotation could efficiently be suppressed by increasing charge separation, thus strengthening the coordination of the OH-group to the Ru centre, the only remaining pathway would be β-hydroxide elimination leading to the desired allylarene products. The key hurdle to this pathway, the low leaving-group ability of hydroxide, might be overcome by its solvent stabilisation. We believed that by adjusting the proton activity within the solvent and the charge at the metal centre, it should be possible to steer the catalyst towards the desired pathway despite these obstacles.
Results and discussion
In search for an effective catalyst system, we used the reaction between 2-methylbenzoic acid 1a and the secondary allylic alcohol 2a as the model and systematically investigated various catalysts, additives and solvents (Table 1). We were pleased to see that a combination of [Ru(p-cymene)Cl2]2 with substoichiometric inorganic bases led to the formation of allylation products. The solvent turned out to be the critical parameter.21 Aprotic solvents (toluene or CH3CN) gave low yields and insufficient selectivity for the desired allylarene 3aa over the β-H elimination by-product 3aa′ (entries 1 and 2). In protic solvents, in contrast, the reaction was highly selective for the desired product 3aa. Yields were optimised by adjusting the pKa of the solvents and bases (entries 3–9). A combination of the acidic alcohol 2,2,2-trichloroethanol (TCE, pKa = 12.24)22 with potassium phosphate and a reaction temperature of 50 °C were found to be optimal (entry 11). Evaluation of Ru pre-catalysts showed the cymene-ligated [Ru(p-cymene)Cl2]2 to be uniquely effective (entries 11–13). The presence of ligands dramatically reduced the yields and selectivities (entries 14 and 15). All findings are in agreement with our mechanistic blueprint, which relies on a coordinatively unsaturated metal centre and facile interactions between the hydroxyl group and ruthenium. Under the optimal conditions (2 mol% [Ru(p-cymene)Cl2]2, 0.5 equiv. K3PO4, TCE, 50 °C), allylarene 3aa was obtained in 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 E/Z ratio, along with only traces of the vinylarene double-bond isomer.
:1 E/Z ratio, along with only traces of the vinylarene double-bond isomer.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Base | Solvent | T (°C) | 3aa (%) | 3aa′ (%) |
| a Reaction conditions: 0.5 mmol 1a, 0.75 mmol 2a, 2 mol% [Ru], 0.25 mmol base, 0.5 mL solvent, 60 °C, 16 h, yields determined by 1H NMR spectroscopy using dibenzyl ether as internal standard, E/Z ratios in parentheses. b 2 mol% [Ru(C6Me6)Cl2]2. c 2 mol% Ru(cod)Cl2. d 4 mol% Ph3P. e 2 mol% dppb. [Ru] = [Ru(p-cymene)Cl2]2. tAmOH = 2-methylbutan-2-ol. HFIP = hexafluoro-2-propanol. TFE = 2,2,2-trifluoroethanol. TCE = 2,2,2-trichloroethanol. | |||||
| 1 | K3PO4 | Toluene | 60 | 13 (2.5:1) |
6 |
| 2 | K3PO4 | CH3CN | 60 | 8 (2:1) |
3 |
| 3 | K3PO4 | t AmOH | 60 | 16 (1.8:1) |
— |
| 4 | K3PO4 | HFIP | 60 | 27 (2:1) |
— |
| 5 | K3PO4 | TFE | 60 | 68 (1.7:1) |
— |
| 6 | K3PO4 | TCE | 60 | 80 (2:1) |
— |
| 7 | K2CO3 | TCE | 60 | 73 (2:1) |
— |
| 8 | Cs2CO3 | TCE | 60 | 68 (2:1) |
— |
| 9 | K2HPO4 | TCE | 60 | 58 (2:1) |
— |
| 10 | K3PO4 | TCE | 40 | 81 (2:1) |
— |
| 11 | K3PO4 | TCE | 50 | 89 (2:1) |
— |
| 12b | K3PO4 | TCE | 50 | 3 (n.d.) | — |
| 13c | K3PO4 | TCE | 50 | — | — |
| 14d | K3PO4 | TCE | 50 | 30 (2:1) |
6 |
| 15e | K3PO4 | TCE | 50 | — | — |
When starting from unsubstituted allyl alcohol (2t), double-bond migration could not be completely suppressed (Scheme 2). In an attempt to improve this, we also tested ethers as allylation reagents.23 To our delight, not only allyl phenyl ether (2u, pKa (PhOH) = 9.98)24 but also allyl methyl ether (2v, pKa (MeOH) = 15.5)22 cleanly converted 1a to 3at with high selectivity (allyl-to-vinyl ratio >15:1). This is the first time that the strong alkyl ether bond has been cleaved in a sp2 C–H functionalisation. This reaction variant may prove useful particularly in total synthesis, since allyl ether intermediates can now be coupled directly without difficult deprotection and activation steps.
 | ||
| Scheme 2 C–H-allylation with alcohols and ethers as allylating reagents. | ||
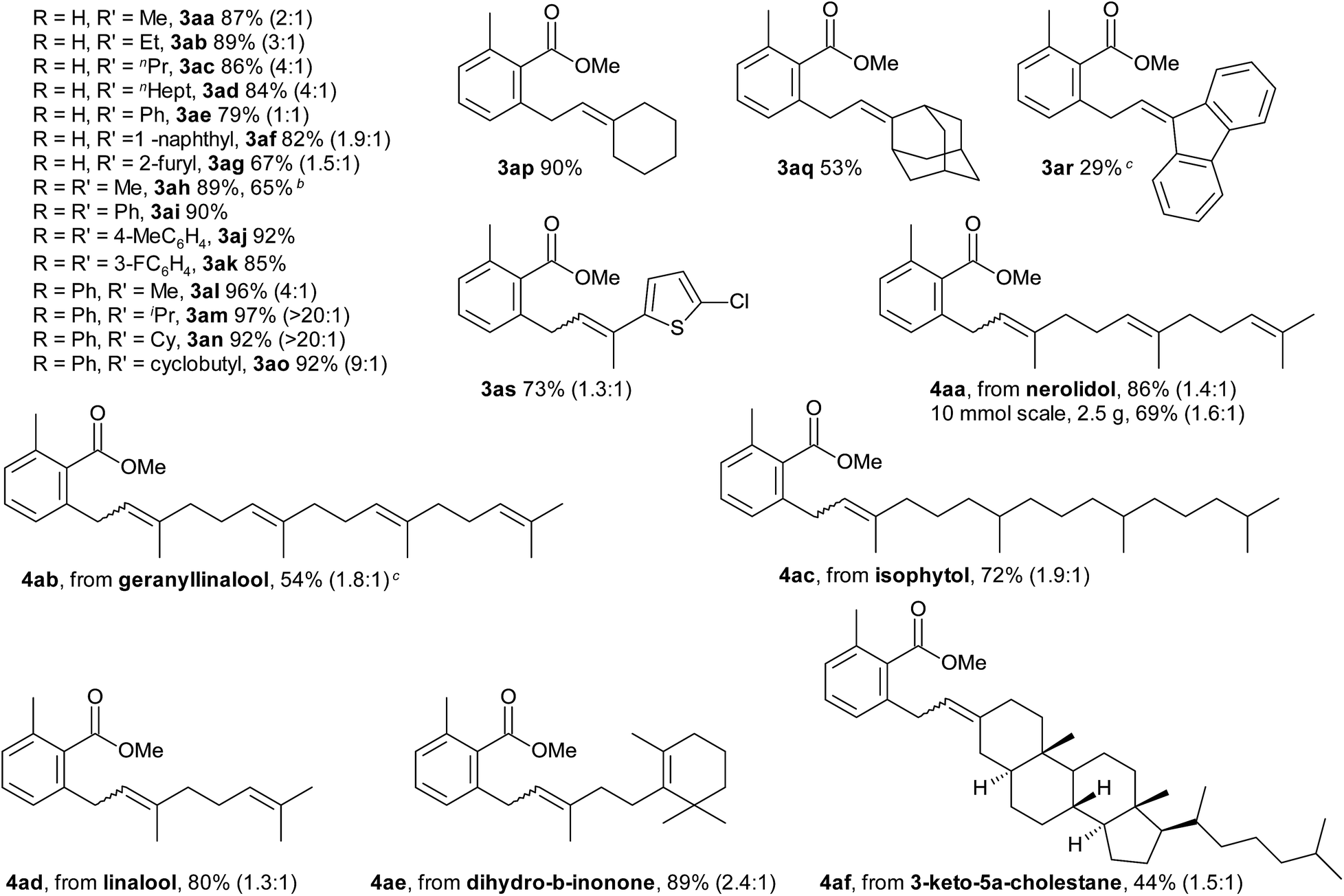
The scope of the C–H allylation with regard to the alcohol substrate is presented Table 2. Various primary, secondary, and even tertiary alcohols were coupled with 2-toluic acid (1a) in good yields. Substrates bearing alkyl, aryl, or heteroaryl groups at the carbinol carbon, e.g. dimethyl, cyclohexyl and even sterically crowded adamantyl alcohols, were successfully coupled. This, as well as the high selectivity for SN2′-type products, is in good agreement with the proposed β-OH elimination pathway. The E/Z selectivity correlates with the magnitude of steric interactions between the two substituents at the carbinol carbon, which is particularly high for 3am and 3an. The reaction is well-suited for naturally occurring allyl alcohols. The sensitive terpene alcohols nerolidol, geranyllinalool, isophytol and linalool were smoothly coupled, as were allylic alcohols obtained by vinylation of dihydro-β-ionone and 3-keto-5α-cholestane. The reaction was successfully performed on gram scale (4aa, 2.5 g, 69% yield). In the presence of potassium fluoride, allyl trimethylsilyl ethers also become effective allylating agents in this reaction. Thus, the TMS analogue of 2h was converted to product 3ah in 65% yield. A remaining limitation of this prototype protocol is that no substituents are tolerated at the double bond.
|
|
|---|
|
a Reaction conditions: 0.5 mmol 1a, 0.75 mmol 2, 2 mol% [Ru(p-cymene)Cl2]2, 0.25 mmol K3PO4, 0.5 mL TCE, 50 °C, 16 h. After the reaction was complete, 2 mL MeCN, 1.5 mmol K2CO3 and 2.5 mmol MeI were added and the mixture was stirred at 50 °C for 2 h, isolated yields of corresponding methyl esters, E:Z ratios in parentheses.
b Using TMS analogue of 2h in the presence of KF.
c 0.5 mmol K2CO3, 0.5 mL CH3CN, 80 °C, 16 h.
|
|
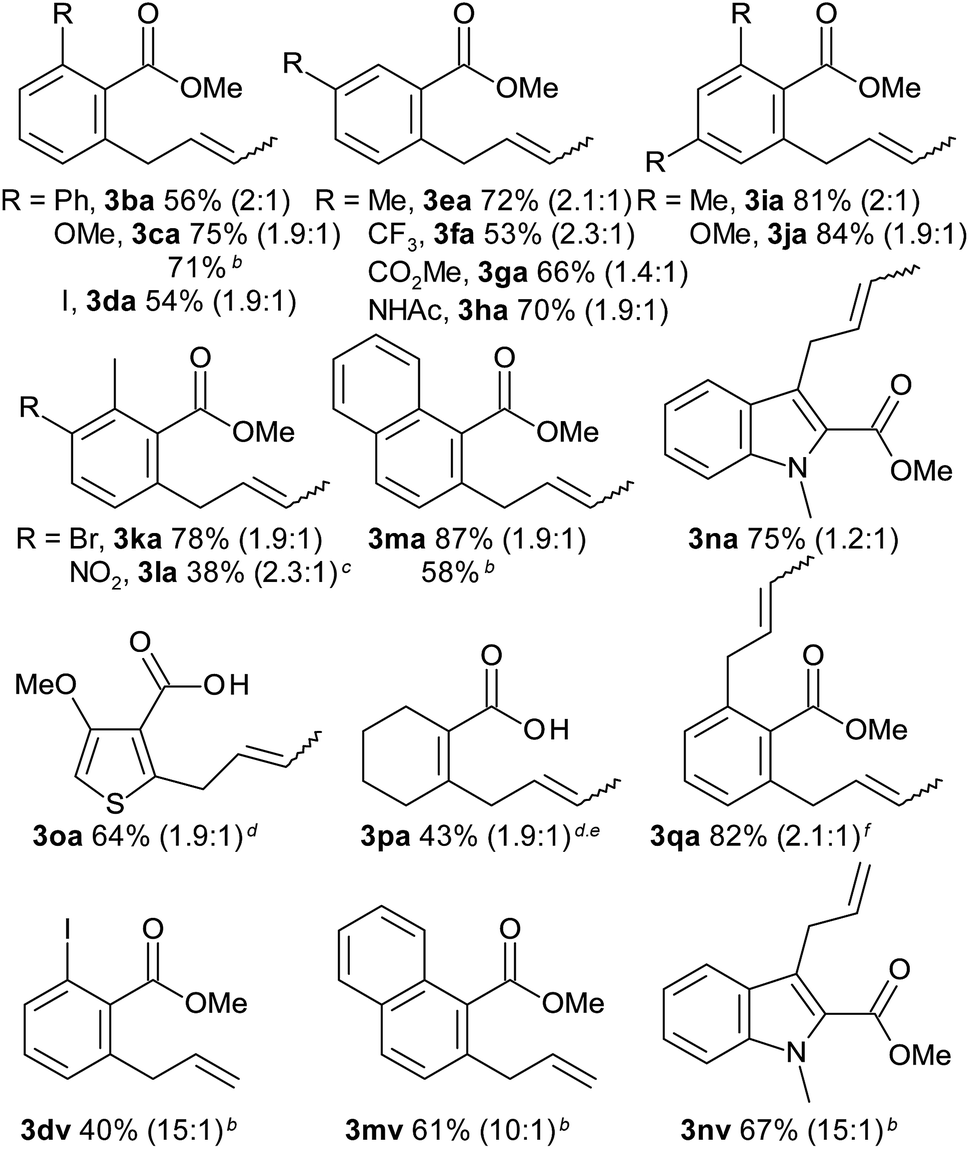
The scope with regard to the carboxylate substrate was investigated using 2a as the coupling partner (Table 3). Benzoic acids bearing electron-donating and electron-withdrawing substituents in ortho-, meta-, and para-positions all afforded comparable yields. Sensitive functionalities, such as ester, nitro and CF3 groups and reactive leaving groups such as bromo and even iodo substituents are left intact. Moreover, functional groups that are efficient directing groups in other C–H functionalisations, such as amide groups, were tolerated, opening up opportunities for orthogonal C(sp2)–H di-functionalisations. The scope also extends to heterocyclic carboxylates. The conversion of the vinylic carboxylate 1-cyclohexene-1-carboxylic acid required only minor deviations from the standard conditions. Para- and non-substituted benzoic acids reacted with competing diallylation. The diallylation product was obtained selectively when using 2.5 equivalents of the allyl alcohol (3qa).
|
|
|---|
|
a Reaction conditions: 0.5 mmol 1, 0.75 mmol 2a, 2 mol% [Ru(p-cymene)Cl2]2, 0.25 mmol K3PO4, 0.5 mL TCE, 50 °C, 16 h. After the reaction was complete, 2 mL MeCN, 1.5 mmol K2CO3 and 2.5 mmol MeI were added and the mixture was stirred at 50 °C for 2 h isolated yields of methyl esters, E:Z ratios in parentheses.
b Starting from methyl allyl ether, ratios of terminal to internal alkene in parentheses.
c 60 °C.
d Isolated as acid.
e 0.5 mmol K2CO3, 60 °C.
f 1.25 mmol 2a.
|
|
Starting from allyl methyl ethers, the reaction proceeded similarly well (3ca, 3ma). Their use is advantageous for introducing unsubstituted allyl groups, since double-bond isomerisation is efficiently suppressed (3dv, 3mv, 3nv).
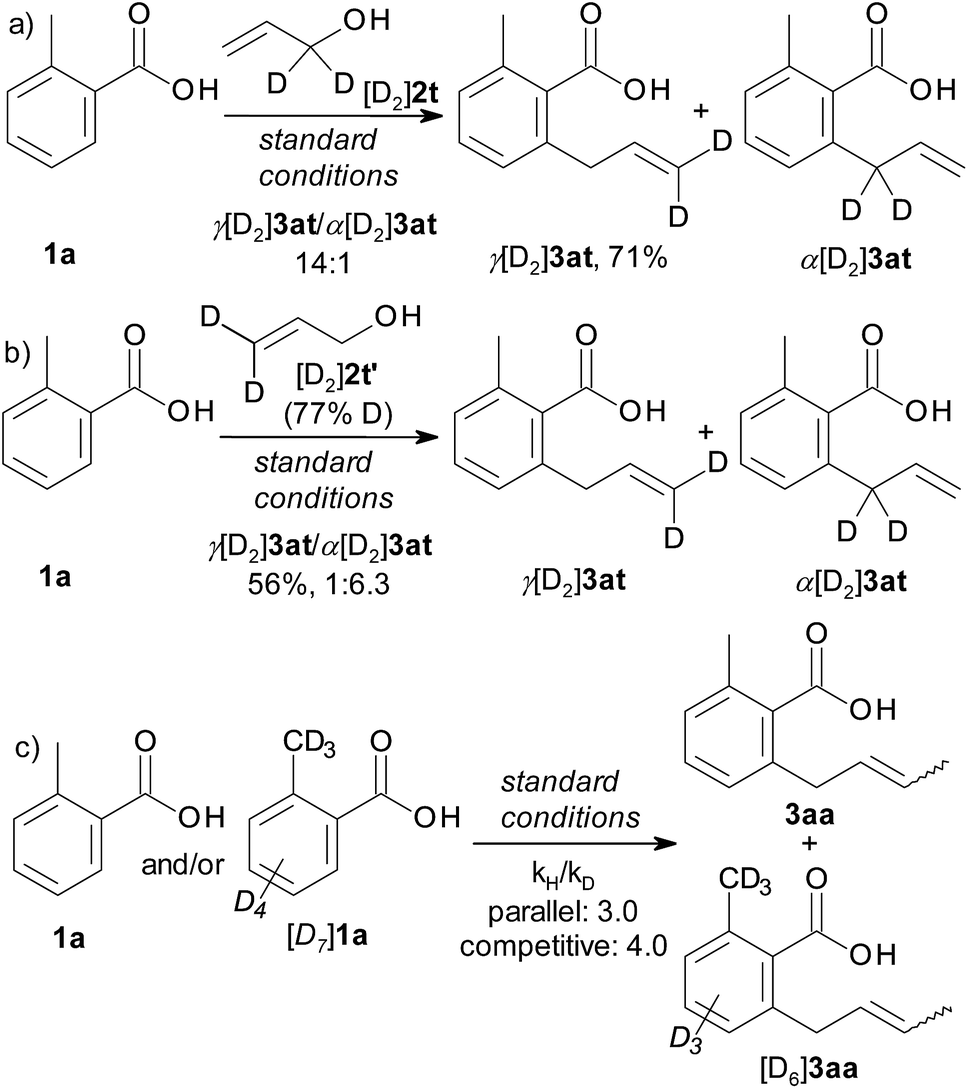
Deuterium-labelling experiments were conducted to elucidate the reaction mechanism. The allylation of 1a with 1,1-dideuterio-allyl alcohol [D2]2t afforded a 14:1 mixture of γ[D2]3at (71% yield) and α[D2]3at (Scheme 3a). When 3,3-dideuterio-allyl alcohol [D2]2t′ (77% D incorporation) was employed, a 1:6.3 mixture of γ[D2]3at and α[D2]3at was observed (Scheme 3b). These results further support that the β-OH elimination is the dominant pathway. The significant kinetic isotope effects in intermolecular competition (kH/kD = 4.0) and parallel experiments (kH/kD = 3.0) indicate that C–H activation rather than β-OH elimination is the rate-determining step (Scheme 3c). These observations agree with the proposed catalytic cycle.
 | ||
| Scheme 3 Mechanistic studies. | ||
Conclusions
In conclusion, this Ru-catalysed C(sp2)–H allylation gives efficient and sustainable access to a wide range of allylarenes from benzoic acids and non-activated allylic alcohols or ethers along with water or methanol as the only by-product.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
We thank Umicore for the donation of chemicals, the CSC (fellowships to Z. H. and G. Z.), and the DFG (EXC 1069 “RESOLV” and SFB-TRR 88 “3MET”) for financial support.Notes and references
- (a) M. Hassam, A. Taher, G. E. Arnott, I. R. Green and W. A. L. van Otterlo, Chem. Rev., 2015, 115, 5462–5569 CrossRef PubMed; (b) G. Ni, Q.-J. Zhang, Z.-F. Zheng, R.-Y. Chen and D.-Q. Yu, J. Nat. Prod., 2009, 72, 966–968 CrossRef PubMed; (c) J. L. Farmer, H. N. Hunter and M. G. Organ, J. Am. Chem. Soc., 2012, 134, 17470–17473 CrossRef PubMed.
- (a) N. K. Mishra, S. Sharma, J. Park, S. Han and I. S. Kim, ACS Catal., 2017, 7, 2821–2847 CrossRef; (b) J. D. Weaver, A. Recio III, A. J. Grenning and J. A. Tunge, Chem. Rev., 2011, 111, 1846–1913 CrossRef PubMed; (c) Z. Lu and S. Ma, Angew. Chem., Int. Ed., 2008, 47, 258–297 CrossRef PubMed; (d) B. M. Trost and D. L. Van Vranken, Chem. Rev., 1996, 96, 395–422 CrossRef PubMed.
- (a) S. Y. Lee and J. F. Hartwig, J. Am. Chem. Soc., 2016, 138, 15278–15284 CrossRef PubMed; (b) S. Oi, Y. Tanaka and Y. Inoue, Organometallics, 2006, 25, 4773–4778 CrossRef; (c) R. Manikandan, P. Madasamy and M. Jeganmohan, Chem.–Eur. J., 2015, 21, 13934–13938 CrossRef PubMed; (d) A. S. Tsai, M. Brasse, R. G. Bergman and J. A. Ellman, Org. Lett., 2011, 13, 540–542 CrossRef PubMed; (e) S. Bae, H.-L. Jang, H. Jung and J. M. Joo, J. Org. Chem., 2015, 80, 690–697 CrossRef PubMed; (f) C. Feng, D. Feng and T.-P. Loh, Chem. Commun., 2015, 51, 342–345 RSC; (g) H. Dai, C. Yu, Z. Wang, H. Yan and C. Lu, Org. Lett., 2016, 18, 3410–3413 CrossRef PubMed.
- (a) S. Fan, F. Chen and X. Zhang, Angew. Chem., Int. Ed., 2011, 50, 5918–5923 CrossRef PubMed; (b) H. Wang, N. Schröder and F. Glorius, Angew. Chem., Int. Ed., 2013, 52, 5386–5389 CrossRef PubMed; (c) W. Liu, S. C. Richter, Y. Zhang and L. Ackermann, Angew. Chem., Int. Ed., 2016, 55, 7747–7750 CrossRef PubMed; (d) D.-G. Yu, T. Gensch, F. de Azambuja, S. Vasquez-Cespedes and F. Glorius, J. Am. Chem. Soc., 2014, 136, 17722–17725 CrossRef PubMed; (e) H. He, W.-B. Liu, L.-X. Dai and S.-L. You, J. Am. Chem. Soc., 2009, 131, 8346–8347 CrossRef PubMed; (f) S.-S. Zhang, J.-Q. Wu, X. Liu and H. Wang, ACS Catal., 2014, 5, 210–214 CrossRef; (g) M. Kim, S. Sharma, N. K. Mishra, S. Han, J. Park, M. Kim, Y. Shin, J. H. Kwak, S. H. Han and I. S. Kim, Chem. Commun., 2014, 50, 11303–11306 RSC; (h) M. R. Sk, S. S. Bera and M. S. Maji, Org. Lett., 2018, 20, 134–137 CrossRef PubMed; (i) A. Cajaraville, S. Lopez, J. A. Varela and C. Saa, Org. Lett., 2013, 15, 4576–4579 CrossRef PubMed.
- (a) T. Yao, K. Hirano, T. Satoh and M. Miura, Angew. Chem., Int. Ed., 2011, 50, 2990–2994 CrossRef PubMed; (b) X. Cong, Y. Li, Y. Wei and X. Zeng, Org. Lett., 2014, 16, 3926–3929 CrossRef PubMed.
- (a) G. Cera, T. Haven and L. Ackermann, Angew. Chem., Int. Ed., 2016, 55, 1484–1488 CrossRef PubMed; (b) Y.-B. Yu, S. Fan and X. Zhang, Chem.–Eur. J., 2012, 18, 14643–14648 CrossRef PubMed; (c) N. Barsu, D. Kalsi and B. Sundararaju, Chem.–Eur. J., 2015, 21, 9364–9368 CrossRef PubMed; (d) Y. Aihara and N. Chatani, J. Am. Chem. Soc., 2013, 135, 5308–5311 CrossRef PubMed.
- (a) R. Zeng, C. Fu and S. Ma, J. Am. Chem. Soc., 2012, 134, 9597–9600 CrossRef PubMed; (b) B. Ye and N. Cramer, J. Am. Chem. Soc., 2013, 135, 636–639 CrossRef PubMed; (c) Y. J. Zhang, E. Skucas and M. J. Krische, Org. Lett., 2009, 11, 4248–4250 CrossRef PubMed; (d) S. Nakanowatari, T. Müller, J. C. A. Oliveira and L. Ackermann, Angew. Chem., Int. Ed., 2017, 56, 15891–15895 CrossRef PubMed.
- (a) N. A. Butt and W. Zhang, Chem. Soc. Rev., 2015, 44, 7929–7967 RSC; (b) B. Sundararaju, M. Achard and C. Bruneau, Chem. Soc. Rev., 2012, 41, 4467–4483 RSC; (c) S. Krautwald, D. Sarlah, M. A. Schafroth and E. M. Carreira, Science, 2013, 340, 1065–1068 CrossRef PubMed; (d) J. Qu and G. Helmchen, Acc. Chem. Res., 2017, 50, 2539–2555 CrossRef PubMed; (e) A. Lumbroso, M. L. Cooke and B. Breit, Angew. Chem., Int. Ed., 2013, 52, 1890–1932 CrossRef PubMed.
- (a) L. Huang, Q. Wang, J. Qi, X. Wu, K. Huang and H. Jiang, Chem. Sci., 2013, 4, 2665–2669 RSC; (b) Z. Shi, M. Boultadakis-Arapinis and F. Glorius, Chem. Commun., 2013, 49, 6489–6491 RSC; (c) S. H. Han, M. Choi, T. Jeong, S. Sharma, N. K. Mishra, J. Park, J. S. Oh, W. J. Kim, J. S. Lee and I. S. Kim, J. Org. Chem., 2015, 80, 11092–11099 CrossRef PubMed; (d) R. Yan and Z.-X. Wang, Asian J. Org. Chem., 2018, 7, 240–247 CrossRef; (e) R. Manoharan and M. Jeganmohan, Chem. Commun., 2015, 51, 2929–2932 RSC.
- (a) Y. Suzuki, B. Sun, K. Sakata, T. Yoshino, S. Matsunaga and M. Kanai, Angew. Chem., Int. Ed., 2015, 54, 9944–9947 CrossRef PubMed; (b) D. Kalsi, R. A. Laskar, N. Barsu, J. R. Premkumar and B. Sundararaju, Org. Lett., 2016, 18, 4198–4201 CrossRef PubMed.
- Y. Bunno, N. Murakami, Y. Suzuki, M. Kanai, T. Yoshino and S. Matsunaga, Org. Lett., 2016, 18, 2216–2219 CrossRef PubMed.
- G. S. Kumar and M. Kapur, Org. Lett., 2016, 18, 1112–1115 CrossRef PubMed.
- (a) L. J. Gooßen, G. Deng and L. M. Levy, Science, 2006, 313, 662–664 CrossRef PubMed; (b) Y. Wei, P. Hu, M. Zhang and W. Su, Chem. Rev., 2017, 117, 8864–8907 CrossRef PubMed; (c) G. J. P. Perry and I. Larrosa, Eur. J. Org. Chem., 2017, 3517–3527 CrossRef PubMed; (d) N. Rodriguez and L. J. Gooßen, Chem. Soc. Rev., 2011, 40, 5030–5048 RSC; (e) J. Xuan, Z.-G. Zhang and W.-J. Xiao, Angew. Chem., Int. Ed., 2015, 54, 15632–15641 CrossRef PubMed; (f) L. J. Gooßen and K. Gooßen, in Inventing Reactions, Decarboxylative Coupling Reactions, Springer-Verlag, Berlin Heidelberg, 2012, pp. 121–141 Search PubMed; (g) W. I. Dzik, P. P. Lange and L. J. Gooßen, Chem. Sci., 2012, 3, 2671–2678 RSC.
- (a) M. Font, J. M. Quibell, G. J. P. Perry and I. Larrosa, Chem. Commun., 2017, 53, 5584–5597 RSC; (b) M. Pichette Drapeau and L. J. Gooßen, Chem.–Eur. J., 2016, 22, 18654–18677 CrossRef PubMed; (c) G. Shi and Y. Zhang, Adv. Synth. Catal., 2014, 356, 1419–1442 CrossRef.
- (a) H. A. Chiong, Q.-N. Pham and O. Daugulis, J. Am. Chem. Soc., 2007, 129, 9879–9884 CrossRef PubMed; (b) L. Huang, D. Hackenberger and L. J. Gooßen, Angew. Chem., Int. Ed., 2015, 54, 12607–12611 CrossRef PubMed; (c) A. Biafora, T. Krause, D. Hackenberger, F. Belitz and L. J. Gooßen, Angew. Chem., Int. Ed., 2016, 55, 14752–14755 CrossRef PubMed; (d) D.-H. Wang, T.-S. Mei and J.-Q. Yu, J. Am. Chem. Soc., 2008, 130, 17676–17677 CrossRef PubMed; (e) J. Cornella, M. Righi and I. Larrosa, Angew. Chem., Int. Ed., 2011, 50, 9429–9432 CrossRef PubMed; (f) J. Luo, S. Preciado and I. Larrosa, J. Am. Chem. Soc., 2014, 136, 4109–4112 CrossRef PubMed; (g) Y. Zhang, H. Zhao, M. Zhang and W. Su, Angew. Chem., Int. Ed., 2015, 54, 3817–3821 CrossRef PubMed; (h) K. Takamatsu, K. Hirano and M. Miura, Angew. Chem., Int. Ed., 2017, 56, 5353–5357 CrossRef PubMed.
- (a) P. Mamone, G. Danoun and L. J. Gooßen, Angew. Chem., Int. Ed., 2013, 52, 6704–6708 CrossRef PubMed; (b) J. Miao and H. Ge, Org. Lett., 2013, 15, 2930–2933 CrossRef PubMed; (c) X.-Y. Shi, A. Renzetti, S. Kundu and C.-J. Li, Adv. Synth. Catal., 2014, 356, 723–728 CrossRef.
- (a) G. Cheng, T.-J. Li and J.-Q. Yu, J. Am. Chem. Soc., 2015, 137, 10950–10953 CrossRef PubMed; (b) B. R. Rosen, L. R. Simke, P. S. Thuy-Boun, D. D. Dixon, J.-Q. Yu and P. S. Baran, Angew. Chem., Int. Ed., 2013, 52, 7317–7320 CrossRef PubMed; (c) G. Zhang, F. Jia and L. J. Gooßen, Chem.–Eur. J., 2018, 24, 4537–4541 CrossRef PubMed; (d) R. Shang, L. Ilies and E. Nakamura, J. Am. Chem. Soc., 2016, 138, 10132–10135 CrossRef PubMed.
- (a) L. Huang, A. Biafora, G. Zhang, V. Bragoni and L. J. Gooßen, Angew. Chem., Int. Ed., 2016, 55, 6933–6937 CrossRef PubMed; (b) A. Biafora, B. A. Khan, J. Bahri, J. M. Hewer and L. J. Gooßen, Org. Lett., 2017, 19, 1232–1235 CrossRef PubMed; (c) K. Ueura, T. Satoh and M. Miura, Org. Lett., 2007, 9, 1407–1409 CrossRef PubMed; (d) S. Warratz, C. Kornhaaß, A. Cajaraville, B. Niepötter, D. Stalke and L. Ackermann, Angew. Chem., Int. Ed., 2015, 54, 5513–5517 CrossRef PubMed; (e) N. Y. P. Kumar, A. Bechtoldt, K. Raghuvanshi and L. Ackermann, Angew. Chem., Int. Ed., 2016, 55, 6929–6932 CrossRef PubMed; (f) J. Zhang, R. Shrestha, J. F. Hartwig and P. Zhao, Nat. Chem., 2016, 8, 1144–1151 CrossRef PubMed; (g) J. Kim, S.-W. Park, M.-H. Baik and S. Chang, J. Am. Chem. Soc., 2015, 137, 13448–13451 CrossRef PubMed.
- (a) Y. Kuninobu, K. Ohta and K. Takai, Chem. Commun., 2011, 47, 10791–10793 RSC; (b) A. S. Trita, A. Biafora, M. Pichette Drapeau, P. Weber and L. J. Gooßen, Angew. Chem., Int. Ed., 2018 DOI:10.1002/anie.201712520.
- (a) P. B. Arockiam, C. Bruneau and P. H. Dixneuf, Chem. Rev., 2012, 112, 5879–5918 CrossRef PubMed; (b) In Ruthenium Catalysts and Fine Chemistry, ed. C. Bruneau and P. Dixneuf, Springer, Heidelberg, Germany, 2004 Search PubMed.
- A. Bernhardt, H. Kelm and F. W. Patureau, ChemCatChem, 2018, 10, 1547–1551 CrossRef PubMed.
- P. Ballinger and F. A. Long, J. Am. Chem. Soc., 1960, 82, 795–798 CrossRef.
- S. Asako, L. Ilies and E. Nakamura, J. Am. Chem. Soc., 2013, 135, 17755–17757 CrossRef PubMed.
- M. D. Liptak, K. C. Gross, P. G. Seybold, S. Feldgus and G. C. Shields, J. Am. Chem. Soc., 2002, 124, 6421–6427 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8sc01741g |
| This journal is © The Royal Society of Chemistry 2018 |