
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOrganic emitter integrating aggregation-induced delayed fluorescence and room-temperature phosphorescence characteristics, and its application in time-resolved luminescence imaging†
Fan
Ni‡
ab,
Zece
Zhu‡
a,
Xiao
Tong
c,
Mingjuan
Xie
c,
Qiang
Zhao
 *c,
Cheng
Zhong
a,
Yang
Zou
b and
Chuluo
Yang
*ab
*c,
Cheng
Zhong
a,
Yang
Zou
b and
Chuluo
Yang
*ab
aDepartment of Chemistry, Hubei Key Lab on Organic and Polymeric Optoelectronic Materials, Wuhan University, Wuhan, 430072, P. R. China. E-mail: clyang@whu.edu.cn
bShenzhen Key Laboratory of Polymer Science and Technology, College of Materials Science and Engineering, Shenzhen University, Shenzhen, 518060, P. R. China
cKey Laboratory for Organic Electronics & Information Displays, Institute of Advanced Materials, Nanjing University of Posts and Telecommunications, Nanjing, 210023, P. R. China. E-mail: iamqzhao@njupt.edu.cn
First published on 28th June 2018
Abstract
Thermally activated delayed fluorescence (TADF) with a substantially long lifetime furnishes a new paradigm in developing probes for time-resolved imaging. Herein, a novel TADF fluorophore, namely, PXZT, with terpyridine as the acceptor and phenoxazine (PXZ) as the donor, was rationally designed and synthesized. The new compound shows typical thermally activated delayed fluorescence, aggregation-induced emission and crystallization-induced room-temperature phosphorescence (RTP). The coordination of PXZT with a zinc ion causes the quenching of the fluorescence of PXZT due to the enhanced intramolecular charge transfer of the resulting complex ZnPXZT1. With the dissociation of the ZnPXZT1 to release PXZT and the subsequent in situ hydrophobic aggregation of the free PXZT to resist the influence of oxygen, the TADF emission of PXZT is recovered. This zinc-assisted process is successfully used for time-resolved imaging of HeLa and 3T3 cells. This work presents a simple and effective strategy for time-resolved imaging by in situ forming TADF aggregates to turn on the TADF emission.
Introduction
Time-resolved luminescence imaging (TRLI) has revolutionized contemporary visualization methods by facilitating indispensable advancements in the observation of functional and molecular recognition events in the cell.1 By setting a suitable delay time between the pulsed excitation light and the long-lived luminescence of the exploited luminophore, TRLI presents an elegant solution to the problem of short-lived background signals scattering or vanishing from the observed medium. Consequently, it has made the acquisition of a high signal-to-noise ratio possible during imaging measurements.2In general, to pursue the attractive advantages in TRLI, a basic parameter that should be possessed by the employed luminophore is a relatively long excited-state lifetime. In the past few decades, interest in luminophores with a long-lived triplet state has emerged and witnessed tremendous progress in association with TRLI. Due to the suitable long lifetimes (from microseconds to milliseconds), phosphorescence transition-metal (e.g., Ru(II),3 Ir(III),4 Pt(II),5 and Ln(III)6) complexes have been verified to play a key role in TRLI. In addition, pure organic luminophores that feature long-lived room-temperature phosphorescence (RTP)7 have also been incorporated in TRLI. As a result of their pertinent features, phosphorescence-based luminophores have demonstrated appealing prospects and presented practical channels for designing probes for TRLI.
Very recently, a great breakthrough has been made in the field of long-lived singlet-state emissive luminophores.8 Pure organic thermally activated delayed fluorescence (TADF) materials accompanied by effective singlet-state emission and appropriately long lifetimes can theoretically offer advantages for time-resolved imaging. Aside from the essential long emission lifetimes, the structural diversity and emissive adjustability also make TADF-based luminophores ideal alternatives for use within organisms. However, the design of TADF-based probes for biological imaging, especially for TRLI, remains a big challenge.9 The sensitivity of the excited triplet state in TADF emitters to oxygen is the most significant obstacle for their application. In 2014, by employing BSA to eliminate singlet oxygen and enhance the intensity of delayed fluorescence, Peng et al. revealed a TADF-based time-resolved platform to achieve fluorescence sensing in MCF-7 cancer cells, which proves the feasibility of TADF emitters for TRLI.9a In 2016, as a means to eliminate the influence of oxygen, Huang et al. proposed an efficient strategy of embedding aggregates of TADF dyes into a diblock polymer matrix.9d The creative approach opened up a new opportunity for TRLI in living cells. Despite the progress, developing reaction-based probes to turn on the delayed fluorescence while achieving the specific detection has become a burning issue.
To solve the problem of the triplet state quenching of TADF-based probes in an oxygen-containing atmosphere,10 herein, we reported a new strategy by in situ aggregation-induced TADF turn-on response. This is unlike the previously reported approach of excluding oxygen with additional assistance or preparing aggregates before the detection to cut off oxygen access.
First, the luminophore PXZT was constructed based on a practical D–A-type structure with phenoxazine (PXZ) and terpyridine as the donor and acceptor units, respectively. The luminophore PXZT demonstrated ideal emission properties and typical TADF features. Next, the coordination of the luminophore PXZT with a zinc ion to form ZnPXZT1 was conducted. The coordination process greatly enhanced the intramolecular charge transfer (ICT), and thus quenched the self-fluorescent emission of PXZT, which can reduce the background signal. Finally, the water-soluble complex of ZnPXZT1 was successfully employed in time-resolved imaging of HeLa and 3T3 cells by ingeniously releasing TADF emitter of PXZTvia the in situ dissociation of ZnPXZT1 in cell. The monitored long-lived emission of PXZT during the TRLI in live cells suggested ZnPXZT1 is a practical probe for cellar imaging (Scheme 1).
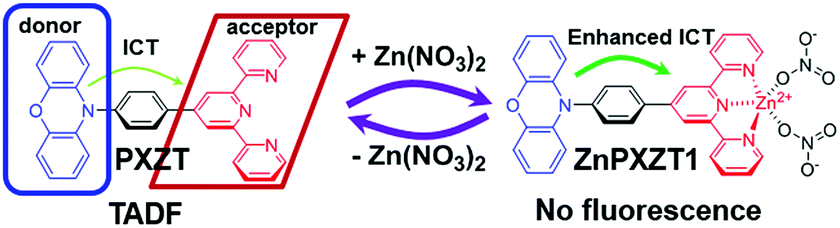 | ||
| Scheme 1 Proposed mechanism for zinc ion-assisted regulation of thermally activated delayed fluorescence. | ||
Results and discussion
1 TADF features of PXZT
Serving as a D–A-type molecule, PXZT shows obvious intra-molecular charge transfer (ICT) characteristics. The strong absorption bands at approximately 275 and 315 nm can be attributed to the π–π* electronic transition, while the broad absorption band from 360 nm to 450 nm can be ascribed to the ICT transition from the PXZ unit to the terpyridine unit (Fig. S3†). The emission spectra of PXZT displayed the significant red shift with the increasing solvent polarity (Fig. S4†), demonstrating positive solvatochromism. Due to the strong charge transfer effect, employing highly polar DMSO or ethanol as the solvent led to quenching of the emission.8e,f These results provided a paradigm for the design of a turn-on-type probe with regulation of the ICT intensity in PXZT.The emission spectra of PXZT in toluene under degassed and aerated conditions were measured to evaluate the sensitivity of PXZT emission to oxygen. The fluorescence intensities of the PXZT solutions (in toluene and 2-methyltetrahydrofuran) after degassing with argon were stronger than those under aerated conditions (Fig. 1a and S5a†). The transient photoluminescence decay spectra of PXZT in solution after degassing with argon showed biexponential fluorescence decays (Fig. 1b and S5b†). When exposed to air, the dilute solution only showed a short-lived emission. The obvious quenching of the emission of the PXZT solution by oxygen implied that the luminophore PXZT can hardly be used for TRLI in oxygen-containing environment.
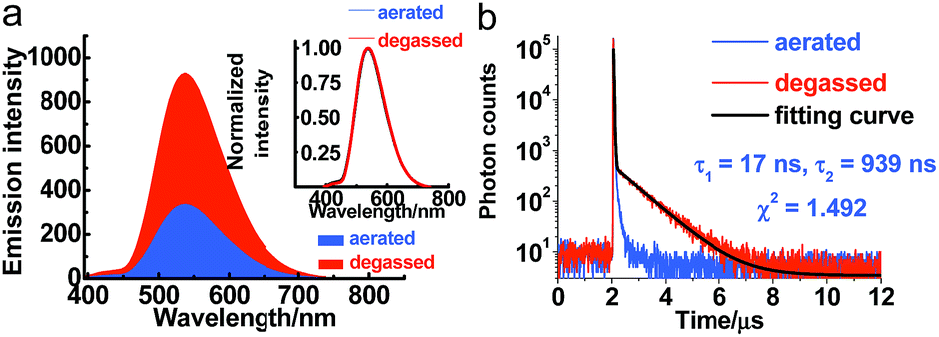 | ||
| Fig. 1 Steady-state emission spectra (a) and transient photoluminescence decay spectra (b) of PXZT in 2-methyltetrahydrofuran at room temperature with and without degassing with argon, λex = 377 nm, λem = 540 nm. | ||
A time-gated measurement of the emission of PXZT in a 2-methyltetrahydrofuran solution at room temperature after degassing with argon was conducted to determine the origins of the prompt and delayed emissions. The normalized spectrum with a delay time of 595 ns overlapped well with the spectrum with a delay time of 10 ns (Fig. S6†), which directly demonstrated that the emission of PXZT resulted from its radiative relaxation from the excited singlet state.
The transient emission of PXZT in 2-methyltetrahydrofuran at different temperatures after degassing was carefully measured. With the rising temperature, the steady-state emission intensity and the ratios of the delayed components of the PXZT emission increased obviously (Fig. 2a and b), proving that thermal energy accelerates the process of reverse intersystem crossing.
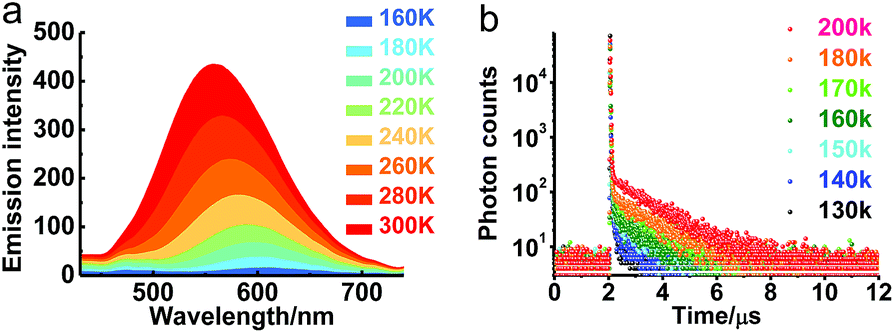 | ||
| Fig. 2 Steady-state emission spectra (a) and transient photoluminescence decay spectra (b) of PXZT in 2-methyltetrahydrofuran at different temperatures after degassing with argon, λex = 377 nm, λem = 540 nm. | ||
With PXZT in degassed toluene and 2-methyltetrahydrofuran solutions owing TADF features, the emissive characteristics of PXZT in solid state were also recorded. Even in air, the doped PMMA film containing 5 wt% PXZT showed a long-lived emission in the microsecond range (Fig. S7†). This result implied that dense packing in solid state may restrain the quench of air to the triplet state of PXZT, which can be utilized for TRLI in oxygen-containing atmosphere. The delay lifetime and emission intensity of PXZT in the PMMA film were reasonably longer in an argon atmosphere than those in air. In the time-gated measurements, the normalized spectrum at a delaytime of 0.1 ms overlapped well with the steady-state spectrum (Fig. S8†). This indicated that the delayed emission in the doped PMMA film also resulted from the radiative relaxation from the exited singlet state.
As the aggregation of PXZT can also form dense packing, the steady-state emission spectra and transient photoluminescence decay spectra of PXZT in THF/water mixtures with different volume ratios were investigated in air. The steady-state emission in air demonstrated typical twisted intramolecular charge transfer (TICT) at a low proportion of water and aggregation-induced emission enhancement behavior with the continuously increasing the proportion of water (fw) (Fig. 3a and b). With the increase of fw or the concentration of PXZT, the lifetimes of the prompt fluorescence showed no obvious change, while the lifetimes and ratios of the delayed components obviously increased, demonstrating aggregation-induced TADF emission enhancement (Fig. 3c and d, Tables S1 and S2†). These positive results presented a practical channel for turning on the TADF of PXZT by its in situ aggregation for TRLI in oxygen-containing environment.
 | ||
| Fig. 3 (a) Steady-state emission spectra of 20 μM PXZT in THF and water mixtures at different fw in air, λex = 340 nm. (b) The emission intensity at 506 nm with the change of fw, λex = 340 nm. (c) Transient photoluminescence decay spectra of PXZT in THF/water mixtures with different fw in air, λex = 377 nm. (d) Transient photoluminescence decay spectra of different concentration of PXZT in THF/water mixtures (fw = 90) in air, λex = 377 nm, λem = 506 nm. | ||
2 Luminescent properties of the PXZT in regularly arranged solid state
To further understand the photophysical properties of solid PXZT in the regular packing state, the emission of a single crystal of PXZT was measured. Unlike the emission in solutions or film doped in PMMA, a much longer emission lifetime in the millisecond range was observed. In time-gated measurements with a delay time of 0.5 ms, PXZT showed an emission peak at approximately 560 nm, which presented a remarkable redshift of approximately 100 nm with respect to its steady state spectrum (Fig. 4a). The emission lifetime at room temperature was approximately 11 ms (Fig. S11†), which is much longer than that in solution or in doped PMMA film. Based on above observations, the crystallization-induced room temperature phosphorescence of PXZT may be a possible avenue for exploration.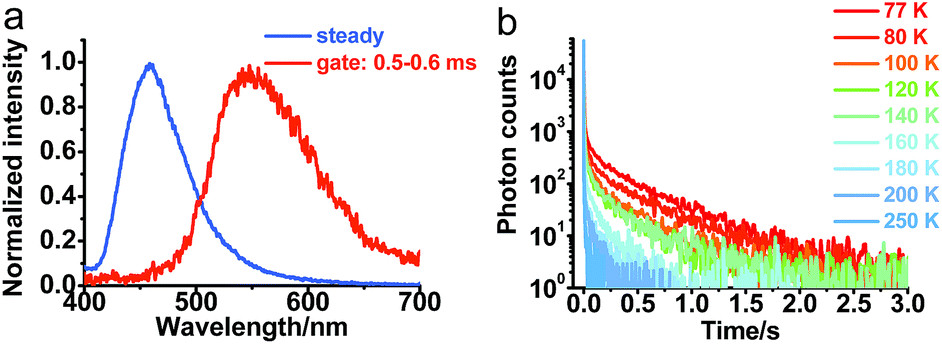 | ||
| Fig. 4 (a) Normalized steady-state and time-gated emission spectra of the crystal of PXZT at room temperature in air. The energy of first excited singlet state ES = 3.00 eV, the energy of first excited triplet state ET = 2.55 eV, and the singlet-triplet splitting energy ΔEST = 0.45 eV. (b) Transient photoluminescence decay spectra of the crystal of PXZT at different temperatures under Ar, λex = 377 nm, λem = 560 nm. | ||
To confirm the mechanism of the phosphorescence emission, the transient photoluminescence decay spectra of crystalline PXZT were measured at different temperatures. With the decreasing temperature, the emission decay became slower (Fig. 4b). At 77 K, the lifetime at 560 nm was 0.4 s (Fig. S11†). The stable T1 state at 77 K radiated more slowly and phosphoresced over a longer lifetime.
The single-crystal analysis of PXZT revealed the face-to-face antiparallel packing mode between the two planes of adjacent PXZT molecules (Fig. S12a†). The obvious π–π stacking (3.42 Å) between the moiety of the electron-poor terpyridine in the two stacking molecules occurred in the opposite direction. The electron-rich PXZ in two neighbouring molecules also stacked with each other (3.59 Å) (Fig. S12b†). C–H⋯O (2.59 Å) and C–H⋯N (2.74 Å) hydrogen bonds existed in the crystal structure can further stabilize the crystal structure (Fig. S12c†). Similar to the conditions observed in many organic crystals,7b,c the intermolecular interaction in PXZT can stabilize the triplet state and decrease its decay rate with the room-temperature luminescence lasting for tens of milliseconds.
To identify the role of π–π stacking in the crystal state and to explain the above phenomena, natural transition orbital (NTO) calculations were carried out. The calculated NTO pairs for the first excited singlet state were localized on a phenoxazine and a terpyridine unit, while the NTO pairs of the first triplet excited state were localized on the aggregated terpyridine units (Fig. S13b†). According to the calculations, the first excited singlet state of the crystal was a charge-transfer state, while the first triplet state was a localized electronic state. The localized electronic state delocalized significantly as terpyridine formed π–π stacking in the crystalline state, which may decrease the triplet state energy.
Then the fluorescence and phosphorescence emission in PMMA film and crystalline state were compared to understand the reason why the TADF was switched to RTP in regularly arranged crystalline state. An obvious blueshift of about 40 nm of the steady-state fluorescence peak in the crystalline state (Fig. S9 and S10†) meant a rise of 0.12 eV of ES. What's more, a significant redshift of about 55 nm of the phosphorescence peak was observed in the crystalline form relative to that in the PMMA film (Fig. S9 and S10†), and thus ET was directly reduced by 0.15 eV. Consequently, the ΔEST in crystalline PXZT reached 0.45 eV, which is improper for reverse intersystem crossing (RISC) processing to produce effective TADF emission. And the stabilized triplet state in crystalline state can be account for the room-temperature phosphorescence emission.
3 The TADF quenching of PXZT by zinc ions
Though PXZT exhibited long-lived luminescence, its poor solubility in water limited its further application in biological imaging. To achieve a fluorescence and lifetime dual turn-on-type detection, quenching of the TADF emission was imperative. To solve the key issue, a strategy by using the coordination of terpyridine unit of PXZT with a zinc ion to enable an enhanced ICT was proposed.11 Upon the addition of Zn(NO3)2·6H2O to a 1,4-dioxane solution of PXZT, the fluorescence of the PXZT was quenched gradually (Fig. 5). The change in the fluorescence intensity fitted well with a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 binding mode,12 and the dissociation constant (Kd) was estimated to be 9.3 nM.
:1 binding mode,12 and the dissociation constant (Kd) was estimated to be 9.3 nM.
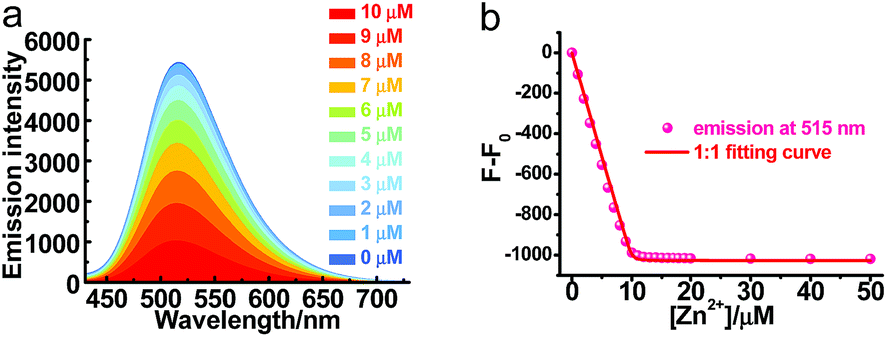 | ||
| Fig. 5 (a) Steady-state emission spectra of 10 μM PXZT upon the addition of Zn(NO3)2·6H2O in dioxane at room temperature, λex = 330 nm. (b) The decrease in the emission intensity vs. the concentration of zinc ions. | ||
The formation of ZnPZXT1 was also monitored by UV titration. The UV titration of PXZT upon coordination with a zinc ion in ethanol solution showed the appearance and enhancement of a new charge transfer absorption band with a peak at approximately 430 nm, which was a significant redshift of 55 nm compared to that of PXZT (Fig. S14†). Through coordination of PXZT with a zinc ion, the fluorescence of PXZT was totally quenched, which is benefited to decreasing the background signal in imaging. Moreover, the resulting Zn2+ complex also acquired solubility in water. The improved solubility and quenched emission suggested ZnPXZT1 is an ideal candidate for cell imaging.
4 Time-resolved luminescence cell imaging
The fluorescence recovery of PXZT is the key process in time-resolved cell imaging. Since zinc is an essential trace element for eukaryotes13 and many zinc proteins and participates in a variety of cellular activities,14 we supposed ZnPXZT1 may dissociate to free its fluorescent PXZT during the cell imaging process.To investigate the feasibility of fluorescence recovery, ZnPXZT1 was then utilized for TRLI in cells. After incubating the 10 μM complex for 5 h in HeLa cells, obvious green luminescence signals could be detected (Fig. 6). The luminescence signals were distributed in different regions of the HeLa cells, and the lifetimes of the luminescence signals at different regions were significantly different (Fig. 6d). Considering the lifetimes of cellular autofluorescence were no more than 10 ns,15 the short-lived fluorescence signals were completely filtrated by a delay time of 50 ns. Due to the luminescence quenching of ZnPXZT1, the detected long-lived luminescence signals with lifetimes over 50 ns may come from the long lifetime of delay fluorescence of PXZT (Fig. 6e and f), which may be released by the dissociation of ZnPXZT1 during the cell imaging process.
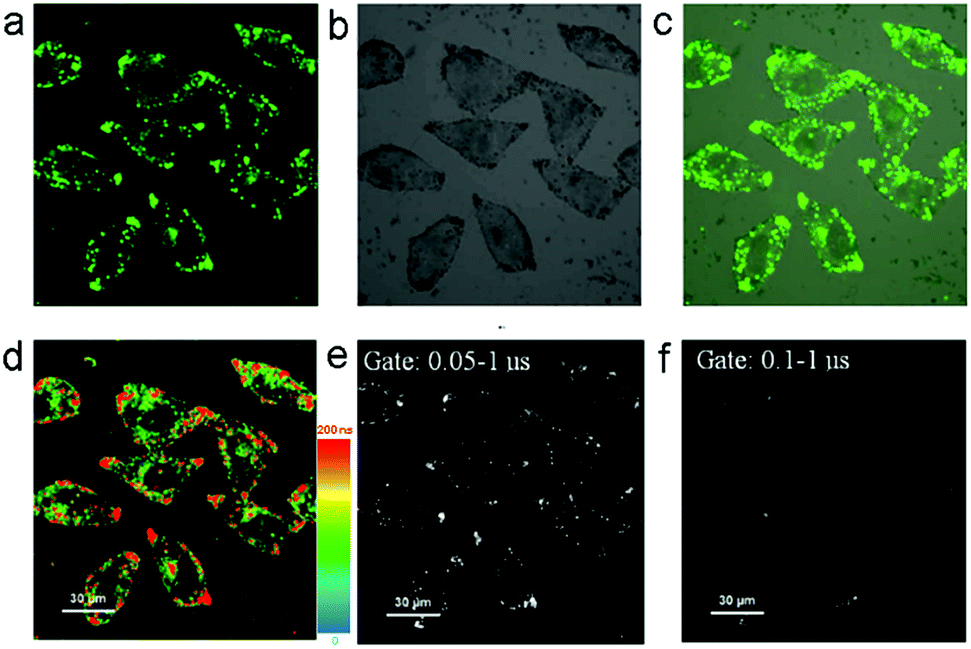 | ||
| Fig. 6 Steady-state (a–c), luminescence lifetime (d) and time-gated (e and f) imaging of HeLa cells incubated with 10 μM ZnPXZT1. λex = 405 nm, λem = 470–570 nm. (a) Darkfield; (b) bright field; (c) merging of (a) and (b). | ||
To further investigate the applicability of the TADF-based technique in other type of cells, the 3T3 cells were incubated with 10 μM complex for ZnPXZT1-based TRLI. In the steady-state imaging process, it can be found that obvious green luminescence signals appeared after 5 h incubating (Fig. S16a†). In the luminescence lifetime (Fig. S16d†) and time-gated (Fig. S16e and f†) imaging, the long-lived luminescence signals with lifetimes over 50 ns can also be detected, similar to the case of HeLa cells.
5 Dissociation of ZnPXZT1in vitro
With the successful application of the TADF-based TRLI, the fluorescence titration was carried out to examine our hypothesis of the dissociation of ZnPXZT1. To a solution of 20 μM ZnPXZT1 in HEPES buffer in air, upon the addition of EDTA (Fig. 7a), the fluorescence intensity of PXZT at 505 nm increased significantly with more than 2000-fold of its initial value, which demonstrated an efficient turn-on detection. A long-lived emission with a lifetime in the microsecond range was detected through the transient luminescent spectrum test (Fig. S17†). Based on the time-gated measurement, the normalized spectrum with a delay time of 20 μs overlapped well with the steady-state spectrum, which suggested that the TADF emission of PXZT was recovered (Fig. 7b). It is probable that EDTA with strong coordinative ability can effectively bind the zinc ion and remove it from the complex of ZnPXZT1, and the ligand of PXZT is then released. Subsequently, the in situ-generated PXZT may aggregate in the buffer due to its insolubility, which inhibits the quenching of PXZT fluorescence by oxygen, consequently the TADF emission of PXZT was recovered in HEPES buffer in air. To get direct evidence of the dissociation of ZnPXZT1 in cell, we attempted to do the same test by using several intracellular active species that coordinate with zinc ion with high affinities to replace EDTA, such as cysteine, pyrophosphate, monohydrogen sulfide (Fig. S18†), unfortunately, we failed. This could be due to the complexity of cell environment where some synergistic effect may result in the dissociation of the zinc complex.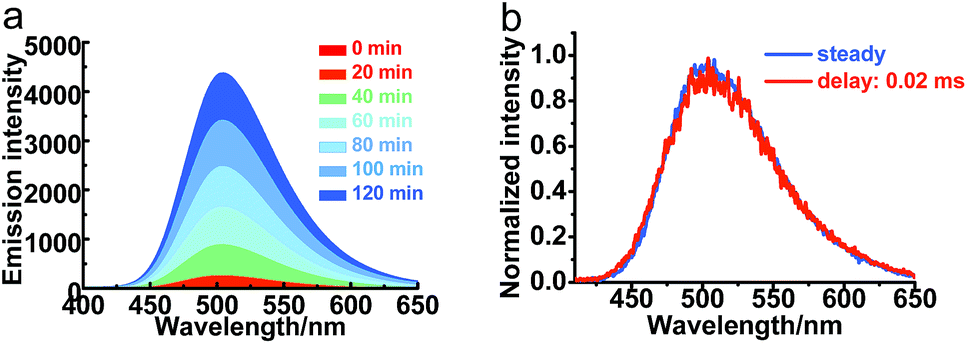 | ||
| Fig. 7 (a) Steady-state emission spectra of a 20 μM ZnPXZT1 solution upon the addition of 20 μM EDTA in 10 mM HEPES buffer (pH = 7) at room temperature in air. (b) Normalized steady-state and time-gated emission spectra of a 20 μM ZnPXZT1 solution upon the addition of 20 μM EDTA in air, λex = 330 nm, λem = 505 nm. | ||
Conclusions
In conclusion, a new D–A-type luminophore (PXZT) featured with typical TADF and crystallization-induced RTP features was developed. By coordination of PXZT with a zinc ion, both the prompt and delayed fluorescence were quenched due to the enhancement of ICT. With the dissociation of ZnPXZT1 to release PXZT and its subsequent hydrophobic aggregation to turn on the aggregation-induced TADF emission, the complex of ZnPXZT1 was rationally used in vitro, which demonstrated an efficient detection of EDTA with more than 2000-fold enhancement of fluorescence intensity. The time-resolved imaging of HeLa and 3T3 cells with ZnPXZT1 was successfully conducted and found to be an efficient approach to eliminate the background signals. Compared with the previous reported methods of excluding oxygen with additional assistance or prearranged aggregates, the newly proposed strategy with dissociation reaction and aggregation process provides an efficient and practical platform for time-resolved cell imaging by the rational regulation of aggregation-induced TADF emission in oxygen-containing atmosphere (Table S4†). We believe that simple TADF emitters with aggregation-induced delayed fluorescence enhancement will become very promising candidates for time-resolved luminescence imaging.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
We gratefully acknowledge financial support from the Innovative Research Groups of the National Natural Science Foundation (No. 21721005), the National Natural Science Foundation of China (No. 51603152 and 91433201) and the Shenzhen Peacock Plan (KQTD20170330110107046).Notes and references
- (a) N. Weibel, L. J. Charbonnière, M. Guardigli, A. Roda and R. Ziessel, J. Am. Chem. Soc., 2004, 126, 4888–4896 CrossRef PubMed; (b) K. Hanaoka, K. Kikuchi, S. Kobayashi and T. Nagano, J. Am. Chem. Soc., 2007, 129, 13502–13509 CrossRef PubMed; (c) D. Jin and J. A. Piper, Anal. Chem., 2011, 83, 2294–2300 CrossRef PubMed; (d) L. Gu, D. J. Hall, Z. Qin, E. Anglin, J. Joo, D. J. Mooney, S. B. Howell and M. J. Sailor, Nat. Commun., 2013, 4, 2326–2332 CrossRef PubMed.
- (a) J. C. G. Bünzli and C. Piguet, Chem. Soc. Rev., 2005, 34, 1048–1077 RSC; (b) G. Muller, Dalton Trans., 2009, 9692–9707 RSC; (c) M. Y. Berezin and S. Achilefu, Chem. Rev., 2010, 110, 2641–2684 CrossRef PubMed; (d) Z. Zhu, W. Li and C. Yang, Sens. Actuators, B, 2016, 224, 31–36 CrossRef.
- (a) M. R. Gill and J. A. Thomas, Chem. Soc. Rev., 2012, 41, 3179–3192 RSC; (b) D. L. Ma, V. P. Y.Ma, D. S. H. Chan, K. H. Leung, H. Z. He and C. H. Leung, Coord. Chem. Rev., 2012, 256, 3087–3113 CrossRef.
- (a) D. L. Ma, D. S. H. Chan and C. H. Leung, Acc. Chem. Res., 2014, 47, 3614–3631 CrossRef PubMed; (b) Y. Yang, Q. Zhao, W. Feng and F. Li, Chem. Rev., 2013, 113, 192–270 CrossRef PubMed; (c) H. Shi, H. Sun, H. Yang, S. Liu, G. Jenkins, W. Feng, F. Li, Q. Zhao, B. Liu and W. Huang, Adv. Funct. Mater., 2013, 23, 3268–3276 CrossRef.
- T. Zou, J. Liu, C. T. Lum, C. Ma, R. C. Chan, C. N. Lok, W. M. Kwok and C. M. Che, Angew. Chem., Int. Ed., 2014, 126, 10283–10287 CrossRef.
- (a) E. A. Weitz, J. Y. Chang, A. H. Rosenfield and V. C. Pierre, J. Am. Chem. Soc., 2012, 134, 16099–16102 CrossRef PubMed; (b) M. Delbianco, V. Sadovnikova, E. Bourrier, G. Mathis, L. Lamarque, J. M. Zwier and D. Parker, Angew. Chem., Int. Ed., 2014, 53, 10718–10722 CrossRef PubMed; (c) Z. Dai, L. Tian, B. Song, Z. Ye, X. Liu and J. Yuan, Anal. Chem., 2014, 86, 11883–11889 CrossRef PubMed; (d) Z. Zhu, B. Song, J. Yuan and C. Yang, Adv. Sci., 2016, 3, 1600146 CrossRef PubMed.
- (a) M. Palner, K. Pu, S. Shao and J. Rao, Angew. Chem., Int. Ed., 2015, 54, 11477–11480 CrossRef PubMed; (b) X. Zhen, Y. Tao, Z. An, P. Chen, C. Xu, R. Chen, W. Huang and K. Pu, Adv. Mater., 2017, 29, 1606665 CrossRef PubMed; (c) W. Z. Yuan, X. Y. Shen, H. Zhao, J. W. Y. Lam, L. Tang, P. Lu, C. Wang, Y. Liu, Z. Wang and Q. Zheng, et al. , J. Phys. Chem. C, 2010, 114, 6090–6099 CrossRef; (d) O. Bolton, K. Lee, H. J. Kim, K. Y. Lin and J. Kim, Nat. Chem., 2011, 3, 205–210 CrossRef PubMed; (e) Q. Miao, C. Xie, X. Zhen, Y. Lyu, H. Duan, X. Liu, J. Jokerst and K. Pu, Nat. Biotechnol., 2017, 35, 1102–1110 Search PubMed.
- (a) M. Y. Wong and E. Zysman-Colman, Adv. Mater., 2017, 29, 1605444 CrossRef PubMed; (b) Z. Yang, Z. Mao, Z. Xie, Y. Zhang, S. Liu, J. Zhao, J. Xu, Z. Chi and M. P. Aldred, Chem. Soc. Rev., 2017, 46, 915–1016 RSC; (c) Y. Im, M. Kim, Y. J. Cho, J. A. Seo, K. S. Yook and J. Y. Lee, Chem. Mater., 2017, 29, 1946–1963 CrossRef; (d) Z. Zhu, D. Tian and X. Shu, Sens. Actuators, B, 2018, 260, 289–294 CrossRef; (e) R. Ishimatsu, S. Matsunami, K. Shizu, C. Adachi, K. Nakano and T. Imato, J. Phys. Chem. A, 2013, 117, 5607–5612 CrossRef PubMed; (f) T. J. Penfold, F. B. Diasb and A. P. Monkman, Chem. Commun., 2018, 54, 3926–3935 RSC.
- (a) X. Xiong, F. Song, J. Wang, Y. Zhang, Y. Xue, L. Sun, N. Jiang, P. Gao, L. Tian and X. J. Peng, J. Am. Chem. Soc., 2014, 136, 9590–9597 CrossRef PubMed; (b) X. Xiong, L. Zheng, J. Yan, F. Ye, Y. Qian and F. Song, RSC Adv., 2015, 5, 53660–53664 RSC; (c) J. Zhang, R. Chen, Z. Zhu, C. Adachi, X. Zhang and C. Lee, ACS Appl. Mater. Interfaces, 2015, 7, 26266–26274 CrossRef PubMed; (d) T. Li, D. Yang, L. Zhai, S. Wang, B. Zhao, N. Fu, L. Wang, Y. Tao and W. Huang, Adv. Sci., 2017, 4, 1600166 CrossRef PubMed; (e) Y. Wu, F. Song, W. Luo, Z. Liu, B. Song and X. Peng, ChemPhotoChem, 2017, 1, 79–83 CrossRef; (f) S. Gan, J. Zhou, T. A. Smith, H. Su, W. Luo, Y. Hong, Z. Zhao and B. Z. Tang, Mater. Chem. Front., 2017, 1, 2554–2558 RSC; (g) J. Huang, H. Nie, J. Zeng, Z. Zhuang, S. Gan, Y. Cai, J. Guo, S. J. Su, Z. Zhao and B. Z. Tang, Angew. Chem., Int. Ed., 2017, 56, 12971–12976 CrossRef PubMed.
- F. Ni, Z. Wu, Z. Zhu, T. Chen, K. Wu, C. Zhong, K. An, D. Wei, D. Ma and C. Yang, J. Mater. Chem. C, 2017, 5, 1363–1368 RSC.
- H. Hofmeiera and U. S. Schubert, Chem. Soc. Rev., 2004, 33, 373–399 RSC.
- K. A. Conners, Binding constants, the measurement of molecular complex stability, John Wiley and Sons, New York, 1987, vol. 25, pp. 565–566 Search PubMed.
- J. E. Coleman, Annu. Rev. Biochem., 1992, 61, 897–946 CrossRef PubMed.
- S. Iuchi, Cell. Mol. Life Sci., 2001, 58, 625–635 CrossRef PubMed.
- K. Zhang, Q. Yu, H. Wei, S. Liu, Q. Zhao and W. Huang, Chem. Rev., 2018, 118, 1770–1839 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthesis and additional figures details. CCDC 1497984–1497986. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8sc01485j |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2018 |