
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceControllable fabrication of bio-bar codes for dendritically amplified sensing of human T-lymphotropic viruses†
Li-juan
Wang‡
a,
Ming
Ren‡
a,
Li
Liang‡
b and
Chun-yang
Zhang
 *a
*a
aCollege of Chemistry, Chemical Engineering and Materials Science, Collaborative Innovation Center of Functionalized Probes for Chemical Imaging in Universities of Shandong, Key Laboratory of Molecular and Nano Probes, Ministry of Education, Shandong Provincial Key Laboratory of Clean Production of Fine Chemicals, Shandong Normal University, Jinan 250014, China. E-mail: cyzhang@sdnu.edu.cn; Fax: +86 0531 82615258; Tel: +86 0531 86186033
bDepartment of Tumor Chemotherapy and Radiation Sickness, Peking University Third Hospital, Beijing 100191, China
First published on 10th May 2018
Abstract
Human T-lymphotropic virus type II (HTLV-II) is an important type-C retrovirus, closely related to a variety of human diseases. Here, we demonstrate for the first time the controllable fabrication of bio-bar codes for dendritically amplified sensing of low-abundant HTLV-II DNA by the integration of terminal deoxynucleotidyl transferase (TdT)-catalyzed template-free polymerization extension with bio-bar-code amplification (BCA). HTLV-II DNA hybridizes with magnetic microparticle (MMP)-modified capture probe 1, forming a stable DNA duplex with a protruding 3′-hydroxylated sequence which may function as a primer to initiate the TdT-catalyzed first-step polymerization extension for the generation of a poly-thymidine (T) sequence. The resultant poly-T products may hybridize with poly-adenine (A) capture probe 2, inducing the self-assembly of multiple capture probe 2-/reporter probe-functionalized Au nanoparticles (AuNPs) onto the MMP. Subsequently, the reporter probes may act as the primers to initiate the TdT-catalyzed second-step polymerization extension, producing large numbers of G-rich DNAzymes for the generation of an enhanced chemiluminescence signal. Taking advantage of the efficient polymerization extension reaction catalyzed by TdT, the high amplification efficiency of BCA, and the intrinsically high sensitivity of G-rich DNAzyme-driven chemiluminescence, this method exhibits ultrahigh sensitivity with a limit of detection of as low as 0.50 aM and a large dynamic range of 9 orders of magnitude from 1 aM to 1 nM. Moreover, this method can be applied for the discrimination of a single-base mismatch and the measurement of HTLV-II DNA in both human serum and human T-lymphocytic leukemia cells, holding great potential in biomedical research and clinical diagnosis.
Introduction
Human T-lymphotropic viruses are a family of human type-C RNA tumor viruses (i.e. retroviruses) including HTLV-I, HTLV-II and HTLV-III, which are closely related to a variety of human T-cell malignancies and acquired immunodeficiency syndrome (AIDS).1,2 As a close relative of HTLV-I, HTLV-II has strong association with neurological disorders (e.g., HTLV-associated myelopathy/tropical spastic paraparesis (HAM/TSP)), respiratory symptoms (e.g., pneumonia and bronchitis), inflammatory responses (e.g., arthritis) and increased mortality.2 Injection drug users (IDU) infected with HTLV-II may introduce the virus into the general population and blood donors through secondary sexual transmission, inducing global neurological disability and neuropathy.3 Moreover, HTLV-II DNA is usually present in clinical samples at trace levels (10–100 copy number per μg DNA).4 Taking into account its low abundance, short size, high sequence homology among the HTLV family members and its critical roles in biomedical research, the development of an effective method for sensitive and selective detection of HTLV-II DNA is highly urgent.So far, a variety of amplification strategies have been developed for sensitive detection of target nucleotides including polymerase chain reaction (PCR),5–7 rolling circle amplification (RCA),8–10 loop-mediated isothermal amplification (LAMP),11,12 hybridization chain reaction (HCR),13,14 catalyzed hairpin assembly (CHA) amplification,15 ligase chain reaction (LCR),16,17 and exonuclease-18,19/endonuclease-assisted signal amplification (EASA).20–23 PCR is a thermal cycle-based DNA amplification technique with the involvement of a stringent primer/template design and precise thermal cycling.5–7 RCA8–10 and LAMP11,12 are isothermal amplification techniques that circumvent the cumbersome thermal cycling of PCR. However, RCA involves complicated preparation and isolation of circle templates,8–10 and LAMP requires a complex design of DNA hairpin probes.11,12 In addition, PCR, RCA and LAMP are DNA template-dependent amplification, which inevitably involve amplicon cross-contamination induced by nonspecific amplification. Alternatively, HCR is based on a chain hybridization reaction between two sets of DNA hairpin probes,13,14 and CHA is based on accelerated hybridization between two DNA hairpin probes catalyzed by an input DNA target.15 Both HCR and CHA can offer enzyme-free nucleic acid-based signal amplification, but their performances heavily rely on the precise design of DNA hairpin probes. LCR utilizes thermostable DNA ligase to ligate adjacently hybridized DNA probes for amplification detection of target DNA.16 However, the reaction conditions of LCR (e.g. appropriate ligation temperature, cycle number and ligase concentration) are relatively complicated with the involvement of gel electrophoresis for ligation product separation.17 EASA employs exonucleases (e.g. exonuclease III15,18,24 and lambda exonuclease19) and endonucleases (e.g. Nt.AlwI,20 Nt.BbvCI,21 N.BstNBI22 and Nb.BsmI23) to cyclically digest/cleave specific nucleotide sequences for signal amplification, but the low cleavage efficiency and the non-specific digestion may lead to poor specificity and low sensitivity.
Bio-bar-code amplification (BCA) is a new amplification technique that utilizes short oligonucleotides as the recognition strands and surrogate amplification units for enhanced signal amplification.25,26 Especially, the assembly of multiple oligonucleotides on a single nanoparticle and the subsequent introduction of magnetic separation offer a very clean reaction environment for BCA, enabling improved sensitivity and specificity25–28 for the detection of a variety of proteins and nucleic acids.27,29–31 However, the reported BCA usually involves a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 hybridization ratio for nanoparticles and the target strand, complicated preparation of oligonucleotide-/protein-modified nanoparticles and tedious procedures for distinguishing signal probes from the nanoparticle.25,26,28 Therefore, the achievement of high sensitivity and a wide dynamic range still remains a great challenge and calls for the introduction of new techniques into BCA. Terminal deoxynucleotidyl transferase (TdT) is a template-free intranuclear polymerase, and it can catalyze the incorporation of a highly variable number of mononucleotides into the 3′-hydroxyl (OH) termini of single-stranded DNA (ssDNA) fragments32,33 and may be exploited as a new tool for signal amplification.34–36 With the addition of a deoxynucleotide (dNTP) pool (60% dGTP + 40% dATP), TdT can catalyze the polymerization extension to produce long randomly arrayed guanine (G)-rich sequences which may bind cofactor hemin to form G-quadruplex/hemin complexes with similar catalytic activity to horseradish peroxidase (HRP).37 G-quadruplex HRP-mimicking DNAzyme is a functional nucleic acid that can fold into a four-stranded G-quadruplex structure with the incorporation of hemin into catalytically active structures to catalyze the peroxidase (H2O2)-mediated oxidation of chemical reagents (e.g. ABTS2− and luminol) for the generation of amplified colorimetric and optical signals.38,39 The G-quadruplex DNAzyme has been used as a biocatalytic label for amplified detection of various biomolecules including nucleotides,40,41 proteins,42,43 enzymes39,44 and cations.45,46 To the best of our knowledge, the combination of BCA with TdT-catalyzed formation of G-quadruplex DNAzymes for chemiluminescence detection of target DNA has never been reported so far.
:1 hybridization ratio for nanoparticles and the target strand, complicated preparation of oligonucleotide-/protein-modified nanoparticles and tedious procedures for distinguishing signal probes from the nanoparticle.25,26,28 Therefore, the achievement of high sensitivity and a wide dynamic range still remains a great challenge and calls for the introduction of new techniques into BCA. Terminal deoxynucleotidyl transferase (TdT) is a template-free intranuclear polymerase, and it can catalyze the incorporation of a highly variable number of mononucleotides into the 3′-hydroxyl (OH) termini of single-stranded DNA (ssDNA) fragments32,33 and may be exploited as a new tool for signal amplification.34–36 With the addition of a deoxynucleotide (dNTP) pool (60% dGTP + 40% dATP), TdT can catalyze the polymerization extension to produce long randomly arrayed guanine (G)-rich sequences which may bind cofactor hemin to form G-quadruplex/hemin complexes with similar catalytic activity to horseradish peroxidase (HRP).37 G-quadruplex HRP-mimicking DNAzyme is a functional nucleic acid that can fold into a four-stranded G-quadruplex structure with the incorporation of hemin into catalytically active structures to catalyze the peroxidase (H2O2)-mediated oxidation of chemical reagents (e.g. ABTS2− and luminol) for the generation of amplified colorimetric and optical signals.38,39 The G-quadruplex DNAzyme has been used as a biocatalytic label for amplified detection of various biomolecules including nucleotides,40,41 proteins,42,43 enzymes39,44 and cations.45,46 To the best of our knowledge, the combination of BCA with TdT-catalyzed formation of G-quadruplex DNAzymes for chemiluminescence detection of target DNA has never been reported so far.
Here we demonstrate for the first time the controllable fabrication of bio-bar codes for dendritically amplified sensing of HTLV-II DNA. Taking advantage of the efficient polymerization reaction catalyzed by TdT, the high amplification efficiency of BCA, and the high sensitivity of G-rich DNAzyme-driven chemiluminescence, this method can detect HTLV-II DNA with a limit of detection of as low as 0.50 aM and a large dynamic range of 9 orders of magnitude from 1 aM to 1 nM. Moreover, it can be applied for the discrimination of a single-base mismatch and the measurement of HTLV-II DNA in both human serum and human T-lymphocytic leukemia cells.
Results and discussion
The principle of HTLV-II DNA assay is illustrated in Scheme 1. This assay involves two kinds of streptavidin-coated nanoparticles including 2.8 μm-diameter iron oxide MMPs and 10 nm-diameter AuNPs. Through specific biotin–streptavidin interaction, the MMPs are functionalized with 3′-biotinylated capture probe 1 (Scheme 1, black color) to obtain capture probe 1-functionalized MMPs. Capture probe 1 is complementary to one 17-mer portion (5′-AGC ACC AAC TCA CCT GG-3′) of HTLV-II DNA (Scheme 1, red color). The AuNPs are modified with two kinds of oligonucleotides including capture probe 2 (Scheme 1, orange color) and a reporter probe (Scheme 1, green color). 3′-Biotinylated capture probe 2 with a poly-adenine (A) sequence is complementary to the poly-thymidine (T) product (Scheme 1, blue color) obtained in the TdT-catalyzed first-step polymerization reaction, and the 5′-biotinylated reporter probe may act as a primer for the initiation of TdT-catalyzed second-step polymerization extension to produce G-rich DNAzymes (Scheme 1, pink color). The streptavidin-coated AuNPs are functionalized with capture probe 2 and reporter probes at a 1:100 ratio, respectively, through specific biotin–streptavidin interaction26 to obtain capture probe 2/reporter probe-functionalized AuNPs. As shown in Scheme 1, this assay involves two consecutive steps: (1) HTLV-II DNA-induced first-step enzymatic extension coupled with dendritic self-assembly of bio-bar codes, and (2) the second-step enzymatic extension-induced chemiluminescence in the presence of hemin. In the first-step reaction, target HTLV-II DNA may partly hybridize with MMP-modified capture probe 1 to form a stable double-stranded DNA (dsDNA) duplex with a protruding 3′-hydroxylated sequence of HTLV-II DNA. The protruding ssDNA sequence may function as a primer to initiate TdT-catalyzed template-independent polymerization extension. Upon the addition of a dTTP pool, TdT can catalyze the repeated incorporation of dTTPs into the 3′-OH termini of HTLV-II DNA, generating a long chain of poly-thymidine (T) sequence. The resultant poly-T products may hybridize with poly-A capture probe 2, leading to the self-assembly of multiple capture probe 2-/reporter probe-functionalized AuNPs onto the poly-T product-linked MMPs. In the second-step reaction, the excess TdT enzymes are removed by magnetic separation, and subsequently the capture probe 2-/reporter probe-functionalized AuNPs, TdT, and a dNTP pool (60% dGTP and 40% dATP) are added into the reaction system. With reporter probes as primers, TdT will randomly incorporate dGTPs and dATPs into the 3′-OH ends of the reporter probes, producing large numbers of long chains of G-rich sequences. In the presence of hemin, the G-rich products can bind with the cofactor hemin to form hemin–G-quadruplex nanostructures which can catalyze the H2O2-mediated oxidation of luminol to generate an amplified chemiluminescence signal. Taking advantage of the efficient TdT-catalyzed two-step polymerization reaction, the high amplification efficiency of BCA, and the intrinsically high sensitivity of G-rich DNAzyme-driven chemiluminescence, the proposed method can sensitively detect HTLV-II DNA.
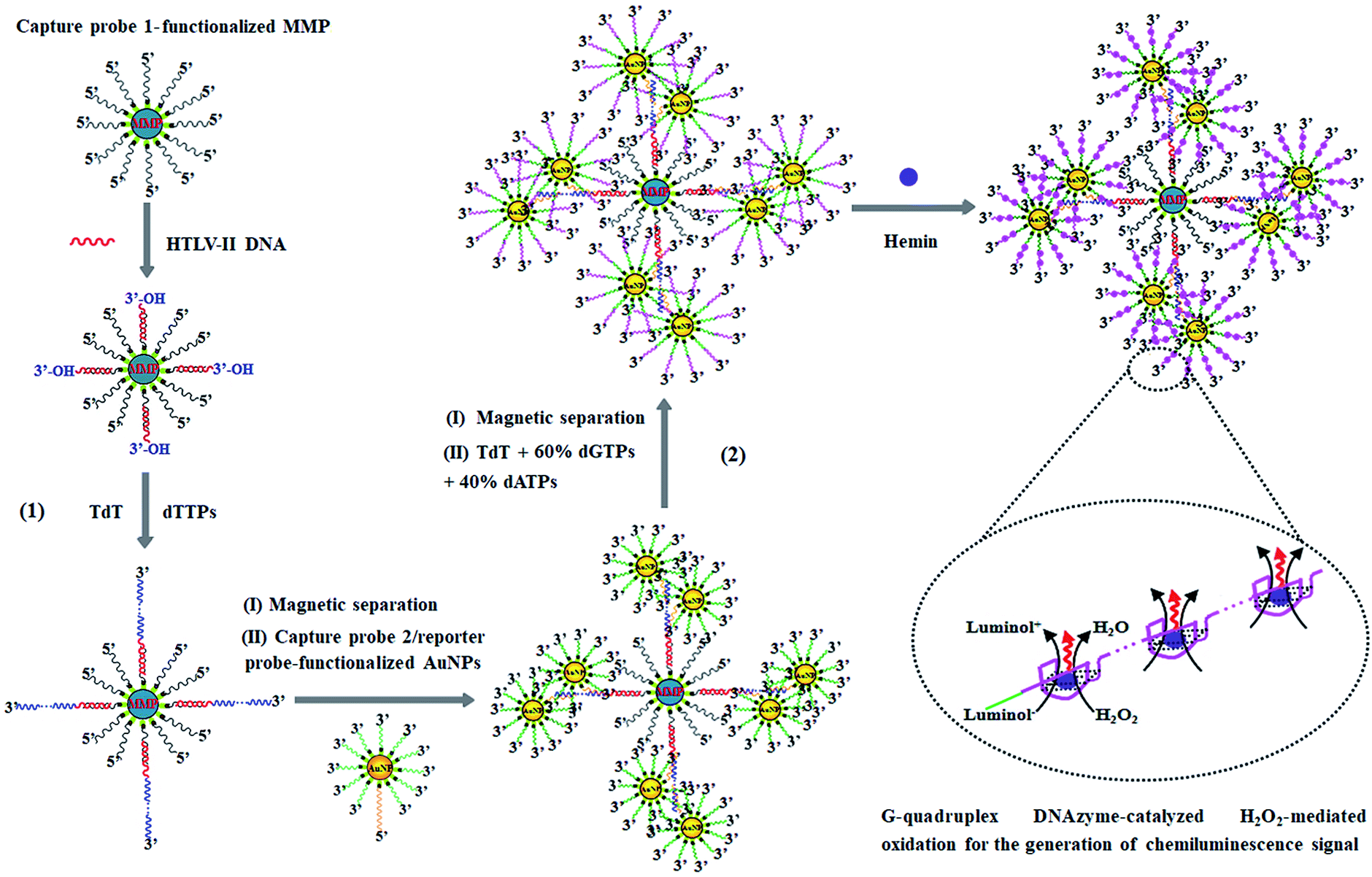 | ||
| Scheme 1 Schematic illustration of controllable fabrication of bio-bar codes for dendritically amplified sensing of HTLV-II DNA. This assay involves two consecutive steps: (1) HTLV-II DNA-induced TdT-catalyzed first-step enzymatic extension coupled with dendritic self-assembly of bio-bar codes, and (2) TdT-catalyzed second-step enzymatic extension-induced chemiluminescence in the presence of hemin. | ||
To characterize the functionalized nanoprobes, we measured the UV-vis spectra of the capture probe 1-functionalized MMPs (Fig. 1A) and the capture probe 2-/reporter probe-functionalized AuNPs (Fig. 1B), respectively. A characteristic absorption peak at 260 nm is observed for the capture probe 1-functionalized MMPs (Fig. 1A, red line), indicating the successful conjugation of capture probe 1 onto the surface of MMPs. However, no visible absorption peak is detected for only MMPs (Fig. 1A, black line). In contrast to only one significant absorbance peak at 520 nm for only AuNPs (Fig. 1B, black line), two characteristic absorbance peaks at 260 and 520 nm are observed for capture probe 2-/reporter probe-functionalized AuNPs (Fig. 1B, red line), suggesting the successful conjugation of capture probe 2 and the reporter probe onto the surface of AuNPs. To verify whether HTLV-II DNA can initiate the TdT-catalyzed polymerization extension and the subsequent assembly of AuNPs–DNA, we employed TEM to characterize the AuNPs and the HTLV-II DNA/poly-T product/AuNPs–DNA nanostructure. In the absence of HTLV-II DNA, the AuNPs show a uniform spherical crystallite with an average diameter of 10 nm (Fig. 1C). In contrast, HTLV-II DNA induces distinct AuNP aggregation (Fig. 1D). In the presence of HTLV-II DNA, TdT-catalyzed polymerization extension is initiated for the generation of a >300 nt chain of poly-T sequence after 30 min reaction.47–50 The resultant poly-T product may hybridize with AuNP-modified poly-A capture probe 2 to form a nanostructure of HTLV II-DNA/poly-T product/AuNPs–DNA, leading to the assembly of more than 30 AuNPs–DNA around each poly-T product (Fig. 1D). In theory, each poly-T product may assemble a maximum of 16 AuNPs when capture probe 2 with an 18 nt poly-A sequence completely hybridizes with the poly-T sequence. In fact, each poly-T product can assemble more than 30 AuNPs (Fig. 1D) due to the partial hybridization of capture probe 2 with the poly-T sequence, facilitating the subsequent dendritic amplification. To validate the feasibility of the proposed method, we measured the chemiluminescence signals under various experimental conditions (Fig. 1E). Significant chemiluminescence enhancement is observed only in the presence of HTLV-II DNA, TdT (added in the first- and second-step reactions, respectively), dTTP, AuNPs and dATP + dGTP (Fig. 1E, red column), demonstrating that only HTLV-II DNA can trigger TdT-catalyzed first-step polymerization extension and the subsequent bio-bar-code-based dendritic amplification for the generation of an amplified chemiluminescence signal. In contrast, no distinct chemiluminescence signal is observed in the absence of either one of HTLV-II DNA, TdT, dTTP, AuNPs and dATP + dGTP (Fig. 1E). These results clearly demonstrate the feasibility of bio-bar codes for chemiluminescence detection of HTLV-II DNA.
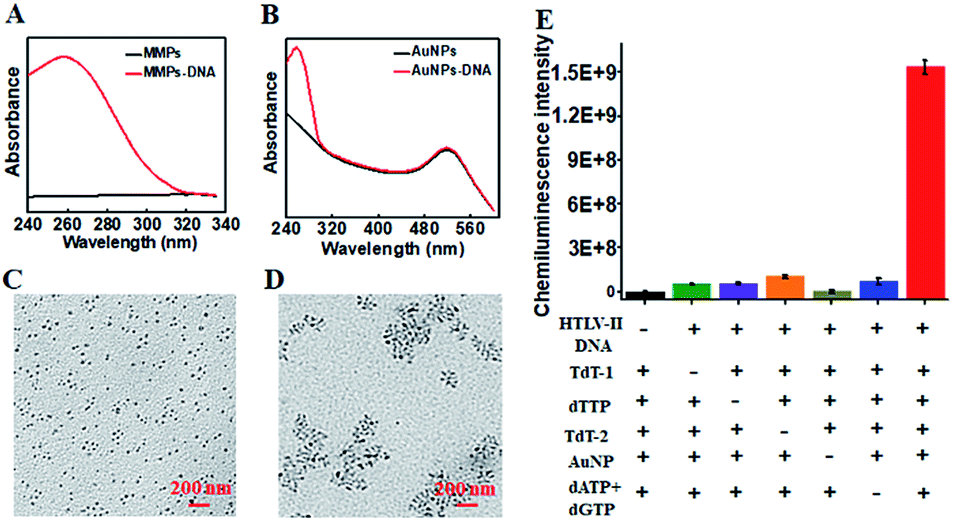 | ||
| Fig. 1 (A) UV-visible spectra of the capture probe 1-functionalized MMPs (red line) and MMPs (black line). (B) UV-visible spectra of the capture probe 2-/reporter probe-functionalized AuNPs (red line) and only AuNPs (black line). (C and D) TEM images of AuNPs (C) and the HTLV II-DNA/poly-T product/AuNPs–DNA nanostructures (D). The scale bar is 200 nm. (E) Measurement of chemiluminescence intensity of the reaction system in the absence of HTLV-II DNA (black column), TdT (TdT-1, added in the first-step reaction, green column; TdT-2, added in the second-step reaction, orange column), dTTP (purple column), AuNPs (brown column), and dATP + dGTP (blue column), and in the presence of all the above reagents (red column). Error bars show the standard deviations of three experiments. | ||
To achieve the best performance, we optimized the experimental conditions including the polymerization time of TdT-catalyzed two-step extension reactions, the concentrations of hemin, luminol, and piperazine-1-ethanesulfonic acid sodium salt (HEPES), and the ratio of dATP to dGTP (see ESI, Fig. S1†). Under the optimal experimental conditions, we measured the chemiluminescence intensity in response to different concentrations of HTLV-II DNA. As shown in Fig. 2, the chemiluminescence intensity enhances with increasing concentration of HTLV-II DNA from 1 aM to 1 nM. Moreover, the chemiluminescence intensity shows a good linear relationship with the logarithm of HTLV-II DNA concentration over a large dynamic range of 9 orders of magnitude from 1 aM to 1 nM (inset of Fig. 2). The correlation equation is I = 1.59 × 108log10C + 3.05 × 109 with a correlation coefficient of 0.9887, where I represents the chemiluminescence intensity and C represents the HTLV-II DNA concentration (M). The limit of detection (LOD) is calculated to be 0.50 aM by evaluating the average response of the negative control plus three times the standard deviation. Notably, the introduction of capture probe 2-/reporter probe-functionalized AuNPs in the current assay induces a 913.30-fold enhancement compared to that without the capture probe 2-/reporter probe-functionalized AuNPs (see ESI, Fig. S2†). In theory, each MMP is modified with ∼3.08 amol capture probe 1 according to the instruction of the manufacturer, resulting in the assembly of ∼3.08 amol HTLV-II DNA around each MMP through specific DNA hybridization and consequently the generation of ∼3.08 amol poly-T polymerization products (>300 nt) by TdT-catalyzed first-step enzymatic extension (see ESI, Fig. S1A,† lane 4). Since each >300 nt poly-T product may couple with more than 30 capture probe 2-/reporter probe-functionalized AuNPs (Fig. 1D), one MMP can assemble more than 5.55 × 107 capture probe 2-/reporter probe-functionalized AuNPs. According to the instructions, each AuNP is theoretically modified with 0.05 zmol reporter probes which may function as the primers for TdT-initiated second-step enzymatic extension, producing 0.05 zmol long G-rich DNAzymes (see ESI, Fig. S1C,† lane 4). As a result, each MMP may be assembled by more than 2.34 × 109 G-rich DNAzymes, which is 1.26 × 103-fold more than that without the involvement of capture probe 2-/reporter probe-functionalized AuNPs (1.85 × 106 G-rich DNAzymes) (see ESI, Fig. S2†). The discrepancy between the theoretically calculated signal enhancement (1.26 × 103-fold) and the experimental result (913.30-fold, Fig. S2†) may be ascribed to that the assembly of a large number of long G-rich DNAzymes on one poly-T sequence may cause steric hindrance for the formation of functional G-quadruplex structures which are proportional to the chemiluminescence intensity. Notably, in comparison with the reported methods for DNA assay (see ESI, Table S1†), this method exhibits the highest sensitivity (0.50 aM) and the largest dynamic range (9 orders of magnitude). The high sensitivity of the proposed method may be ascribed to (1) the highly efficient two-step polymerization extension reactions catalyzed by TdT, (2) the high amplification efficiency of BCA, (3) the intrinsically high sensitivity of G-rich DNA product-driven chemiluminescence, and (4) the near-zero background resulting from the magnetic separation. The large dynamic range is mainly attributed to the involvement of the chemiluminescence detection system which utilizes unique circuitry-mediated photoelectric multiplication and a software-controlled automatic operating system, enabling fast, accurate and sensitive detection of weak optical signals with a wide linear dynamic range.39,43,51 In comparison with the reported methods for DNA assay (see ESI, Table S1†), the proposed method possesses significant advantages of high sensitivity, a large dynamic range, a low background signal and ease of operation, without the involvement of either varying reaction temperature or multiple enzymes for signal amplification.
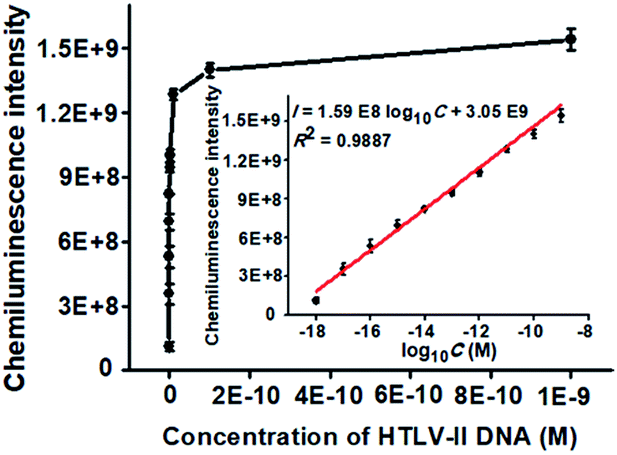 | ||
| Fig. 2 Variance of the chemiluminescence intensity with different concentrations of HTLV-II DNA. Inset shows the linear relationship between the chemiluminescence intensity and the logarithm of HTLV-II DNA concentration. The amount of TdT in the first-step polymerization reactions is 0.4 U, and the amount of TdT in the second-step polymerization reactions is 0.4 U. Error bars show the standard deviations of three experiments. | ||
Due to the high sequence homology among the HTLV family, the selective detection of HTLV-II DNA remains a great challenge.4 To investigate the selectivity of the proposed method, we measured the chemiluminescence intensity in response to HTLV-II DNA, one-base mismatched DNA, three-base mismatched DNA, and noncomplementary DNA under the same experimental conditions. As shown in Fig. 3, high chemiluminescence intensity is detected in response to 1 nM HTLV-II DNA, which is 3.99-fold higher than that in response to one-base mismatched, 5.25-fold higher than that in response to three-base mismatched, and 35.4-fold higher than that in response to noncomplementary DNA. These results demonstrate the high selectivity of the proposed method with the capability of discriminating even a single-base mismatch.
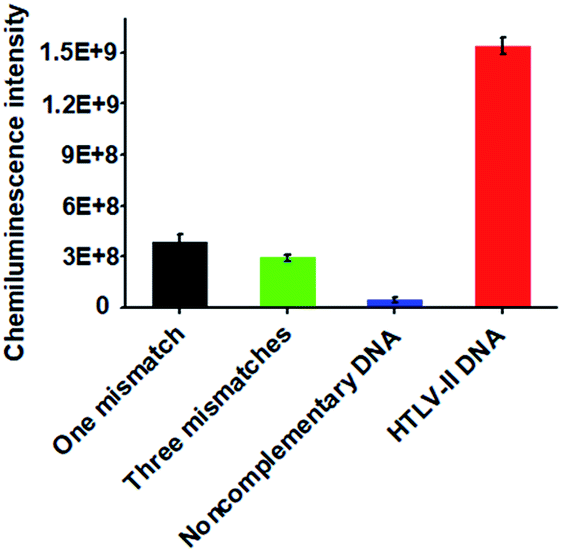 | ||
| Fig. 3 Measurement of chemiluminescence intensity in response to 1 nM one-base mismatched (black column), 1 nM three-base mismatched (green column), 1 nM noncomplementary DNA (blue column), and 1 nM HTLV-II DNA (red column), respectively. Error bars show the standard deviation of three independent experiments. | ||
To verify the feasibility of the proposed method for real sample analysis, we measured the chemiluminescence intensity in response to HTLV-II DNA spiked in 10% human serum (Fig. 4A). In the absence of HTLV-II DNA, no distinct chemiluminescence signal is observed in either the control group without HTLV-II DNA and human serum (Fig. 4A, black column) or 10% human serum group (Fig. 4A, purple column), while a high chemiluminescence signal is detected in response to HTLV-II DNA + 10% human serum (Fig. 4A, green column), with a similar value obtained in response to only HTLV-II DNA (Fig. 4A, red column), suggesting that the performance of this assay is not affected by the interference from human serum. Notably, the chemiluminescence intensity improves with increasing concentration of HTLV-II DNA in human serum, and a linear correlation is obtained between the chemiluminescence intensity and the logarithm of HTLV-II DNA concentration in the range from 10 aM to 1 nM (Fig. 4B). The correlation equation is I = 1.59 × 108log10C + 2.95 × 109 with a correlation coefficient of 0.9857, where I is the chemiluminescence intensity and C is the HTLV-II DNA concentration. The limit of detection is calculated to be 1.0 aM based on the evaluation of the average signal of blank plus three times the standard deviation, which is comparable to the value (0.5 aM) obtained in the absence of human serum (Fig. 2). These results demonstrate that the proposed method can be used to quantify HTLV-II DNA in real samples.
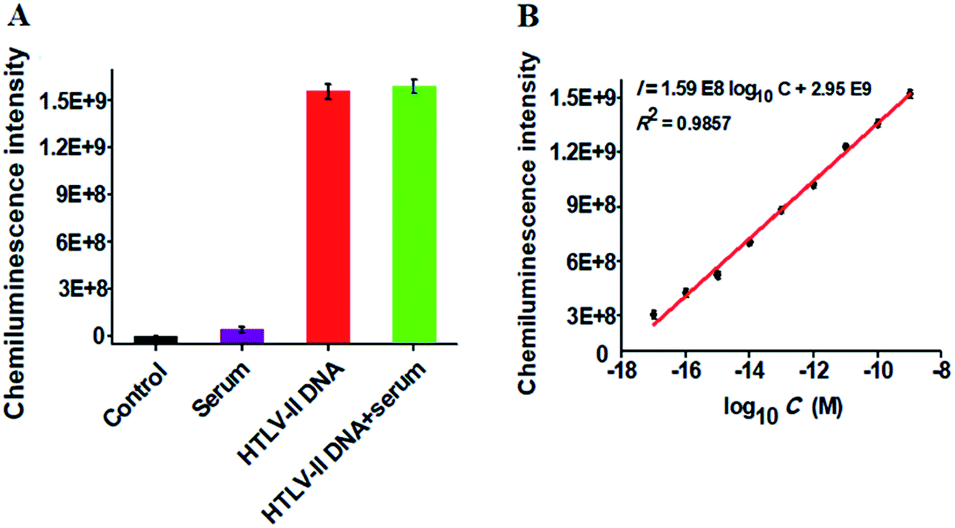 | ||
| Fig. 4 (A) Variance of the chemiluminescence intensity in response to the control group without HTLV-II DNA and human serum (black column), 10% human serum (purple column), 1 nM HTLV-II DNA (red column), and 1 nM HTLV-II DNA + 10% human serum (green column), respectively. (B) Linear relationship between the chemiluminescence intensity and the logarithm of HTLV-II DNA concentration in 10% human serum. Error bars show the standard deviations of three experiments. | ||
We further employed the proposed method to detect HTLV-II DNA in a human T-cell leukemia line (HuT-78 cells) and a human myeloma cell line (U266B1 cells). As shown in Fig. 5A, no distinct chemiluminescence signal is observed in either the control group with only extraction buffer or U266B1 cells due to the lack of HTLV-II DNA in U266B1 cells (non-T cells).2 In contrast, a high chemiluminescence signal is detected in HuT-78 cells, indicating the existence of HTLV-II DNA in HuT-78 cells.2 Moreover, according to the fitted calibration equation in Fig. 2, the amount of HTLV-II DNA in HuT-78 cells is determined to be 2.10 pM. In addition, the genomic DNA samples extracted from HuT-78 cells and U266B1 cells were further measured by real-time quantitative PCR (qPCR) (Fig. 5B). In the control with only extraction buffer, no visible fluorescence signal is observed (Fig. 5B, black curve), while in U266B1 cells (Fig. 5B, green curve) and HuT-78 cells (Fig. 5B, red curve), both the real-time fluorescence signals increase in a sigmoidal fashion, implying that non-target DNA existing in U266B1 cells may induce nonspecific amplification for the generation of a high background signal in the qPCR assay. In contrast, our method exhibits better tolerance to intracellular interference than the qPCR-based assay. According to the fitted calibration equation (see ESI, Fig. S3A and B†), the expression levels of HTLV-II DNA are detected to be 0.28 aM in U266B1 cells and 2.31 pM in HuT-78 cells, consistent with the results (0 aM in U266B1 cells and 2.10 pM in HuT-78 cells) obtained by the proposed method. Notably, taking into account the fact that no HTLV-II DNA is expressed in U266B1 cells,2 our result (0 aM) is more accurate than that of the qPCR assay (0.28 aM).
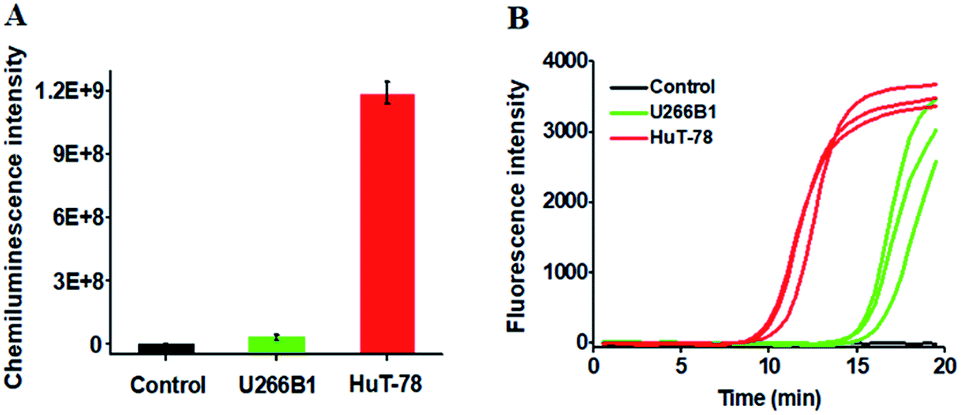 | ||
| Fig. 5 (A) Measurement of chemiluminescence intensity in response to the control group with only extraction buffer (black column), genomic DNA from U266B1 cells (green column) and HuT-78 cells (red column). (B) Quantitative real-time fluorescence monitoring of the PCR amplification reaction in response to the control group with only extraction buffer (black curve), genomic DNA from U266B1 cells (green curve) and HuT-78 cells (red curve). SYBR gold is used as the fluorescent indicator. In A, error bars show the standard deviations of three experiments. In B, the three curves indicate the triplicate measurements of each sample. | ||
Conclusions
We developed for the first time the controllable fabrication of bio-bar codes for dendritically amplified sensing of HTLV-II DNA. Taking advantage of the efficient polymerization extension reaction catalyzed by TdT, the high amplification efficiency of BCA, and the intrinsically high sensitivity of G-rich DNAzyme-driven chemiluminescence, this method exhibits ultrahigh sensitivity with a detection limit of 0.50 aM and a large dynamic range of 9 orders of magnitude, which is superior to the reported methods for DNA assay (see ESI, Table S1†). Moreover, this method can specifically discriminate the single-base mismatch in HTLV-II DNA, and it can be applied to accurately quantify HTLV-II DNA in human serum and human T-lymphocytic leukemia cells. Especially, this method has distinct advantages: (1) the whole experiment is performed at a constant temperature (37 °C) without the involvement of strict temperature cycling; (2) only one enzyme (i.e. TdT) is required to initiate two-step enzymatic extension for dendritic chemiluminescence signal amplification, without the involvement of multiple polymerases, exonucleases and endonucleases, greatly simplifying the operations; (3) all the involved DNA probes are linear strand DNAs, without the involvement of a complicated design of hairpin-structure probes; (4) the magnetic separation may provide a clean reaction environment to significantly improve the signal-to-noise ratio; and (5) the template-free polymerization extension is employed in this strategy, eliminating the preparation of polymerization templates and signal probes. Importantly, this method can be easily extended to sensitive detection of other types of trace-level nucleic acid targets, holding great potential in biomedical research and clinical diagnosis.Experimental section
Chemicals and materials
All oligonucleotides (Table 1) were synthesized by Sangon Biotechnology Co. Ltd. (Shanghai, China). Terminal deoxynucleotidyl transferase (TdT), 10× terminal deoxynucleotidyl transferase reaction buffer (500 mM potassium acetate (KAc), 200 mM Tris–acetate (Tris–Ac), 100 mM magnesium acetate (Mg(Ac)2), pH 7.9), 10× cobalt(II) chloride (CoCl2) (2.5 mM), deoxynucleotide (dNTP) solution set (dATP, dTTP, dGTP and dCTP), Q5® high-fidelity DNA polymerase and 5× Q5® high-fidelity polymerase reaction buffer were purchased from New England Biolabs (Ipswich, MA, USA). The streptavidin-conjugated magnetic microparticles (MMPs, Dynabeads™ M-280 Streptavidin) were obtained from Invitrogen Corporation (Carlsbad, CA, USA). The streptavidin-conjugated Au nanoparticles (AuNPs) were purchased from Nanocs Inc. (New York, NY, USA). SYBR gold was bought from Life Technologies (Carlsbad, CA, USA). Hemin, luminol, peroxidase (H2O2) and 4-(2-hydroxyethyl)-piperazine-1-ethanesulfonic acid sodium salt (HEPES) were obtained from Sigma-Aldrich Company (St. Louis, MO, USA). Human T-cell leukemia lines (HuT-78 cells) and human myeloma cell lines (U266B1 cells) were purchased from the Cell Bank of Chinese Academy of Sciences (Shanghai, China). The hemin solution was prepared by dissolving 0.15 g of hemin in 5 mL of dimethylsulfoxide (DMSO) followed by dilution with 30.5 mL of ultra-pure water, and ultimately stored at −20 °C in the dark. The luminol solution was prepared with 0.1 M NaOH and stored at room temperature in the dark. Other chemicals were of analytical grade and used without further purification. Ultrapure water was obtained through a Millipore filtration system.| Note | Sequences (5′–3′) |
|---|---|
| a In one-base mismatched DNA, the bold “A” base indicates the mismatched base as compared with “C” in HTLV-II DNA. In three-base mismatched DNA, the bold “A” and “C” bases indicate the mismatched bases as compared with “C”, “A” and “C” in HTLV-II DNA, respectively. In capture probes 1 and 2, the 3′ end is modified with biotin, respectively. In the reporter probe, the 5′ end is modified with biotin. | |
| HTLV-II DNA | AGC ACC AAC TCA CCT GGG ACC CCA T |
| One-base mismatched DNA | AGC ACA AAC TCA CCT GGG ACC CCA T |
| Three-base mismatched DNA | AGA ACC ACC TCA ACT GGG ACC CCA T |
| Noncomplementary DNA | GAG GGC CTG CAG GAT CAT TGG CTT T |
| Capture probe 1 | ATG GGG TCC CAG GTG AG-biotin |
| Capture probe 2 | AAA AAA AAA AAA AAA AAA TCT TAT CTT-biotin |
| Reporter probe | Biotin-ACA TGC TTG GAC TGC |
Preparation of capture probe 1-functionalized MMPs and capture probe 2-/reporter probe-functionalized AuNPs
All oligonucleotides were dissolved in 1× Tris–EDTA buffer to prepare the stock solutions. 1 μL of capture probe 1 (10 μM) and 1 μL of MMPs (10 mg mL−1) were added into 10 μL of ultrapure water and incubated at room temperature for 10 min to form capture probe 1-functionalized MMPs through biotin–streptavidin interaction. After magnetic separation, excess capture probe 1 was removed, and capture probe 1-functionalized MMPs were obtained. 1 μL of AuNPs (0.5 mg mL−1), 0.45 μL of capture probe 2 (1 μM) and 4.05 μL of reporter probes (1 μM) were added into 1200 μL of ultrapure water and incubated at room temperature for 10 min to form capture probe 2-/reporter probe-functionalized AuNPs through specific biotin–streptavidin interaction.TdT-catalyzed controllable fabrication of bio-bar codes for dendritic chemiluminescence signal amplification
For the TdT-catalyzed first-step polymerization extension reaction, different concentrations of HTLV-II DNA were added into 20 μL of hybridization solution containing freshly prepared capture probe 1-functionalized MMPs, 750 mM NaCl and 75 mM sodium citrate, followed by incubation at room temperature for 10 min to form dsDNA duplexes with the protruding 3′-hydroxylated ssDNA sequences. After magnetic separation, 0.4 U of TdT was added into 20 μL of polymerization reaction solution consisting of the precipitated products, 2 μL of dTTPs (100 μM), 2 μL of 10× terminal transferase reaction buffer, and 2 μL of CoCl2 (2.5 mM), and incubated at 37 °C for 30 min to perform the polymerization reaction with the protruding 3′-end sequence of HTLV-II DNA as a primer. After the polymerization reaction, 0.15 μL of freshly prepared capture probe 2-/reporter probe-functionalized AuNPs were added into the hybridization solution containing the poly-T products, 750 mM NaCl and 75 mM sodium citrate, followed by incubation at room temperature for 10 min. The excess TdT, dTTPs and capture probe 2-/reporter probe-functionalized AuNPs were removed through magnetic separation. For the TdT-catalyzed second-step polymerization extension reaction, 0.4 U of TdT was added into 20 μL of polymerization reaction system consisting of the precipitated products, 0.8 μL of dATPs (100 μM), 1.2 μL of dGTPs (100 μM), 2 μL of 10× terminal transferase reaction buffer, and 2 μL of CoCl2 (2.5 mM), followed by incubation at 37 °C for 20 min. After magnetic separation, 63 μL of deionized water was added to suspend the precipitated products. After incubation at 90 °C for 3 min, the supernatants were transferred into fresh tubes and subjected to chemiluminescence measurements.Characterization of MMPs, oligonucleotide-functionalized MMPs, AuNPs, and HTLV II DNA/poly-T product/AuNP–DNA nanostructures
The UV absorption spectra of MMPs, capture probe 1-functionalized MMPs, AuNPs and capture probe 2-/reporter probe-functionalized AuNPs were measured by using a NanoDrop 2000/2000C spectrophotometer (Thermofisher, Friendship, ME, USA). The morphologies of AuNPs and the HTLV II DNA/poly-T product/AuNP–DNA nanostructures were examined by using a Hitachi HT-7700 transmission electron microscope (TEM, Tokyo, Japan).Gel electrophoresis analysis
After the TdT-catalyzed polymerization reaction, the reaction products were analyzed by 3% agarose gel electrophoresis in 1× TAE buffer (40 mM Tris–acetic acid, 2 mM EDTA, pH 8.0) at a 110 V constant voltage for 40 min at room temperature. The gels were stained with 1× SYBR gold and imaged using a ChemiDoc MP Imaging System (Hercules, California, USA).Chemiluminescence measurements
The freshly prepared luminol solution (0.5 mM) and hemin solution (750 nM) were added into the mixture containing 63.1 μL of reaction products and 30 μL of incubation buffer (40 mM HEPES, 300 mM NaCl, 20 mM KCl, pH 8.0), and incubated at room temperature for 30 min to make the G-rich polymerization products to fold into four-stranded G-quadruplex structures. Upon the addition of 15 μL of H2O2 (100 mM) into the mixture, the chemiluminescence signals were measured by using a GloMax 96 Microplate Luminometer (Promega, Madison, WI, USA) with a time interval of 1.5 s.Cell culture and preparation of genomic DNA
HuT-78 cells and U266B1 cells were cultured in Roswell Park Memorial Institute (RPMI) medium 1640 (Gibco, USA) with 20% fetal bovine serum (FBS, Gibco, USA) and 1% penicillin–streptomycin (PS, Gibco, USA) in a humidified chamber containing 5% CO2 at 37 °C. At the exponential phase of growth, the cells were collected and counted using a Countstar automated cell counter (Inno-Alliance Biotech Inc., Wilmington, DE, USA). Genomic DNA from HuT-78 cells (1 × 107) and U266B1 cells (1 × 107) was extracted with a universal genomic DNA extraction kit Ver.5.0 (TaKaRa Biotechnology Co., Ltd., Dalian, China). After extraction, 200 ng of genomic DNA was taken for the measurement of HTLV II DNA by the proposed method and the qPCR-based method, respectively. For the qPCR experiment, 200 ng of the extracted genomic DNA was added into 25 μL of polymerization buffer containing 200 μM dNTPs, 500 nM forward primer (5′-TGT CCA GAG CAC CAA CTC AC-3′) and 500 nM reverse primer (5′-GTC CAT CGA TGG GGT CCC AG-3′), 0.5 U of Q5® high-fidelity DNA polymerase, and 5 μL of 5× Q5 high-fidelity polymerase reaction buffer. The polymerization reaction was performed on a Bio-Rad CFX Connect™ thermocycler (Hercules, CA, USA) under the following conditions: 98 °C for 30 s followed by 40 cycles at 98 °C for 10 s, 64 °C for 20 s, 72 °C for 15 s and 72 °C for 2 min. The obtained PCR products were subjected to HTLV-II DNA analysis.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant No. 21325523, 21527811, 21735003, and 21705097), and the Award for Team Leader Program of Taishan Scholars of Shandong Province, China.Notes and references
- S. K. Arya, C. Guo, S. F. Josephs and F. Wong-Staal, Science, 1985, 229, 69–74 Search PubMed.
- V. S. Kalyanaraman, M. G. Sarngadharan, M. Robert-Guroff, I. Miyoshi, D. Blayney, D. Golde and R. C. Gallo, Science, 1982, 218, 571–573 Search PubMed.
- E. L. Murphy, K. Watanabe, C. C. Nass, H. Ownby, A. Williams and G. Nemo, J. Infect. Dis., 1999, 180, 1777–1783 CrossRef PubMed.
- D. D. Ho, T. Moudgil and M. Alam, N. Engl. J. Med., 1989, 321, 1621–1625 CrossRef PubMed.
- H. A. Erlich, D. Gelfand and J. J. Sninsky, Science, 1991, 252, 1643–1651 CrossRef PubMed.
- C. Chen, D. A. Ridzon, A. J. Broomer, Z. Zhou, D. H. Lee, J. T. Nguyen, M. Barbisin, N. L. Xu, V. R. Mahuvakar, M. R. Andersen, K. Q. Lao, K. J. Livak and K. J. Guegler, Nucleic Acids Res., 2005, 33, e179 CrossRef PubMed.
- C.-Y. Yu, B.-C. Yin, S. Wang, Z. Xu and B.-C. Ye, Anal. Chem., 2014, 86, 7214–7218 CrossRef PubMed.
- M. M. Ali, F. Li, Z. Zhang, K. Zhang, D.-K. Kang, J. A. Ankrum, X. C. Le and W. Zhao, Chem. Soc. Rev., 2014, 43, 3324–3341 RSC.
- Y. Cheng, X. Zhang, Z. Li, X. Jiao, Y. Wang and Y. Zhang, Angew. Chem., Int. Ed., 2009, 121, 3318–3322 CrossRef.
- H. Liu, L. Li, L. Duan, X. Wang, Y. Xie, L. Tong, Q. Wang and B. Tang, Anal. Chem., 2013, 85, 7941–7947 CrossRef PubMed.
- C. Li, Z. Li, H. Jia and J. Yan, Chem. Commun., 2011, 47, 2595–2597 RSC.
- T. Notomi, H. Okayama, H. Masubuchi, T. Yonekawa, K. Watanabe, N. Amino and T. Hase, Nucleic Acids Res., 2000, 28, e63 CrossRef PubMed.
- C. Li, H. Wang, J. Shen and B. Tang, Anal. Chem., 2015, 87, 4283–4291 CrossRef PubMed.
- W. Ren, Z. F. Gao, N. B. Li and H. Q. Luo, Biosens. Bioelectron., 2015, 63, 153–158 CrossRef PubMed.
- C. Tao, Y. Yan, H. Xiang, D. Zhu, W. Cheng, H. Ju and S. Ding, Chem. Commun., 2015, 51, 4220–4222 RSC.
- J. Yan, Z. Li, C. Liu and Y. Cheng, Chem. Commun., 2010, 46, 2432–2434 RSC.
- P. Zhang, J. Zhang, C. Wang, C. Liu, H. Wang and Z. Li, Anal. Chem., 2014, 86, 1076–1082 CrossRef PubMed.
- F. Xuan, X. Luo and I. M. Hsing, Anal. Chem., 2012, 84, 5216–5220 CrossRef PubMed.
- S. Liu, T. Liu and L. Wang, Chem. Commun., 2015, 51, 176–179 RSC.
- N. Li, Z. F. Gao, B. H. Kang, N. B. Li and H. Q. Luo, RSC Adv., 2015, 5, 20020–20024 RSC.
- J. Liu, L. Chen, P. Lie, B. Dun and L. Zeng, Chem. Commun., 2013, 49, 5165–5167 RSC.
- L. Yuan, W. Tu, J. Bao and Z. Dai, Anal. Chem., 2015, 87, 686–692 CrossRef PubMed.
- B. Zou, X. Cao, H. Wu, Q. Song, J. Wang, T. Kajiyama, H. Kambara and G. Zhou, Biosens. Bioelectron., 2015, 66, 50–54 CrossRef PubMed.
- C. Zhao, L. Wu, J. Ren and X. Qu, Chem. Commun., 2011, 47, 5461–5463 RSC.
- J.-M. Nam, C. S. Thaxton and C. A. Mirkin, Science, 2003, 301, 1884–1886 CrossRef PubMed.
- J.-M. Nam, S. I. Stoeva and C. A. Mirkin, J. Am. Chem. Soc., 2004, 126, 5932–5933 CrossRef PubMed.
- Y. P. Bao, T.-F. Wei, P. A. Lefebvre, H. An, L. He, G. T. Kunkel and U. R. Müller, Anal. Chem., 2006, 78, 2055–2059 CrossRef PubMed.
- H. Dong, C. Wang, Y. Xiong, H. Lu, H. Ju and X. Zhang, Biosens. Bioelectron., 2013, 41, 348–353 CrossRef PubMed.
- C. S. Thaxton, H. D. Hill, D. G. Georganopoulou, S. I. Stoeva and C. A. Mirkin, Anal. Chem., 2005, 77, 8174–8178 CrossRef PubMed.
- N. Jaffrezic-Renault, C. Martelet, Y. Chevolot and J.-P. Cloarec, Sensors, 2007, 7, 589–614 CrossRef.
- H. D. Hill, R. A. Vega and C. A. Mirkin, Anal. Chem., 2007, 79, 9218–9223 CrossRef PubMed.
- T. Komori, A. Okada, V. Stewart and F. W. Alt, Science, 1993, 261, 1171–1175 Search PubMed.
- L.-J. Wang, M.-L. Luo, Q. Zhang, B. Tang and C.-Y. Zhang, Chem. Commun., 2017, 53, 11016–11019 RSC.
- V. Tjong, H. Yu, A. Hucknall and A. Chilkoti, Anal. Chem., 2013, 85, 426–433 CrossRef PubMed.
- Y. Yuan, W. Li, Z. Liu, Z. Nie, Y. Huang and S. Yao, Biosens. Bioelectron., 2014, 61, 321–327 CrossRef PubMed.
- Y. Hu, Q. Shen, W. Li, Z. Liu, Z. Nie and S. Yao, Biosens. Bioelectron., 2015, 63, 331–338 CrossRef PubMed.
- K. Shi, B. Dou, J. Yang, R. Yuan and Y. Xiang, Biosens. Bioelectron., 2017, 87, 495–500 CrossRef PubMed.
- X. Zhu, Y. Cao, Z. Liang and G. Li, Protein Cell, 2010, 1, 842–846 CrossRef PubMed.
- L.-j. Wang, Y. Zhang and C.-y. Zhang, Anal. Chem., 2013, 85, 11509–11517 CrossRef PubMed.
- S. Nakayama and H. O. Sintim, J. Am. Chem. Soc., 2009, 131, 10320–10333 CrossRef PubMed.
- Y. Weizmann, Z. Cheglakov and I. Willner, J. Am. Chem. Soc., 2008, 130, 17224–17225 CrossRef PubMed.
- D. Li, B. Shlyahovsky, J. Elbaz and I. Willner, J. Am. Chem. Soc., 2007, 129, 5804–5805 CrossRef PubMed.
- F. Ma, Y. Yang and C.-y. Zhang, Anal. Chem., 2014, 86, 6006–6011 CrossRef PubMed.
- Y.-p. Zeng, J. Hu, Y. Long and C.-y. Zhang, Anal. Chem., 2013, 85, 6143–6150 CrossRef PubMed.
- B.-C. Yin, B.-C. Ye, W. Tan, H. Wang and C.-C. Xie, J. Am. Chem. Soc., 2009, 131, 14624–14625 CrossRef PubMed.
- T. Li, S. Dong and E. Wang, J. Am. Chem. Soc., 2010, 132, 13156–13157 CrossRef PubMed.
- C.-P. D. Tu and S. N. Cohen, Gene, 1980, 10, 177–183 CrossRef PubMed.
- J.-B. Boulé, F. Rougeon and C. Papanicolaou, J. Biol. Chem., 2001, 276, 31388–31393 CrossRef PubMed.
- L. M. S. Chang, F. J. Bollum and R. C. Gallo, Crit. Rev. Biochem., 1986, 21, 27–52 CrossRef PubMed.
- R. Roychoudhury, E. Jay and R. Wu, Nucleic Acids Res., 1976, 3, 863–878 CrossRef PubMed.
- R. Nie, X. Deng, L. Feng, G. Hu, Y. Wang, G. Yu and J. Xu, Small, 2017, 13, 1603260–1603270 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8sc01641k |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2018 |