
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Deciphering the working mechanism of aggregation-induced emission of tetraphenylethylene derivatives by ultrafast spectroscopy†
Yuanjing
Cai‡
 abc,
Lili
Du‡
dg,
Kerim
Samedov‡
e,
Xinggui
Gu‡
a,
Fei
Qi‡
f,
Herman H. Y.
Sung
a,
Brian O.
Patrick
e,
Zhiping
Yan
d,
Xiaofang
Jiang
b,
Haoke
Zhang
a,
Jacky W. Y.
Lam
a,
Ian D.
Williams
a,
David
Lee Phillips
*d,
Anjun
Qin
*b and
Ben Zhong
Tang
*abc
abc,
Lili
Du‡
dg,
Kerim
Samedov‡
e,
Xinggui
Gu‡
a,
Fei
Qi‡
f,
Herman H. Y.
Sung
a,
Brian O.
Patrick
e,
Zhiping
Yan
d,
Xiaofang
Jiang
b,
Haoke
Zhang
a,
Jacky W. Y.
Lam
a,
Ian D.
Williams
a,
David
Lee Phillips
*d,
Anjun
Qin
*b and
Ben Zhong
Tang
*abc
aDepartment of Chemistry, Hong Kong Branch of Chinese National Engineering Research Center for Tissue Restoration and Reconstruction, The Hong Kong University of Science & Technology, Clear Water Bay, Kowloon, Hong Kong SAR, China. E-mail: tangbenz@ust.hk
bCenter for Aggregation-Induced Emission, NSFC Center for Luminescence from Molecular Aggregates, SCUT-HKUST Joint Research Institute, State Key Laboratory of Luminescent Materials and Devices, South China University of Technology, Guangzhou 510640, China. E-mail: msqinaj@scut.edu.cn
cHKUST Shenzhen Research Institute, No. 9 Yuexing 1st RD, South Area, Hitech Park Nanshan, Shenzhen 518057, China
dDepartment of Chemistry, The University of Hong Kong, Pokfulam Road, Hong Kong SAR, China. E-mail: phillips@hku.hk
eDepartment of Chemistry, University of British Columbia, 2036 Main Mall, Vancouver, British Columbia, Canada V6T 1Z1
fInstitute of Computational and Theoretical Studies & Department of Physics, Hong Kong Baptist University, Hong Kong SAR, China
gInstitute of Life Sciences, Jiangsu University, Zhenjiang 212013, China
First published on 24th April 2018
Abstract
Aggregation-induced emission (AIE) is the long-sought solution to the problem of aggregation-caused quenching that has hampered efficient application of fluorescent organic materials. An important goal on the way to fully understand the working mechanism of the AIE process was, for more than a decade, and still remains obtaining more comprehensive insights into the correlation between the ultrafast excited-state dynamics in tetraphenylethylene (TPE)-based molecules and the AIE effect in them. Here we report a number of TPE-based derivatives with varying structural rigidities and AIE properties. Using a combination of ultrafast time-resolved spectroscopy and computational studies, we observe a direct correlation between the state-dependent coupling motions and inhibited fluorescence, and prove the existence of photocyclized intermediates in them. We demonstrate that the dominant non-radiative relaxation dynamics, i.e. formation of intermediate or rotation around the elongated C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond, is responsible for the AIE effect, which is strongly structure-dependent but not related to structural rigidity.
C bond, is responsible for the AIE effect, which is strongly structure-dependent but not related to structural rigidity.
Introduction
In less than two decades, the seminal discovery of aggregation-induced emission (AIE)1,2 has revolutionized the research fields of bioimaging,3–5 chemo/biosensing,6–8 and optoelectronic materials sciences.9,10 AIE describes the phenomenon of molecules with low fluorescence quantum yields in solution “lighting up” with enhanced fluorescence emission upon aggregation. The AIE effect has proven to be an effective molecular-level remedy for the aggregation-caused quenching effect,11,12 which has been a major obstacle in the way of many practical applications of most fluorescent materials for decades. However, the working mechanism behind the AIE phenomenon remains a subject of much debate and intense elucidation efforts.13–27 Among several hypotheses concerning the mechanism of the AIE effect, the restriction of intramolecular rotation (RIR)15–19 and restriction of intramolecular vibration (RIV)20–22 subsumed under the overarching concept of restriction of intramolecular motion (RIM)2,21 are the most frequently cited ones, with photocyclization23–25 as well as E–Z isomerization18,26,27 being invoked in a number of cases. Although the discourse nowadays appears to lend much support to the general mechanism of RIM due to an extensive body of experimental evidence substantiated in part by computational studies, a clear, consistent, and deep fundamental understanding of different elemental relaxation processes associated with the basic molecular motions in non-radiative excited-state dynamics even in the simplest archetypal AIE-active compounds such as tetraphenylethylene (TPE) is yet to emerge.The excited-state dynamics in TPE and TPE derivatives, including the role of phenyl rotation,15,28,29 CC twisting30–33 and photocyclization,24,25,34 have been under debate and investigation for almost four decades. Certain aspects of it, such as how the non-radiative relaxation takes place, what photophysical processes are involved, what the relationships between the intramolecular motions and their respective relative contributions to the AIE mechanism are, and whether they involve photochemical reactions, have not been sufficiently, if at all, addressed neither in previous nor in contemporary subject-related reports. It is thus of fundamental importance to develop a detailed understanding of the ultrafast processes that dominate the radiative and non-radiative excited-state dynamics in TPE derivatives. It would furthermore be a significant step towards perfecting the rational molecular design of highly efficient AIE luminogens (AIEgens).35
In our work to restrict certain relaxation channels in the excited state and investigate their role in the AIE effect, we synthesized a set of TPE-based derivatives, 1–6 (Scheme 1), with varying degrees of rigidity, studied their photophysical and AIE properties, and applied a combination of DFT calculations and ultrafast time-resolved spectroscopy to probe and resolve the excited-state dynamics and photocyclization processes in them. We have found state-dependent coupling of the two major dynamic motions (phenyl torsion and CC twisting) in 1–6, with the formation of a photocyclized intermediate in all compounds on different timescales, and the dominant relaxation channels responsible for the AIE effect (or its absence) in every particular case being strongly structure-dependent. The implications of these observations for the working mechanism of AIE are fully explained.
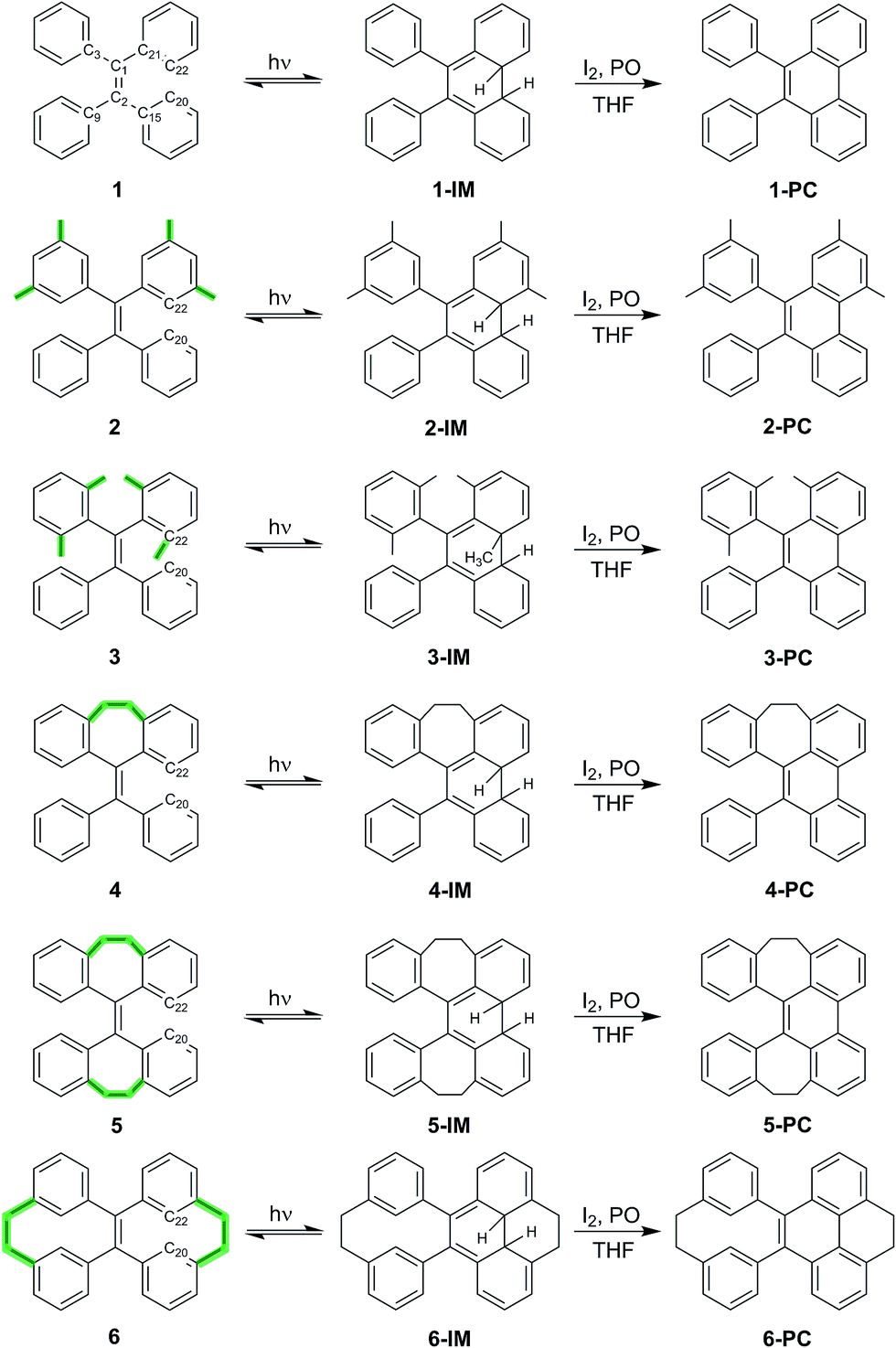 | ||
| Scheme 1 TPE derivatives 1–6 with increased structural rigidity and their transformation upon UV irradiation into the corresponding intermediates (1-IM–6-IM) and subsequent oxidation to isolable and fully characterized photocyclized phenanthrene derivatives 1-PC–6-PC (PO: (±)-propylene oxide). | ||
Results and discussion
Synthesis and characterization of TPE derivatives
TPE derivatives 1–6 with increased structural rigidity and their corresponding photocyclized phenanthrene derivatives 1-PC–6-PC were synthesized according to the synthetic routes shown in Scheme 1. The isolated compounds were characterized by 1H-NMR, 13C-NMR and high resolution mass spectrometry. The structures were determined by single-crystal X-ray diffraction (see the ESI† for details), which confirms their correct structures.Structural rigidity, AIE properties and photocyclization
Scheme 1 shows the structures of TPE derivatives 1–6 in the order of increasing structural rigidity. While the molecular structures of 1–3 and 6 in their respective single crystal unit cells have two different enantiomers with different propeller-like orientations of phenyl groups, molecules of 4 and 5 with ethylene tethers (–CH2–CH2–) between geminal phenyl groups display only one type of phenyl orientation (Fig. S26†). To explore and quantify their different structural rigidities, we have investigated computationally the geometry changes36 in 1–6 upon transition from the ground state to the first excited state as shown in Table 1 (see ESI† Section 7.5 for more details). Upon photoexcitation in solution and in the solid state, the central ethylenic C1–C2 bond in 1–6 elongates (i.e. dC1–C2), giving rise to what we will refer to from now on as a quasi CC bond. In contrast, the bonds connecting the peripheral phenyl groups with the C1–C2 bond shorten (e.g. dC2–C15) to fall into the bond length range that is between that of a typical single C–C bond and a normal CC double bond. The twisting of the CC bond and torsion of the phenyl rings in 1–6 are analyzed metrically in terms of the dihedral angles τC21–C1–C2–C15 and τC1–C2–C15–C20, respectively. For 1–6 in solution, each molecule has its unique set of restricted degrees of freedom with varying degrees of rigidity. The absolute change of the dihedral angle around the C1–C2 bond (|ΔτC21–C1–C2–C15|) in TPE derivatives in solution upon excitation decreases in the order 1–3 and 5–6 (Table 1), which is consistent with the increase of structural rigidity in that order. The smallest structural changes upon photoexcitation with only 0.03 Å C1–C2 bond elongation and less than 9° twisting are predicted for 6, making it the most rigid structure, indicating that two vicinal ethylene bridges (–CH2–CH2–) do effectively restrict the motion of the CC bond. We note that this trend persists for the changes of the dihedral angles of the phenyl torsion (|ΔτC1–C2–C15–C20|) in 1–3 after photoexcitation. Two ethylene tethers in geminally locked structure 5 and vicinally locked structure 6 have similar restriction effects on the phenyl torsion with the smallest change of the dihedral angles (ca. 20°).
| Geometry changes between S0 and S1 in solna | Absbλmax (nm) | PL λem (nm) | QY (%) | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| d C1–C2 | d C2–C15 | ΔτC21–C1–C2–C15 | ΔτC1–C2–C15–C20 | d C20–C22 | Solnb | Film | Solnb | Solid | ||||||
| S 0 | S 1 | S 0 | S 1 | |Δ(S0 − S1)| | |Δ(S0 − S1)| | S 0 | S 1 | |||||||
| a Geometry changes in THF solution were calculated using DFT with the M062X functional and 6-311G (d) basis set. Bond lengths (d) are given in Å and angles in °. The ethylenic C1–C2 bond twisting angle is defined as the dihedral angle τC21–C1–C2–C15. The phenyl torsion angle is defined as the dihedral angle τC1–C2–C15–C20. The distance between the two carbon atoms dC20–C22 is analyzed to investigate the possibility of the formation of photocyclized species. Despite multiple trials, the geometry optimization of 4 in the first excited state in solution did not converge. For more information, see ESI Section 7.5. b All measurements of compounds 1–6 were done in THF solution (10−5 M) in open air. The UV/vis absorption spectra of 1, 3 and 5 in solution, the photoluminescence (PL) spectra of 1 and 3 in the solid state, and the fluorescence quantum yields (QY) of 1 and 3 in solution have already been published elsewhere.16,21,37 For the sake of consistency, we repeated the measurements for these compounds on the same spectrometer. See ESI Section 5 for details. c The extra peaks in the PL spectra of 1 and 2 upon first time excitation. | ||||||||||||||
| 1 | 1.35 | 1.47 | 1.49 | 1.44 | 56.80 | 29.04 | 3.25 | 3.64 | 1 | 239; 309 | (358, 375, 396),c 474 | 445 | 0.8 | 24.1 |
| 2 | 1.35 | 1.47 | 1.49 | 1.44 | 39.59 | 25.19 | 3.23 | 3.31 | 2 | 242; 311 | (383),c 472 | 466 | 0.6 | 30.2 |
| 3 | 1.36 | 1.46 | 1.49 | 1.45 | 33.96 | 20.77 | 3.32 | 3.49 | 3 | 244; 314 | 473 | 460 | 60.0 | 97.6 |
| 5 | 1.35 | 1.47 | 1.49 | 1.46 | 11.57 | 20.35 | 3.44 | 2.96 | 4 | 262 | 391 | 393 | 1.0 | 1.3 |
| 6 | 1.37 | 1.40 | 1.50 | 1.45 | 8.96 | 22.37 | 2.60 | 1.70 | 5 | 268 | 389 | 384 | 0.9 | 16.5 |
| 6 | 262; 368 | 397 | 462 | 0.5 | 0.7 | |||||||||
The RIM mechanism dictates a reciprocal correlation between the rigidification of the structure and possible AIE properties. For molecules 1–6, compounds 1 and 2 are archetypal TPE-based AIEgens showing intense turn-on fluorescence upon aggregation (see the fluorescence quantum yields (QY) in Table 1), while 3 shows unexpectedly highly fluorescence in solution, and becomes even more fluorescent upon aggregation making it AEE (aggregation-enhanced emission)-active. Contrary to our expectations, the significantly more rigid structures 4–6 have low fluorescence quantum yields in solutions, among which 5 is an AIEgen, while 4 and 6 are not AIE-active. These findings clearly demonstrate that structural rigidity is not directly correlated with the AIE properties and the RIM paradigm needs to be revisited.
The UV-vis and fluorescence emission maxima of 1–6 in dilute solutions and film are shown in Table 1. While 1–3 in solution display similar UV absorption peaks at around 310 nm, the UV absorption maxima of 4 and 5 are markedly blue shifted (<270 nm), and the absorption maximum of 6 is notably red shifted (368 nm). All compounds 1–6 in dilute solutions undergo photocyclization upon UV excitation (ESI† Section 5), during which the bond formation between atoms C20 and C22 takes place (Scheme 1). Compared to the emission peaks of 1–6 in film, extra peaks at around 358, 375, and 396 nm in the PL spectrum of a dilute solution of 1 and around 383 nm for 2 as well as discernable asymmetry of the emission peaks in dilute solutions of 4–6 upon first-time photoexcitation indicate the formation of new species which could be further identified as photocyclized (diphenylphenanthrene) derivatives.38 In dilute solution upon prolonged excitation, 3 undergoes photocyclization, as evidenced by the emergence of similar new emission peaks at 361, 378 and 400 nm (ESI† Section 5), suggesting that the steric hindrance of the methyl groups in the ortho-positions of the phenyl rings cannot preclude photocyclization. The above observations reveal that photocyclization in TPE-based derivatives 1–6 is an important event in the relaxation process after photoexcitation.
The coupling relationship of molecular motions
To differentiate between several possible dominant relaxation channels (i.e. quasi CC bond twisting, phenyl torsion and photocyclization) in 1–6 upon excitation, and to determine their contribution to the photophysics of the AIE mechanism, probing the molecular dynamics in the excited state is necessary. Prior to this, developing a quantitative understanding of the coupling relationship between the relaxation channels directly related to the molecular motions is imperative. To that end, we have constructed the 3D potential energy surface (PES) of 1 in solution as a function of the aforementioned modes using DFT calculations (Fig. 1). In the ground state, the absolute minimum of 1 (S0,min, denoted as (8, 50, 0)) has a molecular geometry corresponding to ca. 8° twisting angle along the CC bond twisting mode, ca. 50° torsion along the phenyl torsional coordinate, and potential energy whose value is set to 0 kcal mol−1. The coupling between the CC bond twisting and phenyl torsion along the minimum energy path (MEP) of 1 on the ground state PES (Fig. 1b) is clearly exemplified by the increase of the dihedral angle of phenyl torsion from 50° to 90° actuating slight changes in the CC bond twisting (<9°) and potential energy (<7 kcal mol−1), indicating that under standard conditions the torsion of the phenyl rings dominates the ground state dynamics in 1. This is further corroborated by the thorough analysis of the intrinsic reaction coordinate (IRC) calculations (ESI† Section 7.2.1) showing that the change of the dihedral angle of phenyl torsion is linearly coupled with the small change of the CC bond twisting (Fig. S56†).
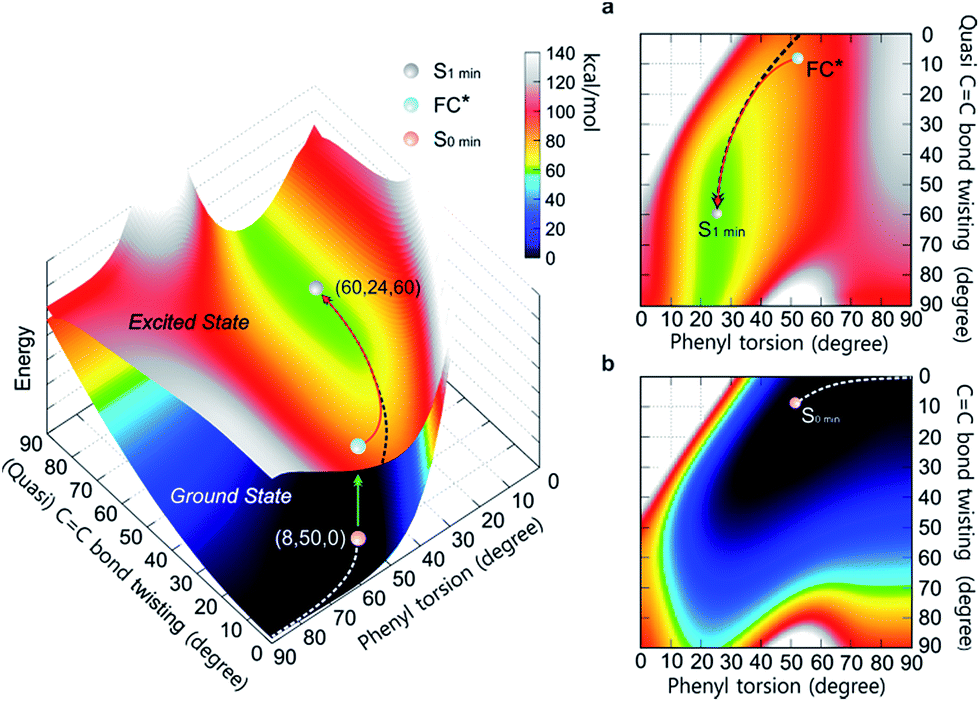 | ||
| Fig. 1 The PES of 1 in the ground state and excited state as a function of the (quasi) CC bond twisting and phenyl torsion dihedral angles (defined in Table 1). The lower surface shows the ground state PES, and the upper one depicts the first excited state PES. (a) Top view of the first excited state PES. (b) Top view of the ground state PES. The MEPs in S0 (white dashed line) and S1 (black dashed line) are marked on the PES. | ||
Following the MEP along the CC bond twisting mode in 1 in the ground state from one minimum energy geometry through the highly twisted transition state to the other minimum energy geometry (ESI† Section 7.2.2), we note that the CC bond twisting is associated with significant changes in the CC bond length (∼0.07 Å), and a marked increase in potential energy (∼60 kcal mol−1). In the excited state, the elongation (∼0.12 Å) and partial loss of the double bond character of the CC bond in 1 triggers the CC bond twisting as the dominant motion which is coupled with the phenyl torsion (Fig. 1a). The MEP along the CC bond twisting mode in 1 on the first excited state PES reveals that on the way from the Frank–Condon (FC*) geometry to the minimum energy geometry (S1,min), the quasi CC bond twists ca. 50°, which is accompanied by the phenyl torsion with an amplitude of less than 25°. Although their coupling relationship in the excited state is well fitted by the quadratic function Δ twisting = 0.0671 (Δ torsion)2 − 3.9048 Δ torsion − 2.7952, the PES around the S1,min is rather shallow (Fig. 1a, green area at ca. 60 kcal mol−1 potential energy) and simple harmonic oscillations39 of the quasi CC bond twisting may easily take place with the damping process, resulting in the relaxation to the S1 min (ESI† Section 7.3).
The observation of intermediates
A characteristic ultrafast transient absorption (TA) spectrum corresponds to the statistically most probable geometry of the molecules on a specific timescale in the excited state. Thus, the photoinduced excited state structural dynamics associated with molecular motions is reflected in the evolution of TA spectra.Upon UV light irradiation, the electron density in 1–6 flows mainly from the central CC bond to the adjacent C–C (Ph) bonds (Fig. S54†), concomitantly resulting in the elongation of the CC bond and shortening of peripheral bonds, which is accompanied by CC bond twisting coupled with phenyl torsion. This process in 1 is reflected in the excited state absorption spectra shown in Fig. 2a. On the sub-picosecond timescale (0.6–1.3 ps), 1 transitions from Sn to the emissive state S1 due to the weak stimulated emission as evidenced by the band at 500 nm with a negative amplitude at 1.3 ps.40,41 The build-up of the band at 600 nm with its red-shift to 613 nm and the growing intensity of the band at 430 nm are attributed to the central CC bond elongation41 associated with the quasi CC bond twisting. On the picosecond timescale between 1.3 ps and 3.79 ps (Fig. 2b), sequential depopulation of the band at 613 nm with the increasing band at 428 nm is attributed to the quasi CC bond twisting,41,42 meaning that the transients in the emissive state revert back to the state in which the shortening of the elongated CC bond took place. After 3.79 ps (Fig. 2c), the decay of the band at 422 nm assigned to phenyl torsion43 takes place, during which a new species forms, and the band at 465 nm is clearly observed after 105 ps. Global fitting analysis of the decay kinetics at all wavelengths yields three time constants: 0.39 ps, 1.2 ps and 18.9 ps (Fig. 2d), which correspond to the lifetimes of the initially dominant motion of CC bond elongation (τ1), the sequential dominant motion of the quasi CC bond twisting (τ2), and the last motion dominated by the phenyl torsion (τ3), respectively. It should be noted that both processes corresponding to τ1 and τ2 are coupled with phenyl torsion, as proved by the band at 430 nm and predicted by the calculation (Fig. 1). The monoexponential fitting of the decay of UV/vis absorption at 465 nm yields a lifetime of 159 s (Fig. 2d inset and S45†), which is attributed to the lifetime of the photocyclized intermediate 1-IM (τ4) having a new bond connecting atoms C20 and C22 directly (Fig. 2e). The longevity of 1-IM with two characteristic absorption bands at 320 nm and 465 nm indicates that it is in its singlet ground state. This is further confirmed by ns-TA spectroscopy, the UV/vis absorption spectrum of the quasi-steady state at 5 s and the consistent calculated UV/vis spectrum (Fig. 2e), indicating that the formation of the photocyclized intermediate is a possible non-radiative relaxation channel. Further characterization of 1-IM was carried out by transient Raman spectroscopy which confirmed the match between the experimentally obtained and computed Raman spectra of 1-IM (Fig. S36†).
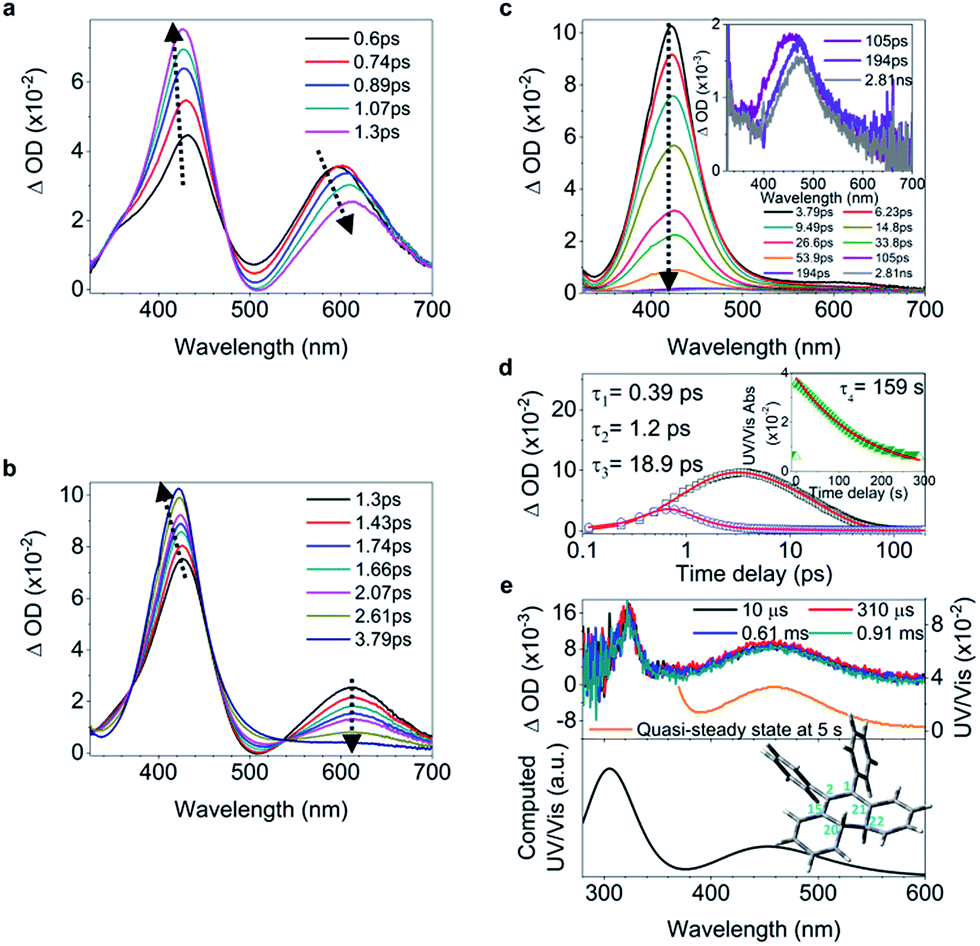 | ||
| Fig. 2 Excited state dynamics of 1 in solution observed by ultrafast time-resolved spectroscopy. (a–c) Femtosecond transient absorption (fs-TA) spectra. (d) Kinetic traces at 430 nm (black square) and 600 nm (blue circle) with the respective fits (red lines, solid) determined using a global analysis that used three exponential functions convoluted with a 120 fs (full-width at half maximum) Gaussian-shaped pulse. The inset shows the lifetime of the photocyclized intermediate 1-IM analyzed by monoexponential fitting of the decay of UV/vis absorption (ESI† Section 6.1.7). (e) Nanosecond transient absorption (ns-TA) spectra (upper panel). UV/vis spectrum of 1-IM in MeCN acquired after excitation of 1 in MeCN at 254 nm for 1 min with a hand-held UV lamp and then without UV/vis excitation for 5 s (upper panel, orange line). The inset shows the calculated electronic absorption spectrum (lower panel) of the optimized structure of 1-IM. | ||
Fluorescence prevails as the rotation of the elongated C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/h3_char_e001.gif) C bond slows down
C bond slows down
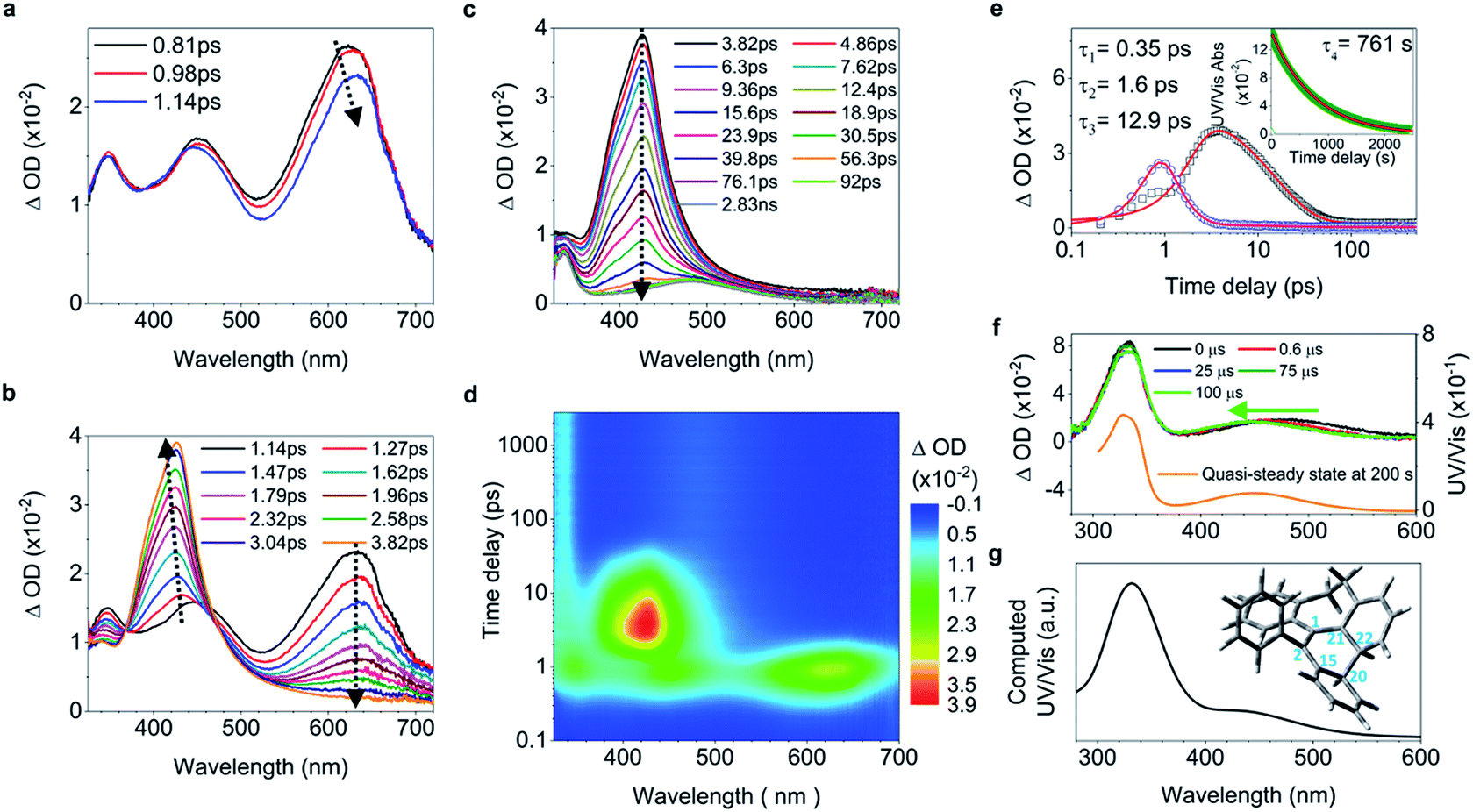
While 2 undergoes the formation of a photocyclized intermediate and has similar ultrafast dynamic features as observed in 1 (ESI† Section 6.1.2), 3 begins to fluoresce on the subpicosecond timescale and its fluorescence intensity reaches its maximum at 1.19 ps (Fig. 3a inset and S40c†). In accordance with the transient fluorescence spectra (Fig. 3a inset, peak at around 480 nm), the dip at 477 nm in the fs-TA spectra on the timescale between 0.63 ps and 1.2 ps (Fig. 3a) originates from the stimulated emission, and the red shift of the band at 600 nm to 610 nm is attributed to the CC bond elongation corresponding to the population of transients in the emissive state (most likely S1,min). In contrast to 1–2, the decay of the band at 610 nm with growing intensity of the band at 430 nm accompanied by a blue shift (Fig. 3b) takes a long time (between 1.2 ps and 48.9 ps), with a time constant of 11.2 ps derived from the global fitting analysis (ESI† Section 6.1.3), indicating that the emissive state of the molecule is relatively stable and thus fluorescence prevails as the rotation (larger degree of twisting, Table 1) of the elongated CC bond slows down. This is probably due to the steric hindrance from the ortho-position substituents in 3. The depopulation of the band at 428 nm (Fig. 3c) with a time constant of 4.07 ns (Fig. S40b†) indicates that the molecules of 3 relaxing to the ground state either revert back to the original ground state structure, or form the photocyclized intermediate (Fig. 3d, upper panel). Although the stabilization of the emissive state of 3 with a twisted structure in the excited state increases the distance between atoms C20 and C22 (Table 1) and thus inhibits the formation of the intermediate, the photocyclized intermediate 3-IM still forms as observed in the ns-TA spectra, and its structure is further confirmed by comparison with the computed UV/vis spectrum (Fig. 3d).
 | ||
| Fig. 3 Excited state dynamics of 3 in solution observed by ultrafast time-resolved spectroscopy. (a–c) Fs-TA spectra and femtosecond time-resolved fluorescence spectra (inset in (a)). (d) Ns-TA spectra (upper panel). The inset shows the calculated electronic absorption spectrum (lower panel) of the optimized structure of the photocyclized intermediate 3-IM. | ||
Ultralong-lived intermediates
The sequentially dominant motions (CC elongation and quasi CC bond twisting) of 4 upon excitation have similar dynamics and time constants to those of 1–2 (Fig. 4a, b, and e, and ESI† Section 6.1.8). After 3.82 ps (Fig. 4c and d), the band at 485 nm emerges during the depopulation of the band at 430 nm, meaning that a new species forms during the decay process of phenyl torsion. The decay process has a shorter time constant of 12.9 ps as compared to 1–2, indicating that the ethylene bridge in 4 restricts the freedom of torsion for the two phenyl rings and thus shortens the timescale of the bond formation between C20 and C22. On the nanosecond timescale and beyond, the band at 485 nm continues to blue-shift to 450 nm until 1.0 μs (Fig. S41a†and 4f, upper panel) due to the structural relaxation of the photocyclized species (Fig. S63†). The intermediate 4-IM has an ultralong lifetime of 761 s as analyzed by the decay of the UV/vis absorption of its quasi-steady state at 450 nm (Fig. 4e and f, and S46†) and its structure is further confirmed by the transient Raman spectrum (Fig. S41b†).
 | ||
| Fig. 4 Excited state dynamics of 4 in solution observed by ultrafast time-resolved spectroscopy. (a–c) Fs-TA spectra. (d) Contour plots of the time-resolved absorption spectroscopic responses. (e) Kinetic traces at 426 nm (black square) and 630 nm (blue circle), and the corresponding fits (red lines, solid) determined using a global analysis making use of three exponential functions convoluted with a 120 fs (full-width at half maximum) Gaussian-shaped pulse. The inset shows the lifetime of the photocyclized intermediate 4-IM analyzed by monoexponential fitting of the decay of UV/vis absorption (ESI† Section 6.1.7). (f) Ns-TA spectra (upper panel, solid lines). UV/vis spectrum of 4-IM in MeCN acquired after excitation of 4 in MeCN at 254 nm for 30 s with a hand-held UV lamp and then without UV/vis excitation for 200 s (orange line and Fig. S31C†). (g) The calculated electronic absorption spectrum of the optimized structure of the photocyclized intermediate 4-IM. | ||
Ultrafast formation of intermediates
In the ultrafast spectra, the initial CC bond elongation in 5 upon excitation in solution is reflected by the population of the 628 nm band with its red shift before 1.03 ps (Fig. 5a). Thereafter, both bands at 462 nm and 630 nm (Fig. 5a, inset) depopulate, meaning that the decay of the quasi CC bond twisting and phenyl torsion take place at the same time. Concomitantly, the intensified red-shifted band at 330 nm indicates that a new species is formed, which is further proved by the emergence of a band at 438 nm (Fig. 5b) attributed to the long-lived photocyclized intermediate (Fig. S42, S43 and S47†). While the process of the CC elongation in 6 upon excitation was not observed in the fs-TA spectra because of its negligible increase of only 0.03 Å (Table 1), the formation of the photocyclized intermediate takes place directly on the subpicosecond timescale during the initial depopulation of the whole TA spectra with a time constant of 0.4 ps (Fig. 5c and S44†). This is further evidenced by the build-up of the band at 510 nm at 1.08 ps (Fig. 5c and e) and the computational prediction that 6 upon excitation has a short dC20–C22 distance (1.70 Å). This means that the vicinally bi-locked structure of 6 restricts the CC elongation and the freedom of torsion for all phenyl rings with short distances, which facilitates the ultrafast formation of the photocyclized intermediate on the subpicosecond timescale, and thus 6 barely fluoresces in solution. After 4.68 ps, the photocyclized intermediate of 6 undergoes structural relaxation (Fig. 5d), and its quasi-steady state structure, 6-IM, is confirmed by the experimentally observed ns-TA and Raman spectra and computationally predicted spectra (Fig. 5f and g), in which bands at 345 nm and 525 nm are identified as characteristic absorption peaks of 6-IM, with the vibrational feature at 1641 cm−1 mainly attributed to the stretching motion of C20–C22, and those at 1304 cm−1 and 1259 cm−1 mainly ascribed to the rocking and wagging modes of C22–H and C20–H. The identity of all intermediate species was further indirectly confirmed by isolating and characterizing their oxidized analogues (ESI† Section 2.4).
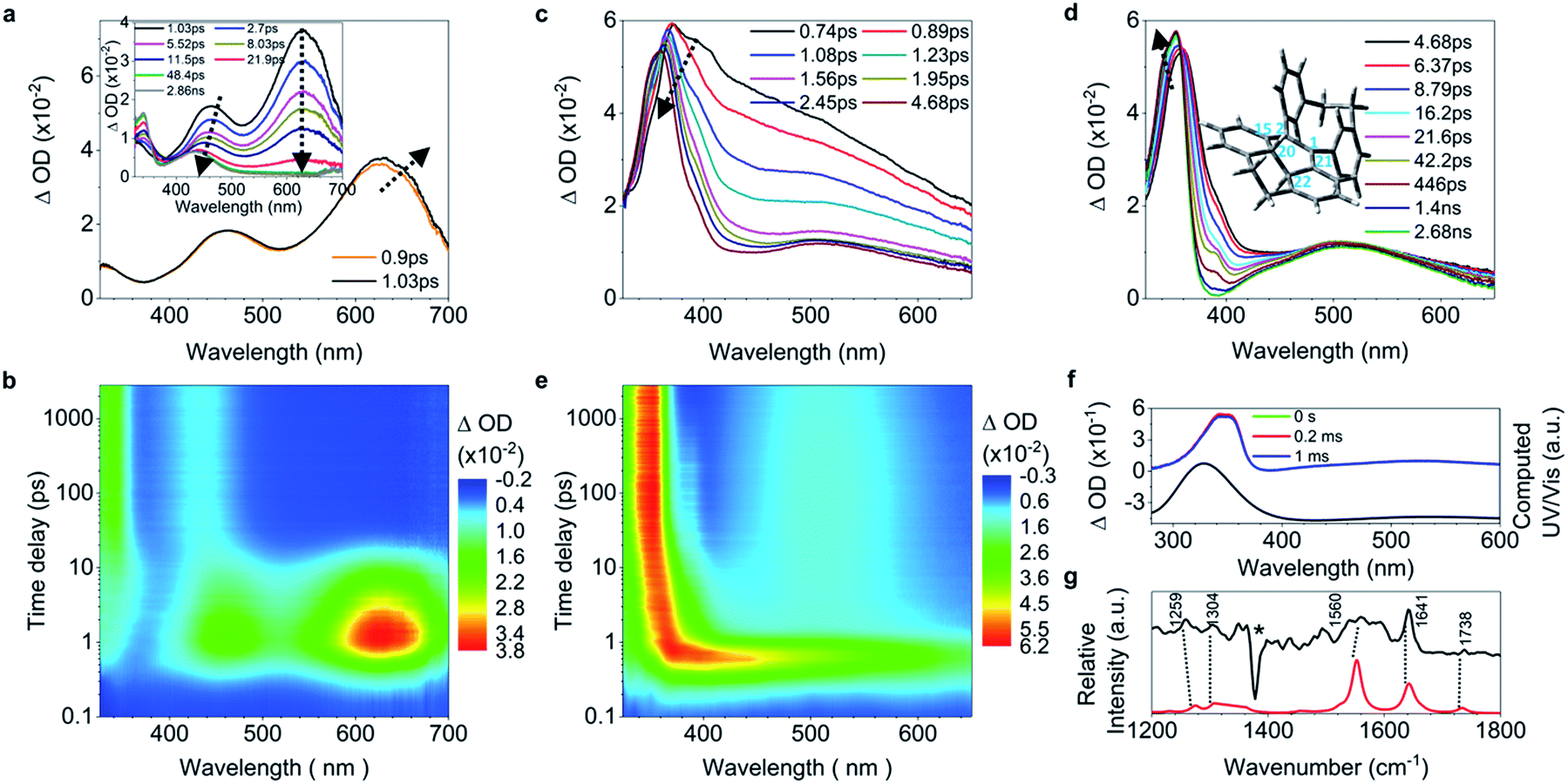 | ||
| Fig. 5 Excited state dynamics of 5 and 6 in solution observed by ultrafast time-resolved spectroscopy. (a) Fs-TA spectra of 5. (b) Contour plots of the time-resolved absorption spectroscopic responses of 5. (c and d) Fs-TA spectra of 6. The optimized structure of the photocyclized intermediate (6-IM) is shown in the inset in (d). (e) Contour plots of the time-resolved absorption spectroscopic responses of 6. (f) Ns-TA spectra of 6 (green, red and blue lines) and the calculated electronic absorption spectrum (black line) of 6-IM. (g) The nanosecond transient resonance Raman (ns-TR2) spectrum of 6-IM (black line) is compared to the computed Raman spectrum (red line). The star represents the bands caused by solvent subtraction artifacts. | ||
AIE mechanism through the lens of ultrafast spectroscopy
After photoexcitation, 1–6 in solution reach the hot emissive excited state S* (most likely S1,min, Fig. 6) upon relaxation via the dominant motion of the CC bond elongation with a time constant of τ1 < 0.4 ps (Table S6†). Due to the structural rigidity, the τ1 components in 1–6 undergo relaxation via three possible channels with one of them being the dominant relaxation pathway (Fig. 6). They either do not fluoresce due to (a) the quasi CC bond twisting with a time constant of 1–2 ps (e.g. flexible structures 1, 2, and 4), or (b) the ultrafast formation of the photocyclized intermediate (IM) on the sub-picosecond timescale (e.g. rigid structures 5, 6), or they do fluoresce (c) due to the structural stability of the emissive state of the molecule on a relatively longer timescale (3). Thus, the dynamics of the quasi CC bond twisting (τ2) with the possibility of intermediate formation in S* is the key factor that determines the fluorescence properties of TPE derivatives in solution. When the non-radiative relaxation pathways for TPE derivatives in solution are blocked in the solid state (ESI† Section 6.2), and no other non-radiative relaxation channels open up, enhanced fluorescence and thus the AIE properties will emerge. After τ2, transients of all compounds finally revert back to the ground state through the phenyl torsion (τ3) via two different routes: relaxation to the original ground state structure and formation of a singlet ground state IM with a long-lived lifetime (τ4) that either (1) reverts back to the original compound or (2) is oxidized to form the corresponding photocyclized product (PC) (ESI† Section 7.4). Intermediate formation from 1–6 upon excitation in solution was experimentally confirmed by the outcome of the photochemically induced reactions (ESI† Section 2.4).
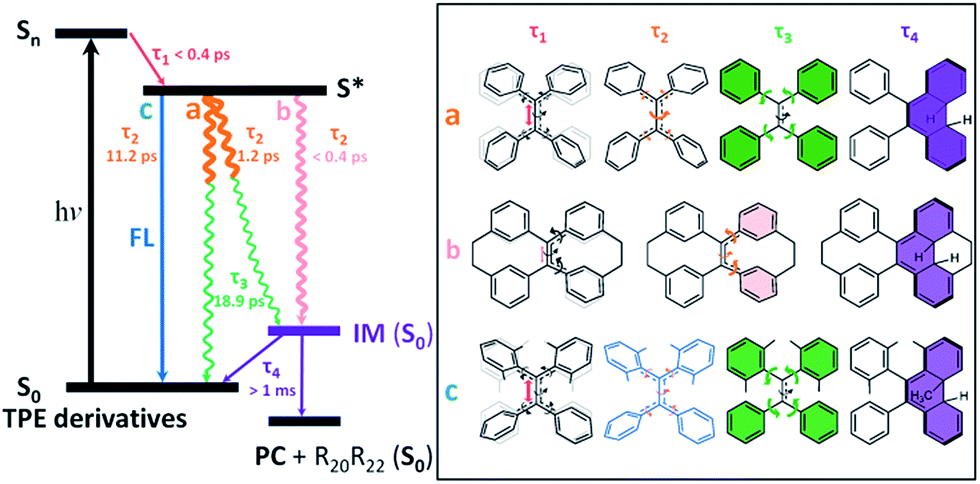 | ||
| Fig. 6 Schematic representation of ultrafast processes responsible for the AIE effect in TPE derivatives. The timescales of the dominant motions of various parts of the molecule are color-coded in red (subpicosecond), blue (subpico- to nanosecond), pink (subpico- to picosecond), brown (picosecond), green (pico- to nanosecond) and purple (millisecond to second/minute). Three different pathways for the decay of S* are represented by non-radiative decay channels (a) and (b), and a fluorescence decay channel (c). Examples of the molecules with a dominant decay channel are shown in the box on the right. R20R22 represents substituents on atoms C20 and C22 in TPE derivatives. τ1 is the time constant of the motion dominated by the elongation of the CC bond associated with quasi CC bond twisting. τ2 is the time constant of the motion dominated by the quasi CC bond twisting coupled with phenyl torsion. τ3 is the time constant of the motion dominated by the phenyl torsion. τ4 is the lifetime of IM. | ||
Experimental
Synthesis, photophysics and computational calculations
Detailed information on the synthesis and characterization of 1–6 and their photoinduced cyclization products 1-PC–6-PC is given in ESI† Sections 1–4. The UV/vis absorption spectra were measured on a UV/vis spectrometer (Shimadzu, UV-2600, Japan). The steady state PL spectra were collected on a Horiba Fluoromax-4 spectrofluorometer. The absolute fluorescence quantum yields were measured by using a Hamamatsu quantum yield spectrometer, C11347 Quantaurus_QY. ESI† Section 5 gives more details on the photophysical properties. The MO calculations were carried out using the Gaussian 16 software package; all other computational calculations, including the electron density difference between S1-FC and S0, the intrinsic reaction coordinates through the transition states of phenyl torsion and ethylenic CC bond twisting, the potential energy hypersurface, the Gibbs free energy, the geometry optimization of TPE derivatives in solution and in the solid state (both in the ground state (S0) and excited state (S1)), and the UV/vis and Raman spectra of photocyclized intermediates 1-IM–6-IM, were performed at the DFT level of theory using the M062X functional44 and 6-311G (d) basis set45 as implemented in the D0.1 version of the Gaussian 09 software package. The scaling factor for all UV/vis and Raman calculations was 1.000. DFT computational details are given in ESI† Sections 6 and 7.
The ultrafast time-resolved spectroscopy
Femtosecond transient absorption (fs-TA), nanosecond transient absorption (ns-TA), femtosecond time-resolved fluorescence (fs-TRF), nanosecond transient resonance Raman (ns-TR2) and time-resolved resonance Raman (ns-TR3) experiments were carried out using the same experimental setups and methods as described previously.46 The pump wavelength employed for the fs-TA and fs-TRF measurements was 267 nm, and the pump wavelength employed for the ns-TA and ns-TR3 measurements was 266 nm. Two probe wavelengths (355 nm and 309.1 nm) were used for the ns-TR3 experiment. The ns-TR2 data were obtained by using the difference between the Raman spectra obtained at different power values of the 266 nm pump laser. Compounds 1–6 in MeCN solution were studied in a flow-through 2 mm path-length cuvette with an absorbance of 0.5 at 267 nm throughout the data acquisition. More details are available in ESI† Section 6.Conclusions
TPE-based derivatives 1–6 with varying rigidities and photophysical properties have been synthesized and their ultrafast excited-state relaxation dynamics and its role in the AIE process were studied. We found that CC bond elongation, quasi CC bond twisting, phenyl torsion and photocyclization are the sequentially dominant processes in TPE derivatives upon photoexcitation in solution. Their state-dependent coupling motions derived from ultrafast time-resolved spectroscopy show that (a) the lifetime of the species with electronic configuration conducive to fluorescence (mostly likely the lowest vibrational state of S1) is the determining factor for the observed fluorescence quantum yields, (b) in less rigid structures, the rotation of the elongated CC bond is the dominant motion that eventually leads to the non-radiative decay (e.g. AIE-active TPE), and (c) in the more rigid structures, the torsion of phenyl rings will dominate the relaxation dynamics and, if possible, lead to the formation of singlet ground state photocyclized intermediates (e.g. AIE-active or AIE-inactive bi-locked TPE derivatives). Thus, while the dominant intramolecular motion that serves as a non-radiative relaxation channel being restricted upon aggregation in every particular case is strongly structure-dependent, counter-intuitively, the AIE effect (or absence thereof) is not directly related to rigidity as seemingly implied by the RIM paradigm.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (21788102), the National Basic Research Program of China (973 Program, 2013CB834701 and 2013CB834702), the Research Grants Council of Hong Kong (16308016, 16305015, C2014-15G, N_HKUST604/14, A_HKUST605/16, and HKU17301815), the Areas of Excellence Scheme (Grant AoE/P-03/08), the UGC Special Equipment Grant (SEG-HKU-07), the University of Hong Kong Development Fund 2013–2014 project “New Ultrafast Spectroscopy Experiments for Shared Facilities”, the Innovation and Technology Commission (ITC-CNERC14SC01 and ITCRD/17-9), and the Technology Plan of Shenzhen (JCYJ20160229205601482). Y. Cai gratefully acknowledges the China Postdoctoral Science Foundation Grant (2016 M602463) and the Fundamental Research Funds for the Central Universities (Grant No. 2017MS015). Y. Cai and X. Gu thank the National Science Foundation for Young Scientists (51703066 and 21702016) for financial support. Y. Cai would also like to thank Prof. R. West for providing access to supercomputing facilities for the theoretical calculations at the UW-Madison Department of Chemistry financially supported by the National Science Foundation of USA (NSF Grant No. CHE-0840494). Y. Cai thanks W. Luo and X. Zheng for the discussion on the calculation of geometry changes. We also acknowledge the technical support received from AIEgen Biotech Co., Ltd.Notes and references
- J. Luo, Z. Xie, J. W. Y. Lam, L. Cheng, H. Chen, C. Qiu, H. S. Kwok, X. Zhan, Y. Liu, D. Zhu and B. Z. Tang, Chem. Commun., 2001, 1740 RSC.
- J. Mei, N. L. C. Leung, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, Chem. Rev., 2015, 115, 11718 CrossRef CAS PubMed.
- F. Hu and B. Liu, Org. Biomol. Chem., 2016, 14, 9931 CAS.
- Q. Wan, M. Liu, D. Xu, L. Mao, H. Huang, P. Gao, F. Deng, X. Zhang and Y. Wei, Polym. Chem., 2016, 7, 4559 RSC.
- J. Shi, Y. Li, Q. Li and Z. Li, ACS Appl. Mater. Interfaces, 2018, 10, 12278 CAS.
- X. Xu, J. Huang, J. Li, J. Yan, J. Qin and Z. Li, Chem. Commun., 2011, 47, 12385 RSC.
- M. Shellaiah, Y.-H. Wu, A. Singh, M. V. R. Raju and H.-C. Lin, J. Mater. Chem. A, 2013, 1, 1310 CAS.
- R. Zhang, M. Gao, S. Bai and B. Liu, J. Mater. Chem. B, 2015, 3, 1590 RSC.
- J. Yang, J. Huang, Q. Li and Z. Li, J. Mater. Chem. C, 2016, 4, 2663 RSC.
- J. Huang, R. Tang, T. Zhang, Q. Li, G. Yu, S. Xie, Y. Liu, S. Ye, J. Qin and Z. Li, Chem.–Eur. J., 2014, 20, 5317 CrossRef CAS PubMed.
- R. H. Friend, R. W. Gymer, A. B. Holmes, J. H. Burroughes, R. N. Marks, C. Taliani, D. D. C. Bradley, D. A. D. Santos, J. L. Brédas, M. Lögdlund and W. R. Salaneck, Nature, 1999, 397, 121 CrossRef CAS.
- S. A. Jenekhe and J. A. Osaheni, Science, 1994, 265, 765 CAS.
- H. Qian, M. E. Cousins, E. H. Horak, A. Wakefield, M. D. Liptak and I. Aprahamian, Nat. Chem., 2017, 9, 83 CrossRef CAS PubMed.
- X. Yan, T. R. Cook, P. Wang, F. Huang and P. J. Stang, Nat. Chem., 2015, 7, 342 CrossRef CAS PubMed.
- E. P. J. Parrott, N. Y. Tan, R. Hu, J. A. Zeitler, B. Z. Tang and E. Pickwell-MacPherson, Mater. Horiz., 2014, 1, 251 RSC.
- G.-F. Zhang, Z.-Q. Chen, M. P. Aldred, Z. Hu, T. Chen, Z. Huang, X. Meng and M.-Q. Zhu, Chem. Commun., 2014, 50, 12058 RSC.
- J. Zhao, D. Yang, Y. Zhao, X.-J. Yang, Y.-Y. Wang and B. Wu, Angew. Chem., Int. Ed., 2014, 53, 6632 CrossRef CAS PubMed.
- Z. Yang, W. Qin, N. L. C. Leung, M. Arseneault, J. W. Y. Lam, G. Liang, H. H. Y. Sung, I. D. Willams and B. Z. Tang, J. Mater. Chem. C, 2016, 4, 99 RSC.
- N. Sinha, L. Stegemann, T. T. Y. Tan, N. L. Doltsinis, C. A. Strassert and F. E. Hahn, Angew. Chem., Int. Ed., 2017, 56, 2785 CrossRef CAS PubMed.
- Q. Peng, Y. Yi, Z. Shuai and J. Shao, J. Am. Chem. Soc., 2007, 129, 9333 CrossRef CAS PubMed.
- N. L. C. Leung, N. Xie, W. Yuan, Y. Liu, Q. Wu, Q. Peng, Q. Miao, J. W. Y. Lam and B. Z. Tang, Chem.–Eur. J., 2014, 20, 15349 CrossRef CAS PubMed.
- T. Zhang, H. Ma, Y. Niu, W. Li, D. Wang, Q. Peng, Z. Shuai and W. Liang, J. Phys. Chem. C, 2015, 119, 5040 CAS.
- U. Mazzucato and A. Spalletti, J. Phys. Chem. A, 2009, 113, 14521 CrossRef CAS PubMed.
- A. Prlj, N. Došlić and C. Corminboeuf, Phys. Chem. Chem. Phys., 2016, 18, 11606 RSC.
- Y.-J. Gao, X.-P. Chang, X.-Y. Liu, Q.-S. Li, G. Cui and W. Thiel, J. Phys. Chem. A, 2017, 121, 2572 CrossRef CAS PubMed.
- N.-W. Tseng, J. Liu, J. C. Y. Ng, J. W. Y. Lam, H. H. Y. Sung, I. D. Williams and B. Z. Tang, Chem. Sci., 2012, 3, 493 RSC.
- K. Garg, E. Ganapathi, P. Rajakannu and M. Ravikanth, Phys. Chem. Chem. Phys., 2015, 17, 19465 RSC.
- D. A. Shultz and M. A. Fox, J. Am. Chem. Soc., 1989, 111, 6311 CrossRef CAS.
- N. B. Shustova, T.-C. Ong, A. F. Cozzolino, V. K. Michaelis, R. G. Griffin and M. Dincã, J. Am. Chem. Soc., 2012, 134, 15061 CrossRef CAS PubMed.
- P. F. Barbara, S. D. Rand and P. M. Rentzepis, J. Am. Chem. Soc., 1981, 103, 2156 CrossRef CAS.
- E. Lenderink, K. Duppen and D. A. Wiersma, J. Phys. Chem., 1995, 99, 8972 CrossRef CAS.
- K. Kokado, T. Machida, T. Iwasa, T. Taketsugu and K. Sada, J. Phys. Chem. C, 2018, 122, 245 CAS.
- J.-B. Xiong, Y.-X. Yuan, L. Wang, J.-P. Sun, W.-G. Qiao, H.-C. Zhang, M. Duan, H. Han, S. Zhang and Y.-S. Zheng, Org. Lett., 2018, 20, 373 CrossRef CAS PubMed.
- R. J. Olsen and R. E. Buckles, J. Photochem., 1979, 10, 215 CrossRef CAS.
- G. Feng and B. Liu, Small, 2016, 12, 6528 CrossRef CAS PubMed.
- G. Sun, Y. Zhao and W. Liang, J. Chem. Theory Comput., 2015, 11, 2257 CrossRef CAS PubMed.
- J. Luo, K. Song, F. L. Gu and Q. Miao, Chem. Sci., 2011, 2, 2029 RSC.
- G. Huang, B. Ma, J. Chen, Q. Peng, G. Zhang, Q. Fan and D. Zhang, Chem.–Eur. J., 2012, 18, 3886 CrossRef CAS PubMed.
- J. Conyard, K. Addison, I. A. Heisler, A. Cnossen, W. R. Browne, B. L. Feringa and S. R. Meech, Nat. Chem., 2012, 4, 547 CrossRef CAS PubMed.
- S. Kayal, K. Roy and S. Umapathy, J. Chem. Phys., 2018, 148, 024301 CrossRef PubMed.
- R. W. J. Zijlstra, P. T. van Duijnen, B. L. Feringa, T. Steffen, K. Duppen and D. A. Wiersma, J. Phys. Chem. A, 1997, 101, 9828 CrossRef CAS.
- B. I. Greene, Chem. Phys. Lett., 1981, 79, 51 CrossRef CAS.
- G.-J. Zhao, K.-L. Han, Y.-B. Lei and Y.-S. Dou, J. Chem. Phys., 2007, 127, 094307 CrossRef PubMed.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215 CrossRef CAS.
- R. Krishnan, J. S. Binkley, R. Seeger and J. A. Pople, J. Chem. Phys., 1980, 72, 650 CrossRef CAS.
- L. Du, Y. Qiu, X. Lan, R. Zhu, D. L. Phillips, M.-D. Li, A. S. Dutton and A. H. Winter, J. Am. Chem. Soc., 2017, 139, 15054 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthetic procedures and characterization data (NMR and X-ray) of all newly synthesized compounds, details of the photophysical properties, the ultrafast spectroscopy studies, and the DFT calculations. CCDC 1579022 (2), 1586452 (3), 1579026 (4), 1579025 (6), 1579024 (2-PC), 1579027 (3-PC), 1579021 (4-PC) and 1579023 (6-PC). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8sc01170b |
| ‡ Dr Y. Cai, Dr L. Du, Dr K. Samedov, Dr X. Gu and Dr F. Qi contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2018 |