DOI:
10.1039/C8SC01494A
(Edge Article)
Chem. Sci., 2019,
10, 284-292
Highly efficient gene release in spatiotemporal precision approached by light and pH dual responsive copolymers†
Received
2nd April 2018
, Accepted 3rd October 2018
First published on 4th October 2018
Abstract
Triblock copolymer of poly(ethylene glycol)-b-poly(2-dimethylaminoethyl methacrylate)-b-poly(pyrenylmethyl methacrylate) (PEG-b-PDMAEMA-b-PPy) has been developed for use as an ideal gene delivery system, which showed both high stability under physiological conditions and efficient gene release in a mimetic cancer environment. The siRNA release from this system without external stimulation was 16% in 1 h and then remained steady. However, the photo-triggered siRNA release was 78% within 1 h and was higher than 91% after 24 h. The remarkable contrast between the stability and release efficiency of these siRNA-condensed micelleplexes before and after photo-irradiation has been rationalized by the light- and pH-induced structural transitions of the triblock copolymer micelles. The negligible cytotoxicity, high cellular uptake efficiency, and remarkable knockdown efficiency shown in in vitro tests further revealed the promising potential of these triblock copolymer micelleplexes for use in stimuli-responsive gene therapy.
Introduction
Small-interfering RNA (siRNA) works in an efficacious manner like RNA interference (RNAi) in gene therapy, and therefore, it has been applied in cancer treatments by activating RNA-induced silencing complex (RISC) to degrade the targeted messenger RNA (mRNA) in cytoplasm.1,2 Gene delivery systems using polymeric materials to protect siRNA from degradation by serum and endonuclease have been investigated in the past decade.3–6 In particular, micelleplexes, nanoparticles with positively charged clusters binding to genes, have attracted considerable interest due to their efficient encapsulation, high stability, and low toxicity.7,8 However, the strengthened affinity between siRNA and the carriers leads to insufficient siRNA release in the cytoplasm, and therefore, ineffective activation of RISC.9
To overcome the limitation because of which the enhanced stability of micelleplexes is always accompanied by the lowered efficiency of gene release, approaches based on introducing stimuli responsiveness in the hydrophobic core of micelleplexes were proposed to promote gene release by external stimulation, while the shielding function is still retained in the physiological environment.7 In particular, environmental changes such as pH,10–12 redox potential,13–15 light irradiation,16 sonication,17 and even various biological signals18,19 have been applied to trigger stimuli-responsive gene delivery systems. Photo-responsive systems have received increased attention due to their operational convenience and spatiotemporal precision.20 Most of the photo-responsive drug delivery systems utilize photo-labile groups such as pyrene,21o-nitrobenzyl,22 coumarin,23 and other derivatives24 to absorb the irradiation and elevate the release efficiency of drugs via structural destabilization. However, no efficient design has been designed that can fully unpack the encapsulated siRNA. Therefore, an improved gene delivery system that can liberate siRNA in spatiotemporal precision through charged repulsion upon light stimulus is of considerable interest.25–29
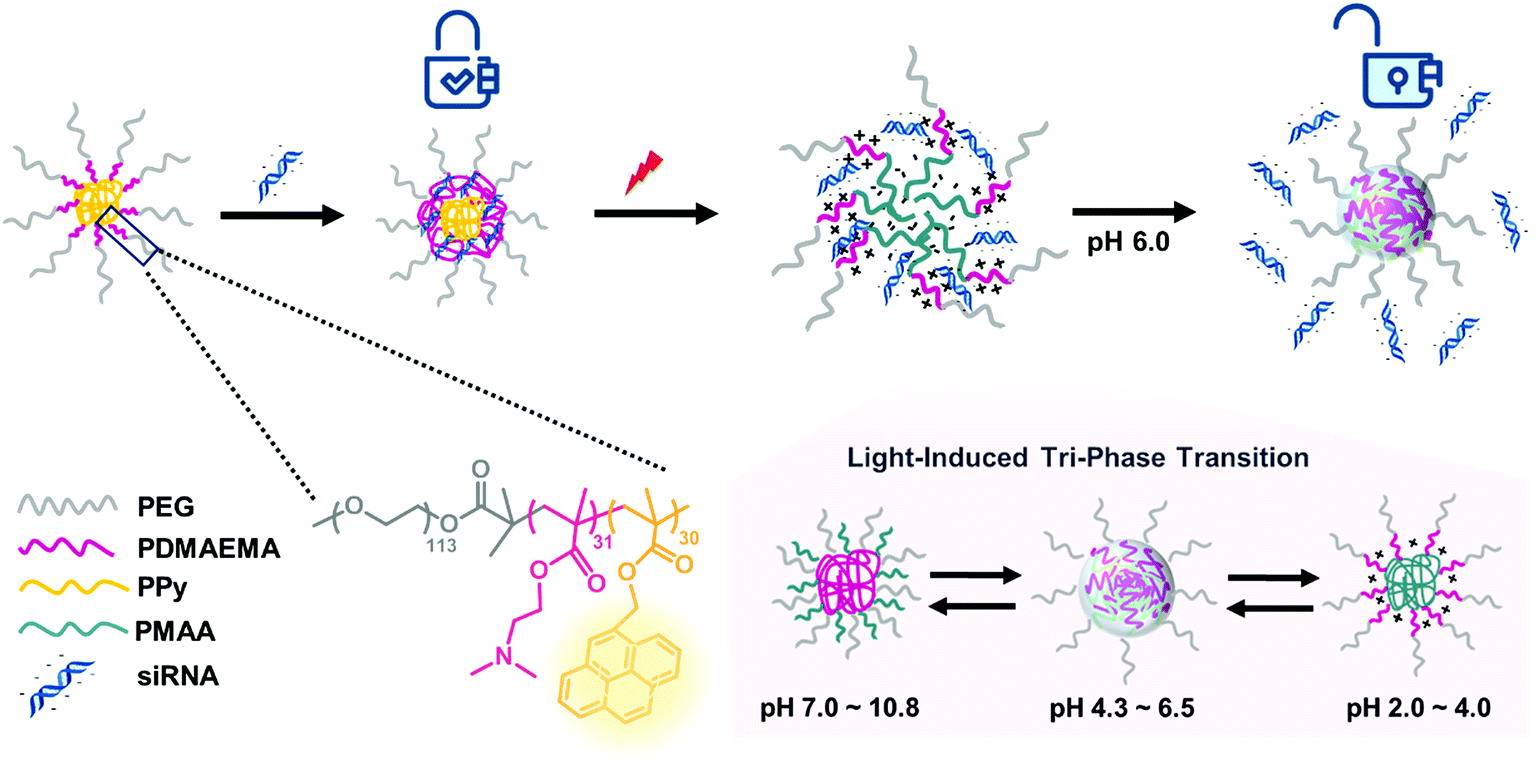
In our group, a triblock copolymer of poly(ethylene glycol)-b-poly(2-dimethylaminoethyl methacrylate)-b-poly(pyrenylmethyl methacrylate) (PEG-b-PDMAEMA-b-PPy) has been designed as the siRNA carrier. The PPy segment was proposed to play the role of triggering siRNA release by disassembling the micelleplexes via the transformation from PPy to poly(methacrylic acid) (PMAA) after light irradiation. In an acidic environment, the PDMAEMA with positive charges can bind to the siRNA using electrostatic interactions. The third segment of the PEG chain provides the biocompatibility for the micelleplexes. This triblock copolymer demonstrates high siRNA condensation efficiency (above 90%) as long as the N/P ratio approaches 5. More importantly, the siRNA carriers formed by the triblock copolymers exhibit increased stability with only 16% cumulative siRNA release within 1 h without irradiation, whereas the photo-triggered releasing efficiency was 78% within 1 h and 91% after 24 h. The substantial increase in the release efficiency before and after the photo-triggering treatment indicated that the triblock copolymer designed in this study is an ideal candidate for gene therapy and can be explained by the combined effects of the photo-triggered structural transformation from PPy to PMAA and tri-phase transition under different pH environments (Scheme 1).
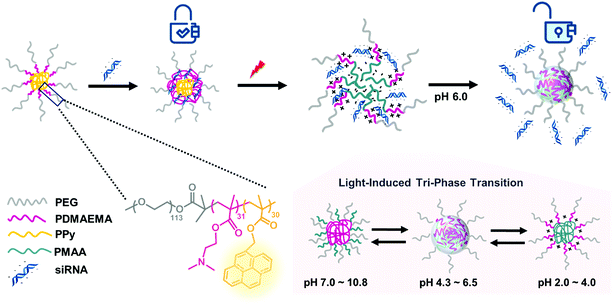 |
| | Scheme 1 Illustration of photo-responsive micelles binding to and releasing siRNA by structural transitions in response to light and pH stimuli. | |
Results and discussion
Synthesis and characterization of PEG-b-PDMAEMA-b-PPy triblock copolymer
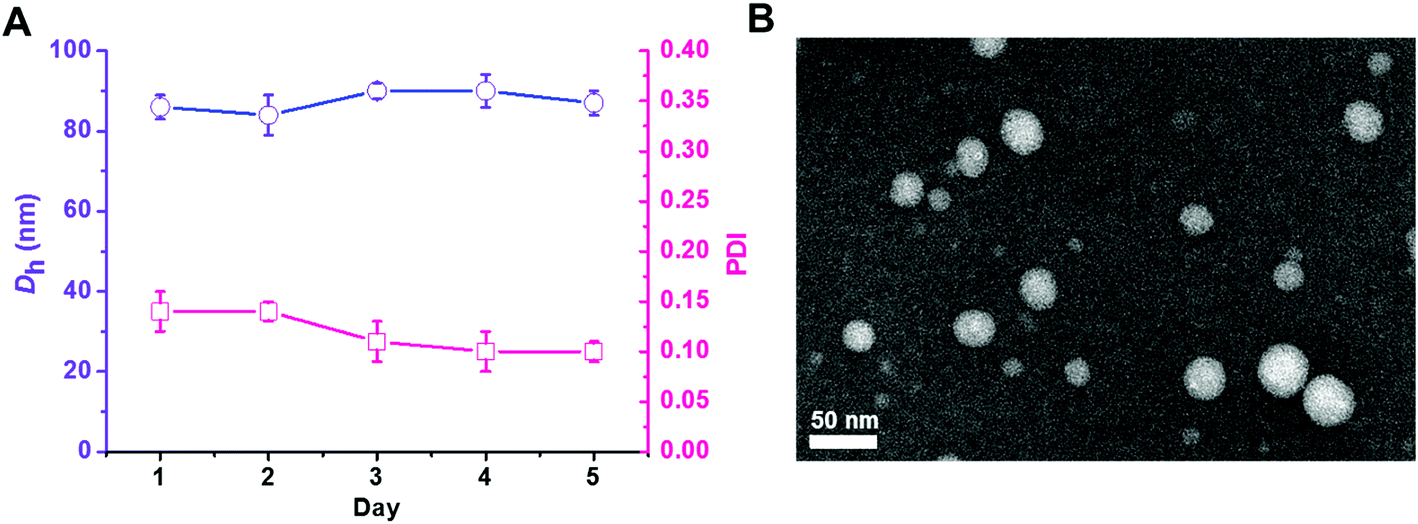
The triblock copolymer PEG113-b-PDMAEMA31-b-PPy30 was prepared by the sequential chain extension from the macro-initiator of PEG113-Br (Mn = 5000 g mol−1, Đ = 1.03) to PDMAEMA (Mn = 9900 g mol−1, Đ = 1.27) and then PPy (Mn = 18![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 900 g mol−1, Đ = 1.38) using the ATRP technique (Table 1).30 The dynamic laser scattering (DLS) analysis revealed that the self-assembly of PEG113-b-PDMAEMA31-b-PPy30 triblock copolymer (50 μg mL−1), which was conducted by typical nanoprecipitation under sonication,31 exhibited a light-intensity-average hydrodynamic diameter of 83 nm with uniform dispersity of 0.11 and retained homogeneous particle dispersity (PDI < 0.14) with a size of around 85 nm in 5 days at 25 °C (Fig. 1A). The high colloidal stability was attributed to the π–π stacking interactions between the pyrene molecules inside the hydrophobic core, which was supported by the transmission electron microscopy (TEM) image. The nano-assemblies negatively stained by phosphotungstic acid (PTA) exhibited a spherical shape with a white core of approximately 40 nm in diameter (Fig. 1B), indicating the formation of core–corona micelles with a hydrophobic core formed by PPy stacking and hydrophilic outer spherical surface composed of PEG and PDMAEMA blocks. The different particle sizes of PEG113-b-PDMAEMA31-b-PPy30 observed by DLS analysis and TEM images could be attributed to the shrinkage of PEG and PDMAEMA segments when water was removed. This is because the DLS analysis was performed in a water solution, whereas the TEM sample was air-dried. Other PEG-b-PDMAEMA-b-PPy block copolymers with various chain lengths of PDMAEMA and PPy were synthesized via a similar process for the optimization of siRNA condensation efficiency (Table S1†).
900 g mol−1, Đ = 1.38) using the ATRP technique (Table 1).30 The dynamic laser scattering (DLS) analysis revealed that the self-assembly of PEG113-b-PDMAEMA31-b-PPy30 triblock copolymer (50 μg mL−1), which was conducted by typical nanoprecipitation under sonication,31 exhibited a light-intensity-average hydrodynamic diameter of 83 nm with uniform dispersity of 0.11 and retained homogeneous particle dispersity (PDI < 0.14) with a size of around 85 nm in 5 days at 25 °C (Fig. 1A). The high colloidal stability was attributed to the π–π stacking interactions between the pyrene molecules inside the hydrophobic core, which was supported by the transmission electron microscopy (TEM) image. The nano-assemblies negatively stained by phosphotungstic acid (PTA) exhibited a spherical shape with a white core of approximately 40 nm in diameter (Fig. 1B), indicating the formation of core–corona micelles with a hydrophobic core formed by PPy stacking and hydrophilic outer spherical surface composed of PEG and PDMAEMA blocks. The different particle sizes of PEG113-b-PDMAEMA31-b-PPy30 observed by DLS analysis and TEM images could be attributed to the shrinkage of PEG and PDMAEMA segments when water was removed. This is because the DLS analysis was performed in a water solution, whereas the TEM sample was air-dried. Other PEG-b-PDMAEMA-b-PPy block copolymers with various chain lengths of PDMAEMA and PPy were synthesized via a similar process for the optimization of siRNA condensation efficiency (Table S1†).
Table 1 Characterization of PEG-b-PDMAEMA and PEG-b-PDMAEMA-b-PPy copolymers
| Entrya |
M
n,NMR
(g mol−1) |
Đ
|
Sizee |
|
Degree of polymerization calculated by 1H-NMR.
Molecular weight determined by 1H-NMR.
Polydispersity measured by GPC using DMF/LiBr as the eluent.
Polydispersity measured by GPC using THF as the eluent.
Particle size distribution measured by dynamic laser scattering (DLS, Malvern Nano S), PDI = (σ/d)2, whereas σ is the standard deviation and d is the measured diameter. The DLS analysis of micelles (50 μg mL−1) was conducted in triplicate.
|
| PEG113-Br |
5000 |
1.03 |
— |
| PEG113-b-PDMAEMA31 |
9900 |
1.27c |
— |
| PEG113-b-PDMAEMA31-b-PPy30 |
18900 |
1.38d |
83 |
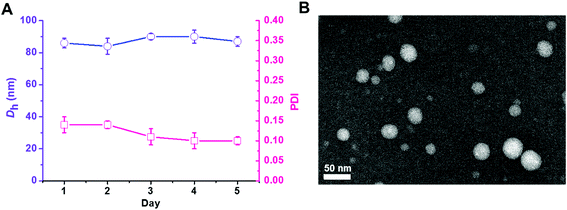 |
| | Fig. 1 Self-assembling of PEG113-b-PDMAEMA31-b-PPy30 triblock copolymer. (A) Time-dependent hydrodynamic diameter (Dh) and particle dispersity (PDI) of PEG113-b-PDMAEMA31-b-PPy30 self-assemblies (50 μg mL−1) measured by dynamic light scattering (DLS) in distilled water. (B) Transmission electron microscope (TEM) image of PEG113-b-PDMAEMA31-b-PPy30 self-assemblies solution (0.5 mg mL−1) cast on carbon/Formvar-coated copper TEM grid and negatively stained by 1% phosphotungstic acid (PTA). | |
Stimuli-responsiveness of PEG-b-PDMAEMA-b-PPy triblock copolymer
The triblock copolymer of PEG113-b-PDMAEMA31-b-PPy30 was expected to be responsive to a change in the pH value because the PDMAEMA segment is a weak polyelectrolyte with pH responsiveness.32–34 The effective pKa value was determined to be 6.76 by titrating PEG113-b-PDMAEMA31-b-PPy30 (50 μg mL−1 in water) with 0.1 M NaOH solution, which falls within the pH range with a weak buffer capacity (Fig. S5†). The pH-responsive behavior of PEG113-b-PDMAEMA31-b-PPy30 was characterized by the observed change in the particle size, which increased from 70 nm to 110 nm when the pH decreased and was most pronounced when the pH value dropped below the pKa value (Fig. S6A†). This phenomenon could be explained by the electrostatic repulsion between the protonated amines of the PDMAEMA block that leads to the increase in the particle size. This fact was further supported by the zeta potential analysis, where the surface charge of the self-assembled PEG113-b-PDMAEMA31-b-PPy30 micelles was approximately 14 mV when pH was above pKa, but it increased drastically to 24 mV once the pH value decreased below pKa (Fig. S6B†).35 Collectively, another contributing factor for the size change could be associated with the increased osmotic pressure.33
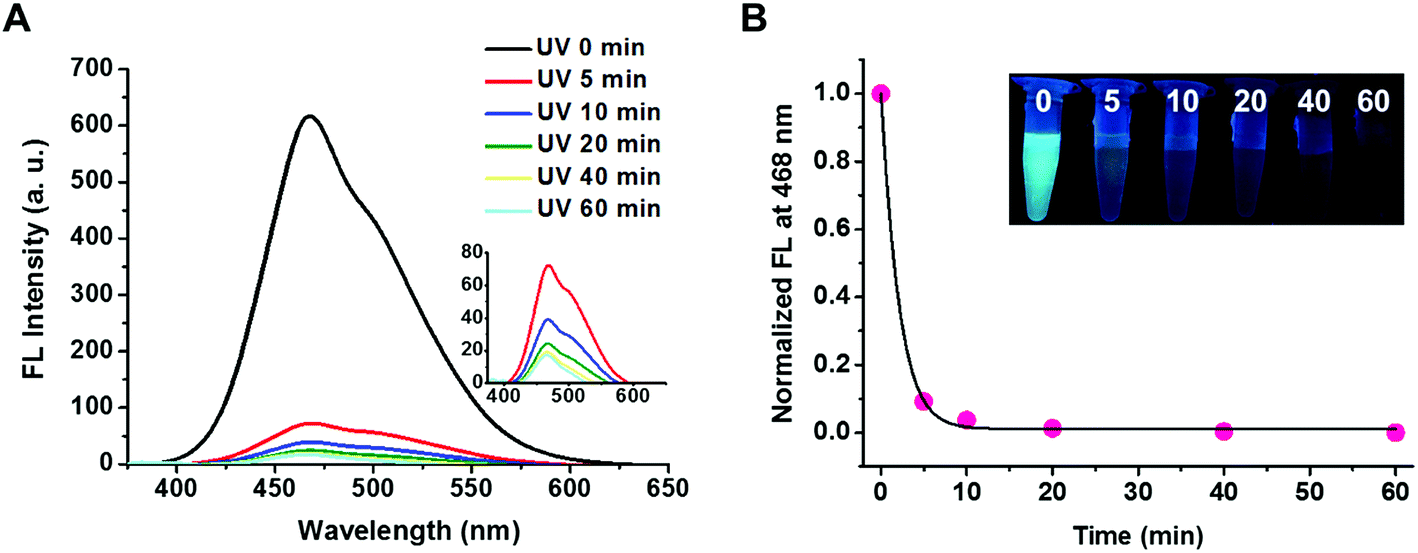
The photo-responsiveness of PEG113-b-PDMAEMA31-b-PPy30 was first characterized by the electronic absorption and emission spectrum. UV light with a wavelength of 365 nm can cleave the ester bond on the PPy block, thereby converting poly(pyrenylmethyl methacrylate) to poly(methacrylic acid) (PMAA). The characteristic absorption bands of the pyrene pendant was identified at a wavelength above 300 nm by using the UV-vis spectroscopy.21 The fluorescence spectrum with excitation at 365 nm revealed the typical peak for excimers at 468 nm within the range from 400 to 600 nm (Fig. S7†), which was attributed to the stacking of pyrene molecules in a constrained space.36 The fluorescent intensity of PEG113-b-PDMAEMA31-b-PPy30 at 468 nm decreased as the UV irradiation time increased (Fig. 2A) and dropped rapidly to 4% of the original intensity within 10 min, after which it stayed relatively constant (Fig. 2B). As a result, the cleavage of the photo-labile ester bond on the PPy segment was considered efficient. In addition, the water insolubility of the detached pyrene derivatives, mainly pyrenemethanol, also contributed to the drastic reduction in the excimer fluorescence, as well as the absence of monomeric pyrene emissions. This fact was supported by conducting a control study using the THF/water (v/v = 1/1) co-solvent instead of DI water, where the fluorescent intensity of PEG113-b-PDMAEMA31-b-PPy30 at 468 nm decreased slower in an increased stepwise manner with irradiation time. The corresponding peak for monomeric pyrene at 393 nm was also observed after UV irradiation (Fig. S8†).
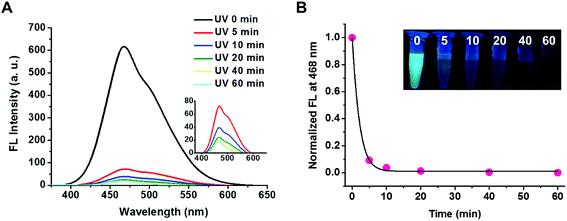 |
| | Fig. 2 PEG113-b-PDMAEMA31-b-PPy30 in distilled water upon UV irradiation (λ = 365 nm). (A) Time-dependent fluorescence spectra of PEG113-b-PDMAEMA31-b-PPy30. (B) Normalized time-dependent fluorescent intensity at 468 nm of PEG113-b-PDMAEMA31-b-PPy30. Inset: visual expressions of blue fluorescence decreasing upon UV irradiation. | |
The photo-responsiveness of PEG113-b-PDMAEMA31-b-PPy30 caused by the cleavage of PPy ester bond affects not only the fluorescent intensity but also the nanostructure. The particle size of PEG113-b-PDMAEMA31-b-PPy30 micelles in water decreased from 88 nm to 65 nm in diameter within 10 min under UV irradiation and subsequently reaching 60 nm in 30 min (Fig. S9†). The consistent light scattering intensity indicated that this structural transformation occurred between two stable phases, which will be discussed later. The significant changes in the fluorescent intensity and particle size both occurred within 10 min under UV irradiation, suggesting that 10 min of UV irradiation was sufficient to convert PPy to PMAA segments along with the disassembly of the hydrophobic core. PMAA formation was then confirmed using the titration curve with NaOH solution as the titrant (Fig. S10†). The enhanced buffering capacity of the triblock copolymer after UV irradiation was attributed to PMAA, a weak electrolyte with a pKa value of 5.5,37 which was converted from PPy after light irradiation.
Condensation and release of siRNA with micelles
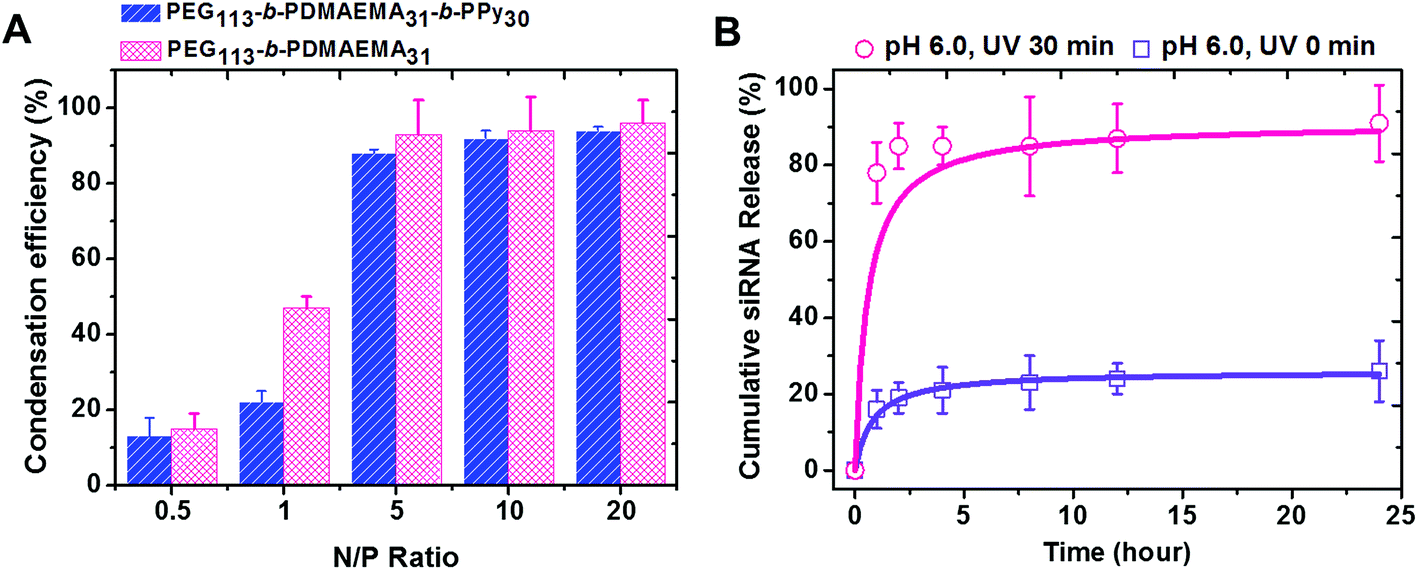
PEG-b-PDMAEMA-b-PPy self-assembled micelles were expected to efficiently carry the siRNA via the electrostatic interaction between positively charged amine groups on the PDMAEMA segment and negatively charged phosphate groups on the siRNA. The ratio of amino groups to the phosphate groups was defined as the N/P ratio, which was used to assess the condensation efficiency of the designed siRNA carrier.38,39 The solutions with different concentrations of PEG-b-PDMAEMA-b-PPy micelles were added into the siRNA solution at the stock concentration to yield micelleplexes with different N/P ratios. The diblock copolymer of PEG-b-PDMAEMA that was reported to show high siRNA condensation efficiency was also used to formulate the samples as a control study.39 The siRNA condensation efficiency of these samples was then evaluated by using ethidium bromide assay (EBA), as shown in Fig. S11.†28 It was clear that more siRNA is wrapped inside the triblock and diblock copolymers when the N/P ratio increased, where the efficiency reached a plateau around 90% when N/P = 5. Once the N/P ratio increases beyond 5, the siRNA condensation efficiency of PEG-b-PDMAEMA-b-PPy and PEG-b-PDMAEMA became fairly comparable. Since the influence of the chain length of the triblock copolymers to siRNA condensation efficiency is still ambiguous, PEG113-b-PDMAEMA31-b-PPy30 was, therefore, selected as the model carrier in this study (Fig. 3A). A decrease in the particle size from 87 nm to 72 nm was observed after siRNA condensation (Fig. S12†) and can be attributed to the electrostatic interaction between the PDMAEMA segment and siRNA, which contributed to the contraction of the PDMAEMA shell (Scheme S3A†). The low N/P ratio and comparable condensation efficiency of siRNA to PEG113-b-PDMAEMA31 demonstrated that the PEG113-b-PDMAEMA31-b-PPy30 could be the ideal carrier for siRNA delivery.
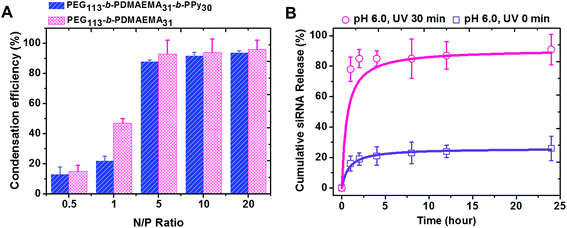 |
| | Fig. 3 (A) siRNA condensation efficiency of PEG113-b-PDMAEMA31 and PEG113-b-PDMAEMA31-b-PPy30 micelles determined by ethidium bromide assay at various N/P ratios. (B) Cumulative siRNA release of micelleplexes (N/P = 5) at pH 6.0 with and without light irradiation. | |
The micelleplexes formed by siRNA and PEG113-b-PDMAEMA31-b-PPy30 showed considerable stability with high condensation efficiency in the stock solution for 1 week (Fig. S13†) and low siRNA release efficiency in distilled water (<10% of the siRNA amount released in 24 h, Fig. S14†). This could be explained by the existence of the hydrophobic core and the strong ionic interaction between siRNA and PDMAEMA. To assess siRNA delivery, the cumulative siRNA release efficiency of PEG113-b-PDMAEMA31-b-PPy30 micelleplexes was further evaluated in a phosphate buffer solution under a controlled pH environment. At pH 6.0, the micelleplexes released around 16% siRNA within 1 h, and then, the cumulative release efficiency reached a plateau at 26% after 24 h (Fig. 3B). When the pH was increased to 7.4, the releasing rate approached 34% in 24 h (Fig. S14†) because the environment of higher pH value reduces the protonation of PDMAEMA, which, in turn, weakens the binding between siRNA and the triblock copolymer. Nevertheless, the micelleplexes of PEG113-b-PDMAEMA31-b-PPy30 and siRNA still effectively minimize the uncontrolled release of siRNA,7,38,40,41 and therefore, should possess higher stability under physiological conditions. Contrary to general conditions in previous studies, wherein the siRNA carrier with high stability accompanies low release efficiency,9,42,43 the micelleplexes of PEG113-b-PDMAEMA31-b-PPy30 designed in this study allowed the highly efficient release of siRNA after 30 min of UV irradiation. In particular, the cumulative release of siRNA was 78% in 1 h and further approached 91% within 24 h (Fig. 3B), implying that the negatively charged carboxylic groups formed from the PPy segment after photo-degradation could contribute toward the release of encapsulated siRNA (Scheme S3B†). The significant contrast of the siRNA releasing rate before and after UV irradiation (26% versus 91%) guarantees the use of PEG113-b-PDMAEMA31-b-PPy30 as an ideal siRNA carrier for gene therapy.
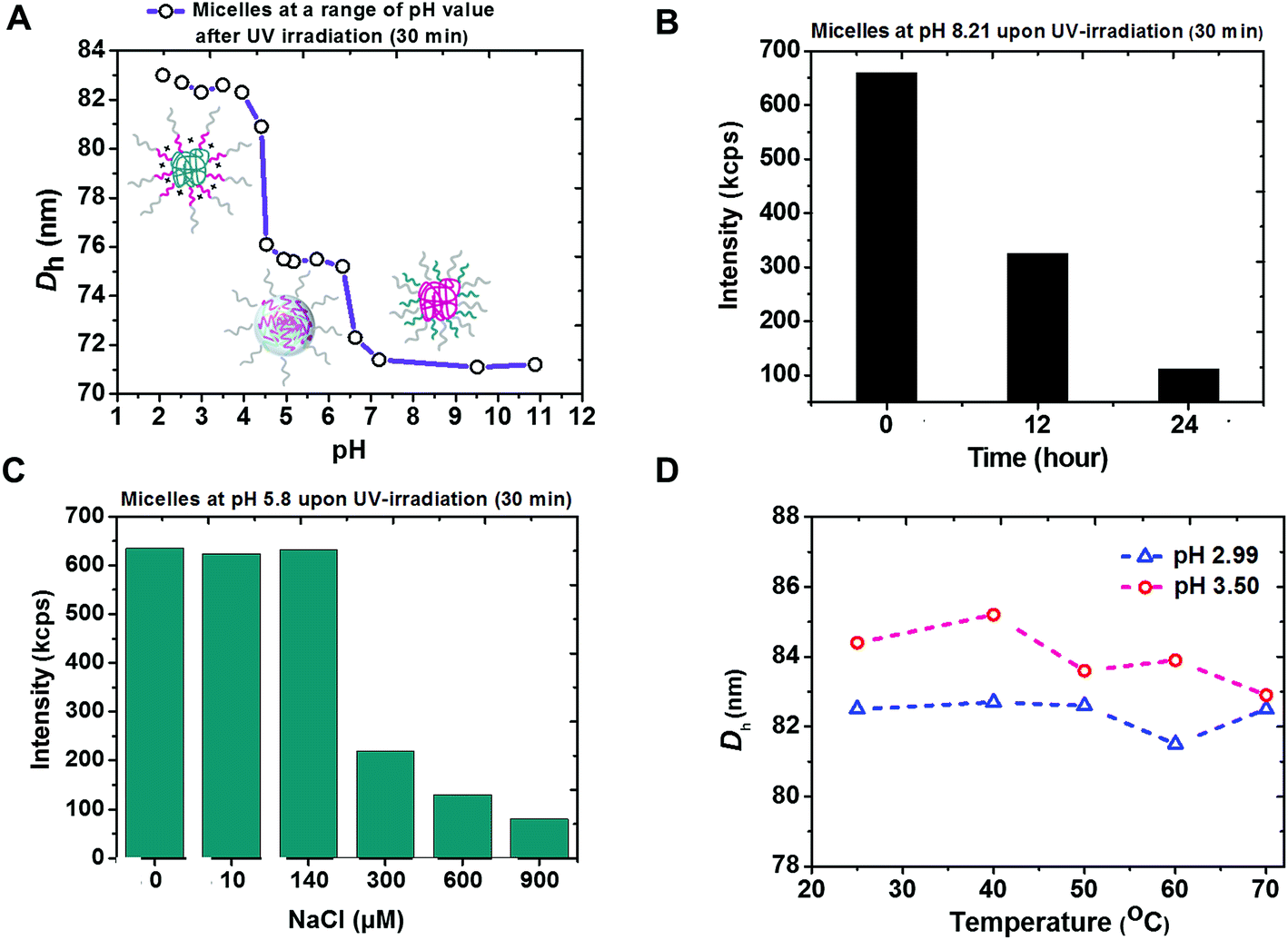
Tri-phase transition of PEG113-b-PDMAEMA31-b-PPy30 micelles
The coexistence of remarkably high stability and siRNA releasing rate shown by micelleplexes of PEG113-b-PDMAEMA31-b-PPy30 and siRNA was attributed to not only the effectively photo-triggered conversion from PPy to PMAA but also the phase transition of PEG113-b-PDMAEMA31-b-PMAA30 particles in different pH environments. According to the pH-dependent DLS measurement, three self-assembled morphologies with hydrodynamic diameters equal to 71 nm, 75 nm, and 83 nm were observed (Fig. 4A). At the basic condition, the particle was proposed to have a PDMAEMA core surrounded by PEG and PMAA chains. However, the decreased light intensity in DLS measurement at pH 8.2 (Fig. 4B) indicated the instability of these particles, possibly due to the weak hydrophobicity of unprotonated PDMAEMA44 and could be attributed to the observed smallest particle size. The attempt to further confirm the particle structure by recognizing the superficial PMAA chains using zeta potential measurements was not successful because of the unstable morphology.
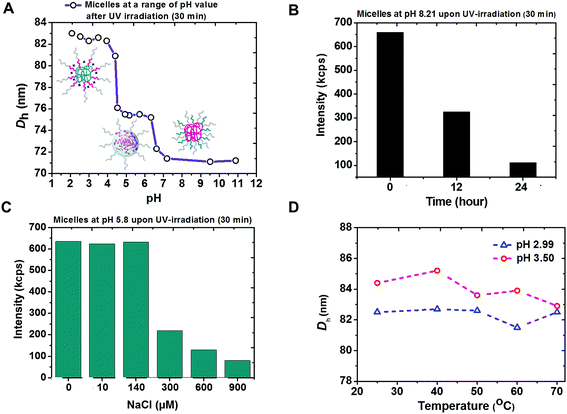 |
| | Fig. 4 Characterization of PEG113-b-PDMAEMA31-b-PMAA30 particles generated from PEG113-b-PDMAEMA31-b-PPy30via UV irradiation (30 min) in an aqueous solution. (A) Change in light-intensity-average hydrodynamic diameter of PEG113-b-PDMAEMA31-b-PMAA30 under different pH environments. (B) Decrease in the light-scattering intensity of PEG113-b-PDMAEMA31-b-PMAA30 at pH 8.21 with time. (C) Variation in light-scattering intensity of PEG113-b-PDMAEMA31-b-PMAA30 with various ionic strengths at pH 5.8. (D) Effect of temperature on the size of PEG113-b-PDMAEMA31-b-PMAA30 particles at pH 2.99 (blue line) and pH 3.50 (red line). | |
The core of PEG113-b-PDMAEMA31-b-PMAA30 particle should be formed by positively charged PDMAEMA and negatively charged PMAA via intra-micellar electrostatic interactions (Scheme S3B†) to provide a consistent size of 75 nm at pH 4.3 to 6.5, which covers the range of pH 5.8 to 6.1 as the isoelectric point45 estimated by the measured pKa values of PDMAEMA and reported pKa values of PMAA.37 The low zeta potential (less than 5 mV, Fig. S15†) also supported the mutual shielding of cationic PDMAEMA and anionic PMAA. The structure of PDMAEMA and PMAA core surrounded by PEG was further confirmed by the chart of ionic-strength-dependent light scattering intensity (Fig. 4C). With the increasing ionic strength because of the addition of NaCl, the light scattering intensity showed a significant drop when the NaCl concentration reached 300 μM. This is because the electrostatic interaction between PDMAEMA and PMAA was susceptible to the high ionic strength that causes the disassembling of particles. The characterization of particle structure rationalized the burst release of siRNA as high as 91% at pH 6.0 because the photo-triggered conversion from PPy to PMAA not only removes the hydrophobic core to destabilize the triblock copolymer carrier but also forms the negatively charged carboxyl groups to knock-off the siRNA from PDMAEMA by inducing competition between the positively charged amine groups and siRNA.
When the pH went below 4.3, the self-assembled micelles of PEG113-b-PDMAEMA31-b-PMAA30 showed a sharp increase in the particle size, up to 83 nm. The most possible structure at this stage should be a PMAA core associated with the corona composed of PEG and PDMAEMA chains due to the hydrophobicity of the undissociated PMAA segment. The increased zeta potential indicated that the protonated PDMAEMA chains were located in the outer shell (Fig. S15†). Although the particle core formed by PEG and PMAA via the hydrogen bonding has also been reported,46 the consistence in the particle size under varying temperatures from 25 °C to 70 °C (Fig. 4D) excluded this possibility because the particles should have become unstable at higher temperatures if the hydrogen bond were the dominant interaction to form the micelles.
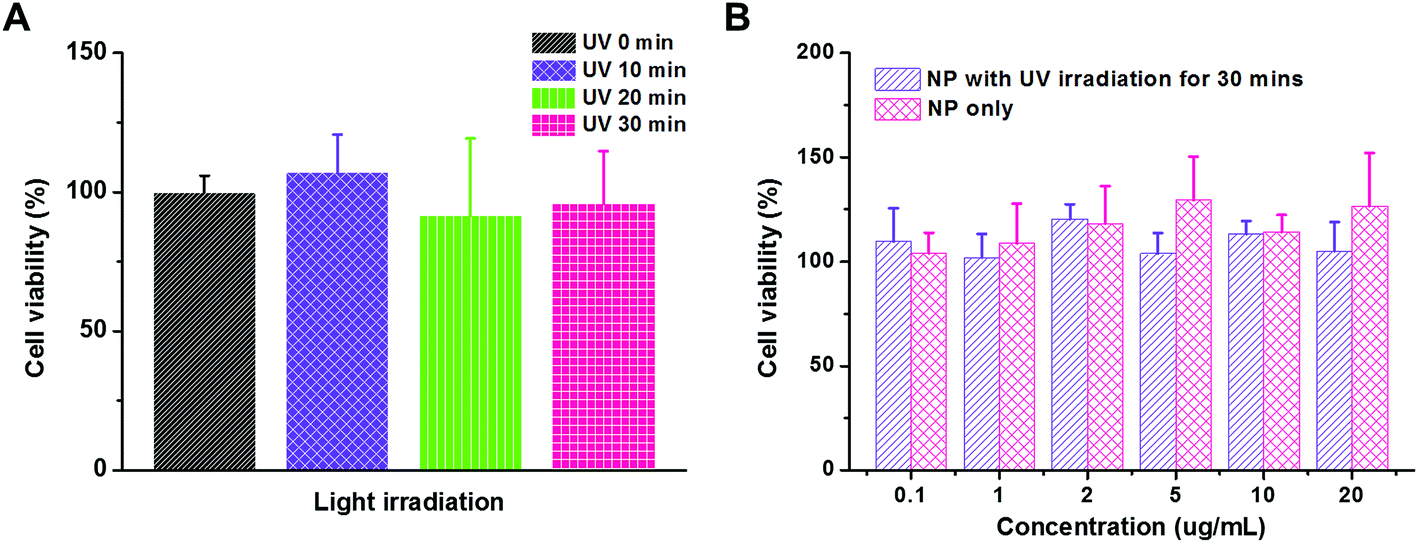
Material cytotoxicity
Cytotoxicity is a crucial issue in the development of a gene delivery system. For example, the systems with densely cationic charges that induce cell membrane disruption have shown significant cytotoxicity, and therefore, have limited applications.47 The in vivo experimental or clinical use of polyethylenimine, an effective transfection vector, is hindered due to its significant cytotoxicity, too.25 However, material cytotoxicity could be reduced by micellization or PEGylation that shields the cationic charges.40 In our study, both UV irradiation and functional groups on polymeric carriers would raise the concern of cytotoxicity. Therefore, we first evaluated the influence of UV irradiation to the cell viability using MTT assay (Fig. 5A). The MDA-MB-231 cancer cells were exposed to UV irradiation for 10, 20, and 30 min. As compared to the control study without UV irradiation, the cell viability was comparable in all the trials, which indicated that the cytotoxicity caused by the UV irradiation is negligible in our study. Then, the PEG113-b-PDMAEMA31-b-PPy30 self-assembled micelles were tested for their cytotoxicity (Fig. 5B). The range of micelle concentration was selected from 0.1 μg mL−1 to 20 μg mL−1 because 20 μg mL−1 of micelles with N/P = 5 is able to condense 160 nM of siRNA, which is much higher than the effective dose of siRNA (30 nM) for activating RISC in vitro.38 The MDA-MB-231 cancer cells were treated with the micelles of PEG113-b-PDMAEMA31-b-PPy30 for 6 h and then further incubated for another 24 h. According to the cell viability, neither the micelles nor the micelles exposed to UV irradiation significantly induced cytotoxicity, demonstrating that the micelles of PEG113-b-PDMAEMA31-b-PPy30 or PEG113-b-PDMAEMA31-b-PMAA30 and pyrenemethanol, the major products after irradiation, are not harmful to cells under the conditions of this study.
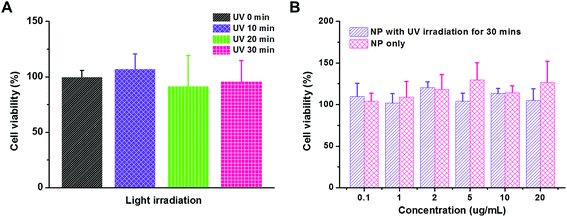 |
| | Fig. 5 (A) Cell viability of MDA-MB-231 cancer cells upon UV irradiation for 10, 20, and 30 min. (B) Material cytotoxicity of micelles with or without UV irradiation for 30 min. The MDA-MB-231 cancer cells were further incubated for 24 h and cell viability was evaluated using MTT assay. | |
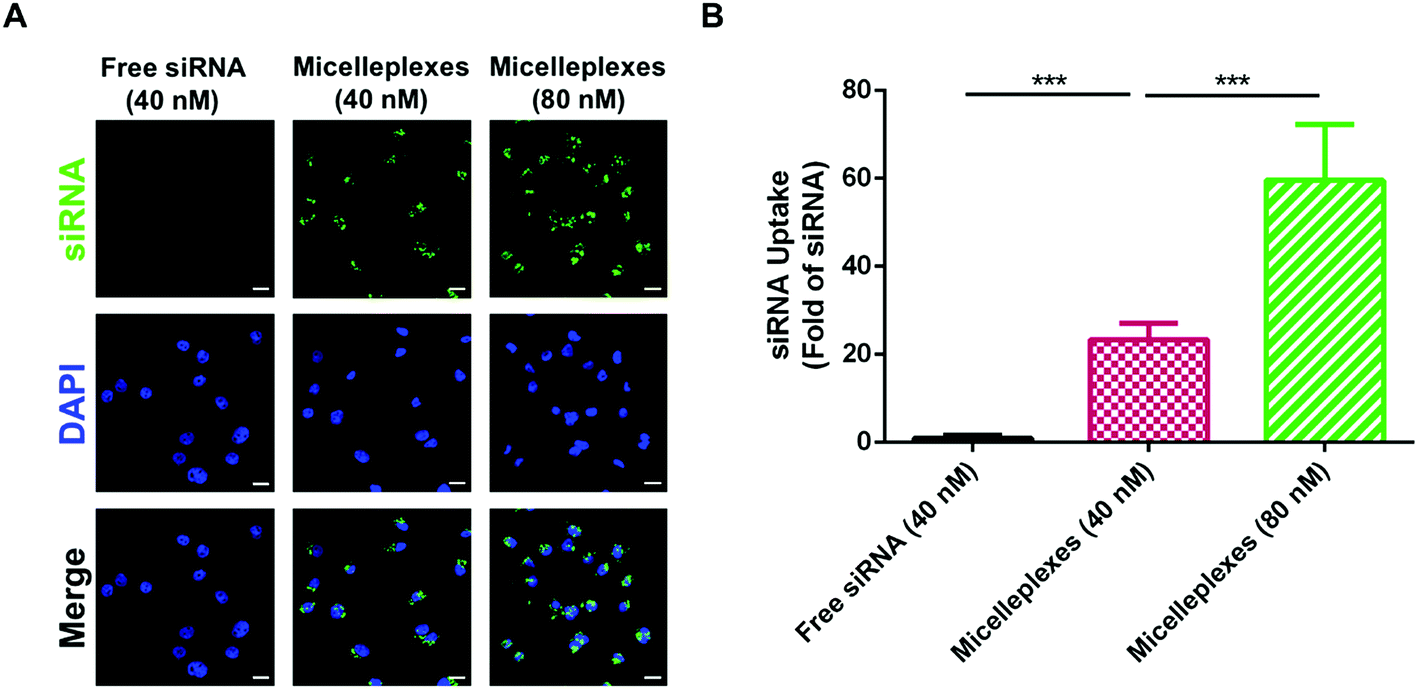
In vitro cellular uptake of micelleplexes
The cellular uptake of free siRNA and siRNA carried by PEG113-b-PDMAEMA31-b-PPy30 in MDA-MB-231 cells was performed by using FAM-labeled control siRNA with 4 h of cellular incubation and evaluated using the average optical density (AOD) calculations of FAM-labeled control siRNA, which was detected by using confocal laser scanning microscopy (CLSM) (Fig. 6A). As expected, free siRNA exhibited very low uptake efficiency because the electrostatic repulsion between the negatively charged cell membrane and siRNA could interfere with cellular internalization. In contrast, the negative charges of siRNA would be shielded by the complexation with triblock copolymers, and therefore, siRNA carried by micelleplexes demonstrated a significantly enhanced uptake efficiency in MDA-MB-231 cells, which was almost 23-fold higher than that of free siRNA. The increasing siRNA concentration from 40 nM to 80 nM could further elevate the uptake efficiency to 60 folds (Fig. 6B), revealing a proper correlation between the number of micelleplexes and the quantity of siRNA engulfed by cancer cells. The enhanced cellular internalization of siRNA carried by PEG113-b-PDMAEMA31-b-PPy30 should be rationalized by caveolar endocytosis, which was reported as the pathway for micelleplexes to be engulfed by the cells.38,40,48–50 Notably, 5 μg mL−1 of triblock copolymer micelles, which was demonstrated to be nontoxic to MDA-MB-231 cells, is sufficient to carry 40 nM of siRNA. Therefore, this condition was used in the evaluation of knockdown efficiency.
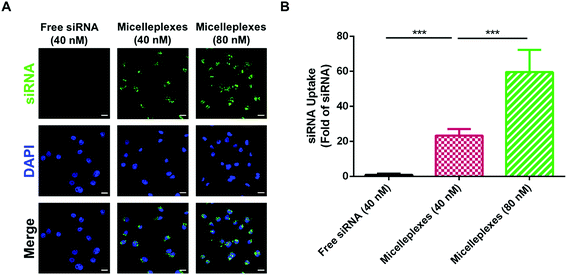 |
| | Fig. 6 Cellular uptake of FAM-labeled siRNA in MDA-MB-231 cancer cells. (A) Images observed by a Zeiss LSM 780 confocal microscope for MDA-MB-231 cells treated with 40 nM free siRNA, micelleplexes carrying 40 nM siRNA, and micelleplexes carrying 80 nM siRNA for 4 h of incubation. The FAM-labeled siRNA was represented in green and the nucleus stained by DAPI was represented in blue. Scale bar = 20 μm. (B) Quantitative data of cellular uptake of free siRNA and siRNA carried by micelleplexes evaluated by the average optical density (AOD) of FAM-labeled siRNA in confocal laser scanning microscopy (CLSM) images. Data are the mean values ± SEM; ***p < 0.001. | |
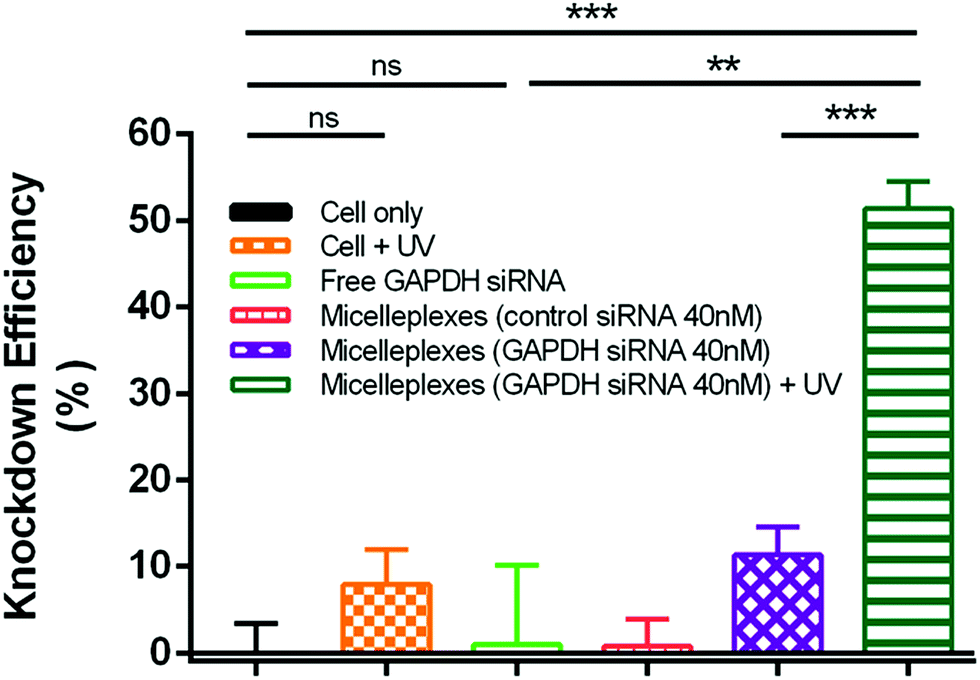
GAPDH knockdown efficiency
The silencing effect of siRNA delivered by PEG113-b-PDMAEMA31-b-PPy30 in MDA-MB-231 cells was evaluated by measuring the GAPDH enzyme activity with a KDalert GAPDH assay kit, which is assessed by the fluorescence increment with predetermined time in cell lysates. The knockdown efficiency was calculated by the ratio of the remaining expressions of GAPDH only between the conditions with a given transfection and the cell (Fig. 7). The detailed calculations were performed by following the reported method12,51–54 and shown in the Experimental section of the ESI.† As compared to the control study with only MDA-MB-231 cancer cells, the free GAPDH siRNA and control siRNA carried by micelleplexes showed almost no knockdown efficiency because free GAPDH siRNA could not penetrate the cell membrane and the control siRNA cannot target the GAPDH mRNA. Although UV treatment showed 8% knockdown efficiency in MDA-MB-231 cancer cells, this value is insignificantly different to that of the control study from the statistical viewpoint. The 40 nM of GAPDH siRNA delivered by micelleplexes showed 11% knockdown efficiency, possibly due to the leakage of siRNA from micelleplexes. However, the knockdown efficiency of micelleplexes with 40 nM of GAPDH siRNA approached 51% when the sample was treated with UV irradiation for 30 min after cellular internalization, indicating that UV irradiation successfully liberated the GAPDH siRNA from micelleplexes, and therefore, activating RISC to degrade the targeted GAPDH mRNA as indicated by the reduced GAPDH enzyme activity. The remarkable difference in the knockdown efficiency of GAPDH siRNA encapsulated in the micelleplexes before and after UV irradiation demonstrated that PEG113-b-PDMAEMA31-b-PPy30 can achieve effective gene delivery and photo-triggered gene release for cancer treatment in spatiotemporal precision.
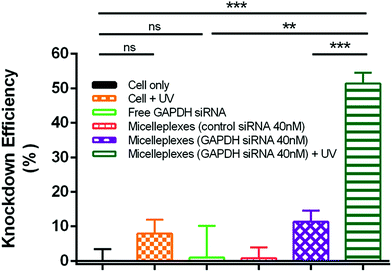 |
| | Fig. 7 Knockdown efficiency of GAPDH enzyme in MDA-MB-231 cells evaluated by using KDalert GAPDH assay after 4 h of cellular internalization and 44 h of incubation. All the experiments were executed in triplicate. Data are mean values ± SEM; **p < 0.01, ***p < 0.001, ns: not significant. | |
Conclusions
The function of each block in PEG113-b-PDMAEMA31-b-PPy30 and the mechanism of its burst siRNA release became evident after the characterization of particle structure before and after UV irradiation. The PEG segment simply provides water solubility. The protonated PDMAEMA was used to bind the siRNA via the ionic force at pH 6.0 to approach 90% condensation efficiency at N/P ratio as low as 5. The high stability of these siRNA-encapsulated micelleplexes was illustrated by a low leakage rate (26%) in 24 h and attributed to the stable hydrophobic core formed by PPy and the interaction between PDMAEMA and siRNA. The siRNA release was triggered by UV irradiation that converted PPy to PMAA. The formation of PMAA segments at pH 4.3 to 6.5 not only removed the hydrophobic core but also induced competition between the protonated amine groups and siRNA using the anionic carboxylic groups to push out the siRNA, causing an ultrahigh release efficiency of 91%. The in vitro tests indicated that PEG113-b-PDMAEMA31-b-PPy30 self-assembled micelles have shown negligible cytotoxicity before and after UV irradiation, as well as the micelleplexes of siRNA and PEG113-b-PDMAEMA31-b-PPy30 exhibit an enhanced siRNA uptake efficiency associated with high protein knockdown efficiency, which are crucial for further in vivo transfection. The triblock copolymer of PEG113-b-PDMAEMA31-b-PPy30, which is a photo- and pH-responsive material, has therefore been demonstrated as an ideal siRNA carrier having not only the properties of remarkable siRNA condensation, high stability, and efficient siRNA release but also great performance in in vitro tests of cytotoxicity, siRNA uptake efficiency, and protein knockdown efficiency.
Conflicts of interest
There are no conflicts to declare
Acknowledgements
We thank the research funding supported by Ministry of Science and Technology, Taiwan, (MOST 104-2113-M-007-012-MY3). Dr Chen's research is supported by MOST 105-2628-E-007-007-MY3 and by the National Institute for Health Research (NHRI-EX106-10609BC). This work was also supported by the “Frontier Research Center on Fundamental and Applied Sciences of Matters” from The Featured Areas Research Center Program within the framework of the Higher Education Sprout Project by the Ministry of Education (MOE 107QR001I5) in Taiwan. We are also grateful to Professor Hsin-Lung Chen for assisting the dynamic light scattering (DLS) measurement. Malvern Instrument (Nano S) is supported by the Department of Chemical Engineering in National Tsing Hua University.
References
- R. Kanasty, J. R. Dorkin, A. Vegas and D. G. Anderson, Nat. Mater., 2013, 12, 967–977 CrossRef CAS PubMed.
- J. Soutschek, A. Akinc, B. Bramlage, K. Charisse, R. Constien, M. Donoghue, S. Elbashir, A. Geick, P. Hadwiger, J. Harborth, M. John, V. Kesavan, G. Lavine, R. K. Pandey, T. Racie, K. G. Rajeev, I. Röhl, I. Toudjarska, G. Wang, S. Wuschko, D. Bumcrot, V. Koteliansky, S. Limmer, M. Manoharan and H.-P. Vornlocher, Nature, 2004, 432, 173–178 CrossRef CAS PubMed.
- H. Yin, R. L. Kanasty, A. A. Eltoukhy, A. J. Vegas, J. Robert Dorkin and D. G. Anderson, Nat. Rev. Genet., 2014, 15, 541–555 CrossRef CAS PubMed.
- E. Wagner, Acc. Chem. Res., 2012, 45, 1005–1013 CrossRef CAS PubMed.
- M. A. Behlke, Mol. Ther., 2006, 13, 644–670 CrossRef CAS PubMed.
- J. M. Layzer, A. P. McCaffrey, A. K. Tanner, Z. Huang, M. A. Kay and B. A. Sullenger, RNA, 2004, 10, 766–771 CrossRef CAS PubMed.
- G. Navarro, J. Pan and V. P. Torchilin, Mol. Pharmaceutics, 2015, 12, 301–313 CrossRef CAS.
- M. Elsabahy, G. S. Heo, S.-M. Lim, G. Sun and K. L. Wooley, Chem. Rev., 2015, 115, 10967–11011 CrossRef CAS PubMed.
- Y. J. Kwon, Acc. Chem. Res., 2012, 45, 1077–1088 CrossRef CAS.
- M. S. Shim and Y. J. Kwon, Bioconjugate Chem., 2009, 20, 488–499 CrossRef CAS PubMed.
- X. Xu, J. Wu, Y. Liu, M. Yu, L. Zhao, X. Zhu, S. Bhasin, Q. Li, E. Ha, J. Shi and O. C. Farokhzad, Angew. Chem., Int. Ed., 2016, 55, 7091–7094 CrossRef CAS.
- Y.-L. Lin, G. Jiang, L. K. Birrell and M. E. H. El-Sayed, Biomaterials, 2010, 31, 7150–7166 CrossRef CAS.
- K. Miyata, Y. Kakizawa, N. Nishiyama, A. Harada, Y. Yamasaki, H. Koyama and K. Kataoka, J. Am. Chem. Soc., 2004, 126, 2355–2361 CrossRef CAS PubMed.
- S. Matsumoto, R. J. Christie, N. Nishiyama, K. Miyata, A. Ishii, M. Oba, H. Koyama, Y. Yamasaki and K. Kataoka, Biomacromolecules, 2009, 10, 119–127 CrossRef CAS PubMed.
- H. Y. Cho, A. Srinivasan, J. Hong, E. Hsu, S. Liu, A. Shrivats, D. Kwak, A. K. Bohaty, H. Paik, J. O. Hollinger and K. Matyjaszewski, Biomacromolecules, 2011, 12, 3478–3486 CrossRef CAS PubMed.
- M. A. Kostiainen, D. K. Smith and O. Ikkala, Angew. Chem., Int. Ed., 2007, 119, 7744–7748 CrossRef.
- B. Chertok, R. Langer and D. G. Anderson, ACS Nano, 2016, 10, 7267–7278 CrossRef CAS.
- H. Li, M. Miteva, K. C. Kirkbride, M. J. Cheng, C. E. Nelson, E. M. Simpson, M. K. Gupta, C. L. Duvall and T. D. Giorgio, Biomacromolecules, 2015, 16, 192–201 CrossRef CAS.
- M. S. Shim and Y. J. Kwon, Adv. Drug Delivery Rev., 2012, 64, 1046–1059 CrossRef CAS.
- J. Olejniczak, C.-J. Carling and A. Almutairi, J. Controlled Release, 2015, 219, 18–30 CrossRef CAS.
- J. Jiang, X. Tong and Y. Zhao, J. Am. Chem. Soc., 2005, 127, 8290–8291 CrossRef CAS.
- J. Jiang, X. Tong, D. Morris and Y. Zhao, Macromolecules, 2006, 39, 4633–4640 CrossRef CAS.
- J. Babin, M. Pelletier, M. Lepage, J. Allard, D. Morris and Y. Zhao, Angew. Chem., Int. Ed., 2009, 48, 3329–3332 CrossRef CAS.
- M. J. Hansen, W. A. Velema, M. M. Lerch, W. Szymanski and B. L. Feringa, Chem. Soc. Rev., 2015, 44, 3358–3377 RSC.
- M. D. Green, A. A. Foster, C. T. Greco, R. Roy, R. M. Lehr, T. H. Epps III and M. O. Sullivan, Polym. Chem., 2014, 5, 5535–5541 RSC.
- A. A. Foster, C. T. Greco, M. D. Green, T. H. Epps III and M. O. Sullivan, Adv. Healthcare Mater., 2015, 4, 760–770 CrossRef CAS.
- C. T. Greco, T. H. Epps III and M. O. Sullivan, ACS Biomater. Sci. Eng., 2016, 2, 1582–1594 CrossRef CAS.
- L. Yin, H. Tang, K. H. Kim, N. Zheng, Z. Song, N. P. Gabrielson, H. Lu and J. Cheng, Angew. Chem., Int. Ed., 2013, 52, 9182–9186 CrossRef CAS.
- C. T. Huynh, M. K. Nguyen, G. Y. Tonga, L. Longé, V. M. Rotello and E. Alsberg, Adv. Healthcare Mater., 2016, 5, 305–310 CrossRef CAS.
- K. Matyjaszewski and J. Xia, Chem. Rev., 2001, 101, 2921–2990 CrossRef CAS.
- K. Landfester, Angew. Chem., Int. Ed., 2009, 48, 4488–4507 CrossRef CAS.
- H. Lee, S. H. Son, R. Sharma and Y.-Y. Won, J. Phys. Chem. B, 2011, 115, 844–860 CrossRef CAS.
- J. E. Laaser, Y. Jiang, D. Sprouse, T. M. Reineke and T. P. Lodge, Macromolecules, 2015, 48, 2677–2685 CrossRef CAS.
- D. Sprouse, Y. Jiang, J. E. Laaser, T. P. Lodge and T. M. Reineke, Biomacromolecules, 2016, 17, 2849–2859 CrossRef CAS.
- J. E. Laaser, E. Lohmann, Y. Jiang, T. M. Reineke and T. P. Lodge, Macromolecules, 2016, 49, 6644–6654 CrossRef CAS.
- J. You, J. A. Yoon, J. Kim, C.-F. Huang, K. Matyjaszewski and E. Kim, Chem. Mater., 2010, 22, 4426–4434 CrossRef CAS.
- S. Dai, P. Ravi, K. C. Tam, B. W. Mao and L. H. Gan, Langmuir, 2003, 19, 5175–5177 CrossRef CAS.
- D. J. Gary, H. Lee, R. Sharma, J.-S. Lee, Y. Kim, Z. Y. Cui, D. Jia, V. D. Bowman, P. R. Chipman, L. Wan, Y. Zou, G. Mao, K. Park, B.-S. Herbert, S. F. Konieczny and Y.-Y. Won, ACS Nano, 2011, 5, 3493–3505 CrossRef CAS.
- M. C. Deshpande, M. C. Davies, M. C. Garnett, P. M. Williams, D. Armitage, L. Bailey, M. Vamvakaki, S. P. Armes and S. Stolnik, J. Controlled Release, 2004, 97, 143–156 CrossRef CAS.
- C. E. Nelson, J. R. Kintzing, A. Hanna, J. M. Shannon, M. K. Gupta and C. L. Duvall, ACS Nano, 2013, 7, 8870–8880 CrossRef CAS PubMed.
- T.-M. Sun, J.-Z. Du, Y.-D. Yao, C.-Q. Mao, S. Dou, S.-Y. Huang, P.-Z. Zhang, K. W. Leong, E.-W. Song and J. Wang, ACS Nano, 2011, 5, 1483–1494 CrossRef CAS.
- M. S. Shim and Y. J. Kwon, J. Controlled Release, 2009, 133, 206–213 CrossRef CAS.
- M. S. Shim and Y. J. Kwon, Biomacromolecules, 2008, 9, 444–455 CrossRef CAS.
- Y. Li, T. Zhao, C. Wang, Z. Lin, G. Huang, B. D. Sumer and J. Gao, Nat. Commun., 2016, 7, 13214 CrossRef.
- J.-F. Gohy, S. Creutz, M. Garcia, B. Mahltig, M. Stamm and R. Jérôme, Macromolecules, 2000, 33, 6378–6387 CrossRef CAS.
- Y. Cai and S. P. Armes, Macromolecules, 2004, 37, 7116–7122 CrossRef CAS.
- M. Moghimi, P. Symonds, C. Murray, C. Hunter, G. Debska and A. Szewczyk, Mol. Ther., 2005, 11, 990–995 CrossRef.
- X. Yue, Y. Qiao, N. Qiao, S. Guo, J. Xing, L. Deng, J. Xu and A. Dong, Biomacromolecules, 2010, 11, 2306–2312 CrossRef CAS.
- S. Han, Q. Cheng, Y. Wu, J. Zhou, X. Long, T. Wei, Y. Huang, S. Zheng, J. Zhang, L. Deng, X. Wang, X.-J. Liang, H. Cao, Z. Liang and A. Dong, Biomaterials, 2015, 48, 45–55 CrossRef CAS.
- C. Wang, L. Du, J. Zhou, L. Meng, Q. Cheng, C. Wang, X. Wang, D. Zhao, Y. Huang, S. Zheng, H. Cao, J. Zhang, L. Deng, Z. Liang and A. Dong, ACS Appl. Mater. Interfaces, 2017, 9, 32463–32474 CrossRef CAS.
- P. Kolhar, N. Doshi and S. Mitragotri, Small, 2011, 7, 2094–2100 CrossRef CAS.
- J. Guan, J. Sun, F. Sun, B. Lou, D. Zhang, V. Mashayekhi, N. Sadeghi, G. Storm, E. Mastrobattista and Z. He, Nanoscale, 2017, 9, 9190–9201 RSC.
- B. B. Lundy, A. Convertine, M. Miteva and P. S. Stayton, Bioconjugate Chem., 2013, 24, 398–407 CrossRef CAS.
- X.-X. Zhang, C. A. H. Prata, T. J. McIntosh, P. Barthélémy and M. W. Grinstaff, Bioconjugate Chem., 2010, 21, 988–993 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8sc01494a |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a
*a