
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnantioselective synthesis of amines by combining photoredox and enzymatic catalysis in a cyclic reaction network†
Xingwei
Guo
a,
Yasunori
Okamoto
 b,
Mirjam R.
Schreier
a,
Thomas R.
Ward
*b and
Oliver S.
Wenger
*a
b,
Mirjam R.
Schreier
a,
Thomas R.
Ward
*b and
Oliver S.
Wenger
*a
aDepartment of Chemistry, University of Basel, St. Johanns-Ring 19, 4056 Basel, Switzerland. E-mail: oliver.wenger@unibas.ch
bDepartment of Chemistry, University of Basel, Mattenstrasse 24a, BPR 1096, 4002 Basel, Switzerland. E-mail: thomas.ward@unibas.ch
First published on 17th May 2018
Abstract
Visible light-driven reduction of imines to enantioenriched amines in aqueous solution is demonstrated for the first time. Excitation of a new water-soluble variant of the widely used [Ir(ppy)3] (ppy = 2-phenylpyridine) photosensitizer in the presence of a cyclic imine affords a highly reactive α-amino alkyl radical that is intercepted by hydrogen atom transfer (HAT) from ascorbate or thiol donors to afford the corresponding amine. The enzyme monoamine oxidase (MAO-N-9) selectively catalyzes the oxidation of one of the enantiomers to the corresponding imine. Upon combining the photoredox and biocatalytic processes under continuous photo-irradiation, enantioenriched amines are obtained in excellent yields. To the best of our knowledge, this is the first demonstration of a concurrent photoredox- and enzymatic catalysis leading to a light-driven asymmetric synthesis of amines.
Introduction
Photoredox catalysis has become a very popular research field,1 and while most prior studies reported on the formation of racemic product mixtures, increasing attention is now being devoted to enantioselective photoredox chemistry.2 Dual catalysis methods have often been employed to gain control over stereoselectivity using for example chiral amines,3N-heterocyclic carbenes,4 chiral Brønsted acids,5 chiral Lewis acids,6 or chiral counter-ions as co-catalysts.7 Independently of these photoredox catalysis endeavors, cyclic reaction networks have emerged as an attractive strategy for asymmetric synthesis (Fig. 1a).8 In a typical network of this type, an initial endergonic process converts the substrate into a racemic product, and a subsequent exergonic process selectively converts one of the enantiomers back to the starting material, while the other enantiomer accumulates. Over time, such selective recycling leads to the enrichment of the unreactive enantiomer.8 This strategy, also termed cyclic deracemization, was used for the chemo-enzymatic de-racemization of amines, using an amine oxidase or reductive aminase (in reverse mode) as recycling catalyst combined with a non-enantioselective chemical reduction or an imine reductase.9 More recently, the initial chemical reduction step was performed inside tailored artificial metalloenzymes, and combination with biocatalysts for the recycling step led to multi-enzymatic cascades.10 We were curious whether the enzymatic recycling would be compatible with a light-driven catalytic step, such as typically the case in photoredox catalysis. Compared to small molecular catalysts, biocatalysts frequently offer the advantage of superior selectivity and efficiency, hence the idea of incorporating a photoredox step into a cyclic reaction network together with an enzyme for overall asymmetric catalysis seemed very appealing. Light-driven biocatalysis has gained increasing attention in recent years,11 however, mostly in the context of co-factor regeneration.12 To complement these efforts, Hyster and co-workers reported on an elegant strategy to access highly selective radical-mediated reactions by irradiating nicotinamide-dependent enzymes.13 To the best of our knowledge, there are no prior photoredox studies that have made use of cyclic reaction networks to achieve overall enantioselective catalysis.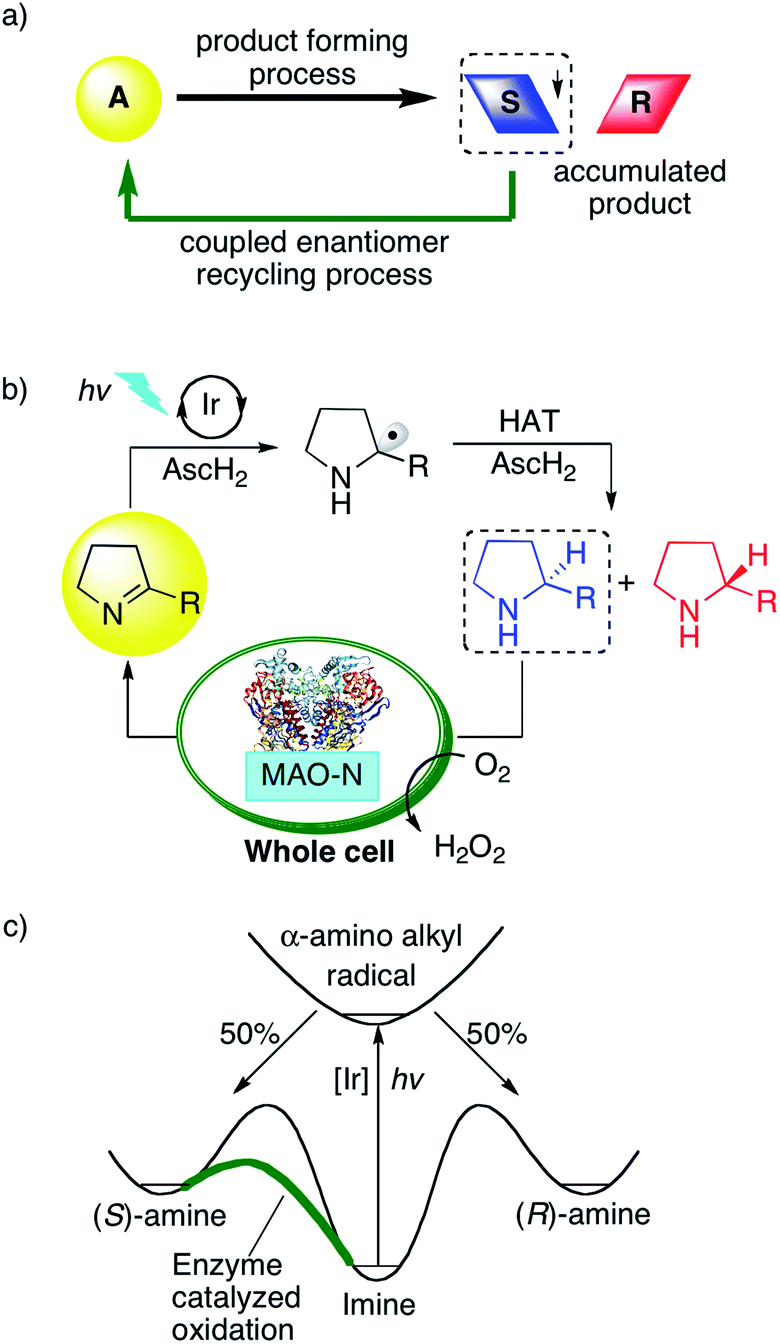 | ||
| Fig. 1 (a) Principle of an enantiomer-recycling reaction network. (b) Photoredox-catalyzed reduction of cyclic imines using an Ir sensitizer, and subsequent re-oxidation of the (S)-amine back to the imine starting material by monoamine oxidase (MAO-N-9) expressed in E. coli cells. The overall two-electron reduction process is driven by visible light and relies on ascorbic acid (AscH2) as a reductant and HAT donor. (c) Energy profile for the light-driven cyclic deracemization. | ||
Direct photoredox-driven reduction of imines to amines does not appear to have been reported previously.14 This may be traced back to the fact that, after initial imine reduction by photoinduced (single) electron transfer, the resulting α-amino alkyl radicals must be intercepted rapidly by (thermal) hydrogen atom transfer (HAT) to achieve the overall two-electron reduction. This may be somewhat challenging as photochemical multi-electron reductions are usually non-trivial to perform.15 In the present case, the identification of suitable HAT donors was of key importance. Thiols and disulfides have recently been identified as suitable co-catalysts to intercept carbon radicals in a variety of different photoredox processes.16 We and others found that thiols react very rapidly with photo-generated α-amino alkyl radicals,17 owing to the principle of polarity-matching.18 We hypothesized that the light-driven reduction of imines to amines with ascorbic acid might be possible using a similar strategy, and we anticipated that this methodology may be coupled with an enzymatic reaction if the photoredox process could be performed in aqueous solution.
Thus, we designed a cyclic reaction network for the enantioselective synthesis of amines from cyclic imines as illustrated in Fig. 1b. A water-soluble photosensitizer serves to reduce iminium ions to α-amino alkyl radicals, which are further converted to the respective amines by HAT from ascorbic acid (AscH2/AscH−). This photoredox-driven formation of amines can only lead to racemic amines. Gratifyingly, monoamine oxidase (MAO-N-9) catalyzes the oxidation of cyclic amines with excellent selectivity for the (S)-enantiomer (Fig. 1c).10 Accordingly, under continuous photo-irradiation, this concurrent enzymatic recycling process should ultimately lead to the accumulation of the (R)-amine. If successful, photoredox catalysis may allow the enantioselective imine-to-amine reduction in a cyclic reaction network. A potential challenge may however be mutual deactivation between the small molecule photoredox catalyst and the enzyme, which is often observed for similar systems.10,19
Results and discussion
We started our investigations with 2-cyclohexyl-1-pyrroline (1a) as a model substrate using [Ru(bpy)3]Cl2 as a photocatalyst (1 mol%) and ascorbic acid (2 eq.) as a reductant in aqueous phosphate buffer solution at pH 8.0 (Scheme 1). Over the course of 20 h of irradiation with a blue LED (455 nm, 3 W) at room temperature, only trace amounts of the desired product rac-2a were formed. We attribute this to insufficient reducing power of [Ru(bpy)3]Cl2,20 as the peak reduction potential of 1a in aqueous solution at pH 8 was determined to be −1.84 V vs. SCE (ESI page S30†). Compared with [Ru(bpy)3]Cl2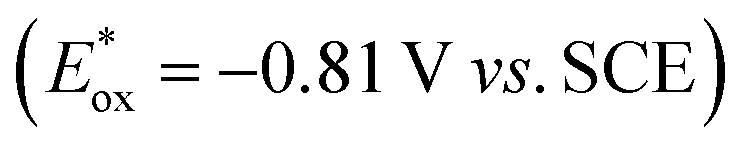 , fac-[Ir(ppy)3]
, fac-[Ir(ppy)3] 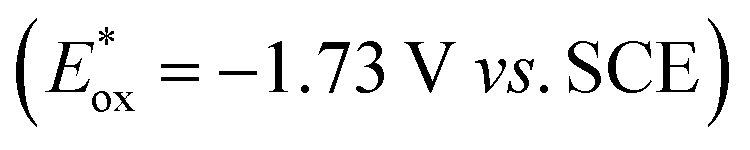 is a much stronger photoreductant.21 To our delight, when using this complex, the reduction of 1a to rac-2a did indeed proceed with 59% yield after 6 h of irradiation (405 nm, 3 W). However, due to the very low solubility of fac-[Ir(ppy)3] in water, the reaction had to be carried out in acetonitrile, and this would be incompatible with enzyme catalysis. We therefore prepared a new tri-sulfonated variant of fac-[Ir(ppy)3] (Scheme 1, right), which turns out to have very similar electrochemical and optical spectroscopic properties as the parent complex (ESI page S5†), despite the electron withdrawing substituents and negative charges at the ligand periphery. Specifically, the excited state oxidation potential of the Na3[Ir(sppy)3] complex is −1.89 V vs. SCE, slightly stronger than its parent complex, and it exhibits 3MLCT luminescence centered at 508 nm with a lifetime of 700 ns in aerated aqueous solution. With Na3[Ir(sppy)3] as a photocatalyst the conversion of 1a to rac-2a in water proceeded in 95% yield after irradiation for 10 hours (Scheme 1). Thus, efficient photoredox-driven reduction of imines to amines in aqueous solution becomes possible, owing to the high reducing power of the water-soluble Na3[Ir(sppy)3] complex and the use of ascorbic acid as a HAT donor that intercepts highly reactive α-amino alkyl radical intermediates formed in the initial light-induced reaction step (Fig. 1b).17 Expectedly, a racemic product mixture was obtained.
is a much stronger photoreductant.21 To our delight, when using this complex, the reduction of 1a to rac-2a did indeed proceed with 59% yield after 6 h of irradiation (405 nm, 3 W). However, due to the very low solubility of fac-[Ir(ppy)3] in water, the reaction had to be carried out in acetonitrile, and this would be incompatible with enzyme catalysis. We therefore prepared a new tri-sulfonated variant of fac-[Ir(ppy)3] (Scheme 1, right), which turns out to have very similar electrochemical and optical spectroscopic properties as the parent complex (ESI page S5†), despite the electron withdrawing substituents and negative charges at the ligand periphery. Specifically, the excited state oxidation potential of the Na3[Ir(sppy)3] complex is −1.89 V vs. SCE, slightly stronger than its parent complex, and it exhibits 3MLCT luminescence centered at 508 nm with a lifetime of 700 ns in aerated aqueous solution. With Na3[Ir(sppy)3] as a photocatalyst the conversion of 1a to rac-2a in water proceeded in 95% yield after irradiation for 10 hours (Scheme 1). Thus, efficient photoredox-driven reduction of imines to amines in aqueous solution becomes possible, owing to the high reducing power of the water-soluble Na3[Ir(sppy)3] complex and the use of ascorbic acid as a HAT donor that intercepts highly reactive α-amino alkyl radical intermediates formed in the initial light-induced reaction step (Fig. 1b).17 Expectedly, a racemic product mixture was obtained.
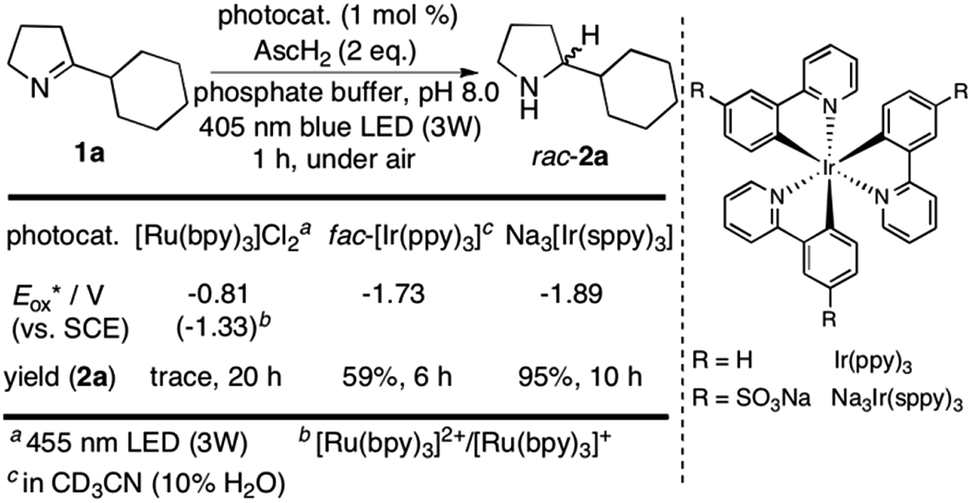 | ||
| Scheme 1 Identification of a suitable photocatalyst for the reduction of 1a. | ||
With these optimized conditions at hand, we attempted to integrate this photoredox step into a cyclic reaction network by combining the light-driven imine reduction with enantioselective amine oxidation catalyzed by MAO-N-9.
Toward this end, imine 1a (10 μmol) was added to a 1 mL phosphate-buffered aqueous suspension at pH 8 containing E. coli cells with recombinantly expressed monoamine oxidase (180 mg, wet weight), Na3[Ir(sppy)3] (1 mol%), and ascorbic acid (0.2 M). Gratifyingly, photo-irradiation by a blue LED (405 nm) for 20 h under air led to 92% conversion of 1a and 91% e.e. (R) (Table 1, entry 1). After a prolonged reaction time (30 h), enantiomerically pure product (R)-2a was obtained (Table 1, entry 2). Control experiments without any biocatalyst (Table 1, entry 3) or using E. coli cells with an empty vector (pET-16b) (Table 1, entry 4) afforded rac-2a in quantitative yield. When using the lysate of E. coli cells containing MAO-N-9, decreased conversion (83%) and lower enantioselectivity (77% e.e.) were observed (Table 1, entry 5). This finding suggests that photoredox and enzymatic catalysis mutually perturb each other in the cell lysate. Independent kinetic resolution experiments of rac-2a using E. coli (MAO-N-9) cell lysate support our hypothesis that the lowered enantioselectivity of the imine to amine reduction in the cell lysate (Table 1, entry 5) is due to partial inactivation of MAO-N-9 by the photocatalyst. Conversely, the lower yield for the overall reaction (83%) is likely caused by a detrimental effect of the biocatalyst (or cellular debris) on the photoredox process (see ESI page S10†). As noted above, such mutual inhibition of chemo- and biocatalysts is a widely-observed phenomenon.19 To circumvent this challenge, the MAO-N-9 was compartmentalized in the cytoplasm of E. coli cells. We hypothesize that the negatively charged [Ir(sppy)3]3− photocatalyst cannot diffuse through the cell membrane due to its anionic nature and high molecular weight, thus warranting the spatial separation of photoredox-related processes and the biocatalyzed reaction steps when using whole cells instead of lysates.
|
|
|||
|---|---|---|---|
| Entry | Biocatalyst | t/h | Yield [e.e.] (%)b |
| a Reaction conditions: 1a (10 mM), Na3[Ir(sppy)3] (0.1 mM), AscH2 (200 mM), biocatalyst (180 or 120 mg, wet weight of cell pellets) in 1.0 mL phosphate buffer solution (pH 8.0) at ambient temperature with 405 nm LED (3 W) irradiation on a shaking agitator (at 200 rpm) under air. b Yields were determined by 1H NMR analysis, enantiomeric excess was determined by GC on a chiral stationary phase (see ESI for details). | |||
| 1 | E. coli (MAO-N-9) whole cell/180 mg | 20 | 92 [91] |
| 2 | E. coli (MAO-N-9) whole cell/180 mg | 30 | 92 [>99] |
| 3 | — | 10 | 95 [0] |
| 4 | E. coli (empty) whole cell/120 mg | 20 | >95 [0] |
| 5 | E. coli (MAO-N-9) lysate/180 mg | 20 | 83 [77] |
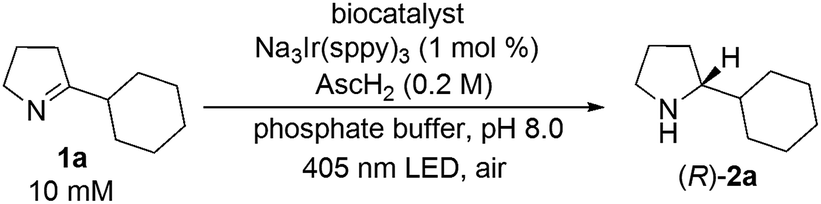
To gain mechanistic insight of this system, we monitored the conversion of imine substrate 1a to the amine product (R)-2a and the enantiomeric excess as a function of time (Fig. 2). It appears that the enzyme-catalyzed amine oxidation is the rate determining step in the overall enantioselective conversion of 1a to (R)-2a. The photoredox-driven reduction of imine to amine is almost complete within 2 hours (black trace), whilst the enantiomeric excess lags much behind (red trace). The slight decrease in the conversion beyond 2 h is likely due to slow deactivation of the photoredox catalyst.
 | ||
| Fig. 2 The conversion of 1a (10 μmol) to (R)-2a with 2 mol% photocatalyst and MAO-N-9 (180 mg wet cells) monitored by chiral GC over time. The data at each time point were determined from the relative peak integrals of starting material and products in a single measurement on chiral phase GC. | ||
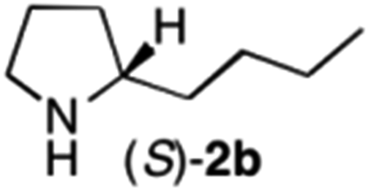
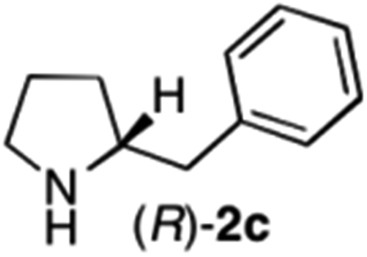
The cyclohexyl residue of 1a is known to fit well into the active site of MAO-N-9,10 and thus it seemed worthwhile to explore the possibility of using imine substrates with other hydrophobic substituents. We found that an imine substrate bearing an n-butyl side chain (substrate 1b) was converted to the respective amine with >95% yield without biocatalyst (Table 2, entry 1), whilst in presence of MAO-N-9 (S)-2b was formed in 95% yield and 98% e.e. (Table 2, entry 2). When installing a benzyl substituent on the 3,4-dihydro-2H-pyrrole core unit (substrate 1c), the photoredox step affords rac-2c quantitatively in the absence of MAO-N-9 (Table 2, entry 3), but only 8% e.e. of (R)-2c was obtained in presence of biocatalyst (entry 4). This low enantioselectivity suggests that substrate 1c cannot be efficiently converted by MAO-N-9.
| Entry | Substrate | Photocat. & reducing agent | Biocat. | Product | Yield [e.e.] (%)b |
|---|---|---|---|---|---|
| a Reaction conditions: 1 (10 mM), E. coli whole cells (MAO-N-9) (180 mg, wet weight), photocatalyst (1 mol%) and reducing agent as specified in each case, in 1.0 mL phosphate buffer solution (pH 8.0) at ambient temperature with 405 or 450 nm LED (3 W) irradiation on a shaking agitator (at 200 rpm, only used for reactions with biocatalyst) for 30 h under air. b Yields were determined by 1H NMR analysis, enantiomeric excess was determined by chiral phase GC or HPLC. c 10 h. d 3 h. e 20 h. | |||||
| 1 |
|
Na3Ir(sppy)3(1 mol%) + AscH2 (0.2 M) | — |
|
>95c |
| 2 | 1b | Na3Ir(sppy)3 (1 mol %) + AscH2 (0.2 M) | MAO-N-9 (180 mg) |
|
>95 [98] |
| 3 |
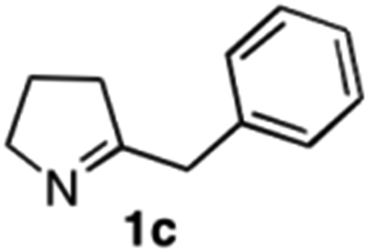
|
Na3Ir(sppy)3 (1 mol %) + AscH2 (0.2 M) | — |
|
>95c |
| 4 | 1c | Na3Ir(sppy)3 (1 mol %) + AscH2 (0.2 M) | MAO-N-9 (180 mg) |
|
>95 [8] |
| 5 |
|
Na3Ir(sppy)3 (1 mol %) + AscH2 (0.2 M) | — |
|
>95c |
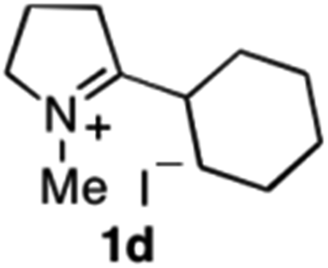
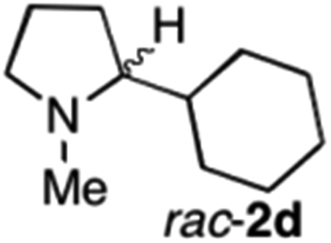
| 6 | 1d | Na3Ir(sppy)3 (1 mol %) + AscH2 (0.2 M) | MAO-N-9 (180 mg) |
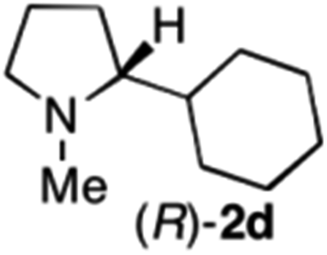
|
>95 [>99] |
| 7 |
|
Ru(bpy)3Cl2(1 mol %) + AscH2 (20 mM) + MPAA-PEG (10 eq.) + MPA-PEG (100 eq.) | — |
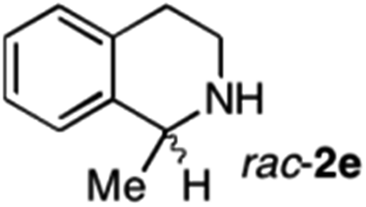
|
>95d |
| 8 | 1e | Ru(bpy)3Cl2(1 mol %) + AscH2 (20 mM) + MPAA-PEG (10 eq.) + MPA-PEG (100 eq.) | MAO-N-9 (180 mg) |
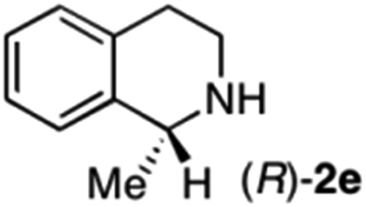
|
>95 [35]e |
To extend the applicability of our concept beyond imine to secondary amine reduction, we investigated whether iminium ion 1d can be converted to tertiary amine product rac-2d (Table 2, entries 5 and 6). With standard conditions, enantiomerically pure tertiary amine (R)-2d was readily obtained.
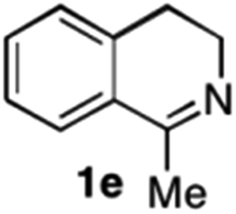
To further extend the substrate scope from aliphatic to aromatic imines, we examined 1-methyl-3,4-dihydroisoquinoline (1e) as a substrate.10 Due to significant stabilization of the respective carbon-centered radical intermediate resulting from initial reduction of the aromatic substrate, HAT is considerably less exergonic than in the case of aliphatic substrates and no product formation is observed as long as only ascorbate is present (ESI, page S15†). Therefore, we attempted to lower the activation barrier for HAT by adding 4-mercaptophenylacetic acid and 3-mercaptopropionic acid derivatives (MPAA-PEG and MPA-PEG, respectively, ESI page S16†), hypothesizing that polarity-matched HAT between these thiol donors and the α-amino alkyl radical intermediates would proceed sufficiently rapidly (full experimental and mechanistic details are given in the ESI on pages S15–S17†).17 Gratifyingly, this strategy provides an excellent yield (>95%) when using a combination of AscH2, MPAA-PEG, MPA-PEG as reductants and HAT donors, respectively, and [Ru(bpy)3]Cl2 as a photocatalyst (Table 2, entry 7),22 and 35% enantiomeric excess of amine (R)-2e was obtained (entry 8). The moderate enantioselectivity suggests that oxidation of (S)-2e by MAO-N-9 is still slow, presumably due to the relatively low oxygen concentration caused by both the photocatalyst and the direct aerobic oxidation of thiols.
Conclusions
In summary, this study provides a proof-of-concept of asymmetric catalysis that results from combining photoredox and biocatalysis in a cyclic reaction network. Thanks to their exquisite catalytic properties, enzymes offer an attractive means to complement previously explored strategies for performing asymmetric synthesis under photoredox conditions. The light-driven reduction of aliphatic and aromatic imines to (racemic) amines in aqueous solution is an important result in itself, because the rapid interception of carbon-centered radical intermediates by HAT is non-trivial. Depending on the substrate type, the combination of ascorbic acid with aliphatic or aromatic thiol additives to enable polarity-matched catalysis for HAT is required. The cooperation between photoredox and enzymatic catalysis for light-driven asymmetric catalysis was first demonstrated for aliphatic and aromatic cyclic imine to enantioenriched amine synthesis. We anticipate that the concepts and methods summarized herein will be widely applicable to a range of other chemical transformations, including different photoredox reactions and biocatalysts.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Funding from the Swiss National Science Foundation through the NCCR Molecular Systems Engineering is gratefully acknowledged. Y. O. acknowledges a JSPS Overseas research fellowship. Prof. Nick Turner is thanked for providing us with the MAO-N-9 plasmid.Notes and references
- (a) K. Zeitler, Angew. Chem., Int. Ed., 2009, 48, 9785–9789 CrossRef PubMed; (b) J. M. R. Narayanam and C. R. J. Stephenson, Chem. Soc. Rev., 2011, 40, 102–113 RSC; (c) J. Xuan and W. J. Xiao, Angew. Chem., Int. Ed., 2012, 51, 6828–6838 CrossRef PubMed; (d) C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322–5363 CrossRef PubMed; (e) D. M. Schultz and T. P. Yoon, Science, 2014, 343, 1239176 CrossRef PubMed; (f) S. A. Morris, J. Wang and N. Zheng, Acc. Chem. Res., 2016, 49, 1957–1968 CrossRef PubMed; (g) T. P. Yoon, Acc. Chem. Res., 2016, 49, 2307–2315 CrossRef PubMed; (h) N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075–10166 CrossRef PubMed.
- (a) A. Bauer, F. Westkämper, S. Grimme and T. Bach, Nature, 2005, 436, 1139–1140 CrossRef PubMed; (b) R. Brimioulle, D. Lenhart, M. M. Maturi and T. Bach, Angew. Chem., Int. Ed., 2015, 54, 3872–3890 CrossRef PubMed; (c) H. Huo, X. Shen, C. Wang, L. Zhang, P. Röse, L.-A. Chen, K. Harms, M. Marsch, G. Hilt and E. Meggers, Nature, 2015, 515, 100–103 CrossRef PubMed; (d) H. Huo, C. Wang, K. Harms and E. Meggers, J. Am. Chem. Soc., 2015, 137, 9551–9554 CrossRef PubMed; (e) Q. M. Kainz, C. D. Matier, A. Bartoszewicz, S. L. Zultanski, J. C. Peters and G. C. Fu, Science, 2016, 351, 681–684 CrossRef PubMed.
- (a) D. A. Nicewicz and D. W. C. MacMillan, Science, 2008, 322, 77–80 CrossRef PubMed; (b) H.-W. Shih, M. N. Vander Wal, R. L. Grange and D. W. C. MacMillan, J. Am. Chem. Soc., 2010, 132, 13600–13603 CrossRef PubMed; (c) M. Neumann, S. Füldner, B. König and K. Zeitler, Angew. Chem., Int. Ed., 2011, 50, 951–954 CrossRef PubMed; (d) M. Cherevatskaya, M. Neumann, S. Füldner, C. Harlander, S. Kümmel, S. Dankesreiter, A. Pfitzner, K. Zeitler and B. König, Angew. Chem., Int. Ed., 2012, 51, 4062–4066 CrossRef PubMed; (e) Q. Yang, L. Zhang, C. Ye, S. Luo, L.-Z. Wu and C.-H. Tung, Angew. Chem., Int. Ed., 2017, 56, 3694–3698 CrossRef PubMed.
- D. A. DiRocco and T. Rovis, J. Am. Chem. Soc., 2012, 134, 8094–8097 CrossRef PubMed.
- K. T. Tarantino, P. Liu and R. R. Knowles, J. Am. Chem. Soc., 2013, 135, 10022–10025 CrossRef PubMed.
- J. Du, K. L. Skubi, D. M. Schultz and T. P. Yoon, Science, 2014, 344, 392–396 CrossRef PubMed.
- P. D. Morse, T. M. Nguyen, C. L. Cruz and D. A. Nicewicz, Tetrahedron, 2018 DOI:10.1016/j.tet.2018.03.052.
- (a) C. Moberg, Acc. Chem. Res., 2016, 49, 2736–2745 CrossRef PubMed; (b) M. Hall and A. S. Bommarius, Chem. Rev., 2011, 111, 4088–4110 CrossRef PubMed; (c) A. D. Lackner, A. V. Samant and F. D. Toste, J. Am. Chem. Soc., 2013, 135, 14090–14093 CrossRef PubMed.
- (a) M. Alexeeva, A. Enright, M. J. Dawson, M. Mahmoudian and N. J. Turner, Angew. Chem., Int. Ed., 2002, 41, 3177–3180 CrossRef; (b) R. Carr, M. Alexeeva, M. J. Dawson, V.-G. Fernández, C. E. Humphrey and N. J. Turner, ChemBioChem, 2005, 6, 637–639 CrossRef PubMed; (c) C. J. Dunsmore, R. Carr, T. Fleming and N. J. Turner, J. Am. Chem. Soc., 2006, 128, 2224–2225 CrossRef PubMed; (d) N. J. Turner, Chem. Rev., 2011, 111, 4073–4087 CrossRef PubMed; (e) J. M. Foulkes, K. J. Malone, V. S. Coker, N. J. Turner and J. R. Lloyd, ACS Catal., 2011, 1, 1589–1594 CrossRef; (f) R. S. Heath, M. Pontini, S. Hussain and N. J. Turner, ChemCatChem, 2015, 8, 117–120 CrossRef; (g) G. A. Aleku, J. Mangas-Sanchez, J. Citoler, S. P. France, S. L. Montgomery, R. S. Heath, M. P. Thompson and N. J. Turner, ChemCatChem, 2018, 10, 515–519 CrossRef.
- (a) V. Köhler, Y. M. Wilson, M. Dürrenberger, D. Ghislieri, E. Churakova, T. Quinto, L. Knörr, D. Häussinger, F. Hollmann, N. J. Turner and T. R. Ward, Nat. Chem., 2013, 5, 93–99 CrossRef PubMed; (b) Y. Okamoto, V. Köhler and T. R. Ward, J. Am. Chem. Soc., 2016, 138, 5781–5784 CrossRef PubMed.
- G. Knör, Coord. Chem. Rev., 2016, 325, 102–115 CrossRef.
- (a) J. A. Maciá-Agulló, A. Corma and H. Garcia, Chem.–Eur. J., 2015, 21, 10940–10959 CrossRef PubMed; (b) J. H. Park, S. H. Lee, G. S. Cha, D. S. Choi, D. H. Nam, J. H. Lee, J.-K. Lee, C.-H. Yun, K. J. Jeong and C. B. Park, Angew. Chem., Int. Ed., 2015, 54, 969–973 CrossRef PubMed; (c) M. Mifsud, S. Gargiulo, S. Iborra, I. W. C. E. Arends, F. Hollmann and A. Corma, Nat. Commun., 2014, 5, 3145 Search PubMed; (d) A. K. Mengele, G. M. Seibold, B. J. Eikmanns and S. Rau, ChemCatChem, 2017, 9, 4369–4376 CrossRef.
- M. A. Emmanuel, N. R. Greenberg, D. G. Oblinsky and T. K. Hyster, Nature, 2016, 540, 414–417 CrossRef PubMed.
- J.-H. Xie, S.-F. Zhu and Q.-L. Zhou, Chem. Rev., 2011, 111, 1713–1760 CrossRef PubMed.
- (a) M. Orazietti, M. Kuss-Petermann, P. Hamm and O. S. Wenger, Angew. Chem., Int. Ed., 2016, 55, 9407–9410 CrossRef PubMed; (b) M. Kuss-Petermann, M. Orazietti, M. Neuburger, P. Hamm and O. S. Wenger, J. Am. Chem. Soc., 2017, 139, 5225–5232 CrossRef PubMed; (c) J. Nomrowski and O. S. Wenger, J. Am. Chem. Soc., 2018, 140, 5343–5346 CrossRef PubMed.
- (a) D. Hager and D. W. C. MacMillan, J. Am. Chem. Soc., 2014, 136, 16986–16989 CrossRef PubMed; (b) A. J. Musacchio, B. C. Lainhart, X. Zhang, S. G. Naguib, T. C. Sherwood and R. R. Knowles, Science, 2017, 355, 727–730 CrossRef PubMed; (c) K. A. Margrey and D. A. Nicewicz, Acc. Chem. Res., 2016, 49, 1997–2006 CrossRef PubMed; (d) F. Wu, L. Wang, J. Chen, D. A. Nicewicz and Y. Huang, Angew. Chem., Int. Ed., 2018, 57, 2174–2178 CrossRef PubMed; (e) X. Hu, G. Zhang, F. Bu and A. Lei, ACS Catal., 2017, 7, 1432–1437 CrossRef.
- (a) X. Guo and O. S. Wenger, Angew. Chem., Int. Ed., 2018, 57, 2469–2473 CrossRef PubMed; (b) Y. Y. Loh, K. Nagao, A. J. Hoover, D. Hesk, N. R. Rivera, S. L.Colletti, I. W. Davies and D. W. C. MacMillan, Science, 2017, 358, 1182–1187 CrossRef PubMed; (c) R. Alam and G. A. Molander, Org. Lett., 2018, 20, 2680–2684 CrossRef PubMed.
- (a) B. P. Roberts and A. J. Steel, J. Chem. Soc., Perkin Trans. 2, 1994, 2, 2155–2162 RSC; (b) B. P. Roberts, Chem. Soc. Rev., 1999, 28, 25–35 RSC; (c) C. Le, Y. Liang, R. W. Evans, X. Li and D. W. C. MacMillan, Nature, 2017, 547, 79–83 CrossRef PubMed.
- (a) R. M. Haak, F. Berthiol, T. Jerphagnon, A. J. A. Gayet, C. Tarabiono, C. P. Postema, V. Ritleng, M. Pfeffer, D. B. Janssen, A. J. Minnaard, B. L. Feringa and J. G. de Vries, J. Am. Chem. Soc., 2008, 130, 13508–13509 CrossRef PubMed; (b) F. G. Mutti, A. Orthaber, J. H. Schrittwieser, J. G. de Vries, R. Pietschnig and W. Kroutil, Chem. Commun., 2010, 46, 8046–8048 RSC.
- D. D. M. Wayner, J. J. Dannenberg and D. Griller, Chem. Phys. Lett., 1986, 131, 189–191 CrossRef.
- (a) K. A. King, P. J. Spellane and R. J. Watts, J. Am. Chem. Soc., 1985, 107, 1431–1432 CrossRef; (b) L. Flamigni, A. Barbieri, C. Sabatini, B. Ventura and F. Barigelletti, Top. Curr. Chem., 2007, 281, 143–203 CrossRef.
- The ground state reduction potential of [Ru(bpy)3]Cl2 (Ered = −1.33 V vs. SCE) is sufficient to reduce the aromatic imine substrate 1e (Ep = −1.27 V vs. SCE, see ESI† page S30).
Footnote |
| † Electronic supplementary information (ESI) available: Complete experimental details, additional reference experiments, spectroscopic and electrochemical results. See DOI: 10.1039/c8sc01561a |
| This journal is © The Royal Society of Chemistry 2018 |