
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInhibition of hydrocarbon autoxidation by nitroxide-catalyzed cross-dismutation of hydroperoxyl and alkylperoxyl radicals†
Kareem A.
Harrison
,
Evan A.
Haidasz
,
Markus
Griesser
 and
Derek A.
Pratt
*
and
Derek A.
Pratt
*
Department of Chemistry and Biomolecular Sciences, University of Ottawa, Ottawa, Ontario, Canada K1N 6N5. E-mail: dpratt@uottawa.ca
First published on 11th June 2018
Abstract
Nitroxides are putative intermediates in the accepted reaction mechanisms of the diarylamine and hindered amine antioxidants that are universally added to preserve synthetic and natural hydrocarbon-based materials. New methodology which enables monitoring of hydrocarbon autoxidations at low rates of radical generation has revealed that diarylnitroxides and hindered nitroxides are far better inhibitors of unsaturated hydrocarbon autoxidation than their precursor amines, implying intervention of a different mechanism. Experimental and computational investigations suggest that the nitroxides catalyze the cross-dismutation of hydroperoxyl and alkylperoxyl radicals to yield O2 and a hydroperoxide, thereby halting the autoxidation chain reaction. The hydroperoxyl radicals – key players in hydrocarbon combustion, but essentially unknown in autoxidation – are proposed to derive from a tunneling-enhanced intramolecular (1,4-) hydrogen-atom transfer/elimination sequence from oxygenated radical addition intermediates. These insights suggest that nitroxides are preferred additives for the protection of hydrocarbon-based materials from autoxidation since they exhibit catalytic activity under conditions where their precursor amines are less effective and/or inefficiently converted to nitroxides in situ.
Introduction
Autoxidation, the archetype free radical chain reaction, limits the lifetime of all hydrocarbon-based materials – natural products such as lipids and terpenes as well as synthetic products such as plastics and lubricants. Unsaturated moieties within these materials render them particularly sensitive to autoxidation; they are activated toward H-atom abstraction by chain-carrying peroxyl radicals and also undergo addition reactions to propagate the chain process (Fig. 1A). Diarylamines and hindered aliphatic amines comprise two of the three key types of radical-trapping antioxidants (RTAs) commonly added to preserve hydrocarbon-based materials.1,2 Extensive investigations since the mid-1950s have established that diarylamines trap 2 peroxyl radicals at ambient temperatures,3,4 but at elevated temperatures trap far more5 by a catalytic mechanism (the “Korcek cycle”, Fig. 1B)6,7 involving the formation of diarylnitroxide intermediates.8,9 In contrast, hindered amines are not RTAs at ambient temperatures, but at elevated temperatures or prolonged irradiation5 they lead to nitroxides which trap radicals in a distinct, but still catalytic manner (the “Denisov cycle”, Fig. 1C).10,11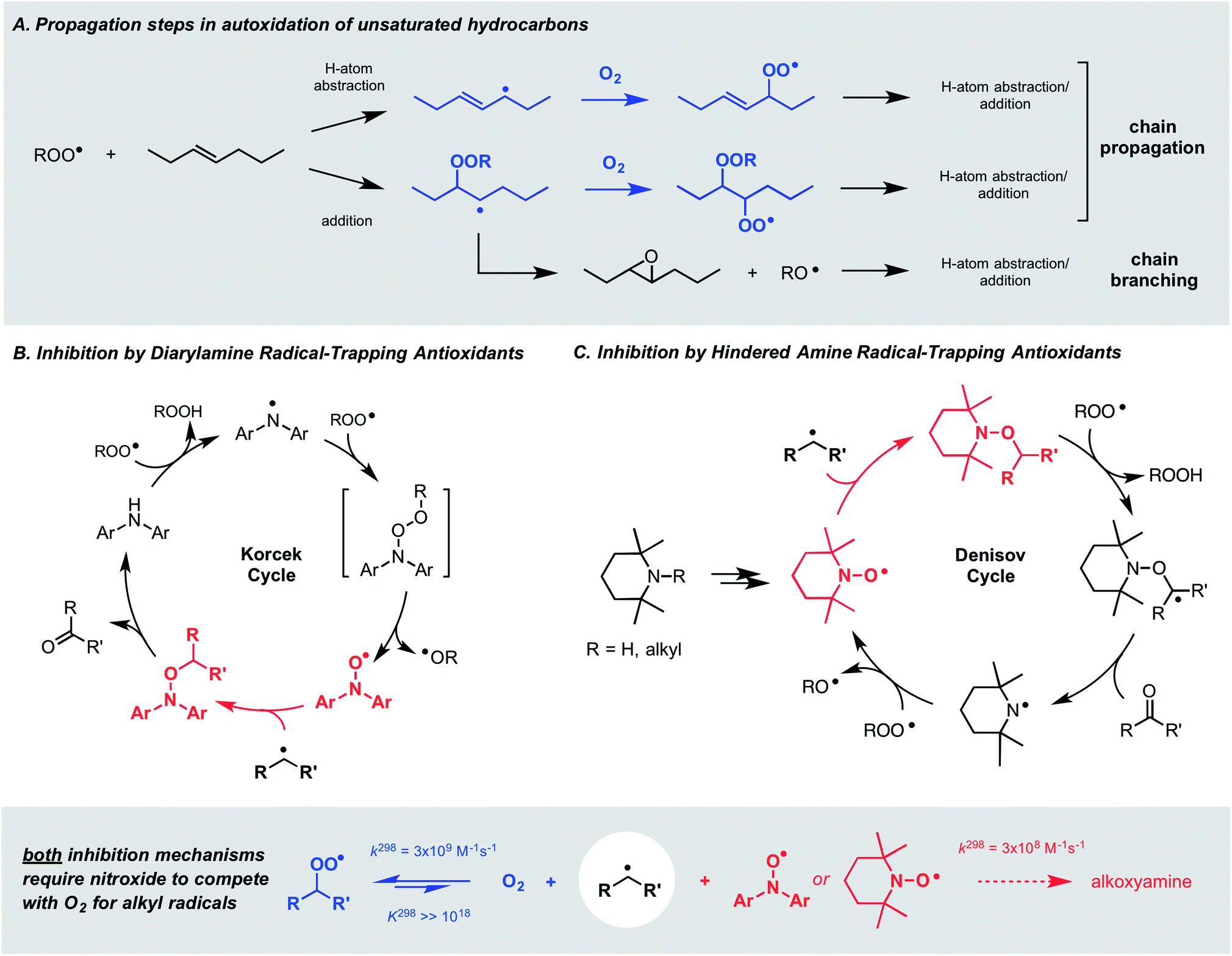 | ||
| Fig. 1 (A) The key propagation steps in the autoxidation of unsaturated hydrocarbons. (B and C) The Korcek and Denisov mechanisms believed responsible for the catalytic inhibition of hydrocarbon autoxidation by diarylamine and hindered amine antioxidants. | ||
Although nitroxides are key intermediates in both the Korcek and Denisov cycles, they do not react with peroxyl radicals directly in these mechanisms – and thereby adhere to the longstanding dogma that nitroxides do not react with oxygen-centered radicals. This belief is based largely on the seminal work of Thomas8 and Ingold,12 who showed that diphenylnitroxide and 4-oxo-2,2,6,6-tetramethylpiperidin-1-oxyl were not particularly effective inhibitors of the autoxidation of cumene and styrene, respectively. Instead, it was proposed that they only retard the autoxidation by competing with O2 for the chain-carrying alkyl radicals – a difficult competition since the reaction of nitroxides with alkyl radicals is at least an order of magnitude slower13,14 than that of O2.15 Paradoxically, this reaction is invoked in both the Korcek and Denisov cycles, implying that catalytic RTA activity arises only when the nitroxide concentration exceeds that of O2.
We recently made the stunning observation that diarylnitroxides and hindered nitroxides are far better inhibitors of unsaturated hydrocarbon autoxidation than are their precursor amines at ambient to moderate temperatures (vide infra). These results were obtained at lower rates of radical generation than those typically employed in laboratory experiments, implying that the mechanism responsible operates at lower steady state concentrations of radicals – a situation representative of most ‘real world’ applications. Moreover, these results suggest a different mechanism of antioxidant activity for the nitroxides than in the Korcek and Denisov mechanisms – insight which could be leveraged for the design and development of badly needed new antioxidant technology. Our observations are presented below, alongside our efforts to elucidate the mechanism and a discussion of the practical implications of these insights: most importantly, that nitroxides should be considered as additives to increase the longevity of both natural and synthetic hydrocarbon-based materials in lieu of currently used amines.
Results
I. Nitroxides are good inhibitors of styrene, but not cumene, autoxidations at low rates of radical generation
We recently reported a method to monitor inhibited autoxidations of hydrocarbons which makes use of highly absorbing and highly autoxidizable probes as signal carriers (e.g. STY-BODIPY, Fig. 2A).18 The consumption of the probe is monitored by conventional spectrophotometry, and since its reactivity toward peroxyl radicals can be independently determined (e.g. kSTY-BODIPY = 4012 M−1 s−1 in styrene/chlorobenzene at 70 °C, see ESI†), the inhibition rate constant (kinh) of an added RTA and the stoichiometry of the inhibition (n, i.e. how many chain-carrying radicals are trapped by each molecule of RTA) can be easily derived from the data as in Fig. 2B. During our efforts to validate this approach – by comparison to measurements using the venerable O2 uptake method19–21 – we obtained unexpected results in autoxidations of styrene inhibited by 4,4′-di-tert-butyldiphenylamine and its corresponding nitroxide (hereafter Ar2NH and Ar2NO, respectively). To our great surprise, the nitroxide was a significantly better RTA than the amine (Fig. 2C) – not just in terms of its kinetics (apparent kinh = 7.9 × 106 M−1 s−1 for Ar2NO compared to kinh = 7.9 × 105 M−1 s−1 for Ar2NH),22 but in that the nitroxide trapped significantly more radicals than the amine (n = 10.6 ± 0.2 for Ar2NO compared to n = 2.1 ± 0.2 for Ar2NH). These results stand in stark contrast to the seminal results obtained by Thomas8 and Ingold.12 Since Thomas had used cumene as the autoxidizable substrate, we carried out autoxidations of cumene as well, again using STY-BODIPY as the signal carrier,18 but under otherwise identical conditions to Thomas (0.29 M cumene in chlorobenzene, initiated by 1.0 mM AIBN at 68.5 °C and inhibited by 50 μM of diphenylamine). Our results were fully consistent with Thomas' observations, in terms of both reactivity and stoichiometry, i.e. n = 2.3 ± 0.1 for Ar2NH and n = 1.1 ± 0.1 for Ar2NO (Fig. S1†).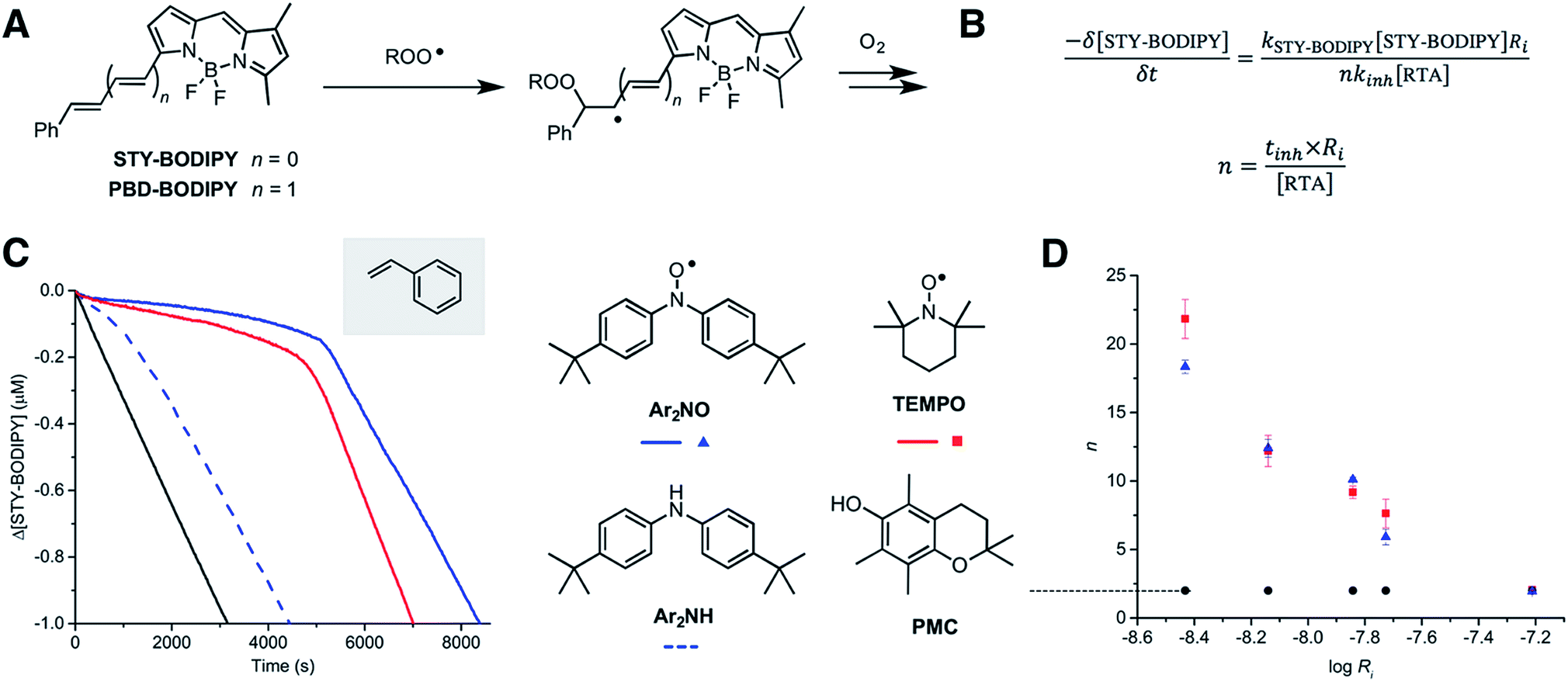 | ||
Fig. 2 Preliminary studies. (A) STY-BODIPY and PBD-BODIPY can be used as signal carriers in the autoxidation of organic substrates. (B) The rate constants (kinh) and stoichiometries (n) for the reactions of inhibitors (RTAs) with chain-carrying peroxyl radicals can be derived from the initial rate of probe consumption and the duration of the inhibited periods (tinh), respectively. (C) Co-autoxidations of styrene (3.5 M) and STY-BODIPY (10 μM) in PhCl initiated with di-tert-butylperoxide (218 mM) at 70 °C (black) and inhibited by Ar2NO, Ar2NH and TEMPO (2 μM). (D) Stoichiometry of peroxyl radical-trapping by Ar2NO, TEMPO and PMC as a function of rate of initiation of styrene autoxidations at 70 °C (obtained using 62.5, 125, 250, 375 and 1000 μM AIBN). Reaction progress was monitored at 571 nm (ε = 97![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 235 M−1 cm−1). 235 M−1 cm−1). | ||
Recognizing that Ingold's styrene autoxidations had been carried out under significantly different conditions from our own, we attempted experiments using Ingold's conditions (7.24 M styrene in o-dichlorobenzene, initiated by 4.3 mM AIBN at 65 °C and inhibited by 4.3 mM antioxidant). Unfortunately, it was impossible to monitor reaction progress by STY-BODIPY consumption due to the absorbance of the massive amounts of antioxidant and/or its oxidation products. However, when the antioxidant and initiator concentrations were reduced to those of Thomas (50 μM and 1 mM, respectively), we observed a relative reactivity that was consistent with what had been reported by Ingold, i.e., Ar2NH was more reactive than Ar2NO (Fig. S2†). Interestingly, the nitroxide still possessed a longer inhibition time than the amine. Ingold wasn't able to report on the stoichiometry of the inhibition due to the length of the inhibited time at the antioxidant concentrations he employed.
Given these results, corresponding data were obtained with 2,2,6,6-tetramethylpiperidine-N-oxyl (hereafter TEMPO), the quintessential nitroxide. The results are shown alongside those for Ar2NO in Fig. 2C (the corresponding tetramethylpiperidine was not investigated as it is known to be unreactive to peroxyl radicals; its N–H BDE is ca. 10 kcal mol−1 higher than that of diphenylamine).23
Consistent with previous work,26 TEMPO was unable to inhibit the autoxidation of cumene (data not shown), but consistent with the foregoing results, it inhibited the autoxidation of styrene with kinetics and stoichiometry similar to those observed for Ar2NO, i.e. apparent kinh = 2.1 × 106 M−1 s−1 and n = 9.3 ± 0.9. Thus, it would appear that the RTA activity is largely independent of the nitroxide structure, but strongly dependent on both the structure of the substrate and the rate of initiation of the autoxidation. This last point can be seen clearly from the results in Fig. 2D, where the stoichiometry determined from a series of nitroxide-inhibited autoxidations of styrene are plotted as a function of the rate of initiation (through an [AIBN] range of 62.5 μM to 1 mM). The variation in n is remarkable, spanning n = 18.3 ± 0.5 and 21.9 ± 1.4 at Ri = 3.7 × 10−9 M s−1 to n = 1.9 ± 0.2 and 2.1 ± 1.4 at Ri = 6.1 × 10−8 M s−1 for Ar2NO and TEMPO, respectively. Laboratory autoxidations are generally carried out at the high end of this range of rates of initiation for practical reasons (ease of measurement by established methods, convenient timescale). In the ‘real world’, autoxidations proceed relatively slowly. It is important to note that the foregoing experiments were carried out at 70 °C to facilitate comparison with Thomas and Ingold's seminal data. Corresponding experiments carried out at 37 °C yielded similar trends, albeit with lower values of n for similar Ri (e.g. n = 10.9 ± 0.1 and 10.1 ± 0.1 at Ri = 2.8 × 10−9 M s−1 for Ar2NO and TEMPO, respectively – see ESI† for additional data).
II. Nitroxides are effective inhibitors of unsaturated, but not saturated, hydrocarbons
To better understand the substrate dependence of this reactivity, investigations were expanded to include autoxidations of other activated hydrocarbons. Since styrene and cumene differ in the presence/absence of a reactive alkene, autoxidizable substrates were selected that either possess an alkene (cyclooctene and norbornene) or do not (ethylbenzene and dioxane). Representative results for these substrates are shown in Fig. 3. In a strikingly consistent fashion, autoxidations of the substrates that possess an unsaturation were very effectively inhibited by the nitroxides whereas those lacking one, were not. Moreover, while the diarylamine expectedly inhibited the autoxidation of all the substrates, it was noticeably more effective in the unsaturated ones, retarding the autoxidations well past the initial inhibited periods corresponding to n = 2. Since diarylamines are known to be converted to diarylnitroxides under autoxidative conditions (i.e.Fig. 1B), this suggests that the retardation of autoxidations of cyclooctene and norbornene beyond the nominal inhibition period result from the in situ formation of nitroxide. Indeed, when the autoxidations were followed by EPR spectroscopy, significant concentrations of nitroxide were observed, reaching a steady-state concentration (ca. 50% and 20% of the initial amine concentration for cyclooctene and norbornene, respectively) around the same time as the autoxidation rate became constant. Taken together, these results suggest that, in unsaturated hydrocarbons, nitroxides must be converted to very efficient RTAs. | ||
| Fig. 3 Substrate dependence. Co-autoxidations of STY-BODIPY (10 μM) and ethylbenzene (3.3 M, (A)), cyclooctene (3.1 M, (B)) norbornene (1.0 M, (C)) and dioxane (2.9 M, (D)) in chlorobenzene initiated with di-tert-butylperoxide (87 mM for ethylbenzene, 218 mM for cyclooctene, norbornene and dioxane) at 70 °C (black) and inhibited by TEMPO, Ar2NO and Ar2NH (2 μM). Reaction progress was monitored at 569 nm in (A) (ε = 123481 M−1 cm−1), 568 nm in (B) (ε = 113982 M−1 cm−1), 568 nm in (C) (ε = 118405 M−1 cm−1) and 569 nm in (D) (ε = 122873 M−1 cm−1). (E) Concentration of Ar2NO determined by EPR (spectrum shown) during Ar2NH-inhibited co-autoxidations. | ||
III. The in situ formation of hydroxylamines accounts for the observed inhibition of autoxidation unsaturated substrates
Since nitroxide-derived alkoxyamines have long been implicated as key intermediates in the RTA activity of diarylamines (Fig. 1B) and hindered amines (Fig. 1C), we sought to determine if they contribute to the observed reactivity in styrene. Thus, O-phenethyl N,N-(4-tert-butylphenyl)hydroxylamine and the corresponding O-phenethylated 2,2,6,6-tetramethylpiperidine-N-ol were synthesized (see ESI†) and their reactivity in the styrene autoxidations was assessed. The phenethyl substituent was chosen due to its similarity to the propagating benzylic radicals that would combine with the nitroxides in inhibited autoxidations of styrene. The data, which show that the alkoxyamines are unreactive, are given in Fig. 4A and B.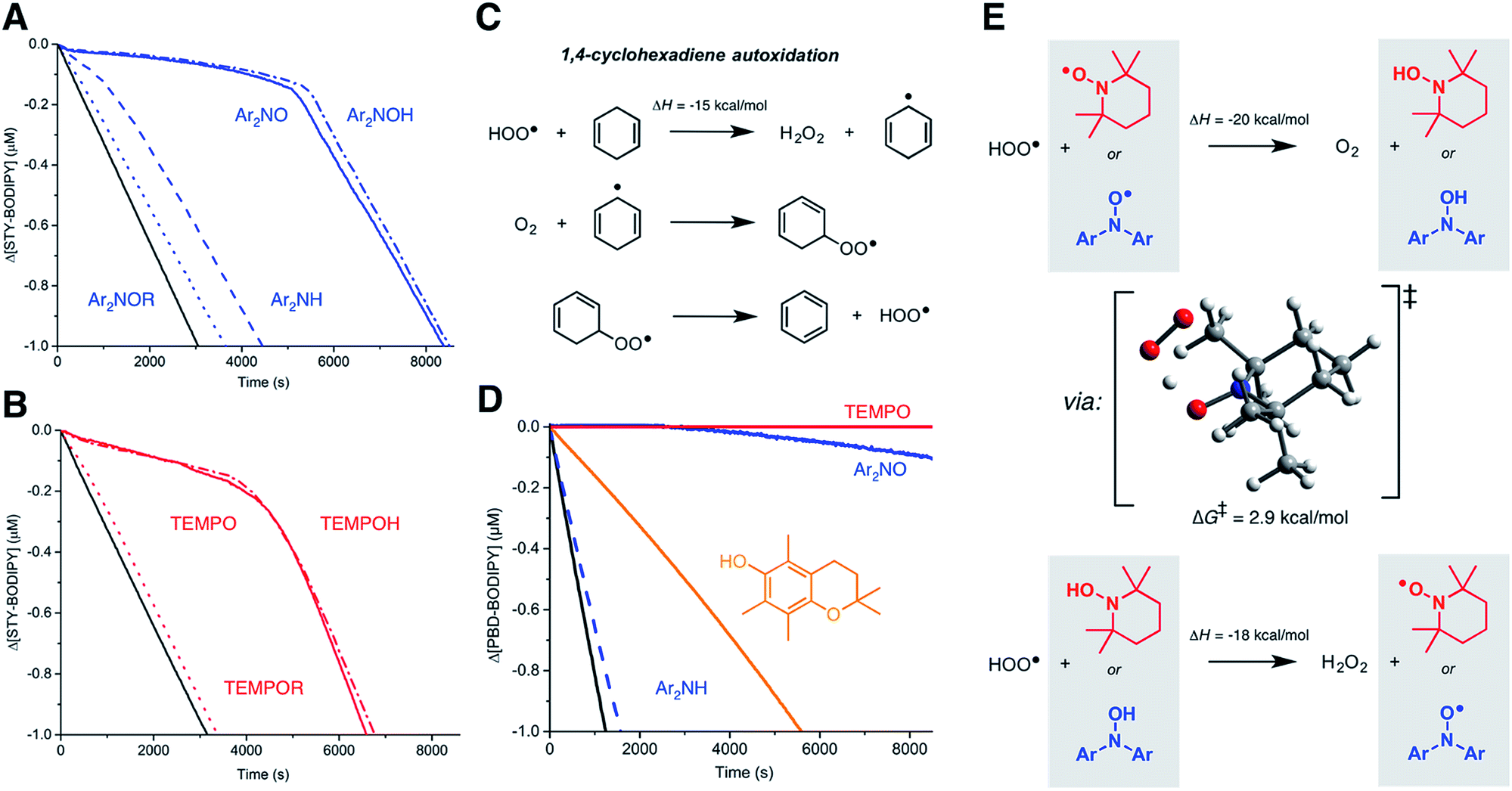 | ||
| Fig. 4 Mechanistic studies. (A) Co-autoxidations of styrene (3.5 M) and STY-BODIPY (10 μM) in PhCl initiated with di-tert-butylperoxide (218 mM) at 70 °C (black) and inhibited by 2 μM of Ar2NOR (dotted line), Ar2NH (dashed line), Ar2NO (solid line) and Ar2NOH (dotted/dashed line). (B) Co-autoxidations carried out under the same conditions and inhibited by TEMPOR (dotted line), TEMPO (solid line) and TEMPOH (dotted/dashed line). Reaction progress was monitored at 571 nm (ε = 97235 M−1 cm−1). (C) Mechanism of 1,4-cyclohexadiene autoxidation. (D) Co-autoxidations of 1,4-CHD (0.26 M) and PBD-BODIPY (10 μM) in chlorobenzene at 30 °C initiated by di-tert-butylperoxide (218 mM, black) and inhibited by 2 μM of TEMPO (red line), Ar2NO (blue line), PMC (orange line) and Ar2NH (dashed line). Reaction progress was monitored at 591 nm (ε = 130797 M−1 cm−1). (E) Nitroxide-catalyzed dismutation of hydroperoxyl radicals showing the CBS-QB3-calculated TS structure for the reaction of TEMPO with HOO˙. | ||
For comparison, we also investigated the corresponding hydroxylamines, which are clearly excellent inhibitors. The initial rates yield kinh = (8.5 ± 1.3) × 106 and (3.0 ± 0.7) × 106 M−1 s−1 for Ar2NOH and TEMPOH, respectively, the latter in excellent agreement with literature precedent (kinh = 3.0 × 106 M−1 s−1 at 30 °C).26 To the best of our knowledge, this is the first time a rate constant has been determined for a diarylhydroxylamine in a non-aqueous solvent; it is slightly greater than for TEMPOH, likely due to lower steric hindrance. The reaction stoichiometry is essentially +1 radical relative to the nitroxide (viz. 11.6 ± 0.1 and 10.0 ± 0.2, respectively), consistent with an initial H-atom transfer from the hydroxylamine to produce the nitroxide, which then reacts as when it is added directly.
The similar initial rates of the autoxidations inhibited by the hydroxylamines and nitroxides prompted us to consider if nitroxides could be reduced to hydroxylamines in situ. Recent work by Litwinienko and Amorati27 has shown that 1,4-cyclohexadiene (hereafter 1,4-CHD) autoxidations are very efficiently inhibited by phenols because the hydroperoxyl radicals (HOO˙) that are known to carry the chain reaction (Fig. 4C)28 can regenerate the phenol. Amazingly, 1,4-CHD/PBD-BODIPY co-autoxidations were perpetually inhibited by either TEMPO or Ar2NO (Fig. 5C). Both nitroxides were demonstrably better than PMC, a truncated analog of vitamin E and one of the most reactive phenolic antioxidants known. (Phenols are the other key type of RTA commonly added to preserve hydrocarbon- based materials.) This is presumably due to the more efficient conversion of nitroxides to chain-breaking hydroxylamines upon reduction by HOO˙ compared to the PMC-derived radical, which can also react via addition to the aryl ring giving inactive peroxyl-phenoxyl adducts.27 TEMPO was observed to be a slightly better inhibitor than Ar2NO, perhaps since the latter can (like the PMC-derived phenoxyl) undergo competitive addition of HOO˙ to the aryl ring. It is interesting that this reactivity trend is opposite of what is observed in styrene and norbornene, where Ar2NO is more effective than TEMPO, perhaps because alkylperoxyl addition is more reversible than HOO˙ addition.
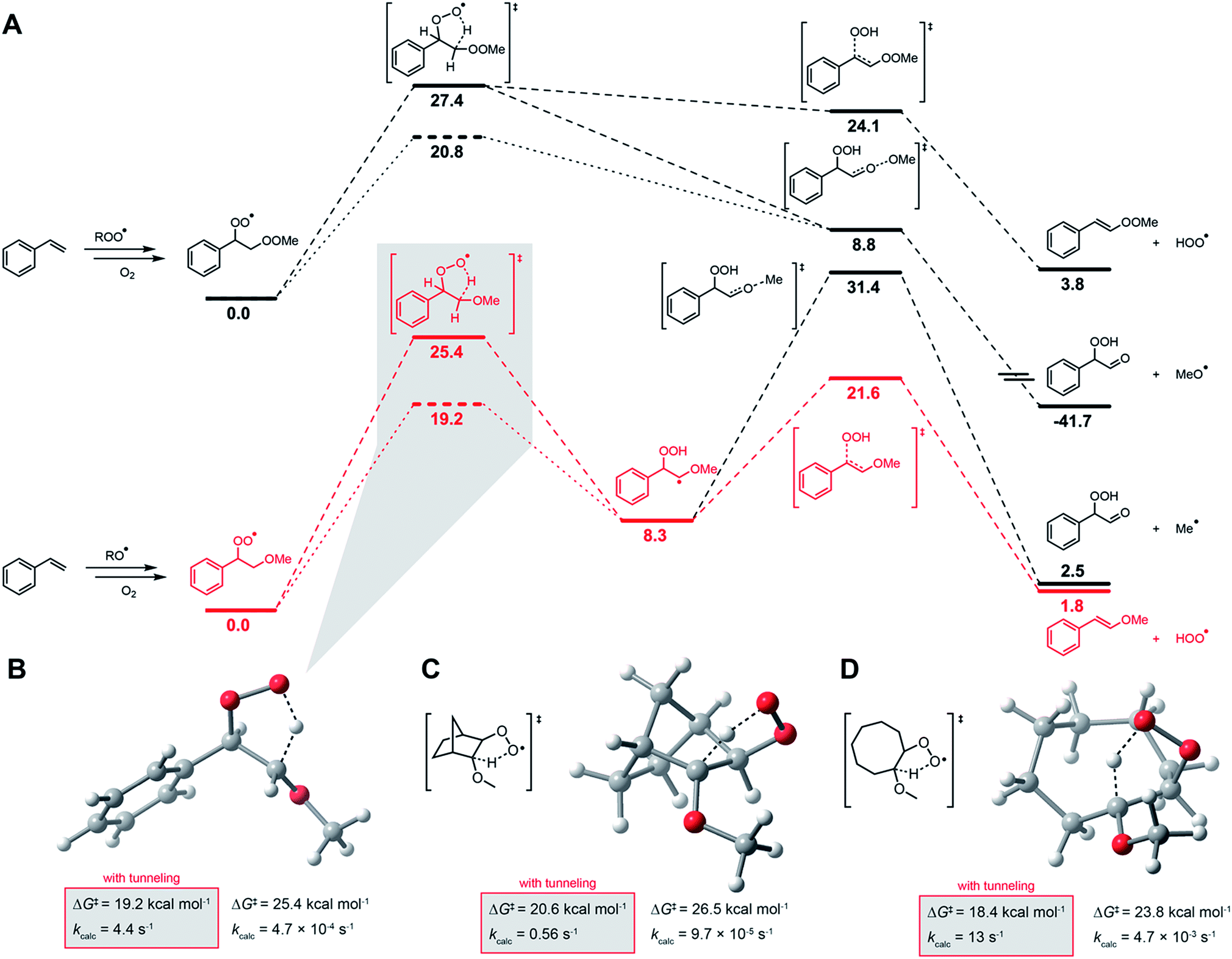 | ||
| Fig. 5 Computational investigations. (A) CBS-QB3-computed free energy profile for HOO˙ formation from peroxyl radical intermediates arising in styrene autoxidation. Energy of the tunneling-enhanced 1,4-HAT TS is given by the dashed line. Methylperoxyl and methoxyl are used as models of chain-propagating alkylperoxyl and alkoxyl radicals, respectively. Transition state structures for the 1,4-HAT in the β-alkoxyalkylperoxyl radicals derived from styrene (B), norborene (C) and cyclooctene (D). | ||
Although the O–H bonds of hydroxylamines are considerably weaker than the O–H bonds of even the most reactive phenols (compare BDEs of 70 and 78 kcal mol−1 in TEMPOH29 and PMC,30 respectively), the reaction is still highly exothermic. The barrier for the H-atom transfer from HOO˙ to TEMPO is predicted by CBS-QB3 (ref. 31) to be ΔG‡ = 2.9 kcal mol−1, corresponding to a rate constant of 2 × 1010 M−1 s−1 upon application of transition state theory. Thus, the reaction is predicted to be diffusion-controlled.32 It is interesting to note the syn orientation of the substituents on the oxygen atoms between which the H-atom is being transferred (Fig. 4E). Although this is unexpected based on steric considerations, it is typical of reactions of peroxyl radicals with H-atom donors that possess an α-heteroatom (e.g. ROO˙ + H–OSR,33 H–OSeR,34 H–SSR35) or unsaturation (e.g. H–OAr,36 H–NHAr37) due to secondary orbital interactions between the substituents.
IV. Computations suggest a tunneling-enhanced 1,4-HAT/elimination sequence leads to hydroperoxyl formation in unsaturated hydrocarbon autoxidation
To the best of our knowledge, there has been no investigation of the intervention of HOO˙ in the propagation of autoxidation of simple monounsaturated hydrocarbons such as styrene, cyclooctene and norbornene. To assess whether HOO˙ could be formed from chain-propagating intermediates in the autoxidation of these substrates, we turned to computation. It has been suggested that hydroperoxyl radicals contribute to the propagation of autoxidation of activated unsaturated substrates, such as acrylates and substituted 1,3-butadienes. Multiple mechanistic possibilities were advanced.73 However, prior to investigating these reactions, we sought to clarify the pathway operating in the autoxidation of 1,4-CHD.The precise mechanism of HOO˙ formation in 1,4-CHD autoxidation has been a matter of some debate. Howard and Ingold had originally proposed a unimolecular elimination from the cyclohexadienylperoxyl radical (as shown in Fig. 4C).28 Hendry and Schuetzle subsequently argued that the reaction must involve direct H-atom transfer from the cyclohexadienyl radical to O2.40 Subsequent reports seem split between the two mechanisms.41–45
Our CBS-QB3 calculations suggest that the cyclohexadienylperoxyl is a bound structure that leads to HOO˙ and benzene via a concerted 1,4-HAT/elimination transition structure that intrinsic reaction coordinate (IRC) calculations confirm connects the two stationary points. No transition state structure was identified for the direct H-atom transfer mechanism.46 The predicted barrier for this process (ΔG‡ = 15.4 kcal mol−1) is strikingly similar to that of the bimolecular propagation reaction of hydroperoxyl and 1,4-cyclohexadiene (ΔG‡ = 15.2 kcal mol−1). Confidence in the calculations comes from the excellent agreement between the predicted rate constant for the latter reaction and the known kp for 1,4-CHD autoxidation (1610 M−1 s−1vs. 1400 M−1 s−1,28 respectively), as well as the thermodynamics of that step (ΔG = −14.2 kcal mol−1), which is fully consistent with the difference in bond dissociation enthalpies (BDEs) between 1,4-CHD (74.5 kcal mol−1)47 and H2O2 (87.5 kcal mol−1).48
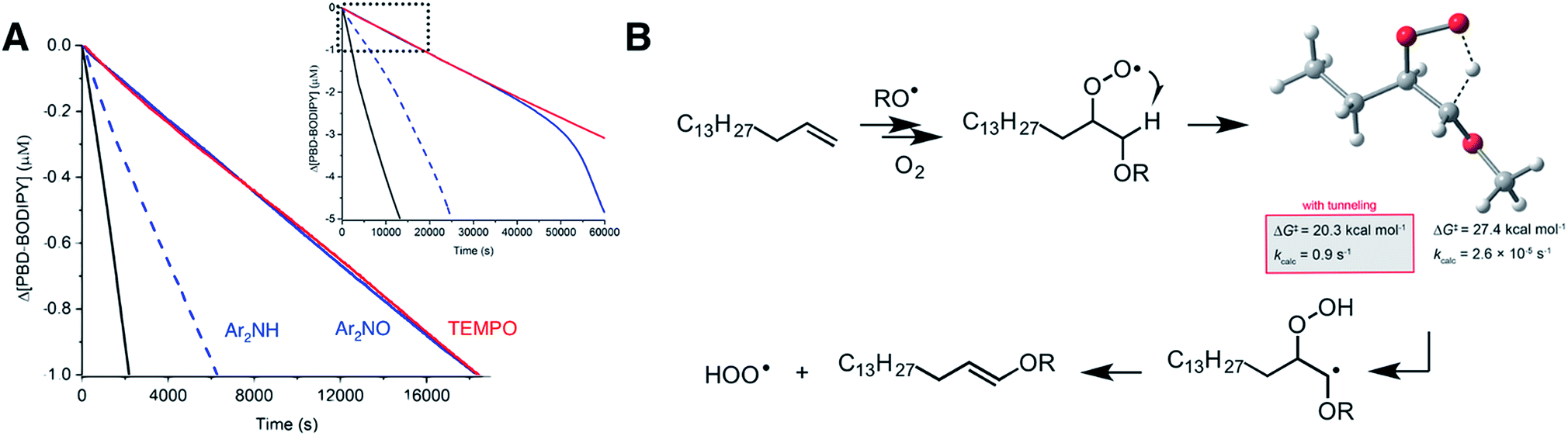
In contrast to 1,4-CHD, styrene is well known to propagate by addition.49 Thus, the analogous 1,4-HAT/elimination process to produce HOO˙ may be expected to come from the propagating peroxyl radical show on the top left in Fig. 5A. A transition state structure for the 1,4-HAT in this intermediate was readily identified, but in lieu of being concerted with HOO˙ elimination as for 1,4-CHD, IRC calculations indicate that no stationary point connects it and the transition state for β-fragmentation of the neighbouring O–O bond. Although a transition state structure could be identified for the elimination of HOO˙, it is 15.3 kcal mol−1 higher in energy.
The propagation of styrene autoxidation by peroxyl radical addition competes with the intramolecular SiH reaction that yields styrene epoxide and an alkoxyl radical which subsequently adds to styrene to continue the chain. Styrene oxide has been reported to account for as much as 39% of the products in a styrene autoxidation,50 and is expectedly influenced by the partial pressure of O2.51 The alkoxyl radicals formed as a result – as well as alkoxyl radicals formed simply from homolysis of the styrene/oxygen copolymer products – add to styrene to yield a benzylic radical that is subsequently oxygenated to yield the chain-propagating peroxyl radical shown on the bottom left of Fig. 5A. A transition state structure for the 1,4-HAT from this intermediate was also readily identified, but this time IRC calculations connect it to a bound α-alkoxyalkyl radical. From this intermediate, HOO˙ elimination proceeds in favour of the β-fragmentation of the alkoxy C–O bond (ΔG‡ = 13.3 kcal mol−1 for the former compared to 23.1 for the latter). The preceding 1,4-HAT has a much higher barrier, but is predicted to be greatly enhanced by quantum mechanical tunneling with k = 4.4 s−1 (70 °C).
Corresponding calculations for cyclooctene and norbornene provide similar insight: elimination from the oxygenated intermediate arising from alkoxyl addition to the double bond is predicted to be a viable route to HOO˙ formation, with similar (tunneling-corrected) rate constants of 13 and 0.56 s−1 (70 °C), respectively. It should be noted that autoxidations of norbornene52,53 and cyclooctene54,55 are known to produce even more epoxide than styrene. Complete potential energy surfaces for the relevant chain-propagating steps of cyclooctene and norbornene autoxidation are given in the ESI.†
V. Kinetic isotope effects support the proposed mechanism
If hydroperoxyl radicals are the stoichiometric reductants in nitroxide-inhibited autoxidations of unsaturated hydrocarbons, a kinetic isotope effect should be observed upon the addition of an acidic deuterium source to the medium due to the HOO˙/DOO˙ equilibrium (the pKa of HOO˙ is 4.9).38 Thus, TEMPO-inhibited and Ar2NO-inhibited styrene/STY-BODIPY co-autoxidations were repeated in the presence of either 1% MeOH or MeOD. The results are shown in Fig. 6A. In both cases, the addition of MeOD had a pronounced effect on both the initial rate of the autoxidation and the duration of the inhibited period. From the initial rates, DKIEs of kH/kD = 4.2 and 3.3 can be calculated for TEMPO and Ar2NO, respectively. The magnitude of these DKIEs are very similar to those reported by Brownlie and Ingold for inhibition of styrene autoxidations by structurally-related hydroxylamines.12 These results are consistent with equilibration of HOO˙ with TEMPO/Ar2NO to produce TEMPOH/Ar2NOH as the chain-breaking antioxidant in situ (Fig. 6B).39,69,71 | ||
| Fig. 6 Kinetic isotope effect studies. (A) Co-autoxidation of styrene (3.5 M) and STY-BODIPY (10 μM) in PhCl initiated with di-tert-butylperoxide (218 mM) at 70 °C (black) and inhibited by either Ar2NO (2 μM, blue) or TEMPO (2 μM, red) in the presence of 1% v/v MeOH (dashed lines) or MeOD (solid lines). Reaction progress was monitored at 571 nm (ε = 97235 M−1 cm−1). (B) Dynamic exchange of the acidic proton in HOO˙ with the acidic deuteron in MeOD is expected to precede the nitroxide-catalyzed cross-dismutation of hydroperoxyl and alkylperoxyl radicals, leading to the observed kinetic isotope effects. (C) Synthesis of 1,2-d2-cyclooctene and the predicted kinetic isotope effect on HOO˙ production. (D) Co-autoxidation of cyclooctene or 1,2-d2-cyclooctene (3.1 M) and STY-BODIPY (10 μM) in PhCl initiated with di-tert-butylperoxide (218 mM) at 70 °C (black line) and inhibited by 2 μM of either Ar2NO (blue line), TEMPO (red) or Ar2NH (dashed blue line). Corresponding traces with deuterated substrate are represented by dotted lines. Inset: expansion of the initial portion of the autoxidation. Reaction progress was monitored at 568 nm (ε = 118405 M−1 cm−1). | ||
Given the computational prediction that HOO˙ can be formed from unsaturated hydrocarbons by a quantum tunneling-enhanced intramolecular 1,4-HAT from a β-alkoxyalkylperoxyl intermediate, we sought to replace the vinylic H-atoms of one of the substrates above with deuterium to suppress the ability of nitroxides to inhibit the autoxidation. We selected cyclooctene for these experiments since 1,2-d2-cyclooctene could be easily accessed simply by catalytic deuteration of cyclooctyne over Lindlar's catalyst (Fig. 6C). The results of STY-BODIPY/1,2-d2-cyclooctene co-autoxidations inhibited by TEMPO and Ar2NO are plotted alongside the corresponding data for cyclooctene in Fig. 6D.
The difference in the cyclooctene and 1,2-d2-cyclooctene data could not be more dramatic. Substitution of the nominally unreactive alkene H-atoms of the substrate with deuterium atoms all but precludes inhibition of the reaction by the nitroxides. The very small effect of the nitroxides on the reaction progress in the autoxidation (which is visible only upon expanding the initial portion of the reaction progress data as in the inset to Fig. 6D) is presumably due to the formation of a small amount of HOO˙ from the residual protiated cyclooctene also undergoing autoxidation. Although catalytic deuteration of cyclooctene is essentially quantitative to prepare 1,2-d2-cyclooctene, the starting cyclooctyne is prepared from cyclooctene, which is difficult to remove (see ESI† for more details). In stark contrast, the initial rates of the Ar2NH-inhibited autoxidations of cyclooctene and 1,2-d2-cyclooctene are very similar (see inset to Fig. 6D) and the reaction progress only diverges once the nitroxide formed in situ is responsible for inhibition of the autoxidation (evident over the longer reaction time shown in Fig. 6D). Overall, these results are fully consistent with the intramolecular 1,4-HAT of the β-alkoxyalkylperoxyl intermediate being key to the activity of the nitroxides, and that the contribution of this reaction to the propagation of autoxidation of these substrates is greatly enabled by quantum mechanical tunneling.
Discussion
Hindered aliphatic nitroxides such as TEMPO have long been considered unreactive to alkylperoxyl radicals, and as such, are not generally considered inhibitors of hydrocarbon autoxidation under most conditions (except at low partial pressures of O2, where alkyl radicals can be trapped). In contrast, diarylnitroxides have been shown to react with peroxyl radicals, albeit it more slowly than their parent amines. Given the difference in reactivity of hindered nitroxides and diarylnitroxides, it was proposed that peroxyl radical addition to one of the aryl rings of the diarylnitroxide is possible (Scheme 1).12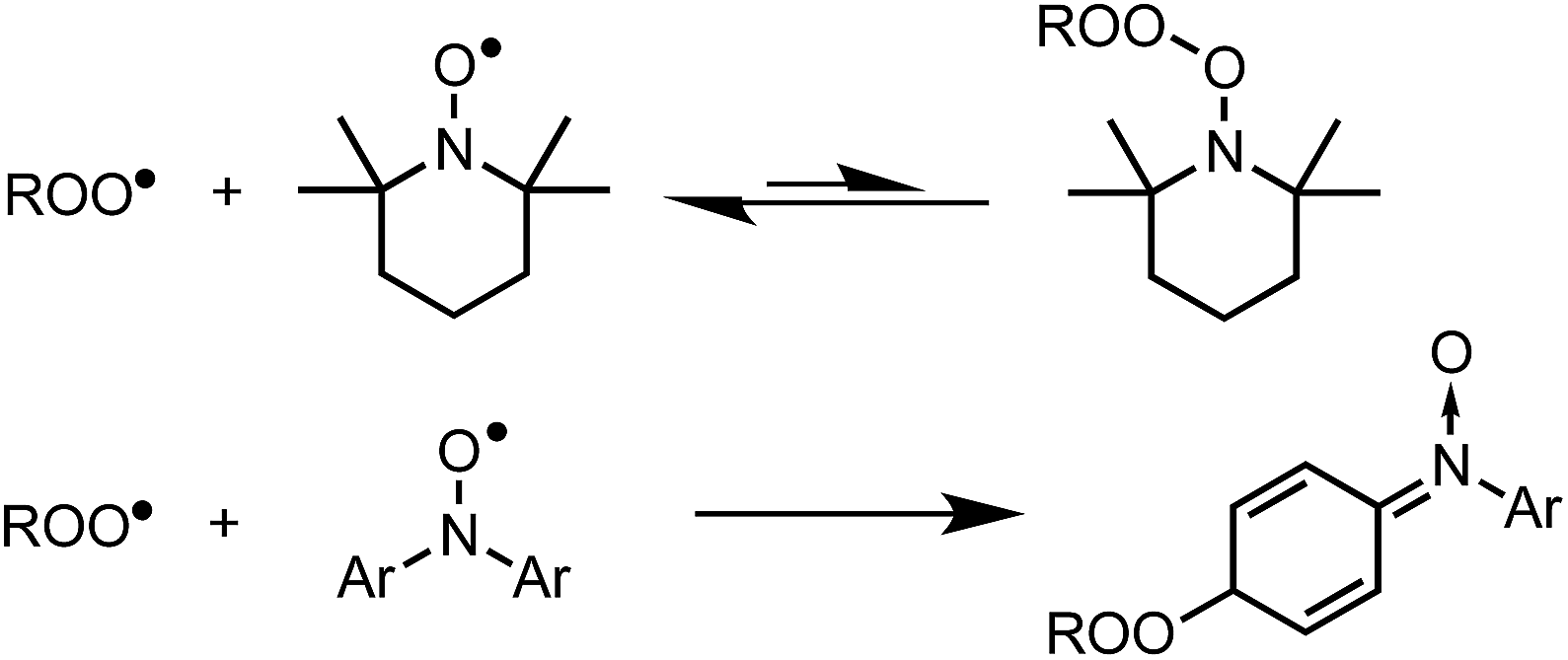 | ||
| Scheme 1 Hindered aliphatic nitroxides do not react with alkylperoxyl radicals; (di)arylnitroxides react by addition to the aryl ring(s). | ||
In recent years, some exceptions to this dogma have begun to appear (i.e. nitroxides undergo electron transfer to peroxyl radicals in aqueous solution,17,56 and nitroxides undergo acid-promoted reactions with peroxyl radicals in organic solution16,26), but they are under specific conditions. As such, we were initially very surprised to obtain the results in Fig. 1, where both hindered aliphatic nitroxides and diarylnitroxides are similarly good inhibitors of styrene autoxidation – better even than the corresponding diphenylamine. The results of subsequent experiments have clarified that nitroxides are excellent inhibitors of unsaturated hydrocarbon autoxidation at low rates of radical generation. Mechanistic studies have enabled the proposition of a mechanism that does not require the reaction of a nitroxide with an alkylperoxyl radical, but rather, a hydroperoxyl radical. Reaction of the resultant hydroxylamine with an alkylperoxyl radical traps a second chain-carrying radical and regenerates the nitroxide. The similarity in the apparent kinh observed for the nitroxides and the kinh observed for the authentic hydroxylamines implies that the reaction of nitroxide with HOO˙ is faster than hydroxylamine + ROO˙, such that hydroxylamine accumulates to serve as the key chain-breaking species.72 Overall, this mechanism can be characterized as a nitroxide-catalyzed cross-dismutation of hydroperoxyl and alkylperoxyl radicals. A similar reaction sequence was once proposed to explain the superoxide dismutase activity of hindered nitroxides in aqueous solution,57 but was subsequently dismissed on kinetic grounds in favour of the half reactions involving conversion of TEMPO to its corresponding oxoammonium ion and back.58
More than a century of research in combustion chemistry has shown that the major initial products formed in the high temperature (>300 °C) gas phase oxidation of simple alkanes are alkenes containing the same number of carbons. This observation has been ascribed to the elimination of HOO˙ from alkylperoxyl intermediates.59 The 1,4-HAT/elimination sequence connecting the alkylperoxyl radicals and alkenes have been the subject of extensive investigations, particularly for ethylperoxyl radicals due to their computational tractability.60 There would appear to be a consensus that, in the case of simple alkylperoxyl radicals, the 1,4-HAT and elimination take place in a concerted fashion. However, as the putative β-hydroperoxyalkyl radical is more stable both pathways are possible,61 or the step-wise process dominates.62 The step-wise path is anticipated to be greatly enhanced by quantum mechanical tunneling since the transition state involves little atomic movement other than the H-atom being transferred. Indeed, kinetic isotope effects derived for the 1,4-HAT in the step-wise formation of hydroperoxyl from n-propylperoxyl are predicted to decrease with temperature from 9.5 at 648 K to 6.2 at 703 K (ref. 63) – implying that at ambient temperatures they would be massive. Indeed, our calculations suggest that elimination of HOO˙ from chain-propagating alkylperoxyl radicals in unsaturated hydrocarbon autoxidation is step-wise and greatly enhanced by tunneling. Furthermore, the lack of inhibition of nitroxides in autoxidations of d2-cyclooctene clearly corroborate the prediction that the reaction is greatly enhanced by tunneling.
In the condensed phase, the elimination of HOO˙ from alkylperoxyl intermediates formation is known only for very specific hydrocarbons, e.g. 1,4-CHD and 1,4-dihydronaphthalene.28 Ingold first demonstrated that the autoxidation of these substrates is propagated primarily by HOO˙; initially due to their extremely rapid termination rate constants (kt = 6.3 × 108 M−1 s−1 in chlorobenzene at 30 °C; >200-fold greater than that measured for cyclohexene), and subsequently confirmed his assertion simply by washing the solutions with water and determining H2O2 by iodometric titration (which corresponded to ca. 95% of all the peroxide formed). The identification of substrates that propagate to only a small extent by hydroperoxyl radicals would have been extremely challenging due to their small contribution to kt and low yields of H2O2. Instead, the foregoing suggest that these substrates can be revealed indirectly by the ability of nitroxides to inhibit their autoxidation.
At first glance, the relative contribution of the 1,4-HAT/elimination pathway that is proposed to yield HOO˙ from chain-propagating alkylperoxyl radicals may be reflected in the relative efficacy of the nitroxides as inhibitors of autoxidation. Indeed, we find that while 1,4-CHD autoxidation is perpetually inhibited by the addition of TEMPO, the nitroxide is relatively less effective in styrene, norbornene and cyclooctene. However, rationalizing the trends observed for styrene, norbornene and cyclooctene is complicated by the fact that styrene and norbornene propagate exclusively by addition (necessary to produce the intermediate peroxyl radical from which the 1,4-HAT/elimination is proposed to take place), whereas cyclooctene propagates largely, but not exclusively, by addition.54,55 Moreover, because the 1,4-HAT and subsequent elimination is competitive only at low rates of initiation where radical–radical reactions are minimized, the differences in termination rate constants are also expected to contribute. Nevertheless, it appears that of the three substrates, the nitroxides are most effective in cyclooctene, perhaps because (as predicted by theory), the rate constant for the key 1,4-HAT is largest. Overall, it seems reasonable to propose that any substrate from which epoxides have been observed in autoxidations – even as minor products – should yield at least some HOO˙ and therefore, be protected by nitroxides.
Since linear unsaturated hydrocarbons such as those which are abundant in petroleum-derived products and biological lipids autoxidize largely by H-atom abstraction, it is unclear from the foregoing if the proposed mechanism contributes to their autoxidation and, thus, that they can be inhibited efficiently by nitroxides. Therefore, we carried out co-autoxidations of 1-hexadecene and PBD-BODIPY to find that not only are they inhibited by the addition of each nitroxide (Fig. 7), but that the inhibited periods correspond to massive stoichiometric factors: n = 117 ± 7 for Ar2NO and even greater for TEMPO. For comparison, Ar2NH is characterized by n = 23 ± 3, which presumably arises since (some of) the amine is converted to the nitroxide in situ (detected by EPR, see ESI†). This is a striking result; the catalytic radical-trapping antioxidant activity of diarylamines is almost universally ascribed to the Korcek cycle (Fig. 1B), but at these temperatures, Ar2NOR is not readily converted to Ar2NH,7 and even if it were, Ar2NH is still significantly less effective than Ar2NO.64
 | ||
| Fig. 7 (A) Co-autoxidation of 1-hexadecene (2.68 M) and PBD-BODIPY (10 μM) in PhCl initiated with dicumyl peroxide (1 mM) at 100 °C (black line) and inhibited by 2 μM of Ar2NO (blue line), TEMPO (red line) or Ar2NH (blue dashed line). Inset: expanded timescale showing the end of the inhibited periods for Ar2NO and Ar2NH. Reaction progress was monitored at 586 nm (ε = 119166 M−1 cm−1). (B) Relevant steps for HOO˙ formation in a 1-hexadecene autoxidation along with the calculated transition state structure and activation parameters for the key 1,4-HAT reaction for comparison with corresponding processes in for styrene, cyclooctene and norbornene (Fig. 5B–D). | ||
It must be emphasized that this is not the only evidence that diarylamines and/or diarylnitroxides catalytically trap radicals via an alternative to the Korcek cycle. For example, an oft-overlooked result of the seminal work of Bolsman et al.65 is that 4′,4-dinitrodiphenylnitroxide is a catalytic inhibitor of paraffin autoxidation at 130 °C, while the corresponding amine is completely ineffective. This could not be explained by the Korcek mechanism, and lead the authors to argue that the amines themselves are not involved in the catalytic cycle of diarylamine antioxidants, but need to be ‘activated’ to the catalytic radical-trapping antioxidant: the nitroxide. It is tempting to suggest that the paraffin oil used by Bolsman et al. (Shell ONDINA 33) contained sufficient unsaturation to produce HOO˙ and enable protection by the nitroxide.
Likewise, this mechanism can account for the ‘non-classical’ stoichiometries (i.e. n > 2) observed for diarylamines in autoxidations carried out at ambient temperatures. For example, some time ago, Lucarini and co-workers reported that phenoxazine was not only among the fastest RTAs ever reported (kinh = 2.9 × 107 M−1 s−1), but that it trapped an amazing 5 radicals in styrene autoxidations (at 50 °C).4 Very recently, we revisited this chemistry66 and found that phenoxazine trapped 10 radicals in styrene at 37 °C – a result which can now be readily explained by the fact that our rate of initiation was lower than Lucarini's, enabling 1,4-HAT/elimination to contribute more to the autoxidation. Interestingly, in our recent studies we were also able to see two distinct phases within the inhibited period of the autoxidation, which can now be understood as an initial phase in which the parent amine traps peroxyl radicals, and a secondary phase wherein the amine-derived nitroxide reacts with HOO˙ and traps peroxyl radicals as the corresponding hydroxylamine.
Conclusions
Autoxidations of unsaturated hydrocarbons are very efficiently inhibited by nitroxides and/or compounds which react readily to produce nitroxides under autoxidative conditions. The utilization of HOO˙ formed in situ as the stoichiometric reductant accounts for the catalytic behaviour of the nitroxides and/or their precursors without invoking a mechanism that requires the formation – and decomposition – of alkoxyamines. Alkoxyamine intermediates are invoked in the Korcek and Denisov mechanisms that have been widely accepted to account for the catalytic active of diarylamines and hindered aliphatic amines, respectively. However, these mechanisms can be expected to apply only when the nitroxide is present at much higher concentrations than O2, since alkoxyamine formation requires that the nitroxide react with an alkyl radical despite the fact that this reaction is characterized by a rate constant that is 10-fold lower than that of the reaction of alkyl radicals with O2. Moreover, elevated temperatures are required to convert the alkoxyamine back to the amine at a relevant rate, making it impossible to explain the catalytic reactivity of nitroxides and/or their precursors at ambient to moderate temperatures. The nitroxide-catalyzed cross-dismutation of HOO˙ and ROO˙ can account for observations that cannot be explained by the Korcek and Denisov cycles. This mechanism may be most relevant for hindered aliphatic amines/nitroxides since they are used in more applications at ambient temperatures where the rate of initiation is low, and radical–radical reactions are minimized.Experimental section
I. Inhibited autoxidations
The co-autoxidations described here were carried out according to our previously published methodology.18 Briefly, the substrate (1–2 mL) was loaded into a 3 mL cuvette along with the appropriate volume of PhCl such that the final volume of the sample was 2.5 mL. The cuvette was placed into the thermostatted sample holder of a UV-vis spectrophotometer and equilibrated at the appropriate temperature. STY-BODIPY or PBD-BODIPY (12.5 μL of a 2.0 mM solution in 1,2,4-trichlorobenzene) was then added followed by 50–100 μL of a solution of initiator in PhCl. The solution was thoroughly mixed. The consumption of the probe was monitored for 10–20 min to ensure that the reaction was proceeding at a constant rate, after which the test compound (10 μL of a 500 μM solution) was added, the solution thoroughly mixed and the absorbance readings resumed. Inhibition rate constants (kinh) and stoichiometries (n) were derived from the reaction progress traces analyzed using the equations in Fig. 2B (see ESI† for representative examples).An analogous procedure was followed when determining nitroxide concentration by EPR in diarylamine-inhibited autoxidations. Reactions were performed under identical conditions in a heat-block at 70 °C and samples taken after the indicated time intervals and transferred to EPR tubes. Spectra were recorded on a Bruker EMXplus (X-band) spectrometer equipped with an ER 4119HS cavity and the radical concentration was determined using the quantitative EPR package of the Bruker Xenon software.
II. Calculations
Calculations were carried out of Skodje, Truhlar and Garrett using the CBS-QB3 complete basis set method31 as implemented in the Gaussian 16 suite of programs.67 The rate constants were calculated via transition state theory at 70 °C. The contribution of quantum mechanical tunneling to the intramolecular hydrogen atom transfer step was estimated using the small curvature tunneling corrections of Skodje, Truhlar and Garrett.68 The evaluation of different computational approaches to address this problem was made using the oxidation of 1,4-cyclohexadiene as a model and are described in the ESI.†III. Synthesis of d2-1,2-cyclooctene
A flame dried round-bottomed flask with magnetic stirrer was flushed with N2, fitted with a rubber septum and charged with cyclooctyne in acetone (1 M). The solution was sparged with N2 for 5 minutes. Lindlar's catalyst (Pd/CaCO3, poisoned with Pb) was added in one portion (10 mol%) and the mixture was sparged for an additional 5 minutes. The atmosphere was evacuated and replaced with D2. The reaction was gently warmed to 30 °C and stirred for 36 h. Once the starting material was consumed (as judged by 1H NMR analysis), the reaction was filtered through a thin pad of Celite/silica gel and the filtrate vacuum distilled. The title product was collected (20 mbar/63 °C) as a clear oil (3 g, 60%) following initial removal of acetone (30 mbar/35 °C).701H NMR (400 MHz; CDCl3): δ (ppm): 5.62 (m, <0.2H), 2.13 (m, 4H), 1.50 (m, 8H). 13C NMR (101 MHz; CDCl3): 130.0, 29.5, 26.6, 25.3. HRMS (EI) m/z for C8H12D2, calc.: 112.1221, found 112.1256.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by grants from the Natural Sciences and Engineering Research Council of Canada and the Canada Foundation for Innovation and through generous access to the computational resources of the Centre for Advanced Computing (cac.queensu.ca). DAP and EAH also acknowledge the support of the Canada Research Chairs and Ontario Graduate Scholarships programs, respectively.Notes and references
- K. U. Ingold, Chem. Rev., 1961, 61, 563 CrossRef.
- K. U. Ingold and D. A. Pratt, Chem. Rev., 2014, 114, 9022 CrossRef PubMed.
- C. E. Boozer, G. S. Hammond, C. E. Hamilton and J. N. Sen, J. Am. Chem. Soc., 1955, 77, 3233 CrossRef.
- M. Lucarini, P. Pedrielli, G. F. Pedulli, L. Valgimigli, D. Gigmes and P. Tordo, J. Am. Chem. Soc., 1999, 121, 11546 CrossRef.
- T. A. B. M. Bolsman, A. P. Blok and J. H. G. Frijns, Recl. Trav. Chim. Pays-Bas, 1978, 97, 310 CrossRef.
- R. K. Jensen, S. Korcek, M. Zinbo and J. L. Gerlock, J. Org. Chem., 1995, 60, 5396 CrossRef.
- E. A. Haidasz, R. Shah and D. A. Pratt, J. Am. Chem. Soc., 2014, 136, 16643 CrossRef PubMed.
- J. R. Thomas and C. A. Tolman, J. Am. Chem. Soc., 1962, 84, 2930 CrossRef.
- K. Adamic, M. Dunn and K. U. Ingold, Can. J. Chem., 1969, 47, 287 CrossRef.
- E. T. Denisov, Polym. Degrad. Stab., 1989, 25, 209 CrossRef.
- G. Gryn'ova, K. U. Ingold and M. L. Coote, J. Am. Chem. Soc., 2012, 134, 12979 CrossRef PubMed.
- I. T. Brownlie and K. U. Ingold, Can. J. Chem., 1967, 45, 2427 CrossRef.
- A. Beckwith, V. W. Bowry and K. U. Ingold, J. Am. Chem. Soc., 1992, 114, 4983 CrossRef.
- V. W. Bowry and K. U. Ingold, J. Am. Chem. Soc., 1992, 114, 4992 CrossRef.
- B. Maillard, K. U. Ingold and J. C. Scaiano, J. Am. Chem. Soc., 1983, 105, 5095 CrossRef.
- E. A. Haidasz, D. Meng, R. Amorati, A. Baschieri, K. U. Ingold, L. Valgimigli and D. A. Pratt, J. Am. Chem. Soc., 2016, 138, 5290 CrossRef PubMed.
- M. Griesser, R. Shah, A. T. Van Kessel, O. Zilka, E. A. Haidasz and D. A. Pratt, J. Am. Chem. Soc., 2018, 140, 3790 CrossRef PubMed.
- E. A. Haidasz, A. T. Van Kessel and D. A. Pratt, J. Org. Chem., 2016, 81, 737 CrossRef PubMed.
- L. Valgimigli and D. A. Pratt, Antioxidants in Chemistry and Biology, John Wiley & Sons, Ltd, Chichester, UK, 2nd edn, vol. 95, 2012 Search PubMed.
- B. Li and D. A. Pratt, Free Radical Biol. Med., 2015, 82, 187 CrossRef PubMed.
- R. Amorati and L. Valgimigli, Free Radical Res., 2015, 49, 633 CrossRef PubMed.
- The inhibition rate constant for Ar2NH is in excellent agreement with reported values for 4,4′-dialkylated diphenylamines in other substrates. For example, values of 1.8 and 2.0 × 105 M−1 s−1 have been determined at 37 °C for n-octyl and t-butyl substituted diphenylamines by the peroxyl radical clock and STY-BODIPY/dioxane co-autoxidation methodologies, respectively. See ref. 37 and R. Shah, K. D. Margison and D. A. Pratt, ACS Chem. Biol., 2017, 12, 2538 CrossRef PubMed.
- The CBS-QB3-calculated N–H BDE in 2,2,6,6,-tetramethylpiperidine and diphenylamine are 96.2 and 86.4 kcal mol−1, respectively. Although there are no reliable experimental values with which the former can be compared, there are ample experimental values for the latter. See, e.g., ref. 24 which yields 85.8 kcal mol−1 after the revision in ref. 25.
- D. A. Pratt, G. A. DiLabio, L. Valgimigli, G. F. Pedulli and K. U. Ingold, J. Am. Chem. Soc., 2002, 124, 11085 CrossRef PubMed.
- P. Mulder, H.-G. Korth, D. A. Pratt, G. A. DiLabio, L. Valgimigli, G. F. Pedulli and K. U. Ingold, J. Phys. Chem. A, 2005, 109, 2647 CrossRef PubMed.
- R. Amorati, G. F. Pedulli, D. A. Pratt and L. Valgimigli, Chem. Commun., 2010, 46, 5139 RSC.
- J. Cedrowski, G. Litwinienko, A. Baschieri and R. Amorati, Chem.–Eur. J., 2016, 22, 16441 CrossRef PubMed.
- J. A. Howard and K. U. Ingold, Can. J. Chem., 1967, 45, 785 CrossRef.
- L. R. Mahoney, G. D. Mendenhall and K. U. Ingold, J. Am. Chem. Soc., 1973, 95, 8610 CrossRef.
- M. Lucarini, G. F. Pedulli and M. Cipollone, J. Org. Chem., 1994, 59, 5063 CrossRef.
- J. A. Montgomery, M. J. Frisch Jr, J. W. Ochterski and G. A. Petersson, J. Chem. Phys., 1999, 110, 2822 CrossRef.
- Litwinienko and Amorati27 estimated that the analogous rate constant for the reaction of HOO˙ and the phenoxyl radical derived from PMC is ca. 109 M−1 s−1 based upon competition with hydroperoxyl addition to the ring, for which they estimated that the rate constant was the same as for an alkylperoxyl radical (ca. 108 M−1 s−1). It is likely that HOO˙ addition is faster than alkylperoxyl radical addition, and that HAT between HOO˙ and phenoxyl radicals is also diffusion-controlled..
- V. Vaidya, K. U. Ingold and D. A. Pratt, Angew. Chem., Int. Ed. Engl., 2009, 48, 157 CrossRef PubMed.
- Z. Zielinski, N. Presseau, R. Amorati, L. Valgimigli and D. A. Pratt, J. Am. Chem. Soc., 2014, 136, 1570 CrossRef PubMed.
- J.-P. R. Chauvin, M. Griesser and D. A. Pratt, J. Am. Chem. Soc., 2017, 139, 6484 CrossRef PubMed.
- L. Valgimigli, R. Amorati, S. Petrucci, G. F. Pedulli, D. Hu, J. J. Hanthorn and D. A. Pratt, Angew. Chem., Int. Ed. Engl., 2009, 48, 8348 CrossRef PubMed.
- J. J. Hanthorn, L. Valgimigli and D. A. Pratt, J. Am. Chem. Soc., 2012, 134, 8306 CrossRef PubMed.
- G. Czapski, Annu. Rev. Phys. Chem., 1971, 22, 171 CrossRef.
- It should be noted that the addition of MeOH to styrene/chlorobenzene enhances the activity of the nitroxides but does so far more so for TEMPO than for Ar2NO; compare n = 26.3 ± 1.0 and 22.0 ± 1.0 in the presence of MeOH to n = 9.3 ± 0.9 and 10.6 ± 0.2 in its absence, respectively.
- D. G. Hendry and D. Schuetzle, J. Am. Chem. Soc., 1975, 97, 7123 CrossRef.
- T. Ohta, Int. J. Chem. Kinet., 1984, 16, 1495 CrossRef.
- F. Berho and R. Lesclaux, Phys. Chem. Chem. Phys., 2001, 3, 970 RSC.
- E. C. Tuazon, S. M. Aschmann, M. V. Nguyen and R. Atkinson, Int. J. Chem. Kinet., 2003, 35, 415 CrossRef.
- E. Estupiñán, E. Villenave, S. Raoult, J. C. Rayez, M. T. Rayez and R. Lesclaux, Phys. Chem. Chem. Phys., 2003, 5, 4840 RSC.
- J. W. Taylor, G. Ehlker, H.-H. Carstensen, L. Ruslen, R. W. Field and W. H. Green, J. Phys. Chem. A, 2004, 108, 7193 CrossRef.
- It must be acknowledged that computations of such a process – should it exist – would likely require a multi-determinantal approach and dynamic considerations.
- F. Agapito, P. M. Nunes, B. J. Costa Cabral, R. M. Borges dos Santos and J. A. Martinho Simões, J. Org. Chem., 2007, 72, 8770 CrossRef PubMed.
- B. Ruscic, R. E. Pinzon, M. L. Morton, N. K. Srinivasan, M.-C. Su, J. W. Sutherland and J. V. Michael, J. Phys. Chem. A, 2006, 110, 6592 CrossRef PubMed.
- F. R. Mayo, A. A. Miller and G. A. Russell, J. Am. Chem. Soc., 1958, 80, 2500 CrossRef.
- W. Suprun and J. Opeida, Chem. Ing. Tech., 2005, 77, 753 CrossRef.
- F. R. Mayo, J. Am. Chem. Soc., 1958, 80, 2465 CrossRef.
- R. G. Naick, W. Pritzkow and J. Rasche, J. Prakt. Chem., 1977, 319, 785 CrossRef.
- E. A. Lazurin, V. V. Voronenkov and Y. G. Osokin, Russ. Chem. Rev., 1977, 46, 915 CrossRef.
- D. E. Van Sickle, F. R. Mayo and R. M. Arluck, J. Am. Chem. Soc., 1965, 87, 4824 CrossRef.
- K. Blau, U. Müller, W. Pritzkow, W. Schmidt-Renner and Z. Sedshaw, J. Prakt. Chem., 1980, 322, 915 CrossRef.
- S. A. Goldstein and A. Samuni, J. Phys. Chem. A, 2007, 111, 1066 CrossRef PubMed.
- A. Samuni, C. M. Krishna, P. Riesz, E. Finkelstein and A. Russo, J. Biol. Chem., 1988, 263, 17921 Search PubMed.
- M. C. Krishna, D. A. Grahame, A. Samuni, J. B. Mitchell and A. Russo, Proc. Natl. Acad. Sci. U. S. A., 1992, 89, 5537 CrossRef.
- J. Zádor, C. A. Taatjes and R. X. Fernandes, Prog. Energy Combust. Sci., 2011, 37, 371 CrossRef.
- J. C. Rienstra-Kiracofe, W. D. Allen and H. F. Schaefer, J. Phys. Chem. A, 2000, 104, 9823 CrossRef.
- E. W. Kaiser, J. Phys. Chem. A, 2002, 106, 1256 CrossRef.
- J. D. Savee, E. Papajak, B. Rotavera, H. Huang, A. J. Eskola, O. Welz, L. Sheps, C. A. Taatjes, J. Zádor and D. L. Osborn, Science, 2015, 347, 643 CrossRef PubMed.
- E. G. Estupiñán, J. D. Smith, A. Tezaki, S. J. Klippenstein and C. A. Taatjes, J. Phys. Chem. A, 2007, 111, 4015 CrossRef PubMed.
- The acid-catalyzed reaction of the nitroxide (as in Fig. 1C) can also be excluded since little acid is formed in hexadecene autoxidations because intermolecular propagation is greatly preferred over the intramolecular propagation that is chiefly responsible for acid formation. See: R. Shah and D. A. Pratt, J. Org. Chem., 2016, 81, 6649 CrossRef PubMed.
- T. A. B. M. Bolsman, A. P. Blok and J. H. G. Frijns, Recl. Trav. Chim. Pays-Bas, 1978, 97, 313 CrossRef.
- L. A. Farmer, E. A. Haidasz, M. Griesser and D. A. Pratt, J. Org. Chem., 2017, 82, 10523 CrossRef PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision A.03, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- R. T. Skodje, D. G. Truhlar and B. C. Garrett, J. Phys. Chem., 2002, 85, 3019 CrossRef.
- Styrene/STY-BODIPY co-autoxidations were also carried out in the presence of an H-bond accepting solvent, 2-octanone (see ESI† for the data). Again, the initial (inhibited) rate increased and the duration of the inhibited period decreased, consistent with the intervention of H-bond donating H-atom donors (i.e. hydroxylamines) as the RTAs formed from the nitroxides in situ.
- H. Lindlar and R. Dubuis, Org. Synth., 1966, 46, 89 CrossRef.
- Although they contribute to the propagation of autoxidation, it is unlikely that reactions of hydroperoxyl and/or alkoxyl radicals with the hydroxylamines are relevant under the reaction conditions. Since both alkoxyl radicals and hydroperoxyl radicals are more reactive than alkylperoxyl radicals, they are less selective for hydroxylamine over the substrate. For example, alkoxyl radicals add/abstract from olefins with k ∼ 106 M−1 s−1 and react with hydroxylamines with k ∼ 108 M−1 s−1 ( E. Abuin, M. V. Encina, S. Diaz and E. A. Lissi, Intl. J. Chem. Kinet., 1978, 10, 677 CrossRef ), whereas alkylperoxyl radicals add/abstract from olefins with k ∼ 1 M−1 s−1, but react with hydroxylamines with k > 106 M−1 s−1. Given the concentration difference (μM vs. M), only the latter competition will be important.
- Since the calculations predict that nitroxide + HOO˙ is diffusion-controlled, inhibition results from the successful competition between the intramolecular 1,4-HAT in the precursor to HOO˙, which is predicted to take place with k ∼ 1 to 10 s−1 depending on the substrate, and hydroxylamine + ROO˙, which we measure to take place with k ∼ 106 M−1 s−1. Although the rate constant for the latter is faster, it depends on [nitroxide] which is 2 μM – suggesting that the elimination is competitive under the reaction conditions.
- E. M. Pliss, V. A. Machtin, A. M. Grobov, R. E. Pliss and A. V. Sirick, Int. J. Chem. Kinet., 2017, 49, 173–181 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8sc01575a |
| This journal is © The Royal Society of Chemistry 2018 |