
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA chiral nickel DBFOX complex as a bifunctional catalyst for visible-light-promoted asymmetric photoredox reactions†
Xiang
Shen
,
Yanjun
Li
,
Zhaorui
Wen
,
Shi
Cao
,
Xinyi
Hou
and
Lei
Gong
 *
*
Department of Chemistry, Key Laboratory of Chemical Biology of Fujian Province, iChEM, College of Chemistry and Chemical Engineering, Xiamen University, Xiamen, 361005, China. E-mail: gongl@xmu.edu.cn
First published on 27th April 2018
Abstract
The enantioselective photoredox reaction of α,β-unsaturated carbonyl compounds and tertiary/secondary α-silylamines was enabled by a readily available single NiII–DBFOX catalyst (DBFOX = 4,6-bis((R)-4-phenyl-4,5-dihydrooxazol-2-yl)dibenzo[b,d]furan) under visible light conditions. The non-precious chiral catalyst is involved in the photochemical process to initiate single electron transfer and at the same time provides a well-organized chiral environment for the subsequent radical transformations. Good to excellent enantioselectivities (80–99% ee) were obtained for the formation of chiral γ-amino carboxylic acid derivatives and γ-lactams.
Introduction
Visible light photoredox catalysis has emerged as a powerful strategy for organic synthesis.1 However, the development of catalytic asymmetric photoredox reactions is still highly desirable and remains a formidable challenge owing to the difficulties in controlling the stereochemistry of highly reactive intermediates such as radicals and radical ions.2,3 Pioneering work by Sibi, Porter, and others has demonstrated that transition-metal-based chiral Lewis acids are capable of governing the enantioselective conjugate addition of radicals produced by stoichiometric reduction of organic halides (Fig. 1a, left).4 These studies inspired chemists to develop cooperative chiral Lewis acid/photoredox catalytic systems for light-induced stereoselective radical transformations, providing a high level of asymmetric induction at mild and convenient reaction conditions (Fig. 1a, right).5–11 For instance, Yoon's group reported the first highly enantioselective intermolecular conjugate addition of α-aminoalkyl radicals to α,β-unsaturated carbonyl compounds by utilizing the combination of a chiral ScIII Lewis acid and RuII tris-bipridine photocatalyst.6 Meggers et al. developed several chiral-at-metal rhodium complexes as chiral Lewis acids together with additional photocatalysts for highly enantioselective conjugate radical additions.7,8 The Kang group utilized a chiral-at-rhodium complex as a single catalyst for a catalytic asymmetric conjugate radical addition.9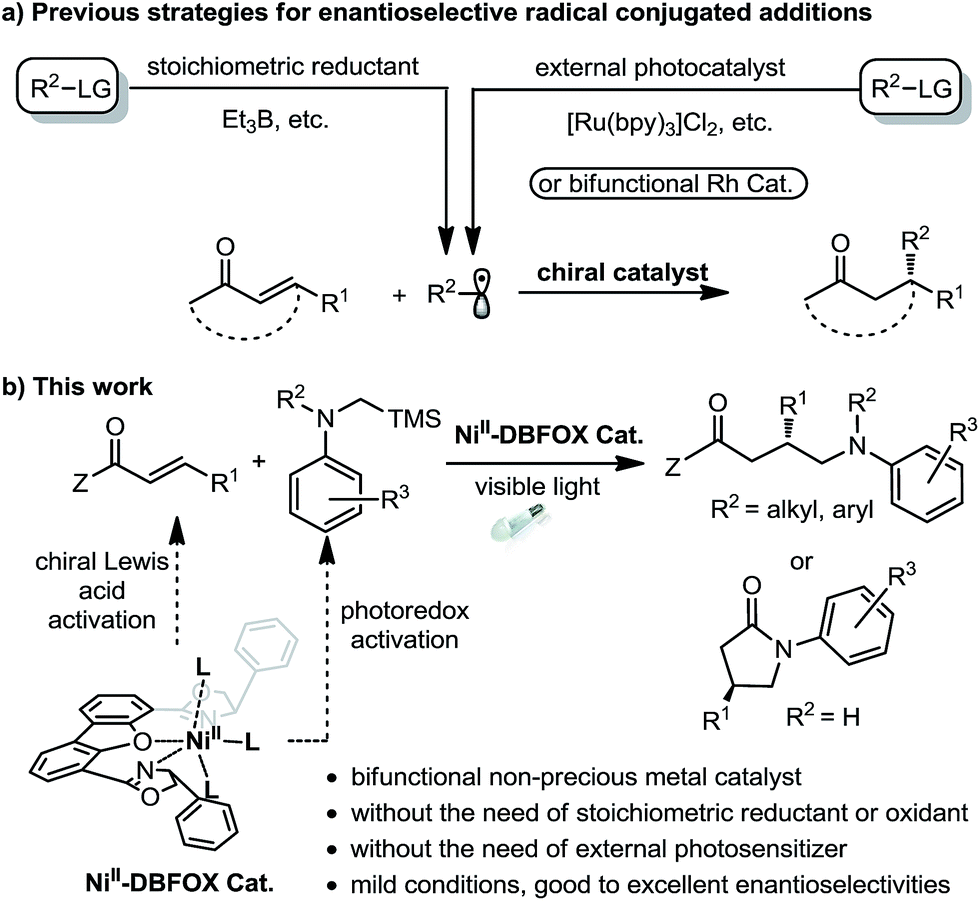 | ||
| Fig. 1 Previous strategies for enantioselective radical conjugate additions and that developed in this study. | ||
Despite the impressive advances, typically two catalysts are required for this type of transformation: one photocatalyst for the radical generation and an additional chiral Lewis acid for controlling the stereoselective radical addition. Furthermore, the photocatalyst and the chiral Lewis acid often contain a precious metal.11,12 Using earth-abundant, first-row transition metal complexes instead as bifunctional single catalysts for asymmetric photoredox reactions opens a new avenue for cheap and green synthesis of chiral molecules, but this has been investigated much less.13 As the only established example, Fu's group demonstrated that a chiral copper complex can catalyze light-induced enantioselective C–N cross-couplings, not only as the asymmetric catalyst but also as the precursor of the photocatalyst.14 Although they are inexpensive and well-compatible in photochemical reactions,15 nickel complexes themselves have only been reported as potential photoredox catalysts very recently.16,17 Herein, we wish to reveal our discovery on using a readily available NiII–DBFOX complex as a bifunctional catalyst for visible-light-promoted enantioselective reactions between α,β-unsaturated carbonyl compounds and α-silylamines (Fig. 1b).
Results and discussion
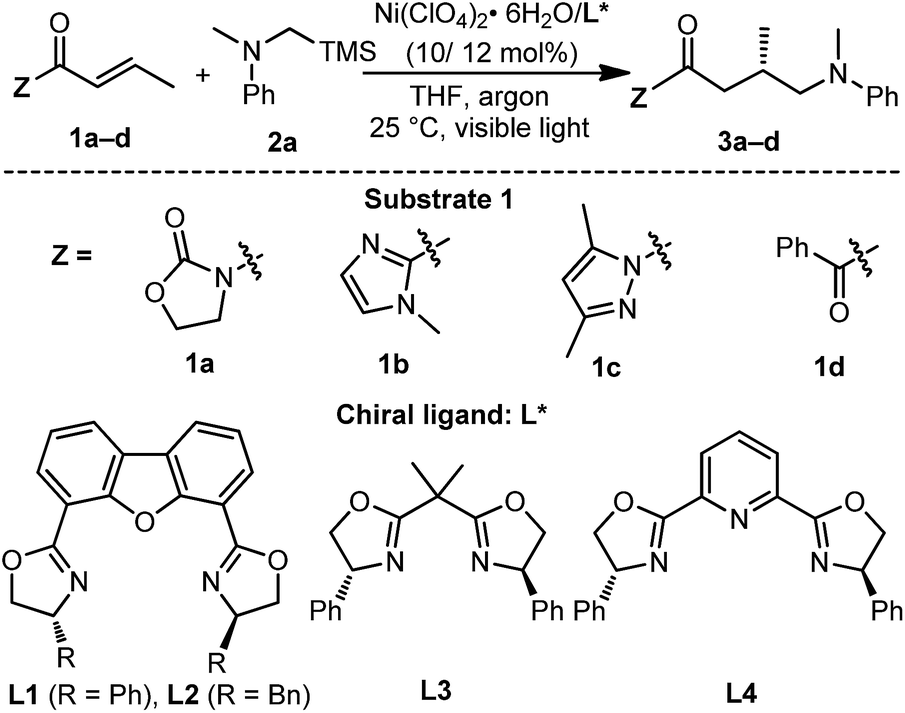
Chiral NiII–DBFOX complexes have been used extensively in catalytic asymmetric nucleophilic additions, halogenation, cycloadditions and other transformations.18 Interestingly, we observed that one member of this class of complexes, Ni–L1, generated in situ by mixing Ni(ClO4)2·6H2O and chiral DBFOX ligand L1 in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio, exhibited obvious blue-green luminescence in THF (Fig. S5 in the ESI†). In combination with its highly organized chiral environment and redox-active metal center, we envisioned that such a complex might be a potential candidate for asymmetric/photoredox bifunctional catalysis.19 With these considerations in mind, we commenced our study with α,β-unsaturated crotonyl oxazolidinone 1a and tertiary α-silylamine 2a as the model substrates. We here chose tertiary α-silylalkylamines as the target substrate because they have low oxidation potentials (for example, Eox (2a˙+/2a) = +0.60 in CH3CN, Fig. S6 in the ESI†) and are well-established to undergo single-electron oxidation followed by rapid desilylation to generate nucleophilic α-aminoalkyl radicals.6,9 However, under irradiation with a 23 W white CFL lamp at 25 °C in the presence of premixed 10 mol% Ni(ClO4)2·6H2O and 12 mol% L1 in THF, no desired product 3a was observed (Table 1, entry 1). Next, we screened α,β-unsaturated carbonyl compounds 1b–d bearing different moieties adjacent to the carbonyl group (entries 2–4). To our delight, the reaction of α,β-unsaturated imidazole 1b led to the formation of 3b in 22% conversion and with 78% ee (entry 2). Most interestingly, α,β-unsaturated N-acyl pyrazole 1c provided significantly improved catalytic outcomes. Adduct 3c was afforded in 95% conversion and with 91% ee within only 6 h (entry 3). Other bisoxazoline ligands, L2–4, were also tested in the reaction, which, however, resulted in a much worse reaction rate and enantioselectivity (entries 5–7). The reaction was further improved by using a 24 W blue LED lamp, providing 3c in 95% conversion and with 91% ee in only 3 h (entry 8). Notably, when we replaced the blue LEDs with 30 W red or yellow LEDs, we failed to obtain the product (entries 9 and 10), while irradiation with a 15 W UV lamp (365 nm) still resulted in the production of 3c in good conversion and with high enantioselectivity (entry 11). These results indicate that the appropriate wavelength range of the light source is very critical for the transformation.
:1 ratio, exhibited obvious blue-green luminescence in THF (Fig. S5 in the ESI†). In combination with its highly organized chiral environment and redox-active metal center, we envisioned that such a complex might be a potential candidate for asymmetric/photoredox bifunctional catalysis.19 With these considerations in mind, we commenced our study with α,β-unsaturated crotonyl oxazolidinone 1a and tertiary α-silylamine 2a as the model substrates. We here chose tertiary α-silylalkylamines as the target substrate because they have low oxidation potentials (for example, Eox (2a˙+/2a) = +0.60 in CH3CN, Fig. S6 in the ESI†) and are well-established to undergo single-electron oxidation followed by rapid desilylation to generate nucleophilic α-aminoalkyl radicals.6,9 However, under irradiation with a 23 W white CFL lamp at 25 °C in the presence of premixed 10 mol% Ni(ClO4)2·6H2O and 12 mol% L1 in THF, no desired product 3a was observed (Table 1, entry 1). Next, we screened α,β-unsaturated carbonyl compounds 1b–d bearing different moieties adjacent to the carbonyl group (entries 2–4). To our delight, the reaction of α,β-unsaturated imidazole 1b led to the formation of 3b in 22% conversion and with 78% ee (entry 2). Most interestingly, α,β-unsaturated N-acyl pyrazole 1c provided significantly improved catalytic outcomes. Adduct 3c was afforded in 95% conversion and with 91% ee within only 6 h (entry 3). Other bisoxazoline ligands, L2–4, were also tested in the reaction, which, however, resulted in a much worse reaction rate and enantioselectivity (entries 5–7). The reaction was further improved by using a 24 W blue LED lamp, providing 3c in 95% conversion and with 91% ee in only 3 h (entry 8). Notably, when we replaced the blue LEDs with 30 W red or yellow LEDs, we failed to obtain the product (entries 9 and 10), while irradiation with a 15 W UV lamp (365 nm) still resulted in the production of 3c in good conversion and with high enantioselectivity (entry 11). These results indicate that the appropriate wavelength range of the light source is very critical for the transformation.
|
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Entry | Substrate | Ligand | Light source | Additives | t (h) | Product | Conv.b (%) | eec (%) |
| a Reaction conditions: 1a–d (0.10 mmol), 2a (0.30 mmol), Ni(ClO4)2·6H2O (10 mol%), ligand L1–4 (12 mol%), THF (0.50 mL), indicated light source, 25 °C, under argon; see more details of the screening of metal salts and solvents in the ESI. b Conversion determined by 1H-NMR. c ee value determined by chiral HPLC. d Reaction performed in the absence of nickel salt. e Reaction performed by replacing Ni(ClO4)2·6H2O with Mg(OTf)2. f Reaction performed by replacing Ni(ClO4)2·6H2O with Ni(COD)2. g Reaction performed in air. n.d. = not determined; n.a. = not applicable. | ||||||||
| 1 | 1a | L1 | White CFL | None | 12 | 3a | 0 | n.a. |
| 2 | 1b | L1 | White CFL | None | 12 | 3b | 22 | 78 |
| 3 | 1c | L1 | White CFL | None | 6 | 3c | 95 | 91 |
| 4 | 1d | L1 | White CFL | None | 12 | 3d | 0 | n.a. |
| 5 | 1c | L2 | White CFL | None | 12 | 3c | 23 | 0 |
| 6 | 1c | L3 | White CFL | None | 12 | 3c | 21 | n.d. |
| 7 | 1c | L4 | White CFL | None | 12 | 3c | 0 | n.a. |
| 8 | 1c | L1 | Blue LEDs | None | 3 | 3c | 95 | 91 |
| 9 | 1c | L1 | Red LEDs | None | 12 | 3c | <5 | n.d. |
| 10 | 1c | L1 | Yellow LEDs | None | 12 | 3c | 0 | n.a. |
| 11 | 1c | L1 | UV (365 nm) | None | 6 | 3c | 90 | 91 |
| 12d | 1c | L1 | Blue LEDs | None | 12 | 3c | 0 | n.a. |
| 13e | 1c | L1 | Blue LEDs | None | 12 | 3c | 0 | n.a. |
| 14f | 1c | L1 | Blue LEDs | None | 12 | 3c | 0 | n.a. |
| 15 | 1c | None | Blue LEDs | None | 12 | 3c | 0 | n.a. |
| 16 | 1c | L1 | None | None | 12 | 3c | <5 | n.a. |
| 17g | 1c | L1 | Blue LEDs | None | 12 | 3c | 0 | n.a. |
| 18 | 1c | L1 | Blue LEDs | 1 eq. TEMPO | 12 | 3c | 0 | n.a. |
| 19 | 1c | L1 | Blue LEDs | 3 eq. BHT | 12 | 3c | 0 | n.a. |
To obtain a better understanding of the system, several control experiments were conducted. As revealed by entries 12–16, the nickel salt, DBFOX ligand L1, and visible light are all essential for the product formation. For example, after removing Ni(ClO4)2·6H2O (entry 12) or replacing Ni(ClO4)2·6H2O with Mg(OTf)2 (entry 13), the reaction did not produce any desired product. Interestingly, the replacement of NiII by Ni0 such as when using Ni(COD)2 as the metal source also led to failure in obtaining 3c, which indicates the possibility of nickel being involved in the redox process (entry 14). Moreover, air (entry 17), radical quencher TEMPO (entry 18) and BHT (entry 19) completely inhibited the transformation of 1c + 2a → 3c. These observations are consistent with a radical pathway.
Next, UV-Vis spectra were recorded to evaluate the light absorptions of the reaction components. The individual substrates 1c and 2a and the chiral ligand L1 do not feature any absorption in the visible light region, while a 1:1 mixture of [Ni(ClO4)·6H2O + L1] and 1:1:1 mixture of [Ni(ClO4)·6H2O + L1 + 1c], leading to the in situ generation of the chiral nickel catalyst Ni–L1 and potential intermediate Ni–L1–1c respectively, exhibit significant absorption enhancement in the range of 400–450 nm (Fig. 2a). The broad peak at ∼600 nm is probably attributed to the nickel center. The spectroscopic analysis indicates that both Ni–L1 and Ni–L1–1c could be the potential photocatalysts. This conjecture was further strengthened by luminescence quenching experiments and Stern–Volmer quenching plots, which revealed obvious intermolecular interactions between Ni–L1 (or Ni–L1–1c) and 2a (Fig. S12–S15 in the ESI†).
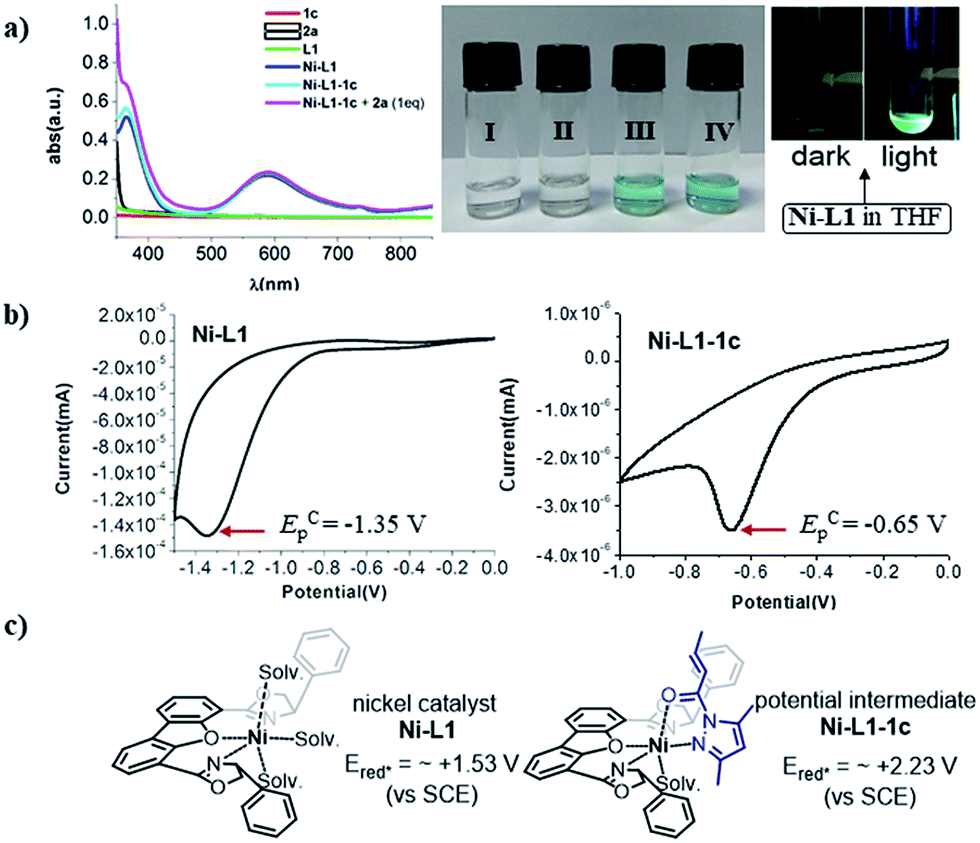 | ||
| Fig. 2 (a) Left: UV-Vis absorption spectra recorded on a Shimadzu UV-2550 in a 10.0 mm quartz cuvette. Middle: I. Substrate 1c in THF (0.030 M). II. Substrate 2a in THF (0.030 M). III. Ni–L1 in THF (0.030 M). IV. Ni–L1–1c in THF (0.030 M). Right: A THF solution of nickel catalyst Ni–L1 (0.030 M) in the dark and in the light. (b) Cyclic voltammogram of nickel catalyst Ni–L1 (0.030 M) and potential intermediate complex Ni–L1–1c (0.030 M) in TBAPF6 (0.10 M) in CH3CN. Sweep rate: 20 mV s−1. A Pt electrode was used as the working electrode, a SCE as the reference electrode, and Pt wire as the auxiliary electrode. EpA is the anodic peak potential. ECp is the cathodic peak potential. (c) Calculated reductive potentials of nickel catalyst Ni–L1 and potential intermediate complex Ni–L1–1c in the excited states. | ||
The cyclic voltammogram of α-silylamine 2a exhibited an irreversible oxidation at +0.60 V (Eox (2a˙+/2a)) in CH3CN (Fig. S6 in the ESI†), while those of Ni–L1 and Ni–L1–1c showed reductive peaks at −1.35 V and −0.65 V respectively (Fig. 2b), most likely corresponding to the reduction of NiII to NiI. The excited-state potentials of the nickel complexes were further calculated by means of the Rehm–Weller formalism (E(I*/I˙−) = E(I/I˙−) + E0,0(I*/I)).19,20 Accordingly, 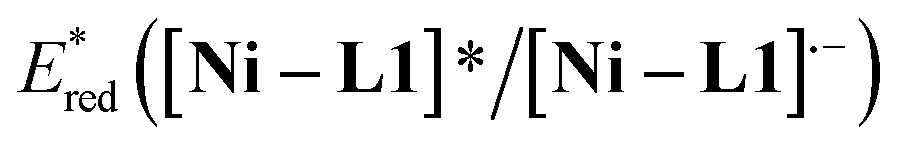 was estimated to be ∼+1.53 V, while
was estimated to be ∼+1.53 V, while 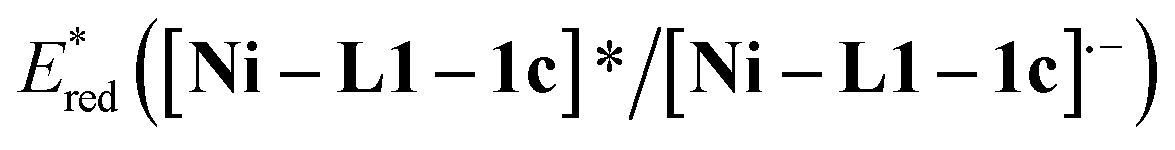 was estimated to be ∼+2.23 V (Fig. 2c). Notably, the calculated reductive potential in the excited state of nickel catalyst Ni–L1 is comparable to that of a NiII complex which was reported as a potential photoredox catalyst by Bach and Hess very recently.17b The electrochemical outcomes suggest that single electron transfer (SET) from the α-silylamine 2a to the excited NiII complexes was thermodynamically favorable.
was estimated to be ∼+2.23 V (Fig. 2c). Notably, the calculated reductive potential in the excited state of nickel catalyst Ni–L1 is comparable to that of a NiII complex which was reported as a potential photoredox catalyst by Bach and Hess very recently.17b The electrochemical outcomes suggest that single electron transfer (SET) from the α-silylamine 2a to the excited NiII complexes was thermodynamically favorable.
Furthermore, the light–dark interval experiments revealed that continuous irradiation with visible light was essential for the reaction (Fig. 3a). The quantum yield (Φ) of the photochemical reaction was calculated to be 0.59, suggesting that radical chain propagation would not be the predominant mechanism (see more details in Section 5.11 of the ESI†).20 In addition, the reaction with the addition of 3 equiv. of ethyl 2-((phenylsulfonyl)methyl)acrylate at the standard conditions led to the observation of product 3r in 13% yield and by-product 4 in 67% yield (Fig. 3b). Isolation of compound 4 further confirms the involvement of an α-amino radical. The homolytic cleavage of α-silylamine 2a by light was excluded by the photostability test (Fig. S16 in the ESI†).
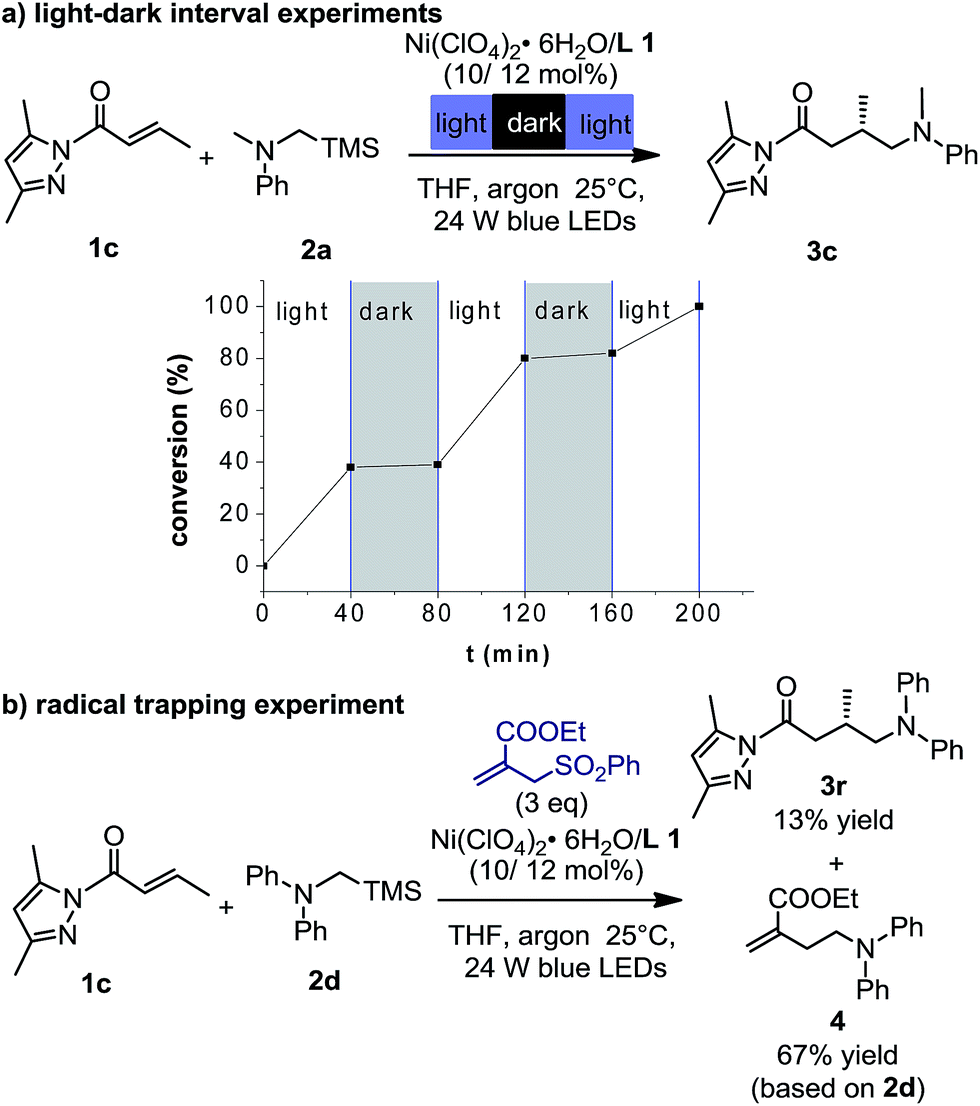 | ||
| Fig. 3 Mechanistic investigation. (a) Intervals of irradiation and dark periods for the nickel-catalyzed reaction 1c + 2a → 3c. (b) Radical trapping experiment in the nickel-catalyzed reaction 1c + 2d → 3r. | ||
On the basis of the initial experiments, mechanistic investigations and related literature,6,10 we propose a plausible reaction mechanism as depicted in Fig. 4. Accordingly, N-acyl pyrazole substrate 1 undergoes fast ligand exchange with the chiral nickel catalyst and affords the intermediate complex A (the above-mentioned potential intermediate Ni–L1–1). On the other hand, the photon-absorbing nickel complex Ni–L1 or Ni–L1–1 is activated by visible light, and then oxidizes α-silylamine 2 by a single electron transfer to generate silylamine cation radical B and the reduced [Ni–L1]˙− (only Ni–L1 as the photocatalyst is shown in the figure). Subsequent desilylation of B led to the formation of the nucleophilic α-aminoalkyl radical C,21,22 which undergoes radical addition to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond of complex A in an enantioselective fashion and transformation to radical species D. Reduction of D by strong reductant [Ni–L1]˙− produces anion E, followed by protonation to afford neutral complex F. Finally, substitution with 1 released chiral product 3 and regenerated intermediate A.
C double bond of complex A in an enantioselective fashion and transformation to radical species D. Reduction of D by strong reductant [Ni–L1]˙− produces anion E, followed by protonation to afford neutral complex F. Finally, substitution with 1 released chiral product 3 and regenerated intermediate A.
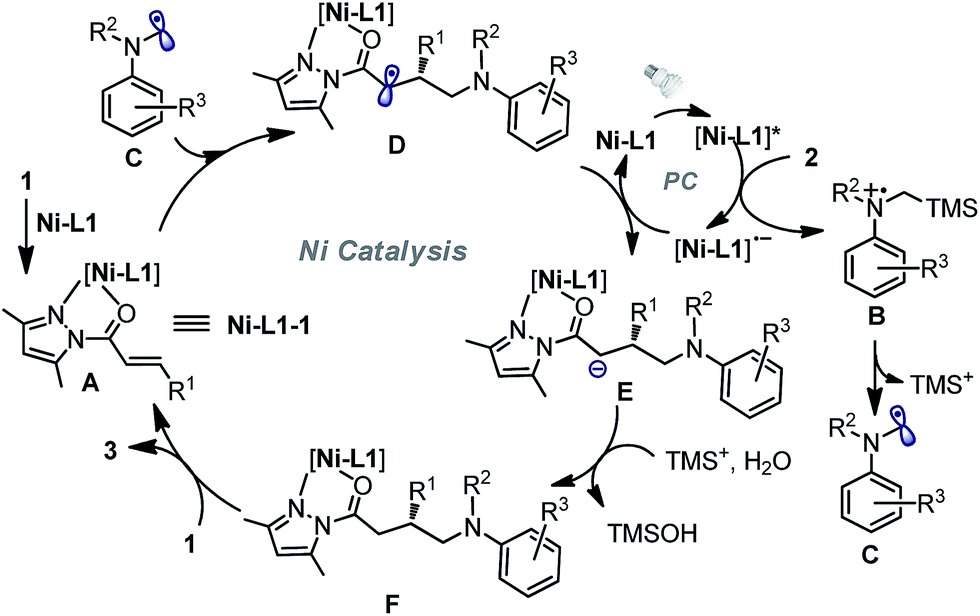 | ||
| Fig. 4 A proposed reaction mechanism for the nickel-catalyzed enantioselective photoredox reaction of α,β-unsaturated carbonyl compounds and α-silylamines. | ||
Considering the steric hindrance created by the tridentate and bidentate coordination, we assume that the nickel center does not interact directly with the free carbon radicals in the nickel catalysis cycle (Fig. 5).15 The metal instead serves as a Lewis acid to activate the electrophilic N-acyl pyrazole through a bidentate coordination.10,16 In the photocatalysis cycle, photon-absorbing nickel complex Ni–L1 or Ni–L1–1 participates in the light-induced single electron transfer process. It has to be mentioned that a radical coupling pathway involving a nickel enolate radical cannot be completely excluded (see more details in Section 5.10 of the ESI†).7c,23 Concerning the stereochemistry, the proposed transition state is fully consistent with an observed S-configuration in the product (Fig. 5).
 | ||
| Fig. 5 proposed transition state for radical addition. | ||
With the optimal conditions and better understanding of the reaction mechanism, we next evaluated the substrate scope of the nickel-catalyzed enantioselective photoredox reaction of α,β-unsaturated carbonyl compounds and α-silylamines. As summarized in Fig. 6, a range of α,β-unsaturated N-acyl pyrazoles containing aliphatic (products 3c and e–h), aromatic (products 3l–p) or electron-donating (product 3i) substituents at the β-position were well tolerated. The products were obtained in 47–81% yields and with 80–99% ee. However, a strong electron-withdrawing β-CF3 or β-CO2Me substituent led to the further cyclized product 3j or 3k. Although the yields and diastereoselectivities were good, the enantioselectivities were both close to zero, which is most likely attributed to uncatalyzed background radical additions.
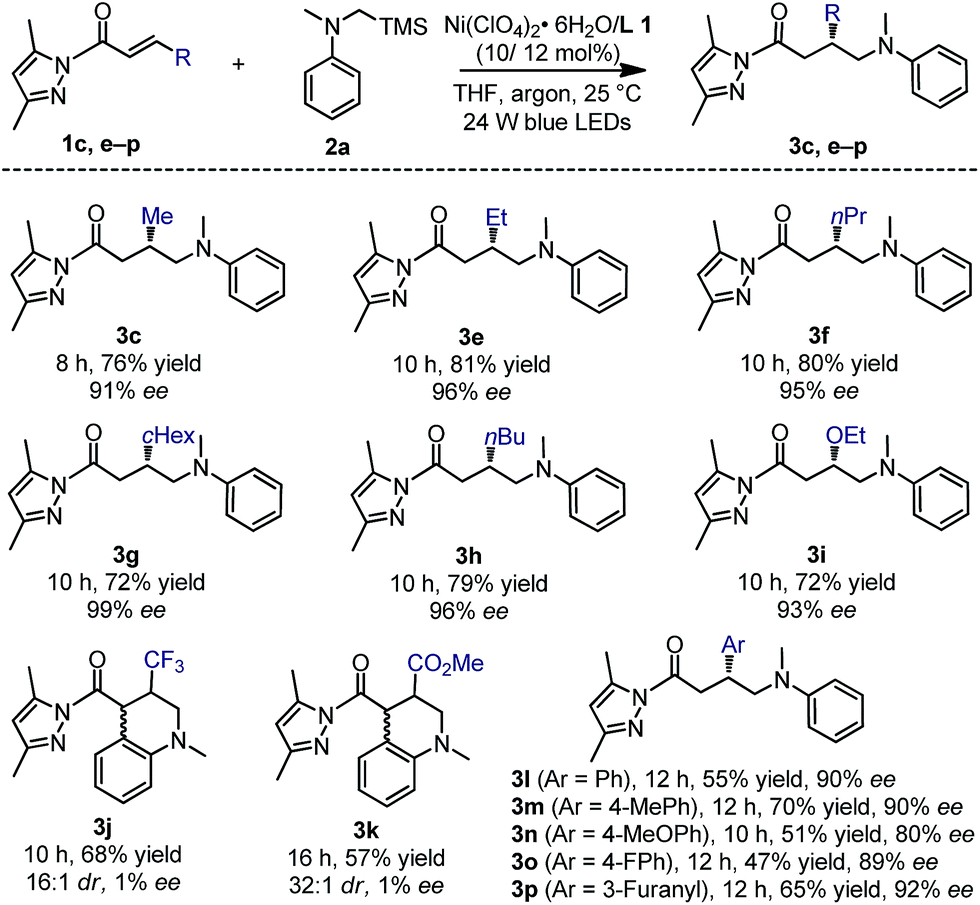 | ||
| Fig. 6 Substrate scope with respect to α,β-unsaturated N-acyl pyrazoles. | ||
The tertiary α-silylamines bearing more sterically demanding N-substituents (products 3q–t), and electron donating (3u–v) or withdrawing groups on the aniline moiety (3x–za) were also compatible (Fig. 7) with respect to the yields (32–86%) and enantioselectivities (87–95% ee). However, a methyl substituent in the 2-position of the aniline ring failed to provide the desired product 3w, perhaps due to steric hindrance.
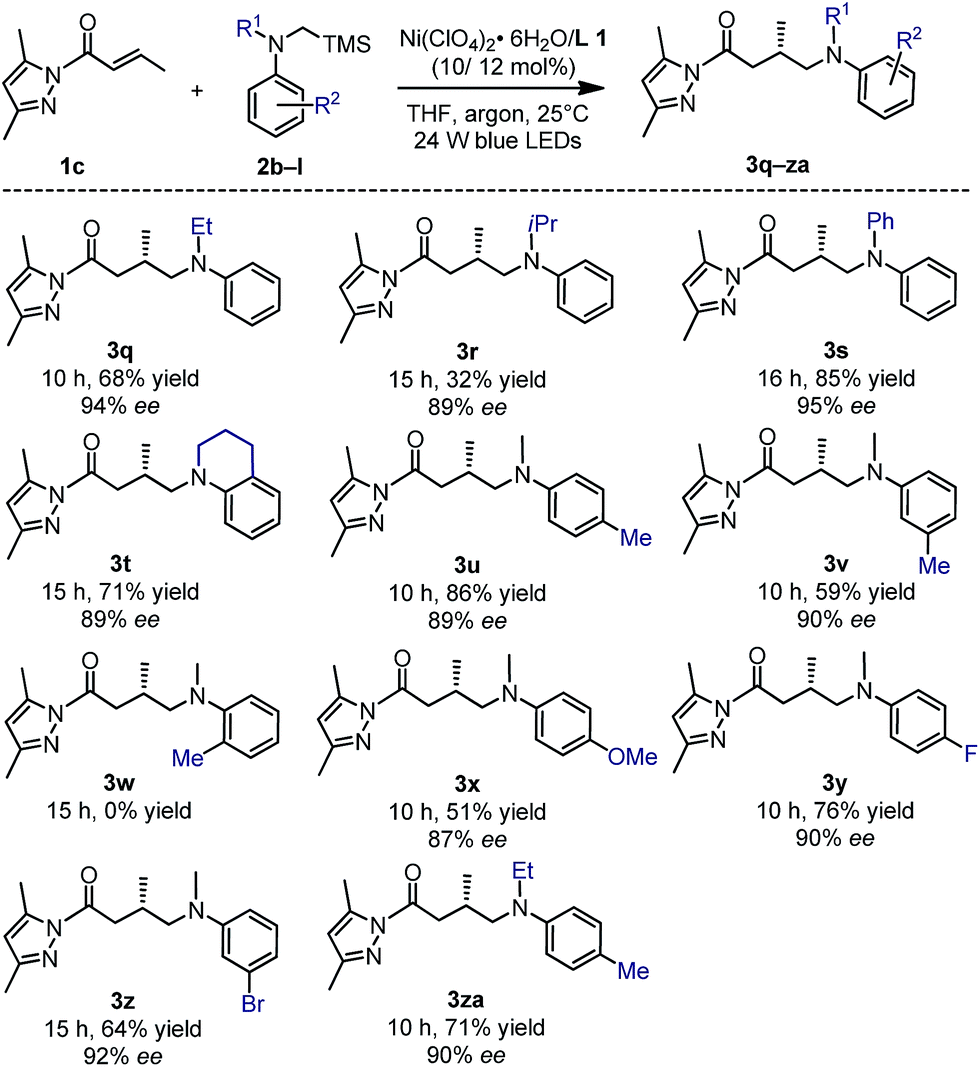 | ||
| Fig. 7 Substrate scope with respect to tertiary α-silylamines. | ||
The N-acyl pyrazole moiety can be readily converted to a range of functional groups under mild conditions.24 As an example, the reaction of 3s (95% ee) with sodium borohydride in a 1:1 mixture of THF and water afforded the corresponding alcohol (S)-5 in a yield of 94% and with 95% ee (Fig. 8a). Accordingly, the absolute configuration of 3s was assigned as S.6 More interestingly, if the tertiary α-silylamines were replaced by secondary α-silylamines in the nickel-catalyzed enantioselective photoredox reaction, further lactamization occurred to afford chiral γ-lactams (3zb–zg, 63–80% yield, 91–93% ee), one class of important building blocks in synthetic chemistry (Fig. 8b).25
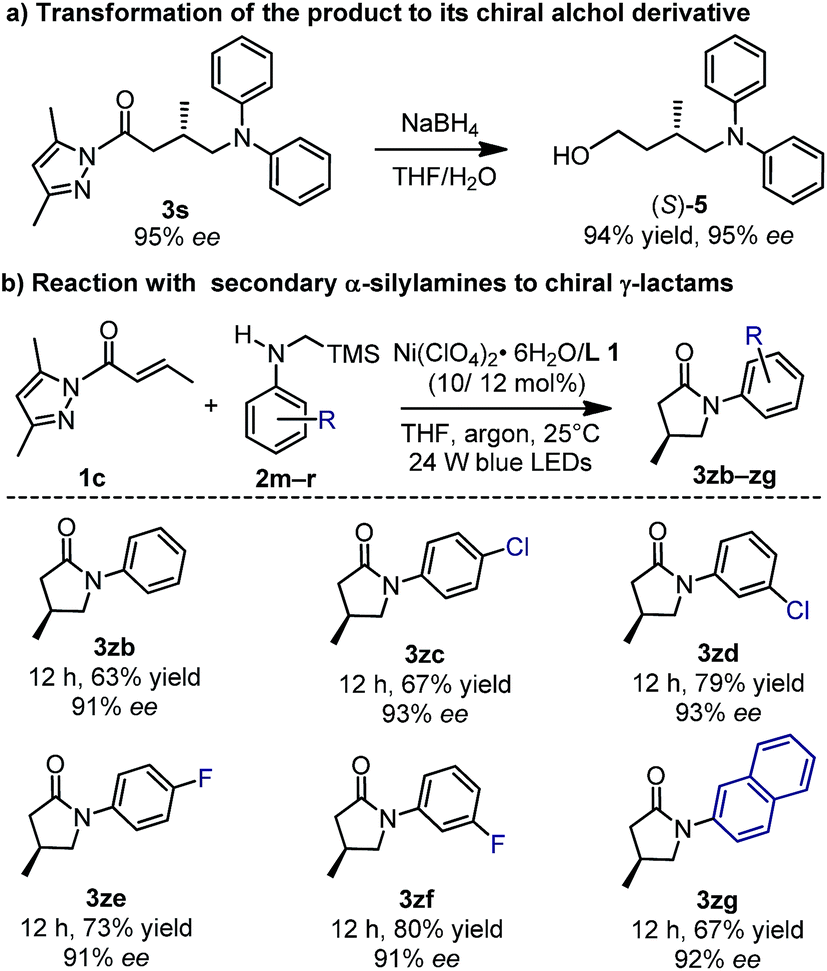 | ||
| Fig. 8 Synthetic applications of the methodology. | ||
Conclusions
In summary, we have revealed that a conventional NiII–DBFOX complex can effectively catalyze the asymmetric photoredox reaction between α,β-unsaturated carbonyl compounds and tertiary/secondary α-silylamines. As a bifunctional catalyst, the chiral nickel complex is not only involved in a photo-induced single electron transfer process to initiate the formation of the radical species, but also activates the α,β-unsaturated carbonyl substrates as a Lewis acid and governs the radical transformation in an enantioselective fashion. Good to excellent yields and enantioselectivities were achieved for the chiral γ-amino carboxylic acid derivatives and γ-lactam products. In view of the non-precious, low-toxic metal salt, readily available chiral ligand, mild reaction conditions and high asymmetric induction, this strategy might provide new opportunities to develop cheap and green synthesis of chiral molecules. Further investigations of reaction mechanisms and applications of the catalytic system in asymmetric photoredox catalysis are ongoing in our laboratory.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge funding from the National Natural Science Foundation of China (grant no. 21572184 and 21472154), the Natural Science Foundation of Fujian Province of China (grant no. 2017J06006), and the Fundamental Research Funds for the Central Universities (grant no. 20720160027). We thank Dr Yan Liu and Mr Xu Yang of Xiamen University for their assistance in HRMS analysis.Notes and references
- For selected reviews on visible-light photoredox catalysis, see: (a) J. M. R. Narayanam and C. R. J. Stephenson, Chem. Soc. Rev., 2011, 40, 102–113 RSC; (b) J. Xuan and W.-J. Xiao, Angew. Chem., Int. Ed., 2012, 51, 6828–6838 CrossRef CAS PubMed; (c) L. Shi and W. Xia, Chem. Soc. Rev., 2012, 41, 7687–7691 RSC; (d) C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS PubMed; (e) Y. Xi, H. Yi and A. Lei, Org. Biomol. Chem., 2013, 11, 2387–2403 RSC; (f) J. Xuan, L.-Q. Lu, J.-R. Chen and W.-J. Xiao, Eur. J. Org. Chem., 2013, 2013, 6755–6770 CrossRef CAS; (g) M. Reckenthaeler and A. G. Griesbeck, Adv. Synth. Catal., 2013, 355, 2727–2744 CrossRef CAS; (h) D. A. Nicewicz and T. M. Nguyen, ACS Catal., 2014, 4, 355–360 CrossRef CAS; (i) S. Fukuzumi and K. Ohkubo, Org. Biomol. Chem., 2014, 12, 6059–6071 RSC; (j) R. A. Angnes, Z. Li, C. R. D. Correia and G. B. Hammond, Org. Biomol. Chem., 2015, 13, 9152–9167 RSC; (k) J.-R. Chen, X.-Q. Hu, L.-Q. Lu and W.-J. Xiao, Chem. Soc. Rev., 2016, 45, 2044–2056 RSC; (l) J. Zhu, W.-C. Yang, X.-D. Wang and L. Wu, Adv. Synth. Catal., 2018, 360, 386–400 CrossRef CAS.
- A. G. Amador and T. P. Yoon, Angew. Chem., Int. Ed., 2016, 55, 2304–2306 CrossRef CAS PubMed.
- For recent reviews on asymmetric photoredox catalysis, see: (a) R. Brimioulle, D. Lenhart, M. M. Maturi and T. Bach, Angew. Chem., Int. Ed., 2015, 54, 3872–3890 CrossRef CAS PubMed; (b) E. Meggers, Chem. Commun., 2015, 51, 3290–3301 RSC; (c) Z.-Y. Cao, W. D. G. Brittain, J. S. Fossey and F. Zhou, Catal. Sci. Technol., 2015, 5, 3441–3451 RSC; (d) C. Wang and Z. Lu, Org. Chem. Front., 2015, 2, 179–190 RSC; (e) H. Huo and E. Meggers, Chimia, 2016, 70, 186–191 CrossRef CAS PubMed; (f) E. Meggers, Angew. Chem., Int. Ed., 2017, 56, 5668–5675 CrossRef CAS PubMed.
- (a) J. H. Wu, G. R. Zhang and N. A. Porter, J. Am. Chem. Soc., 1995, 117, 11029–11030 CrossRef CAS; (b) M. P. Sibi and J. Ji, J. Am. Chem. Soc., 1996, 118, 9200–9201 CrossRef CAS; (c) J. H. Wu, G. R. Zhang and N. A. Porter, Tetrahedron Lett., 1997, 38, 2067–2070 CrossRef CAS; (d) N. A. Porter, H. Feng and I. K. Kavrakova, Tetrahedron Lett., 1999, 40, 6713–6716 CrossRef CAS; (e) M. P. Sibi, J. Ji, J. B. Sausker and C. P. Jasperse, J. Am. Chem. Soc., 1999, 121, 7517–7526 CrossRef CAS; (f) M. Murakata, H. Tsutsui and O. Hoshino, Org. Lett., 2001, 3, 299–302 CrossRef CAS PubMed; (g) M. P. Sibi and J. Chen, J. Am. Chem. Soc., 2001, 123, 9472–9473 CrossRef CAS PubMed; (h) M. P. Sibi and S. Manyem, Org. Lett., 2002, 4, 2929–2932 CrossRef CAS PubMed; (i) M. P. Sibi, J. Zimmerman and T. Rheault, Angew. Chem., Int. Ed., 2003, 42, 4521–4523 CrossRef CAS PubMed; (j) G. K. Friestad, Y. Shen and E. L. Ruggles, Angew. Chem., Int. Ed., 2003, 42, 5061–5063 CrossRef CAS PubMed; (k) M. P. Sibi, G. Petrovic and J. Zimmerman, J. Am. Chem. Soc., 2005, 127, 2390–2391 CrossRef CAS PubMed; (l) S. Lee, C. J. Lim, S. Kim, R. Subramaniam, J. Zimmerman and M. P. Sibi, Org. Lett., 2006, 8, 4311–4313 CrossRef CAS PubMed; (m) M. P. Sibi and J. Zimmermann, J. Am. Chem. Soc., 2006, 128, 13346–13347 CrossRef CAS PubMed; (n) M. P. Sibi, Y.-H. Yang and S. Lee, Org. Lett., 2008, 10, 5349–5352 CrossRef CAS PubMed; for an account, see: (o) M. P. Sibi and N. A. Porter, Acc. Chem. Res., 1999, 32, 163–171 CrossRef CAS.
- For leading reviews on dual catalysis by combining photoredox with organo-, acid, and transition-metal catalysis, see: (a) M. N. Hopkinson, B. Sahoo, J.-L. Li and F. Glorius, Chem.–Eur. J., 2014, 20, 3874–3886 CrossRef CAS PubMed; (b) T. P. Yoon, Acc. Chem. Res., 2016, 49, 2307–2315 CrossRef CAS PubMed; (c) K. L. Skubi, T. R. Blum and T. P. Yoon, Chem. Rev., 2016, 116, 10035–10074 CrossRef CAS PubMed.
- L. R. Espelt, I. S. McPherson, E. M. Wiensch and T. P. Yoon, J. Am. Chem. Soc., 2015, 137, 2452–2455 CrossRef PubMed.
- (a) H. Huo, K. Harms and E. Meggers, J. Am. Chem. Soc., 2016, 138, 6936–6939 CrossRef CAS PubMed; (b) C. Y. Wang, K. Harms and E. Meggers, Angew. Chem., Int. Ed., 2016, 55, 13495–13498 CrossRef CAS PubMed; (c) J. Ma, X. Xie and E. Meggers, Chem.–Eur. J., 2018, 24, 259–265 CrossRef CAS PubMed.
- For a recent account, see: L. Zhang and E. Meggers, Acc. Chem. Res., 2017, 50, 320–330 CrossRef CAS PubMed.
- S.-X. Lin, G.-J. Sun and Q. Kang, Chem. Commun., 2017, 53, 7665–7668 RSC.
- J. Liu, W. Ding, Q.-Q. Zhou, D. Liu, L.-Q. Liu and W.-J. Xiao, Org. Lett., 2018, 20, 461–464 CrossRef CAS PubMed.
- For a sophisticated bifunctional Lewis acid/photocatalyst, see: H. Huo, X. Shen, C. Wang, L. Zhang, P. Röse, L.-A. Chen, K. Harms, M. Marsch, G. Hilt and E. Meggers, Nature, 2014, 515, 100–103 CrossRef CAS PubMed.
- For selected examples of merging photoredox catalysis with asymmetric organocatalysis, see: (a) D. A. Nicewicz and D. W. C. MacMillan, Science, 2008, 322, 77–80 CrossRef CAS PubMed; (b) M. Neumann, S. Füldner, B. König and K. Zeitler, Angew. Chem., Int. Ed., 2011, 50, 951–954 CrossRef CAS PubMed; (c) K. Fidaly, C. Ceballos, A. Falguières, M. S.-I. Veitia, A. Guy and C. Ferroud, Green Chem., 2012, 14, 1293–1297 RSC; (d) D. A. DiRocco and T. Rovis, J. Am. Chem. Soc., 2012, 134, 8094–8097 CrossRef CAS PubMed; (e) L. J. Rono, H. G. Yayla, D. Y. Wang, M. F. Armstrong and R. R. Knowles, J. Am. Chem. Soc., 2013, 135, 17735–17738 CrossRef CAS PubMed; (f) Y. Zhu, L. Zhang and S.-Z. Luo, J. Am. Chem. Soc., 2014, 136, 14642–14645 CrossRef CAS PubMed; (g) P. Riente, A. M. Adams, J. Albero, E. Palomares and M. A. Percàs, Angew. Chem., Int. Ed., 2014, 53, 9613–9616 CrossRef CAS PubMed; (h) G. Bergonzini, C. S. Schindler, C.-J. Wallentin, E. N. Jacobsen and C. R. J. Stephenson, Chem. Sci., 2014, 5, 112–116 RSC; (i) G. Wei, C. Zhang, F. Bures, X. Ye, C.-H. Tan and Z. Jiang, ACS Catal., 2016, 6, 3708–3712 CrossRef CAS; (j) E. Larionov, M. M. Mastandrea and M. A. Pericàs, ACS Catal., 2017, 7, 7008–7013 CrossRef CAS.
- A. Gualandi, M. Marchini, L. Mengozzi, M. Natali, M. Lucarini, P. Ceroni and P. G. Cozzi, ACS Catal., 2015, 5, 5927–5931 CrossRef CAS.
- Q. M. Kainz, C. D. Matier, A. Bartoszewicz, S. L. Zultanski, J. C. Peters and G. C. Fu, Science, 2016, 351, 681–684 CrossRef CAS PubMed.
- For selected examples, see: (a) J. C. Tellis, D. N. Primer and G. A. Molander, Science, 2014, 345, 433–436 CrossRef CAS PubMed; (b) C. P. Johnston, R. T. Smith, S. Almendinger and D. W. C. MacMillan, Nature, 2016, 536, 322–325 CrossRef CAS PubMed; (c) D. R. Heitz, J. C. Tellis and G. A. Molander, J. Am. Chem. Soc., 2016, 138, 12715–12718 CrossRef CAS PubMed; (d) B. Shields and A. G. Doyle, J. Am. Chem. Soc., 2016, 138, 12719–12722 CrossRef CAS PubMed; (e) Z. Zuo, H. Cong, W. Li, J. Choi, G. C. Fu and D. W. C. MacMillan, J. Am. Chem. Soc., 2016, 138, 1832–1835 CrossRef CAS PubMed; (f) P. Zhang, C. C. Le and D. W. C. MacMillan, J. Am. Chem. Soc., 2016, 138, 8084–8087 CrossRef CAS PubMed; (g) S. Zheng, D. N. Primer and G. A. Molander, ACS Catal., 2017, 7, 7957–7961 CrossRef CAS PubMed; (h) C. Remeur, C. B. Kelly, N. R. Patel and G. A. Molander, ACS Catal., 2017, 7, 6065–6069 CrossRef CAS PubMed; (i) K. Lin, R. J. Wiles, C. B. Kelly, G. H. M. Davies and G. A. Molander, ACS Catal., 2017, 7, 5129–5133 CrossRef CAS PubMed; (j) X. Zhang and D. W. C. MacMillan, J. Am. Chem. Soc., 2017, 139, 11353–11356 CrossRef CAS PubMed; (k) H. Huang, X. Li, C. Yu, Y. Zhang, P. S. Mariano and W. Wang, Angew. Chem., Int. Ed., 2017, 56, 1500–1505 CrossRef CAS PubMed; (l) D. N. Primer and G. A. Molander, J. Am. Chem. Soc., 2017, 139, 9847–9850 CrossRef CAS PubMed.
- For a recent study on a nickel complex containing a visible-light-responsive chiral ligand for metal-photocatalytic asymmetric aerobic oxidation reaction, see: W. Ding, L.-Q. Lu, Q.-Q. Zhou, Y. Wei, J.-R. Chen and W.-J. Xiao, J. Am. Chem. Soc., 2017, 139, 63–66 CrossRef CAS PubMed.
- For two very recent studies on establishing nickel(II) complexes as the potential visible-light photocatalyst, see: (a) B. J. Shields, B. Kudisch, G. D. Scholes and A. G. Doyle, J. Am. Chem. Soc., 2018, 140, 3035–3039 CrossRef CAS PubMed; (b) M. Grübel, I. Bosque, P. J. Altmann, T. Bach and C. R. Hess, Chem. Sci., 2018, 9, 3313–3317 RSC.
- For selected examples, see: (a) S. Kanemasa, Y. Oderaotoshi, S.-i. Sakaguchi, H. Yamamoto, J. Tanaka, E. Wada and D. P. Curran, J. Am. Chem. Soc., 1998, 120, 3074–3088 CrossRef CAS; (b) K. Itoh and S. Kanemasa, J. Am. Chem. Soc., 2002, 124, 13394–13395 CrossRef CAS PubMed; (c) N. Shibata, J. Kohno, K. Takai, T. Ishimaru, S. Nakamura, T. Toru and S. Kanemasa, Angew. Chem., Int. Ed., 2005, 44, 4204–4207 CrossRef CAS PubMed; (d) I. Kennosuke, H. Masayuki, T. Junji and K. Shuji, Org. Lett., 2005, 7, 979–981 CrossRef PubMed; (e) J. Esquivias, R. G. Arrayás and J. C. Carretero, J. Am. Chem. Soc., 2007, 129, 1480–1481 CrossRef CAS PubMed; (f) T. Ishimaru, S. Ogawa, E. Tokunaga, S. Nakamura and N. Shibata, J. Fluorine Chem., 2009, 130, 1049–1053 CrossRef CAS; (g) D. S. Reddy, N. Shibata, T. Horikawa, S. Suzuki, S. Nakamura, T. Toru and M. Shiro, Chem.–Asian J., 2009, 4, 1411–1415 CrossRef CAS PubMed; (h) D. S. Reddy, N. Shibata, J. Nagai, S. Nakamura and T. Toru, Angew. Chem., Int. Ed., 2009, 48, 803–806 CrossRef PubMed; (i) X. Yang, F. Cheng, Y.-D. Kou, S. Pang, Y.-C. Shen, Y.-Y. Huang and N. Shibata, Angew. Chem., Int. Ed., 2017, 56, 1510–1514 CrossRef CAS PubMed.
- T. C. Jenks, M. D. Bailey, J. L. Hovey, S. Fernando, G. Basnayake, M. E. Cross, W. Li and M. J. Allen, Chem. Sci., 2018, 9, 1273–1278 RSC.
- (a) E. Arceo, I. D. Jurberg, A. Álvarez-Fernández and P. Melchiorre, Nat. Chem., 2013, 5, 750–756 CrossRef CAS PubMed; (b) M. Silvi, C. Verrier, Y. P. Rey, L. Buzzetti and P. Melchiorre, Nat. Chem., 2017, 9, 868–873 CrossRef CAS PubMed.
- For a recent account on synthetic utilization of α-aminoalkyl radicals in visible-light photoredox catalysis, see: K. Nakajima, Y. Miyake and Y. Nishibayashi, Acc. Chem. Res., 2016, 49, 1946–1956 CrossRef CAS PubMed.
- For a perspective concerning practicality and chemoselectivity of radical transformations, see: M. Yan, J. C. Lo, J. T. Edwards and P. S. Baran, J. Am. Chem. Soc., 2016, 138, 12692–12714 CrossRef CAS PubMed.
- M. P. DeMartino, K. Chen and P. S. Baran, J. Am. Chem. Soc., 2008, 130, 11546–11560 CrossRef CAS PubMed.
- L. Feng, X. Dai, E. Meggers and L. Gong, Chem.–Asian J., 2017, 12, 963–967 CrossRef CAS PubMed.
- For representative reviews, see: (a) C. Nájera and M. Yus, Tetrahedron: Asymmetry, 1999, 10, 2245–2303 CrossRef; (b) S. Hanessian and L. Auzzas, Acc. Chem. Res., 2008, 41, 1241–1251 CrossRef CAS PubMed; (c) A. Stefanucci, E. Novellino, R. Costante and A. Mollica, Heterocycles, 2014, 89, 1801–1825 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8sc01219a |
| This journal is © The Royal Society of Chemistry 2018 |