
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThermodynamic and reactivity studies of a tin corrole–cobalt porphyrin heterobimetallic complex†
Zikuan
Wang
,
Zhengmin
Yao
,
Zeyu
Lyu
,
Qinsi
Xiong
,
Bingwu
Wang
 * and
Xuefeng
Fu
*
* and
Xuefeng
Fu
*
Beijing National Laboratory for Molecular Sciences, College of Chemistry and Molecular Engineering, Peking University, Beijing, 100871, China. E-mail: wangbw@pku.edu.cn; fuxf@pku.edu.cn
First published on 2nd May 2018
Abstract
A heterobimetallic complex, (TPFC)Sn–Co(TAP) (TPFC = 5,10,15-tris(pentafluorophenyl)corrole, TAP = 5,10,15,20-tetrakis(p-methoxyphenyl)porphyrin), was synthesized. The complex featured a Sn–Co bond with a bond dissociation enthalpy (BDE) of 30.2 ± 0.9 kcal mol−1 and a bond dissociation Gibbs free energy (BDFE) of 21.0 ± 0.2 kcal mol−1, which underwent homolysis to produce the (TPFC)Sn radical and (TAP)CoII under either heat or visible light irradiation. The novel tin radical (TPFC)Sn, being the first four-coordinate tin radical observed at room temperature, was studied spectroscopically and computationally. (TPFC)Sn–Co(TAP) promoted the oligomerization of aryl alkynes to give the insertion products (TPFC)Sn–(CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C(Ar))n–Co(TAP) (n = 1, 2, or 3) as well as 1,3,5-triarylbenzenes. Mechanistic studies revealed a radical chain mechanism involving the (TPFC)Sn radical as the key intermediate.
C(Ar))n–Co(TAP) (n = 1, 2, or 3) as well as 1,3,5-triarylbenzenes. Mechanistic studies revealed a radical chain mechanism involving the (TPFC)Sn radical as the key intermediate.
Introduction
Heterobimetallic complexes featuring metal–metal bonds are interesting compounds possessing unconventional bonding patterns, which enable them to act as catalysts,1–3 magnets,4 supramolecular building blocks5 and molecular precursors for chemical vapor deposition (CVD).6 Transition metal (TM)–tin complexes are perhaps one of the most representative classes of such compounds, given the vast scope of their catalytic applications.7,8 Although the introduction of TM–tin bonds often leads to a marked enhancement in the rate of catalysis compared to the parent TM compounds,7,8 in most cases the tin-containing group just acts as a spectator ligand, which modifies the reactivity of the TM center through electronic effects rather than directly participating in the reactions. The strong dative and back-donating bonds between the TM and tin atoms decrease the availability of orbitals of the tin atom towards exogenous substrates, and also diminish the possibility of TM–tin bond dissociation. Rational design of TM–tin complexes featuring weak metal–metal bonds might thus help in revealing the “non-innocent” character of stannyl ligands and possibly lead to novel reaction modes which are not attainable by mononuclear TM systems.Previously, we reported the synthesis of corrole Ge(III) radicals,9 Ge(IV) cation complexes,10 and a corrole Sn(II) complex.11 However, the preparation of high valent corrole tin complexes with well-exposed tin centers, i.e. corrole Sn(III) radicals and Sn(IV) cations, is impeded by the inert pair effect, i.e. the relativistic contraction of the Sn 5s shell and a corresponding lowering of the 5s energy level, which raises the redox potentials of tin complexes relative to their germanium analogs. Cleavage of weak TM–tin bonds bypasses this difficulty and represents a promising route towards corrole tin radicals. Porphyrin cobalt emerges as an ideal candidate for the construction of weak TM–tin bonds due to (1) the similar electronegativities of Sn (1.96) and Co (1.88),12 favoring homolytic Sn–Co bond cleavage, (2) the availability of a Co 3dz2 orbital as the frontier orbital for σ bonding,13 and (3) the known ability of the Co–C bonds of organocobalt(III) porphyrin complexes (bond dissociation enthalpies (BDE) ∼ 20 kcal mol−1)14–16 to undergo homolysis under both thermal14 and photochemical17 conditions. Hence, we envisioned that a complex (cor)Sn–Co(por) (cor = corrole, por = porphyrin) might possess a sufficiently weak Sn–Co bond to be a viable precursor to the hitherto elusive (cor)Sn radical species, featuring a formally trivalent tin center. Moreover, the coproduct (por)CoII might assist in substrate activation promoted by (cor)Sn, resulting in novel reaction pathways.18
Complexes that feature Sn–Co bonds have been documented previously. While formally double bonded SnCo complexes are known, e.g. CpCo(C2H4)Sn(CH(TMS)2)2,19 most Sn–Co complexes feature covalent, single Sn–Co bonds where back-bonding is believed to be relatively unimportant. Although porphyrin,20 dimethylglyoxime derivatives18 and CO21 have been used as the ligands of the cobalt center, practically all heterobimetallic Sn–Co complexes feature alkyl or halogen-substituted tin centers. The proposed (cor)Sn–Co(por) complex, with a nitrogen-ligated pentacoordinate tin atom, might thus reveal novel aspects of the chemistry of such bimetallic complexes.
Herein we report the synthesis of the heterobimetallic complex (TPFC)Sn–Co(TAP) (1, TPFC = 5,10,15-tris(pentafluorophenyl)corrole, TAP = 5,10,15,20-tetrakis(p-methoxyphenyl)porphyrin; Scheme 1). Upon heating or visible light irradiation of 1, the active (TPFC)Sn (2) radical as well as (TAP)CoII (3) were generated. The BDE and bond dissociation free energy (BDFE) of the Sn–Co bond were estimated to be 30.2 ± 0.9 kcal mol−1 and 21.0 ± 0.2 kcal mol−1 at 298 K, respectively. The reactivity was exploited in the reaction with alkynes, revealing 1 as a potential alkyne cyclotrimerization catalyst.
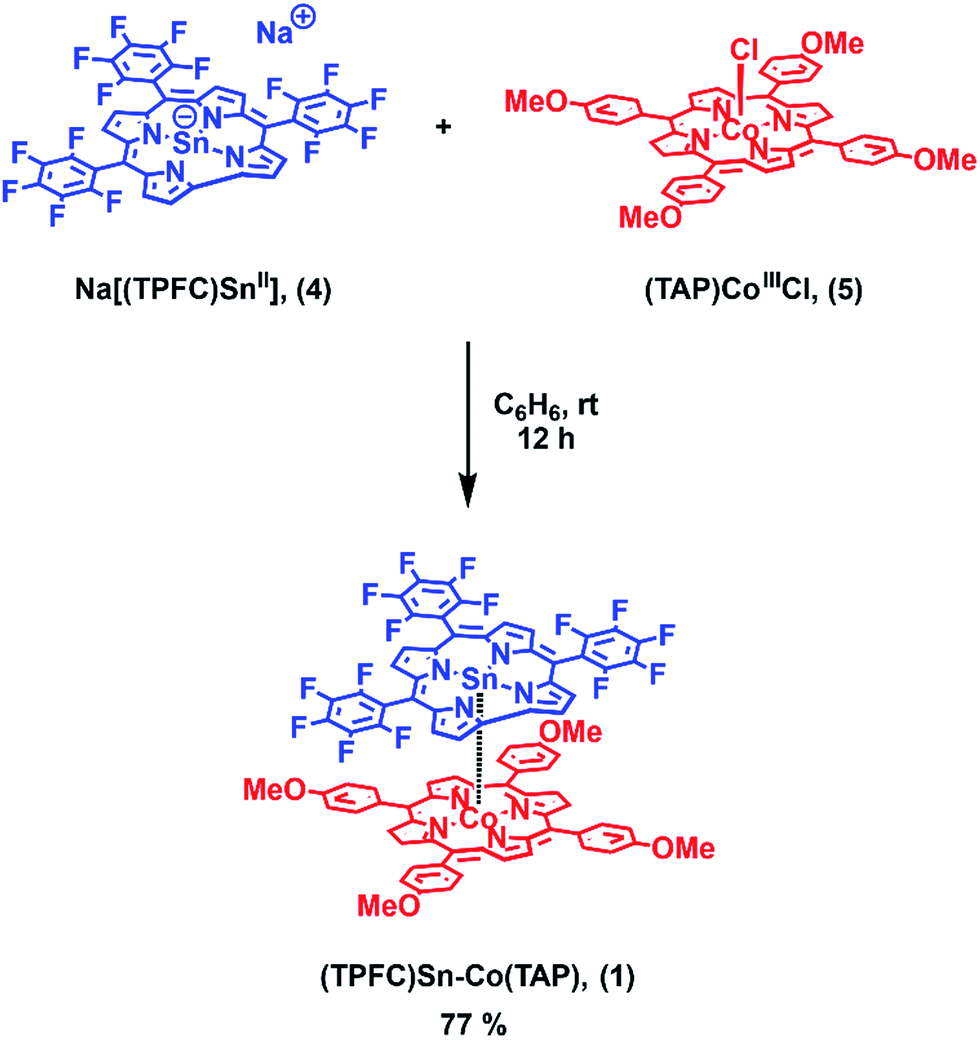 | ||
| Scheme 1 Synthesis of (TPFC)Sn–Co(TAP) (1). | ||
Results and discussion
Synthesis, characterization and DFT studies of 1
1 was synthesized by the reaction of the nucleophilic Na[(TPFC)SnII] (4) with (TAP)Co–Cl (5) in 77% yield (Scheme 1). The complex showed characteristic 1H NMR signals of the corrole and the porphyrin rings (Fig. S2†), which were shifted away from the typical chemical shift ranges of monomeric (TPFC)Sn–X and (TAP)Co–X complexes, respectively, due to the combined ring current effects of the two aromatic macrocyclic ligands. Interestingly, 1 was 119Sn NMR silent, due to the extensive quadrupolar relaxation by the 59Co (I = 7/2) nucleus.Single crystal XRD results unambiguously confirmed the direct Sn–Co bonding (Fig. 1; the Sn–Co bond length was 2.4559(10) Å, which was within the range of typical Sn–Co single bonds8,22). The Sn–Co bond was approximately perpendicular to both the corrole and the porphyrin planes, and the two ligand planes were aligned face-to-face. The Sn atom protruded noticeably (0.872 Å) from the N4-plane of the corrole ligand, comparable with known (TPFC)Sn–X complexes (0.776–0.798 Å),11 while the Co atom was essentially within the porphyrin plane (the distance to the N1–N2–N3–N4 mean plane was 0.102 Å).
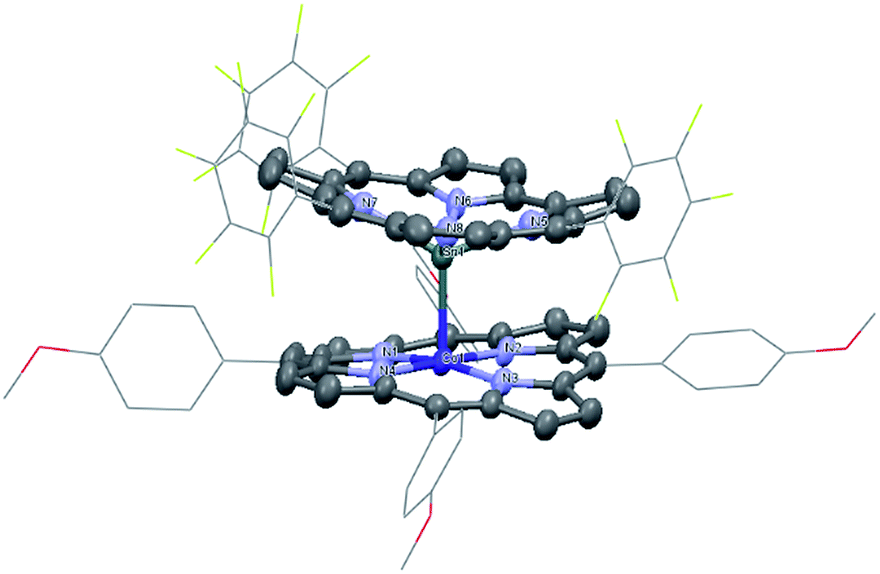 | ||
| Fig. 1 Solid state structure of 1. Hydrogen atoms are omitted for clarity. Selected bond lengths (Å): Sn1–Co1 2.4559(10), Sn1–N5 2.120(5), Sn1–N6 2.107(5), Sn1–N7 2.122(5), Sn1–N8 2.108(5), Co–N1 1.957(4), Co1–N2 1.960(5), Co1–N3 1.967(4), and Co1–N4 1.955(5). | ||
DFT calculations (B3LYP/def2-TZVPP//BP86/def2-TZVP(-f)) were performed to shed light on the bonding between the Sn and Co atoms. The DFT optimized geometry agreed very well with the XRD structure (see Fig. 2 and Table S8†). The Sn–Co bond was a covalent single bond according to natural bonding orbital (NBO) calculations (a Wiberg bond order of 0.60). The Sn–Co σ-bonding NBO comprised 41.5% Sn character and 58.5% Co character, and the sp0.36 and s0.08d hybridizations were adopted by the Sn and Co atoms, respectively (Fig. 2). Thus, the polarity of the bond was best described as Snδ+–Coδ− with a natural ionicity of iSnCo = −0.17, a value comparable to those of aliphatic C–H bonds which typically have iCH = 0.16–0.19.23 Nevertheless, non-Lewis (i.e. ionic) resonance forms contributed significantly to Sn–Co bonding: the second-order perturbative estimate of interaction energy between the bonding and antibonding NBOs of the Sn–Co bond amounted to 31.0 kcal mol−1, leading to a strong occupation (n = 0.463) of the antibonding NBO. Upon formation of the Sn–Co bond from the open-shell (TPFC)Sn and (TAP)Co fragments, a significant electron density depletion was observed at the tin center, accompanied by an electron density accumulation at the cobalt center (Fig. S45†), which confirmed the charge shift as suggested by NBO analysis. Slight density accumulation and depletion were also observed on the corrole and porphyrin ligands, respectively, which were likely due to changes in metal-to-ligand delocalization effects. The Sn 5dπ orbitals were nearly unoccupied (n < 0.003), indicating that Co 3d → Sn 5d back-bonding is negligible. A second-order perturbative estimate yielded a total Co 3d → Sn 5d π back-bonding energy of 1.6 kcal mol−1.
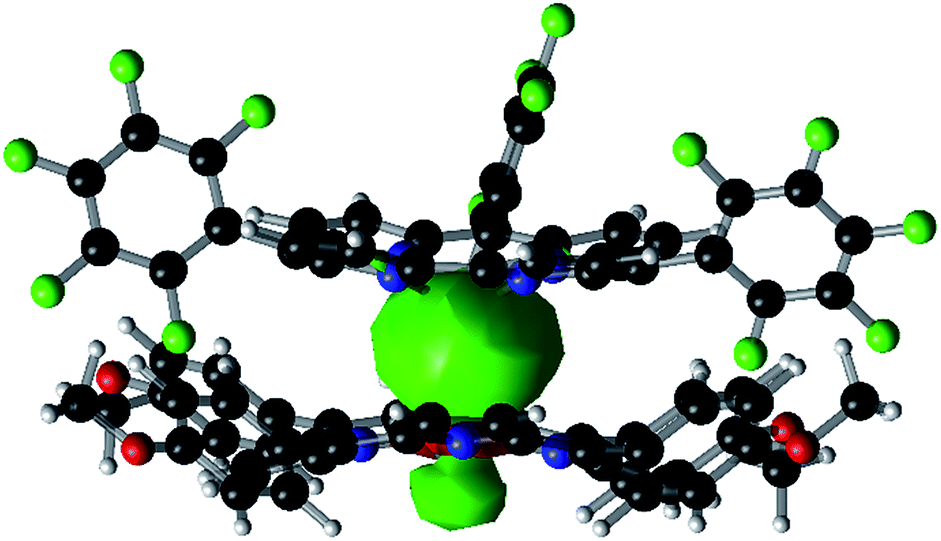 | ||
| Fig. 2 Isosurface plot of the Sn–Co σ-bonding NBO of 1 (isovalue = 0.02). | ||
The energetics of Sn–Co bond cleavage pathways for 1, namely heterolytic formation of SnII + CoIII and SnIV + CoI, and homolytic formation of SnIII + CoII, were subsequently calculated (Table 1). Apparently, the homolysis pathway was the only thermally accessible mode at practical temperatures, and might also be viable under photochemical conditions. In contrast, the heterolysis pathways were so hindered that even visible light (λ > 400 nm, or a photon energy of less than 71 kcal mol−1) was unlikely to overcome the unfavorable thermodynamics.
Photolysis of the Sn–Co bond
Experimentally, the Sn–Co bond of 1 was found to be remarkably photo-labile. An attempt to record the UV-Vis spectrum of 1 in aerated toluene revealed a rapid spectral change within 10 seconds, with isosbestic points at 414, 457, 493 and 610 nm (Fig. 3a). Since 1 was reasonably stable at room temperature under air within a time scale of several hours, the transformation was apparently triggered by light. To elucidate the composition of the photoproducts, we repeated the experiment in C6D6 using a Xe lamp (equipped with a 420–780 nm band pass filter) as the light source, and monitored the reaction course by 1H NMR. When 1 atm of dry O2 was used, the signals of 3 and a new (TPFC)Sn species appeared (Fig. S5a†). The chemical shifts of the latter species (8.88, 8.34, 8.14, and 7.82 ppm) resembled those of (TPFC)Sn–CH2–Sn(TPFC),11 suggesting that the species was the μ-oxo dimer (TPFC)Sn–O–Sn(TPFC) (6). This assignment was confirmed by the independent synthesis of 6 from aerobic oxidation of 4 (see the ESI†) and the single crystal XRD structure (Fig. 4). A comparison of the UV-Vis features of 3 and 6 with that of the reaction mixture further confirmed the composition of the photoproducts (Fig. 3b). When 1 atm of air was used instead, the 1H NMR resonances of 6 were broadened, accompanied by an additional set of signals centered at 8.89, 8.77, 8.57 and 8.50 ppm, characteristic of mononuclear (TPFC)Sn complexes (Fig. S5b†), which was ascribed to the formation of (TPFC)Sn–OH (7) resulting from the reaction of 6 with moisture (Fig. S11†). The hydration reaction was reversible, with K = 0.19 at room temperature (Table S4†), which was very close to that reported for the hydration of ((PhMe2CCH2)3Sn)2O (K = 0.3 at 304 K).24 | ||
| Fig. 3 (a) Photo-oxidation of 1 in aerated toluene over a period of 11 s monitored by stopped-flow UV-Vis absorption spectroscopy. (b) Comparison of the UV-Vis spectra of 3 and 6 with that of the product mixture. | ||
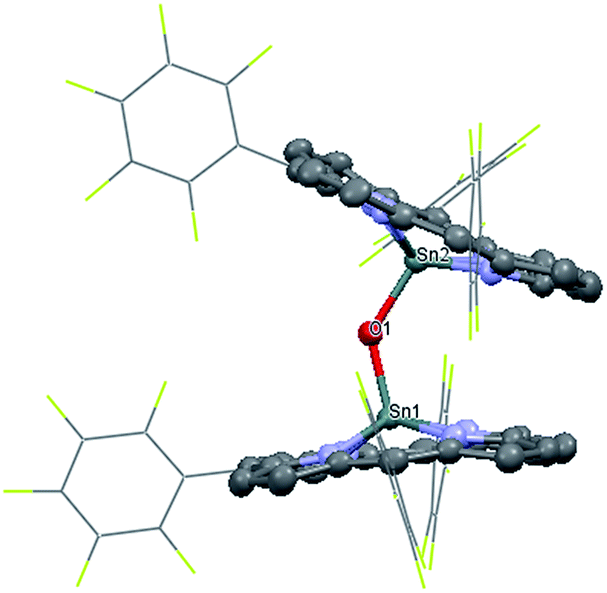 | ||
| Fig. 4 Solid state structure of 6. Hydrogen atoms are omitted for clarity. Selected bond lengths (Å) and angles (°): Sn1–O1 1.924(3), Sn2–O1 1.934(3), and Sn1–O1–Sn2 134.46(14). | ||
A laser flash photolysis study was performed to elucidate the primary photolysis products. Photolyzing a strictly anaerobic toluene solution of 1 with short laser pulses (5 ns) at 413 nm yielded a transient signal with a lifetime of about 1 ms (Fig. 5a). The signal featured a bleach of 1 at 413 nm and in the 550–700 nm region, accompanied by an increase of absorptivity at the absorption maxima of 3 (419 and 534 nm). However, an intriguing peak emerged at 450–500 nm, which could not be explained by 1 and 3 alone. The wavelength was atypical of electronically unperturbed (TPFC)Sn complexes, which have well-defined Soret band absorptions near 420 nm and structured Q band absorptions around 550–600 nm,25 but is in accordance with TDDFT calculated absorptions of 2 (Fig. 5a). Thus, we tentatively attribute the extra signal to complex 2. Such reactivity was also proposed for the photolysis of (py)(dmgH)2Co–SnPh3 (dmg = dimethylglyoximato)18 and (OEP)Co–SnPh3 (OEP = 2,3,7,8,12,13,17,18-octaethylporphyrin),20 but only indirect evidence for the involvement of tin radicals (by halogen abstraction from haloalkanes, for example) was provided.
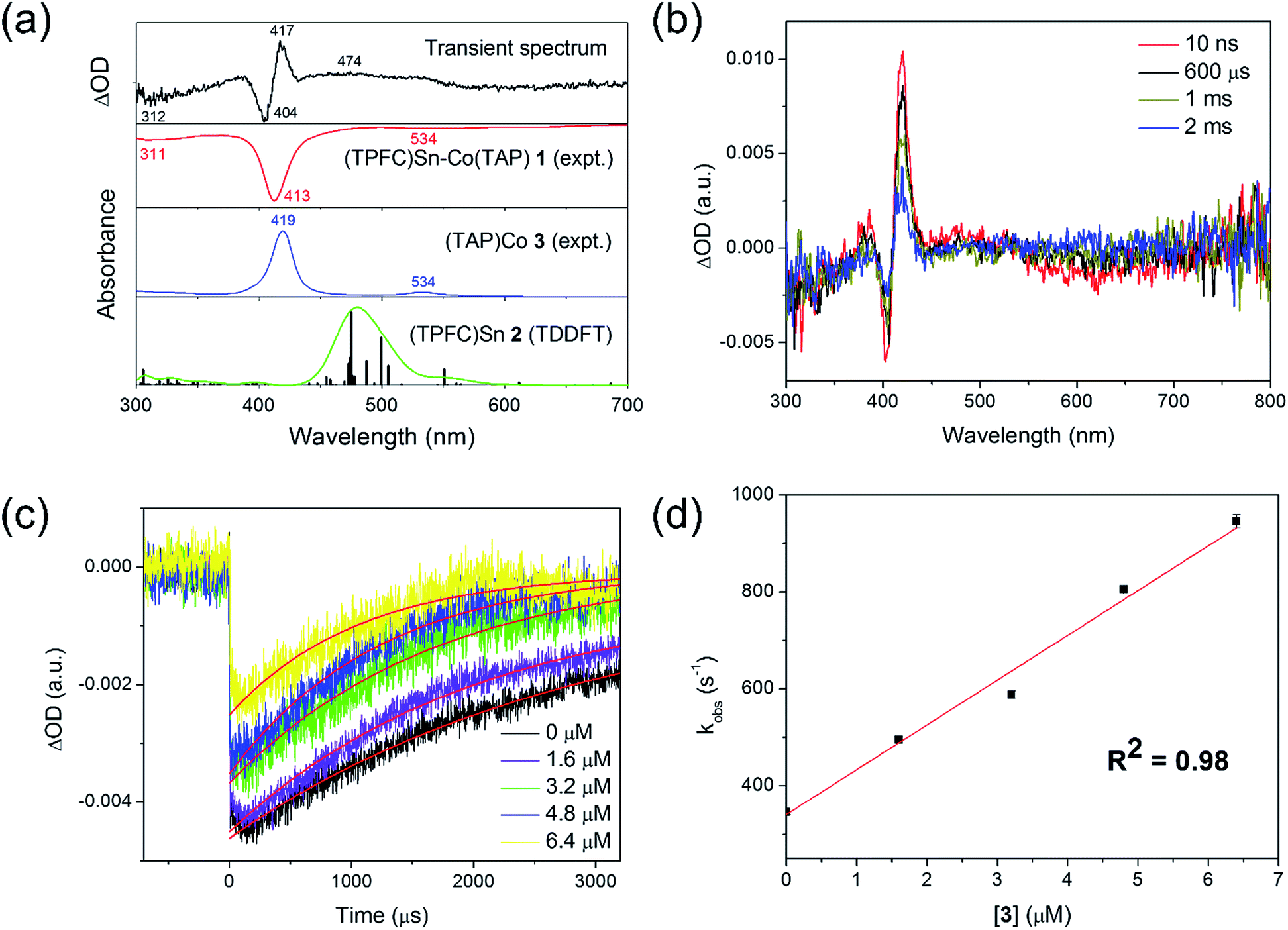 | ||
| Fig. 5 Laser flash photolysis of 1. (a) Transient spectrum of a 5.2 × 10−6 mol L−1 toluene solution of 1, recorded at 10 ns after the laser pulse, compared against the experimental spectra of 1 and 3, and calculated absorptions of 2; (b) time-resolved decay of the transient signal, with [1] = 2.6 × 10−6 mol L−1; (c) transient kinetic decay plots of 2.6 × 10−6 mol L−1 of 1 in the presence of various concentrations of 3, measured at 404 nm; and (d) first-order plot of apparent recombination rate constants against [3] (the non-zero intercept was due to the fact that the amount of 3 formed through photolysis was comparable to that of excess 3; see the ESI†). | ||
Notably, the transient absorption signal uniformly decayed towards zero within a couple of milliseconds (Fig. 5b), suggesting regeneration of 1 through recombination of 2 and 3. The decay kinetics was followed spectroscopically both in the absence and in the presence of various excess amounts of 3. The decay rate was enhanced upon the addition of 3 (Fig. 5c), and exhibited first order dependence on the concentration of the latter (Fig. 5d). The recombination rate constant of 2 and 3 was thus estimated to be (9.2 ± 0.2) × 107 L (mol−1 s−1) at 298 K, which implied a diffusion-controlled process. Under the assumption of strictly diffusion-controlled behavior, the activation enthalpy of recombination could be taken as the activation enthalpy of the viscous flow of toluene,26 which was known accurately to be 1.93 kcal mol−1.27 Thus, the activation entropy of recombination, −15.60 ± 0.04 cal (mol−1 K−1), could be obtained. In case the diffusion-controlled conditions did not hold exactly, slight underestimations of both the activation enthalpy and the activation entropy were expected.
Reported tin radicals mostly feature tricoordinate tin centers with pyramidal or planar conformations, depending on the substituents.28 Tetracoordinate tin radicals, to the best of our knowledge, have been elusive, except for radiogenic radical cations based on organostannanes, which are only stable in solid matrices at low temperatures.29 Calculations revealed that 2 adopted a domed structure in close analogy with (TPFC)GeIII reported by us,9 although the doming effect was more severe for the tin complex due to the larger atomic radius of tin (Fig. 6a and b). The two complexes also shared qualitatively similar electronic structures, where significant spin density was delocalized into the a′(a2u)30 corrole π orbital (Fig. 6c and d). Nevertheless, this effect was much more pronounced for Sn (NPA spin population on the Sn atom = 0.11) than for Ge (spin population on the Ge atom = 0.45). Consequently, in contrast to (TPFC)GeIII, the dominant resonance form of 2 was (TPFC+˙)SnII as indicated by NBO calculations, suggesting significant non-innocent ligand character. The anti-bonding interaction of the Sn sp orbital with the corrole a′(a2u) orbital shifted the latter upwards, explaining the red-shifted Soret band appearing at 450–500 nm (Fig. S46†).
 | ||
| Fig. 6 Side-view of (TPFC)Ge (a) and (TPFC)Sn (b), and spin density plots of (TPFC)Ge (c) and (TPFC)Sn (d). | ||
The above results may suggest that 2 should behave as a ligand-centered radical. Experiments, in contrast, revealed a tin radical-like behavior. Photolyzing a toluene solution of 1 in the presence of N-tert-butyl-α-phenylnitrone (PBN) at −50 °C led to rapid formation of a signal typical of PBN-trapped radicals (Fig. 7a). At −20 °C, the signals were sharpened due to an increase of the molecular tumbling rate and concomitantly better isotropic averaging, revealing pairs of 117,119Sn satellites (Fig. 7b). A spectral fit assuming the isotropic fast-motion regime yielded the parameters g = 2.0048, A(119Sn) = 5.5 G, A(1H) = 14.1 G, and A(14N) = 18.0 G. The A(1H) was considerably larger than most PBN adducts, including adducts of oxygen (2–3 G) and carbon radicals (3–4 G),31 while some heavier group 14 radicals were reported to give A(1H) values of comparable magnitude (e.g. Ph2ClGe˙, 10.9 G).32 Thus, 2 must have been trapped by PBN through the tin atom, rather than one of the ligand atoms, which implied that the tin atom was more reactive than the ligand. Apparently, the favorable sterics and limited structural reorganization energy of the domed tin atom overruled the larger spin density of the ligand. This was also in accord with the observed formation of 6 from photo-oxidation of 1, as opposed to ligand oxidation products. The EPR signal due to 3 was observed upon cooling to −150 °C (Fig. S13†).
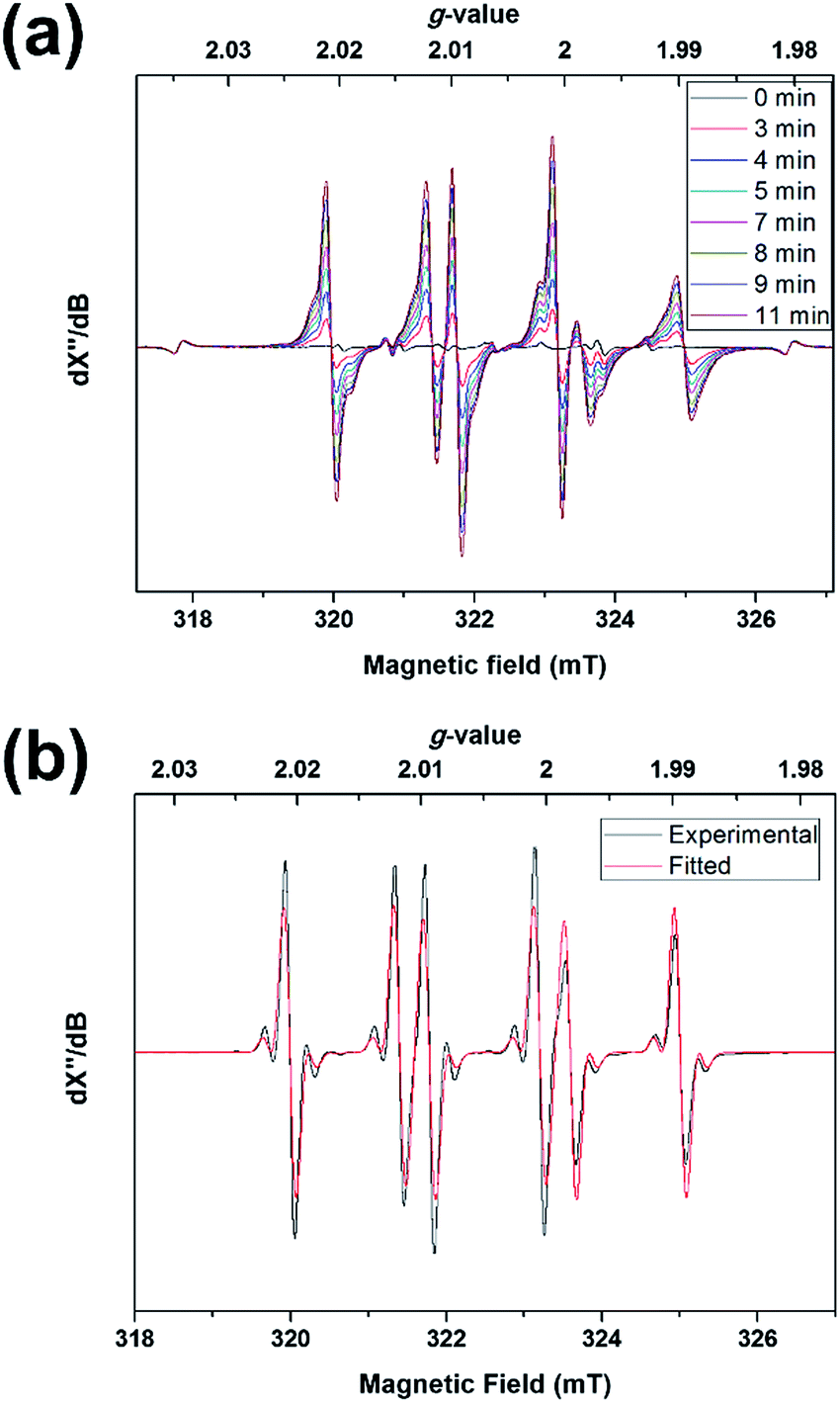 | ||
| Fig. 7 Spin trapping of 2. (a) EPR monitoring of a toluene solution of 1 and PBN at −50 °C, under visible light irradiation (Xe lamp, 420–780 nm), and (b) fitting of the EPR spectrum of the same sample, recorded at −20 °C. | ||
Thermolysis of the Sn–Co bond
With evidence for photolysis in hand, we then investigated the thermal lability of 1. Unfortunately, the PBN trapping experiment was not feasible under thermal conditions because the (TPFC)Sn–PBN adduct was unstable above −20 °C. Thus, the radical trap (2,2,6,6-tetramethylpiperidin-1-yl)oxyl (TEMPO) was used instead. Heating a C6D6 solution of 1 and excess TEMPO at 70 °C gave (TPFC)Sn–TEMPO (8) and 3 as the sole products as evidenced by 1H NMR (Fig. S14†). The reaction rate was first order on 1 and zeroth order on TEMPO (Fig. 8), suggesting homolytic Sn–Co bond dissociation followed by fast trapping of the (TPFC)Sn (2) radical by TEMPO (for further evidence of the homolytic nature of the dissociation, see the ESI†). An activation enthalpy of 32.1 ± 0.9 kcal mol−1 and an activation entropy of 15 ± 3 cal (mol−1 K−1) were determined by kinetic measurements in toluene-d8 at 70–100 °C (Fig. 9). The large activation entropy confirmed the involvement of a dissociative rate-limiting step, and the value was close to that reported for homolytic dissociation of another metal–metal bond complex stabilized by a conjugated planar ligand, (tmtaa)Rh–Rh(tmtaa) (10 ± 1 cal (mol−1 K−1), tmtaa = dibenzotetramethylaza[14]annulene).33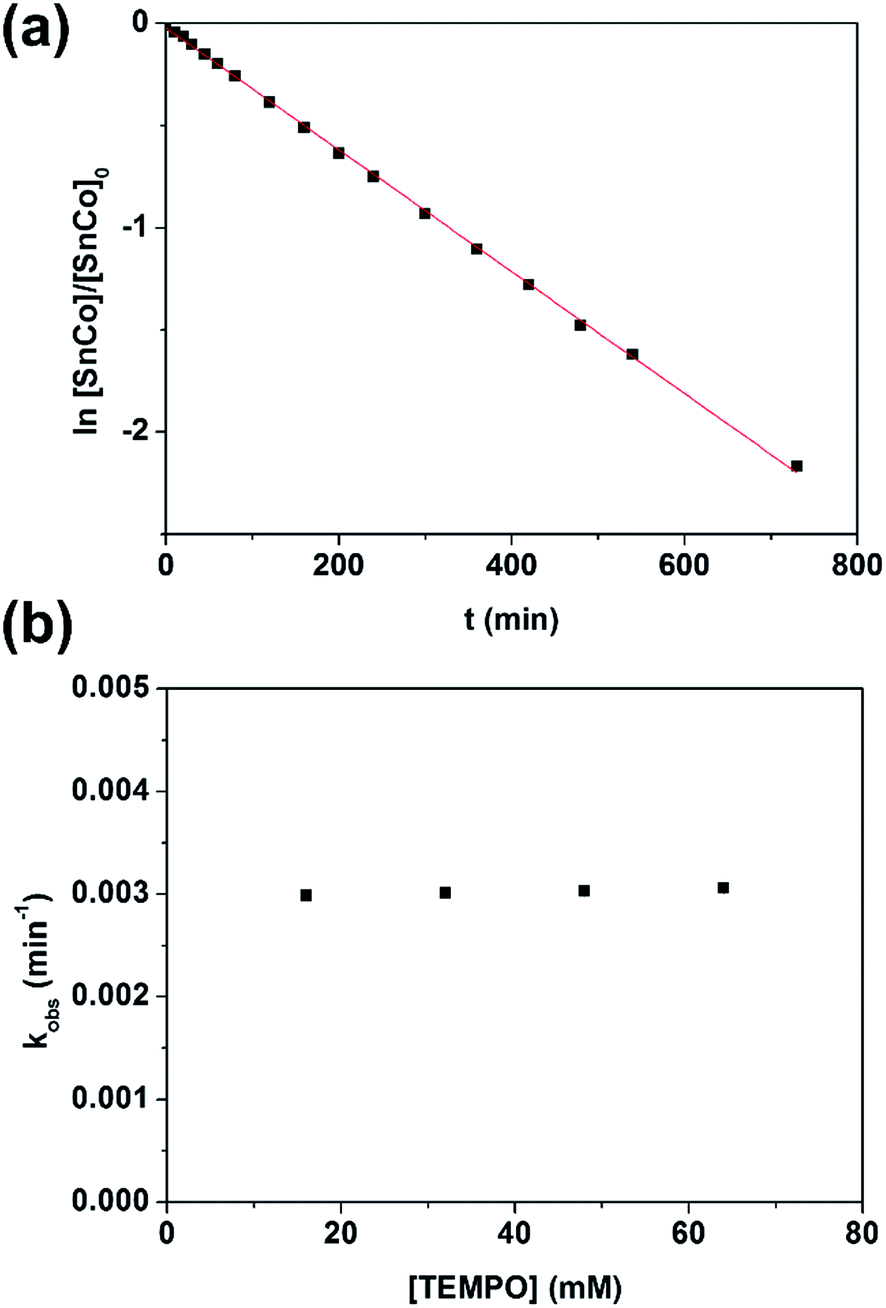 | ||
| Fig. 8 (a) First-order kinetic plot on 1 with 10 equivalents of TEMPO, and (b) zeroth-order kinetic plot on TEMPO. Conditions: 1.6 mmol L−1 of 1, C6D6, 70.0 °C, in the dark and under a N2 atmosphere. | ||
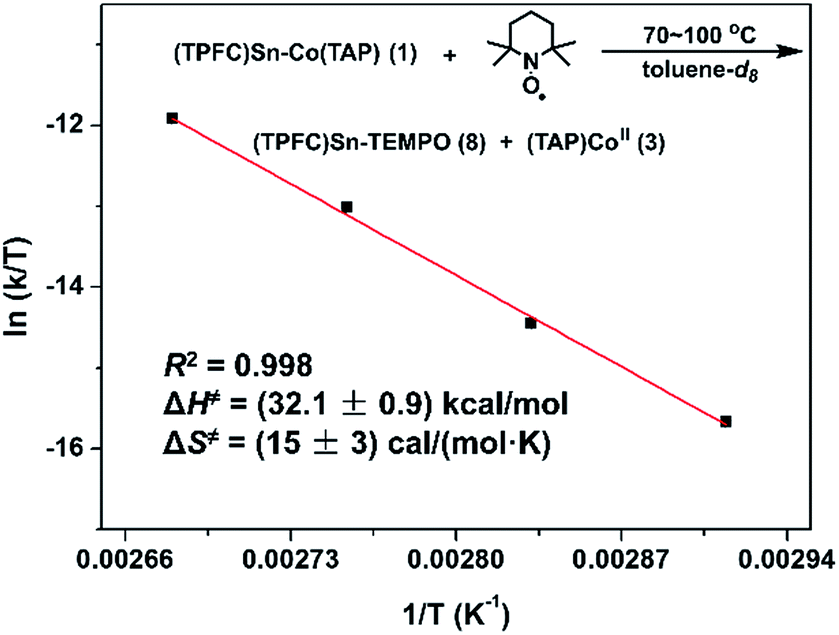 | ||
| Fig. 9 Eyring plot of the reaction of 1 with TEMPO. Conditions: 1.6 mmol L−1 of 1, 16 mmol L−1 of TEMPO, toluene-d8 (containing 15% v/v C6D6), in the dark and under a N2 atmosphere. | ||
Although we are unaware of any existing quantitative kinetic measurements of the homolysis barriers of Sn–Co bonds, the complex (OEP)Co–SnPh3 was reported to have a half-life of ca. 48 h at 120 °C with (TTP)CoII (TTP = 5,10,15,20-tetra(p-tolyl)porphyrin) as a radical trap,20 which indicates a stronger Sn–Co bond than that in 1. This might partly be attributed to the spin delocalization in 2 (Fig. 6), which favored the Sn–Co bond homolysis of 1 compared to (OEP)Co–SnPh3.
Estimation of the Sn–Co bond energy
With the knowledge of the kinetics of both forward and backward directions of the reaction, reliable results on the thermodynamics of Sn–Co bond dissociation could be obtained (Table 2). The bond dissociation enthalpy (BDE) of the Sn–Co bond was estimated to be ΔH = 30.2 ± 0.9 kcal mol−1 by subtracting the backward activation enthalpy, 1.93 kcal mol−1, from the forward activation enthalpy. A bond dissociation entropy of ΔS = 31 ± 3 cal (mol−1 K−1) was obtained similarly. It might be interesting to note that exactly half of the dissociation entropy was released in the transition state. The bond dissociation Gibbs free energy (BDFE) was calculated to be ΔG = 21.0 ± 0.2 kcal mol−1, in excellent agreement with the DFT value (Table 2).34| ΔHa | ΔSb | ΔG298 Ka | |
|---|---|---|---|
| a In kcal mol−1. b In cal (mol−1 K−1). c Obtained by extrapolation of Fig. 9 to 298 K. d Taken as the activation enthalpy of viscous flow (see text) assuming negligible error. e Obtained by subtraction of recombination parameters from those of dissociation. f See ref. 34 for explanations of the discrepancy with experiment. | |||
| Dissociation barrier | 32.1 ± 0.9 | 15 ± 3 | 27.6 ± 0.2c |
| Recombination barrier | 1.93d | −15.60 ± 0.04 | 6.58 ± 0.01 |
| Thermodynamics of dissociation (exp.)e | 30.2 ± 0.9 | 31 ± 3 | 21.0 ± 0.2 |
| Thermodynamics of dissociation (DFT) | 38.9f | 63.8f | 19.9 |
Reaction of 1 with alkynes
It is well known that stannyl radicals readily undergo addition to alkynes, a fact that was exploited in reactions such as radical hydrostannylations as well as cascade ring closing reactions.35 Indeed, 1 was reactive towards p-fluorophenylacetylene under Xe lamp irradiation to form mono-, bis- and tris-insertion products, delivering complexes 9–11 (Scheme 2). The reaction proceeded under either thermal or photochemical conditions as shown by 1H NMR and HR-ESI-MS (Fig. S19 and S20†). Column chromatography of the reaction mixture gave, apart from 9–11, three additional fractions: a mixture of two organocobalt porphyrin complexes (12 and 13), whose structures were confirmed by independent syntheses (see the ESI†); a complex mixture of various organotin corrole complexes in which 14 was identified by HR-ESI-MS; and a cyclotrimerization product, 1,3,5-tris(p-fluorophenyl)benzene (15) (Scheme 2).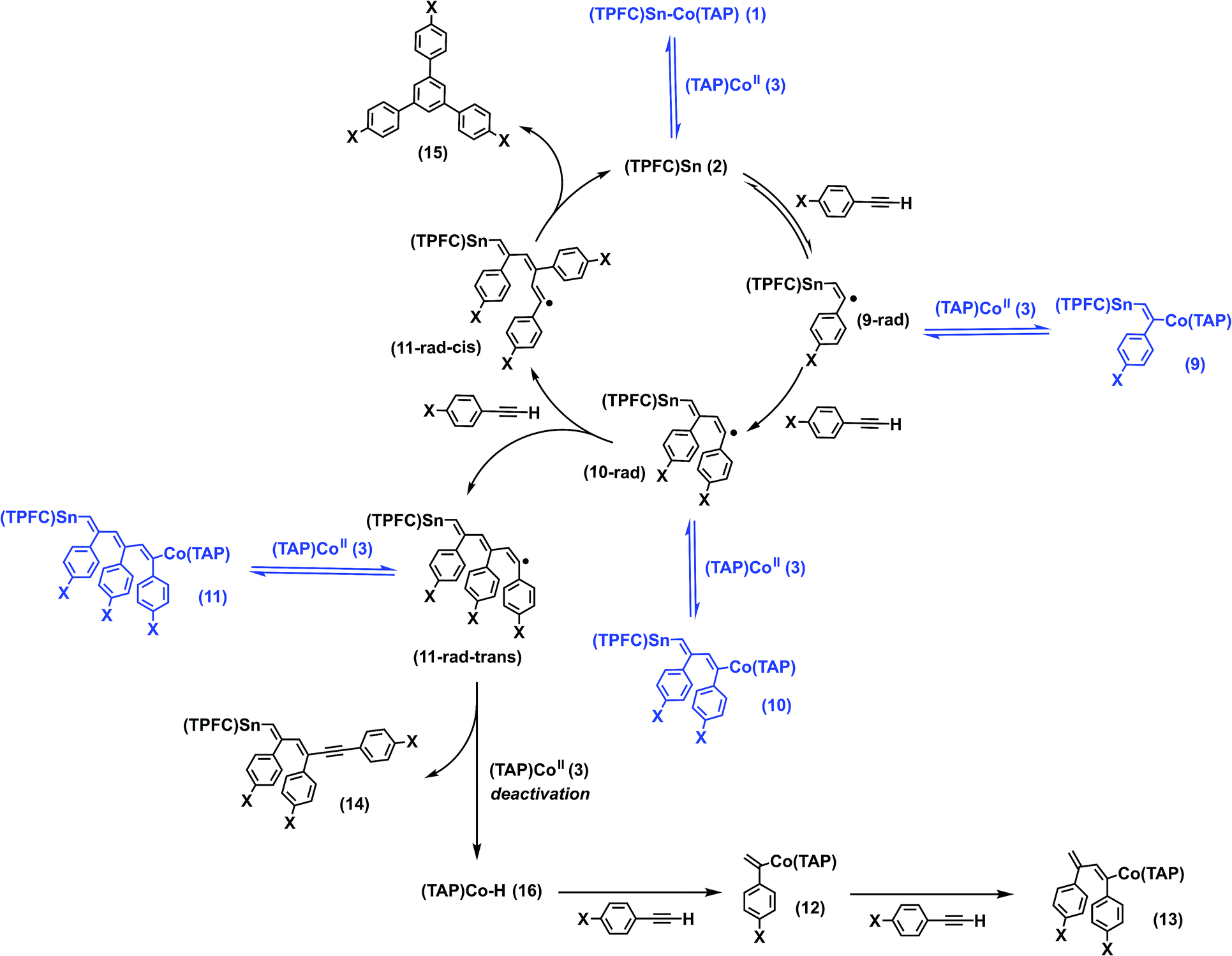 | ||
| Scheme 2 Proposed mechanism for alkyne insertions and cyclization promoted by 1. | ||
To our delight, the turnover number (TON) of the cyclotrimerization product reached 4.3 when p-methoxyphenylacetylene was used as a substrate. The presence of MeO groups also introduced sufficient polarity differences between the insertion products (9′–11′) which allowed us to isolate these complexes. Stoichiometric photolysis of the insertion products revealed that 9′ was cleanly photolyzed to yield 1 and p-methoxyphenylacetylene, while 10′ reacted sluggishly to give 1 and an undefined mixture, where neither p-methoxyphenylacetylene nor the cyclotrimerization product 15′ was found. Unexpectedly, photolysis of 11′, which already possessed an alkyne trimer motif between the metal centers, only gave a small amount (maximum yield 8%) of 15′ after complete conversion, along with 1 and 3 in 41% and 55% yields, respectively.
Based on the evidence above, we propose that the alkyne insertion initially occurred through the sequential addition of alkyne molecules to the stannyl radical 2, followed by reversible capturing of the resulting carbon radicals by 3 (Scheme 2). The latter process regulated the radical concentration in a way similar to that in cobalt-mediated living radical polymerization (CMRP),36 where the chain radicals generated from successive addition of olefin monomers to an initiator radical are reversibly captured by a cobalt(II) complex, reducing the concentration of the former and thereby suppressing biradical termination. The addition of the third alkyne molecule led to two stereoisomers, 11-rad-cis and 11-rad-trans. While back-biting of 11-rad-cis yielded the cyclotrimerization product 15 and regenerated 2, the cyclization of 11-rad-trans necessarily required a trans–cis isomerization. This explains the low yield of 15′ in the stoichiometric photolysis of 11′, which involved 11-rad-trans rather than 11-rad-cis (Fig. S38†). The formation of 14 was due to hydrogen atom transfer (HAT) from 11-rad-trans to 3, which deactivated the catalyst irreversibly. The by-product, (TAP)Co–H (16), underwent fast reaction with excess alkyne to yield 12 and 13, resembling the catalytic chain transfer (CCT) process in CMRP37 where insertion of olefins to the Co–H bond is observed instead.
DFT studies of alkyne cyclotrimerization
To confirm the proposed mechanism as well as to gain more insight into the back-biting process of 11-rad-cis, we performed a computational study of the reaction using p-fluorophenylacetylene as a model substrate (Fig. 10). The addition of the alkyne to 2 was moderately endergonic (ΔG = 12.5 kcal mol−1), consistent with the observed reversibility of the formation of 9′ from 2. Subsequent alkyne additions, however, were exergonic (ΔG = −9.1 and −16.1 kcal mol−1) due to the formation of C–C bonds, which drove the aforementioned monoaddition equilibrium to the product side. It is worth noting that while the second alkyne addition gave exclusively the trans isomer 10-rad due to favorable steric effects of the transition state, the barriers for the formation of 11-rad-trans and 11-rad-cis were almost identical. Accepting 11-rad-trans as a non-productive species, the computed energetics suggested a maximum TON of 2.9 for the alkyne trimerization at 298 K, which was very close to the experimental value (1.6).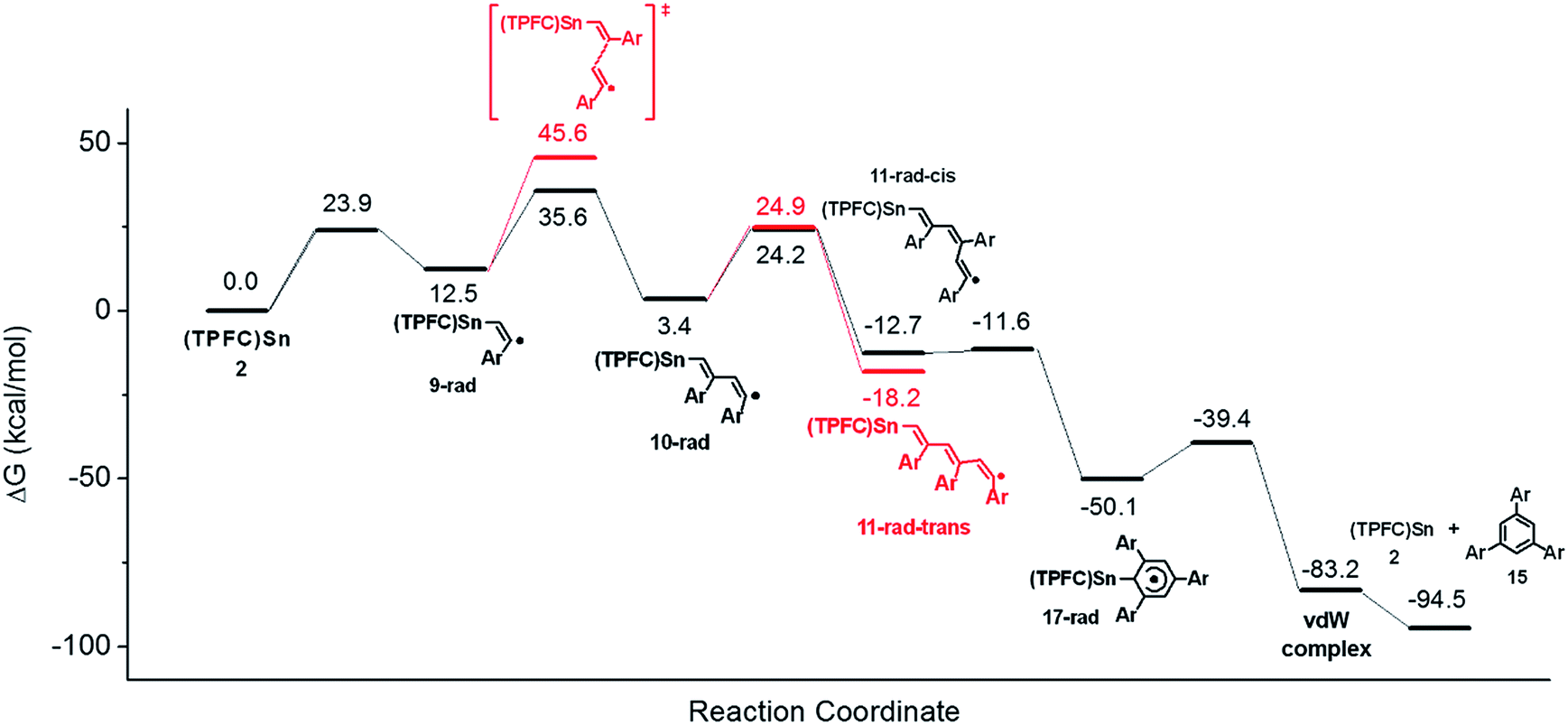 | ||
| Fig. 10 DFT free energy profile of alkyne cyclotrimerization catalyzed by 2. | ||
The back-biting of 11-rad-cis could occur through two possible pathways: (a) a 5-exo-trig cyclization, which is Baldwin allowed but leads to a five-membered ring as opposed to the observed six-membered ring; or (b) a 6-endo-trig cyclization, which is Baldwin disallowed but in which the product 17-rad (Fig. 10) is in accord with the observed structure of the final product 15. In practice, however, the Baldwin disallowed route (b) showed a much lower reaction barrier than that of route (a), thanks to thermodynamic control. Further calculations showed that if route (a) were operative, it would lead to the 1,2,4-cyclotrimerization product through ring expansion (Fig. S47†). Since the 1,2,4-product was not observed experimentally, this further confirmed the predominance of route (b) over route (a). Sn–C bond scission of 17-rad readily afforded the aromatized cyclotrimerization product 15 (Fig. 10).
Since only ESI-MS evidence for 14 was available, the experimentally observed HAT process might as well occur from 11-rad-cis or the cyclized product 17-rad instead of 11-rad-trans, because all three reactions would give products with the same molecular formula. HAT from 11-rad-cis to 3 can be safely ruled out since the barrier of the 6-endo-trig cyclization (1.1 kcal mol−1) was too low for HAT or any bimolecular reaction to be competitive (typical barriers of HAT processes from carbon radicals to porphyrin cobalt are 10–15 kcal mol−1).1617-rad, on the other hand, possessed an HAT-active C–H bond that was so well-shielded as to preclude any possibility for the HAT process (Fig. S48†). This conclusively demonstrated that the observed CCT product originated exclusively from 11-rad-trans, rather than 11-rad-cis or 17-rad.
Conclusions
In conclusion, we have synthesized an unusual heterobimetallic complex, 1, that possesses a covalent, weakly polar Sn–Co bond. The Sn–Co bond undergoes homolysis under thermal and photochemical conditions, releasing an active, four-coordinate stannyl radical 2 in a reversible manner. The thermodynamics and kinetics of the homolysis were studied extensively using a combination of various spectroscopic methods, yielding a Sn–Co BDE of 30.2 ± 0.9 kcal mol−1 and a BDFE of 21.0 ± 0.2 kcal mol−1 at 298 K.Four-coordinate tin radicals such as 2 are rare and unstable due to the reluctance of the less Lewis acidic Sn(III) center to accept a fourth ligand. However, the use of the rigid four-coordinate TPFC ligand forced a four-coordinate situation around the tin atom, leading to the first observation of a four-coordinate tin radical at room temperature, with a lifetime of at least several milliseconds. Moreover, 2 exhibits significant non-innocent ligand character contributing further to its stability, yet with uncompromised and essentially metal-centered reactivity. Taking advantage of the reactivity of 2, we show experimentally and computationally that 1 is catalytically active towards the cyclotrimerization of aryl alkynes.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (21571009 and 21621061). The authors gratefully acknowledge Prof. Ming-Tian Zhang and Ms Han Li for laser flash photolysis experiments, and Mr Jin Xiong for assistance with single crystal structure refinement.Notes and references
- B. G. Cooper, J. W. Napoline and C. M. Thomas, Catal. Rev., 2012, 54, 1 Search PubMed.
- N. P. Mankad, Synlett, 2014, 25, 1197 CrossRef.
- T. Szymańska-Buzar, Coord. Chem. Rev., 2005, 249, 2195 CrossRef.
- J. Černák, I. Kočanová and M. Orendáč, Comments Inorg. Chem., 2012, 33, 2 Search PubMed.
- B. Oelkers, M. V. Butovskii and R. Kempe, Chemistry, 2012, 18, 13566 CrossRef PubMed.
- J. J. Schneider, J. Hagen, D. Blaser, R. Boese and C. Kruger, Angew. Chem., Int. Ed., 1997, 36, 739 CrossRef.
- D. Das, S. S. Mohapatra and S. Roy, Chem. Soc. Rev., 2015, 44, 3666 RSC.
- M. S. Holt, W. L. Wilson and J. H. Nelson, Chem. Rev., 1989, 89, 11 CrossRef.
- (a) H. Fang, Z. Ling, K. Lang, P. J. Brothers, B. de Bruin and X. Fu, Chem. Sci., 2014, 5, 916 RSC; (b) H. Fang, H. Jing, H. Ge, P. J. Brothers, X. Fu and S. Ye, J. Am. Chem. Soc., 2015, 137, 7122 CrossRef PubMed.
- H. Fang, H. Jing, A. Zhang, H. Ge, Z. Yao, P. J. Brothers and X. Fu, J. Am. Chem. Soc., 2016, 138, 7705 CrossRef PubMed.
- L. Yun, H. Vazquez-Lima, H. Fang, Z. Yao, G. Geisberger, C. Dietl, A. Ghosh, P. J. Brothers and X. Fu, Inorg. Chem., 2014, 53, 7047 CrossRef PubMed.
- D. R. Lide, CRC Handbook of Chemistry and Physics, CRC press, Boca Raton, FL, 85th edn, 2005, section 9 Search PubMed.
- M.-S. Liao and S. Scheiner, J. Chem. Phys., 2002, 117, 205 CrossRef.
- S. Fukuzumi, K. Miyamoto, T. Suenobu, E. V. Caemelbecke and K. M. Kadish, J. Am. Chem. Soc., 1998, 120, 2880 CrossRef.
- Y. Zhao, H. Dong, Y. Li and X. Fu, Chem. Commun., 2012, 48, 3506 RSC.
- H. Dong, T. Hou, Y. Zhao, X. Fu and Y. Li, Comput. Theor. Chem., 2012, 1001, 51 CrossRef.
- M. J. Kendrick and W. Al-Akhdar, Inorg. Chem., 1987, 26, 3971 CrossRef.
- (a) G. N. Schrauzer and G. Kratel, Chem. Ber., 1969, 102, 2392 CrossRef; (b) M. Tada and K. Kaneko, J. Org. Chem., 1995, 60, 6635 CrossRef; (c) M. E. Weiss, L. M. Kreis, A. Lauber and E. M. Carreira, Angew. Chem., Int. Ed., 2011, 50, 11125 CrossRef PubMed; (d) M. E. Weiss and E. M. Carreira, Angew. Chem., Int. Ed., 2011, 50, 11501 CrossRef PubMed; (e) L. M. Kreis, S. Krautwald, N. Pfeiffer, R. E. Martin and E. M. Carreira, Org. Lett., 2013, 15, 1634 CrossRef PubMed.
- J. J. Schneider, J. Hagen, O. Heinemann, C. Krüger, F. F. de Biani and P. Zanello, Inorg. Chim. Acta, 1998, 281, 59 CrossRef.
- (a) C. Yang, J. L. Petersen and A. M. Stolzenberg, Inorg. Chem., 1998, 37, 5173 CrossRef; (b) A. M. Stolzenberg, S. R. Workman, J. E. Gutshall, J. L. Petersen and N. Akhmedov, Inorg. Chem., 2007, 46, 6744 CrossRef PubMed.
- D. J. Patmore and W. A. G. Graham, Inorg. Chem., 1967, 6, 981 CrossRef.
- D. Agustin and M. Ehses, C. R. Chim., 2009, 12, 1189 CrossRef.
- F. Weinhold and C. R. Landis, Discovering chemistry with natural bond orbitals, John Wiley & Sons, Hoboken, NJ, 1st edn, 2012, ch. 4 Search PubMed.
- T. P. Lockhart, J. Organomet. Chem., 1985, 287, 179 CrossRef.
- For example, (TPFC)Sn–Cl displays a sharp Soret band at 416 nm and two Q band peaks at 566 and 588 nm: L. Simkhovich, A. Mahammed, I. Goldberg and Z. Gross, Chem.–Eur. J., 2001, 7, 1041 CrossRef.
- (a) I. V. Khudyakov, Res. Chem. Intermed., 2013, 39, 781 CrossRef; (b) F. J. V. Santos, C. A. N. de Castro, J. H. Dymond, N. K. Dalaouti, M. J. Assael and A. Nagashima, J. Phys. Chem. Ref. Data, 2006, 35, 1 CrossRef.
- R. P. Singh and C. P. Sinha, J. Chem. Eng. Data, 1984, 29, 132 CrossRef.
- (a) V. Y. Lee, M. Nakamoto and A. Sekiguchi, Chem. Lett., 2008, 37, 2008 Search PubMed; (b) P. P. Power, Chem. Rev., 2003, 103, 789 CrossRef PubMed.
- (a) A. Hasegawa, S. Kaminaka, T. Wakabayashi, M. Hayashi, M. C. R. Symons and J. Rideout, J. Chem. Soc., Dalton Trans., 1984, 1667 RSC; (b) E. Butcher, C. J. Rhodes, M. Standing, R. S. Davidson and R. Bowser, J. Chem. Soc., Perkin Trans. 2, 1992, 1469 RSC.
- In corrole chemistry it is customary to denote the ligand π orbitals with the irreducible representations of the respective orbitals of porphyrin (D4h): A. Ghosh, Chem. Rev., 2017, 117, 3798 CrossRef PubMed . In the present paper we give the symmetry representations assuming a symmetric domed corrole (Cs), followed by the approximate D4h representations in parentheses.
- G. R. Buettner, Free Radical Biol. Med., 1987, 3, 259 CrossRef PubMed.
- P. Riviere, S. Richelm, M. Riviere-Baudet, J. Satge, M. Gynane and M. Lappert, J. Chem. Res., 1978, 6, 218 Search PubMed.
- G. H. Imler, M. J. Zdilla and B. B. Wayland, Inorg. Chem., 2013, 52, 11509 CrossRef PubMed.
- Note that the SMD solvation model was used in the calculation of ΔH, which already included the effect of solvation entropy. Unfortunately, this contribution could not be separated out, leading to a positive error in ΔH because the solvation entropy discourages dissociation due to an increase of the solvent accessible surface area. On the other hand, the ideal gas model was used in the calculation of ΔS, as is usually done, resulting in a neglect of solvation contribution (which is negative) and thus a positive error. The two errors exactly canceled out each other, and so ΔG was accurately reproduced.
- (a) D. R. Kelly and M. R. Picton, J. Chem. Soc., Perkin Trans. 1, 2000, 1559 RSC; (b) K. Miura and A. Hosomi, J. Synth. Org. Chem., Jpn., 2008, 66, 17 CrossRef.
- C.-H. Peng, T.-Y. Yang, Y. Zhao and X. Fu, Org. Biomol. Chem., 2014, 12, 8580 Search PubMed.
- (a) A. Gridnev, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 1753 CrossRef; (b) A. A. Gridnev and S. D. Ittel, Chem. Rev., 2001, 101, 3611 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis, reactivity, mechanistic studies and DFT calculations, and characterization data of the new compounds. CCDC 1540844–1540848. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8sc01269e |
| This journal is © The Royal Society of Chemistry 2018 |