
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIsolable iodosylarene and iodoxyarene adducts of Co and their O-atom transfer and C–H activation reactivity†
Ethan A.
Hill
,
Margaret L.
Kelty
,
Alexander S.
Filatov
and
John S.
Anderson
 *
*
Department of Chemistry, The University of Chicago, 5735 S. Ellis Ave, Chicago, IL 60637, USA. E-mail: jsanderson@uchicago.edu
First published on 23rd April 2018
Abstract
We report an unusual series of discrete iodosyl- and iodoxyarene adducts of Co. The formation of these adducts was confirmed by a suite of techniques including single crystal X-ray diffraction. The reactivity of these adducts with O-atom acceptors and an H-atom donor has been investigated with particular focus on elucidating mechanistic details. Detailed kinetic analysis allows for discrimination between proposed oxo and adduct mediated mechanisms. In particular, these reactions have been interrogated by competition experiments with isotopically labelled mixtures which shows that all of the studied adducts display a large KIE. These studies suggest different mechanisms may be relevant depending on subtle substituent changes in the adduct complexes. Reactivity data are consistent with the involvement of a transient oxo complex in one case, while the two other systems appear to react with substrates directly as iodosyl- or iodoxyarene adducts. These results support that reactivity typically ascribed to metal-oxo complexes, such as O-atom transfer and C–H activation, can also be mediated by discrete transition metal iodosyl- or iodoxyarene adducts that are frequent intermediates in the generation of oxo complexes. The influence of additional Lewis acids such as Sc3+ on the reactivity of these systems has also been investigated.
Introduction
Iodosylbenzene (PhIO) and its derivative iodosylarenes have been widely used as O-atom donor reagents in transition metal mediated oxidation reactions including C–H hydroxylation and O-atom transfer to substrates.1–4 The mechanisms of these reactions are proposed to proceed via the formation of a metal iodosylarene adduct followed by transfer of an oxygen atom with concomitant oxidation of the metal center to form iodoarene and a high-valent metal-oxo complex as the active oxidant.5–11 Concurrent to the development of this mechanistic paradigm have been studies suggesting that hypervalent iodine adducts themselves are likewise capable of performing atom transfer reactions to substrates.12–24 This alternative mode of activity has motivated efforts at isolating discrete transition metal iodosylarene adducts and studying their reactivity. While rare, there are some examples of well characterized transition metal iodosylarene adducts, including several that have been structurally characterized.25–31 Much of the focus thus far has been on Fe and Mn adducts, as these metals are most frequently featured in oxidation catalysis. More recently, iodosylarenes have been used to generate high-valent Co complexes as well.32–34 Iodosylarene adducts have been cited in these studies as intermediates, but have not yet been isolated or thoroughly characterized to examine their reactivity.Herein, we report the first isolable examples of Co iodosyl- and iodoxyarene adducts [Co(II)TpAd,Me(sPhIO)]+ (1), [Co(II)TptBu(sPhIO)]+ (2), and [Co(II)TptBu(sPhIO2)]+ (3) (Tp = hydrotris(pyrazolyl)borate and sPh = 2-(tert-butylsulfonyl)phenyl; Scheme 1). Complexes 1, 2, and 3 have been crystallographically characterized and detailed kinetic studies reveal a range of reactivity for these three adducts. Complex 1 shows unique behavior that may be consistent with transient oxo formation, but complexes 2 and 3 appear to react as adducts. The observed C–H activation kinetic isotope effect (KIE) is large in all cases, potentially consistent with proton tunnelling transition states.35–38 These observations, in addition to controls with redox-innocent Lewis acids, suggest that Lewis acid activation of iodosylarenes leads to H-atom abstraction with large KIEs.
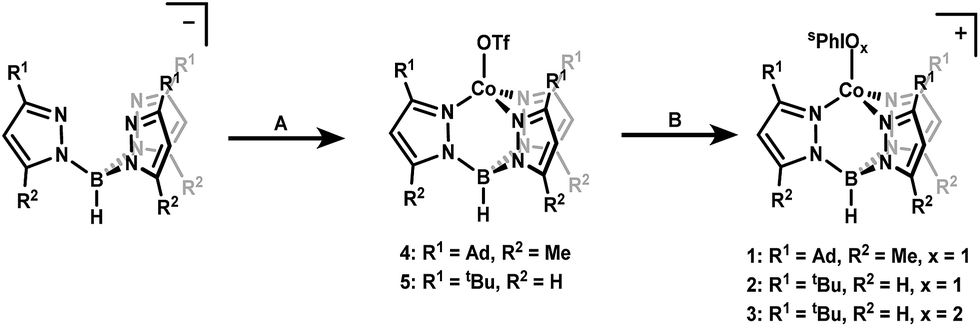 | ||
| Scheme 1 Synthetic routes to generate complexes 1–5 starting from the appropriate Tp ligand. (A) Co(MeCN)6(OTf)2, DCM, N2, r.t., (B) (1) 1.1 equivalents NaBArF4 (2) 1.1 equivalents sPhIOx, Et2O, N2, r.t. | ||
Results and discussion
Synthesis of adduct complexes
Starting materials and ligands were synthesized according to previously reported procedures with slight modifications in some cases (see ESI†).39–45 Treatment of the previously reported NaTpAd,Me or KTptBu ligands with Co(MeCN)6(OTf)2 in dichloromethane (DCM) yielded complexes [Co(II)TpAd,Me(OTf)] (4) and [Co(II)TptBu(OTf)] (5) (OTf = trifluoromethanesulfonate) in 76% and 64% yield as bright blue crystalline solids. The 1H NMR spectra of these complexes confirm overall C3-symmetry in solution with shifted resonances characteristic of paramagnetic species. The 19F NMR spectra of 4 and 5 support the coordination of OTf in solution as demonstrated by a shift of the –CF3 resonance to −8.7 and −30.0 ppm, respectively (the signal from free OTf appears at −78 ppm). Additionally, the EPR spectra of 4 and 5 are consistent with an expected S = 3/2 spin state for high-spin Co(II) centers (see ESI†). Both 4 and 5 display similar electrochemistry, with irreversible oxidations at ∼2 V vs. [FeCp2]0/+ (see ESI†).Synthesis of the cobalt iodosylarene adducts was carried out by treatment of 4 and 5 with 1.1 equivalents of sPhIO and NaBArF4 (BArF4 = tetrakis(3,5-bis(trifluoromethyl)phenyl)borate) in diethyl ether (Et2O). This procedure resulted in the clean formation of new paramagnetic species that maintain C3-symmetry in solution as judged by 1H NMR. A new set of paramagnetically shifted signals consistent with a bound sPhIO ligand are also observed, which suggests the formation of a stable adduct in solution (see ESI†). These species were assigned as the cationic iodosylarene adducts 1 and 2 that can be isolated as blue powders in 76% and 83% yield, respectively (Scheme 1). Complex 3 was first observed as a by-product in 1H NMR spectra from the synthesis of 2. The identity of 3 as an sPhIO2 adduct was suspected due to literature reports that upon standing in solution, sPhIO will disproportionate into sPhI and sPhIO2.41 Treatment of 5 with independently prepared sPhIO2 in the presence of NaBArF4 in Et2O led to the isolation of the targeted sPhIO2 adduct 3 in 67% yield as a light blue powder. Solution 1H NMR spectra showed that complex 3 retains overall C3-symmetry with paramagnetically shifted resonances. The X-band EPR spectra of complexes 1–3 are consistent with S = 3/2 Co(II) centers.
Crystallography
Single crystals of 4 and 5 were grown from concentrated solutions in Et2O layered with petroleum ether and stored at −35 °C for a few days. The X-ray diffraction (XRD) structures of 4 and 5 confirm coordination of the Tp ligand and OTf counter-ion to the Co center (see ESI†). The Co centers of these complexes adopt the expected pseudo-tetrahedral geometry observed for other reported [CoTpX] complexes.46–51 One metric to note is the deviation of the B–Co–O angle from nearly linear in 5 (175.59(4)°) to 161.11(5)° in 4. This distortion is attributed to the steric clashing of the −CF3 group with the adamantyl substituents that is not present for the t-butyl substituted Tp ligand.Crystals of 1 were grown under the same conditions to those of 4 and 5, while crystals of 2 were grown from a concentrated solution of Et2O layered with hexamethyldisiloxane and stored at −35 °C for 7 days. The XRD structures of 1 and 2 confirm the expected pseudo-tetrahedral geometry at Co and furthermore confirm coordination of the sPhIO ligand in the solid state (Chart 1). The average Co–N bond distances are nearly identical to those in 4 and 5, suggesting the oxidation and spin state of Co is unchanged, which is also corroborated by the aforementioned spectroscopic data. The I–O bond lengths in 1 and 2 are 1.878(6) and 1.891(3) Å. When compared to the I–O distance of 1.848(5) Å for free sPhIO there appears to be minimal activation of the iodosylarene by the Co center in these complexes.39 The Co–O bond lengths are 1.920(6) and 1.934(3) Å, which are comparable with other Co(II)–O bonds as well as the OTf starting complexes 4 and 5.52 Complex 3 has also been structurally characterized with crystals grown from a layered Et2O/petroleum ether solution stored at −35 °C. The average Co–N bond lengths of 3 are nearly identical to those of the starting Co–OTf complex 5 and the I–O bond lengths of 1.814(3) and 1.780(3) Å are also similar to those in the structure of free sPhIO2 at 1.822(3) and 1.796(2) Å.41 Isolable iodoxyarene adducts are quite rare, and to our knowledge 3 represents the first structurally characterized example with a transition metal.30 While the I–O bond lengths in this series of complexes are all similar to those reported in the free hypervalent iodine reagents, suggestive of minimal activation, the S![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O⋯I interactions show more sensitivity to Co coordination. The SO⋯I distances are in fact shorter than those found in the structures of the free oxidants, suggesting some effect of binding the Lewis acidic Co(II) center. For 1 and 2 the distances have substantially shortened to 2.481(6) and 2.520(3) Å compared to 2.707(5) Å for free sPhIO.39 For 3, an analogous but smaller contraction from 2.693(2) Å to 2.662(3) Å is observed.41 In the case of 3, there is an unusual close interaction between a solvent THF molecule and the iodine center which may also speak to a more electron deficient adduct (see ESI†).30
O⋯I interactions show more sensitivity to Co coordination. The SO⋯I distances are in fact shorter than those found in the structures of the free oxidants, suggesting some effect of binding the Lewis acidic Co(II) center. For 1 and 2 the distances have substantially shortened to 2.481(6) and 2.520(3) Å compared to 2.707(5) Å for free sPhIO.39 For 3, an analogous but smaller contraction from 2.693(2) Å to 2.662(3) Å is observed.41 In the case of 3, there is an unusual close interaction between a solvent THF molecule and the iodine center which may also speak to a more electron deficient adduct (see ESI†).30
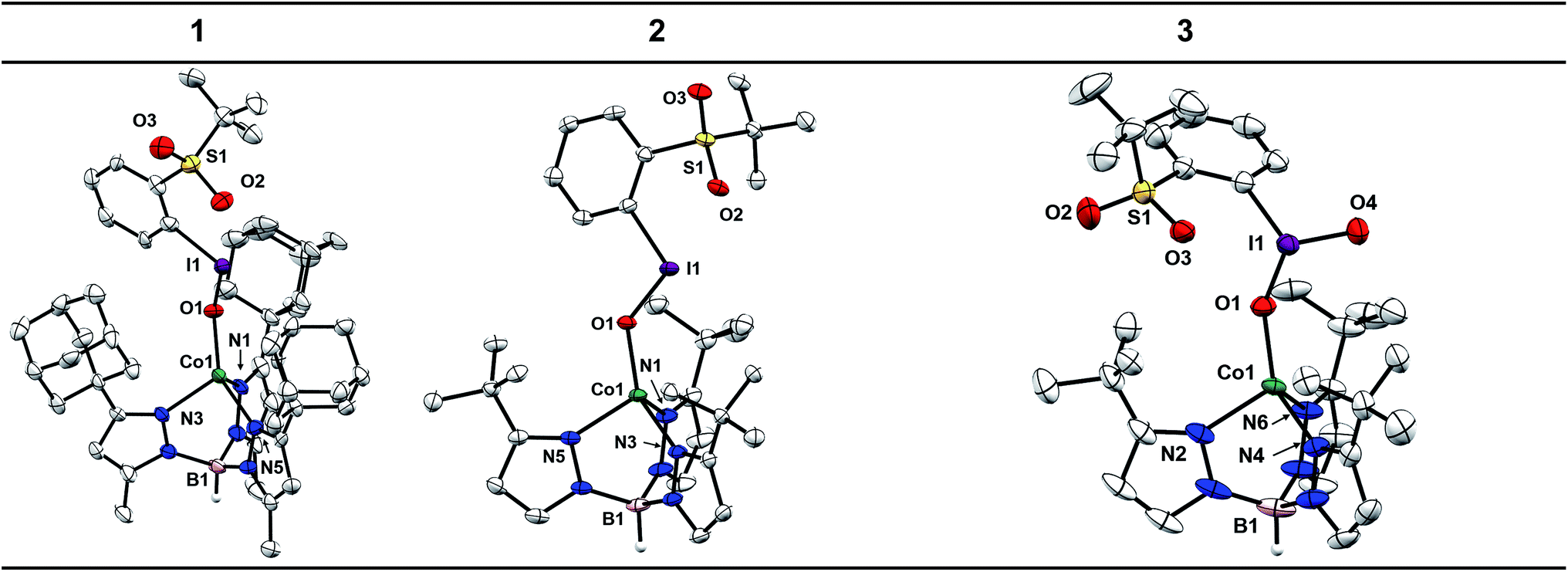 | ||
| Chart 1 XRD structures and selected bond lengths for complexes 1, 2, and 3 (Å). Structures depicted as ellipsoids at 50% probability. Hydrogen atoms (other than B–H), BArF4 counter ions, and solvent molecules omitted for clarity. | ||
The structural data for 1, 2, and 3 support the formation of well-defined adducts with no substantial change in the Co oxidation state, spin state, or activation of the hypervalent iodine unit. This series of complexes represents an unusual family of isolable, well-characterized hypervalent iodine adducts. These species are generally rare and, as mentioned in the introduction, there have been no examples to date of isolated Co adducts despite the use of iodosylarene reagents in Co-mediated oxidation reactivity.32–34
Reactivity studies
The isolability of this series of iodosyl- and iodoxyarene adducts prompted us to investigate their oxidative activity. We were particularly interested in examining the O-atom transfer and C–H activation reactivity of these complexes with a focus on developing an understanding of the mechanism of these transformations. More specifically, we wanted to determine whether high-valent oxo complexes were generated over the course of these reactions or whether the adducts themselves could mediate oxidative reactivity.While complexes 1, 2, and 3 were stable enough to allow for their isolation, they do slowly decompose in solution at room temperature. Complex 1 decays with a rate constant of kobs = 1.4(±0.4) × 10−5 s−1 at room temperature in Et2O (Fig. 1, Table 1). Analysis of the resulting reaction mixture by 1H NMR spectroscopy indicated a complex array of peaks that were not easily assigned (see ESI†). Analysis by ESI-MS shows evidence of incorporation of oxygen into the ligand backbone suggesting ligand-based oxidation as a mode of decay for complex 1 (see ESI†). Attempts to react 1 with external substrates were carried out by addition of either 10 equivalents of thioanisole or up to 100 equivalents of 9,10-dihydroanthracene (DHA). In both cases no increase in the rate of decay was observed by UV-vis spectroscopy (see ESI†). Complex 1 does react rapidly with PMe3 in ∼120 s to generate what is assigned as an OPMe3 adduct. The assignment of the phosphine oxide adduct is supported by independent synthesis from OPMe3 (see ESI†).
 | ||
| Fig. 1 UV-vis spectral changes during the decay of 1 (black, 2.5 mM) in Et2O at 23 °C over 24 h. Inset shows plot of the natural log of the change in concentration of 1 (monitored at 995 nm) vs. time with linear fit (R2 = 0.98). | ||
a Reported rates are 10−6 s−1. Reaction conditions: 2.5 mM cobalt complex, 23 °C, 25 mM substrate (250 mM for DHA).
b Monitored via UV-vis spectroscopy in Et2O.
c Monitored via1H NMR spectroscopy in CDCl3.
d 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of 125 mM each H4-DHA and D4-DHA in DCM. :1 mixture of 125 mM each H4-DHA and D4-DHA in DCM.
|
|||
|---|---|---|---|
| Substrate | 1 | 2 | 3 |
| Self-decay | 14(4) | 0.63(8) | 6.3(4) |
| Thioanisole | No change | 1.12(9) | 16(8) |
| 9,10-DHA | No change | 1.2(3) | 10.3(2) |
| 9,10-DHA KIEd | >12 | 14(5) | 9(1) |
Complexes 2 and 3 were similarly unstable in solution and decayed with markedly slower rate constants than 1. The decay of 2 was monitored via UV-vis spectroscopy to obtain a rate of decay with kobs = 6.3(±0.8) × 10−7 s−1 at room temperature in Et2O. Complex 3 decayed with kobs = 6.3(±0.4) × 10−6 s−1 as monitored with 1H NMR spectroscopy at room temperature in CDCl3. In addition, the decay profiles of both complexes are much less complicated than that observed for 1; complex 2 decays to a single paramagnetic product and 3 decays to this same product while also producing some amount of 2 (see ESI†). Similar to 1, these species show rapid reactivity with phosphines to generate putative phosphine-oxide adducts (also supported by independent syntheses, see ESI†). Unlike 1, both 2 and 3 show distinct reactivity with other substrates. In the presence of 10 equivalents of thioanisole, the rate of disappearance of 2 or 3 increased by roughly two-fold. In the reactions of 2 and 3 with thioanisole, the adduct of the corresponding oxidized substrate, phenylmethylsulfoxide, was observed by 1H NMR spectroscopy. This assignment was again confirmed by independent synthesis of the adduct (see ESI†). Smaller, concentration dependent rate enhancements were observed for 2 and 3 treated with excess DHA (k2 = 2.4 × 10−6 and 1.4 × 10−5 M−1 s−1, Fig. 2 and ESI†). In this case, anthracene was detected by GCMS analysis of the reaction mixtures (see ESI†).
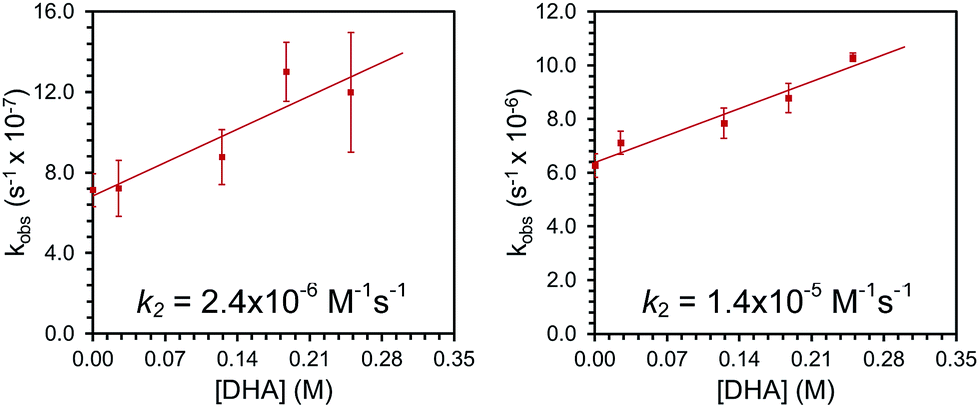 | ||
| Fig. 2 Plots of observed rate constants for the reaction of 2 (left) and 3 (right) with varying concentrations of DHA to determine second order rate constants, k2. | ||
Control reactions were also performed to assess the relative reactivity of 1 and 2 compared to free sPhIO. The complementary study to compare 3 with sPhIO2 was hindered by the extremely poor solubility of sPhIO2 in organic solvents. The rate of decay of sPhIO monitored via1H NMR spectroscopy with either 50 equivalents of DHA or 10 equivalents of thioanisole gave rates of 1.7(±0.2) × 10−5 and 4.3(±0.1) × 10−4 s−1, respectively (see ESI†). The reaction of sPhIO with 10 equivalents of PPh3 was too rapid to monitor by this method; it was complete within 3 minutes of mixing. These data show that the rate of decay of sPhIO with thioanisole is at least an order of magnitude faster than for 1 and 2 under the same conditions and the rate with DHA is on the same order of magnitude as 1 and an order of magnitude faster than for 2 under similar conditions.
It has been previously shown for metal–porphyrin complexes that an equilibrium exists between metal iodosylarene adducts and the corresponding metal–oxo complexes.13,17 This equilibrium can be shifted towards the metal iodosylarene adduct by addition of a sufficient excess of the iodoarene. In order to test whether the Co complexes presented here may reversibly form high-valent Co–oxo species, we tested the effect of added iodoarene on reaction rates. For complex 1, an excess of sPhI (10 equivalents) was added and the self-decay monitored. Under these conditions, no change of the rate of self-decay was observed (see ESI†). The reactions of 2 and 3 with thioanisole or DHA were also investigated with the addition of excess iodoarene. In both cases, no inhibition was observed with additional sPhI. However, in the case of 2, increased rates were observed and for 3, a complex reaction occurred which produced unknown paramagnetic products in addition to the formation of 2 (see ESI†). While we do not have a concrete explanation for these observations, these experiments still oppose a simple reversible oxo formation pathway (see below).
Given a lack of reactivity studies that have been performed with discrete metal iodosyl- and iodoxyarene adducts, we sought to examine more carefully their C–H activation reactivity. As a mechanistic probe of C–H activation, KIEs for the reaction of 1–3 with DHA were measured. Due to the sluggish rates of reaction, we turned to a method reported in the literature for estimating KIE values using GCMS data from the relative ratio of the mass peaks of the H2- and D2-anthracene produced from the competition reaction of the oxidant with a 1:1 mixture of H4- and D4-DHA.53 In these experiments, relatively large KIEs were observed for complexes 1, 2, and 3 (Table 1). With this method, it was difficult to precisely determine the KIE value for 1 because there was no increase in the amount of D2-anthracene detected and instead a value is estimated with a lower bound of 12 using the variability in the instrument response for D2-anthracene in the control mixture (see ESI† for more details). The KIE values for 2 and 3 could be determined more precisely at 14 and 9, respectively. The KIE for free sPhIO was also determined using this method and was found to have a lower bound of 3, which is consistent with the value reported for PhIO.53 Additionally, controls were performed involving redox-innocent, diamagnetic Lewis acids to determine whether the higher KIE values for complexes 1–3 could be due to the influence of the paramagnetic Co ion. Oxidation reactions under the conditions described using sPhIO in the presence of either Sc(OTf)3 or NaBArF4 display a high selectivity for H4-DHA to give KIE values of >66 and >11, respectively. These results suggest that a paramagnetic Lewis acid is not essential for the observation of large KIE values under these conditions. Furthermore, all of the observed KIEs are larger than that measured for free sPhIO, supporting the agency of metal-based intermediates in the observed reactivity as opposed to simple dissociation of sPhIO.
As a final set of experiments to explore the reactivity of the Co iodosyl- and iodoxyarene complexes, we examined the effect of a Lewis acid on their stability and reactivity. Recently, there have been several reports suggesting that in the oxidation of a Co complex with sPhIO, strong Lewis acids such as Sc3+ can stabilize high valent, [Co–O–Sc]5+ moieties.33,34 However, there has been some debate about the nature of these species,54 prompting us to investigate the reactivity of our discrete adducts with Lewis acids. When complexes 2 and 3 were treated with excess Sc(OTf)3, the only observed reaction was slow conversion to complex 5, presumably from the coordination of OTf. When complex 1 was treated with excess Sc(OTf)3 under the same conditions a mixture of products was obtained. We have been unable to purify a single species from this mixture, but we have crystallographically characterized a new asymmetric Co(II) complex which appears to be the major product by 1H NMR analysis (6, see ESI†). Complex 6 shows a single Co center coordinated to two pyrazole arms from the Tp ligand, a free Ad,Me-pyrazole, and OTf with an outer sphere BArF4 ion. Notably, the mass spectrum of this solution did not show evidence of ligand oxidation that is typically observed in the self-decay of 1 (see ESI†). While there was no incorporation of Sc3+ or oxygen into the complex and we do not know the mechanism leading to ligand degradation, this new reaction product demonstrates an alternative pathway is operative for 1 when Sc3+ ions are added. These data do not allow us to definitively support or exclude the formation of transient, high-valent [Co–O–Sc]5+ species.
Discussion of possible mechanism
The key question that remains is the mode of oxidative activity for these complexes. As mentioned in the introduction, the most common paradigm in the literature is a pathway involving adduct decomposition to form a high-valent oxo complex. In our case, we have considered three potential reaction pathways summarized in Fig. 3: (A) irreversible, rate-determining I–O bond scission to form a reactive, transient high-valent Co–oxo complex, (B) reversible I–O bond scission to form a transient Co–oxo complex or (C) direct oxidation of substrate by the Co iodosyl- or iodoxyarene adducts. We have ruled out a mechanism involving dissociation of the hypervalent iodine oxidants on the basis of the observed differences in rates of reactions and KIEs of free sPhIO and the Co adducts (see ESI†). | ||
| Fig. 3 Potential reaction pathways for Co–iodosyl- and iodoxyarene adducts 1–3 involving (A) irreversible Co–oxo formation, (B) reversible Co–oxo formation, or (C) direct substrate oxidation. Where L = Tp ligand, S = generic substrate molecule, and SO = oxidized substrate molecule. | ||
The data for complex 1 are consistent with a rate-determining, irreversible Co–oxo forming step (Fig. 3, pathway A). To be clear, we have no spectroscopic evidence for a Co–oxo species, but its potential involvement is proposed based on the observed reactivity and cannot be ruled out. The rate of decay of complex 1 was not inhibited by addition of excess sPhI, which argues against any equilibrium between 1 and a Co–oxo species as shown in pathway B of Fig. 3. If 1 were to follow pathway C, it is expected that the rate of decay would be dependent on substrate concentration. Instead, the rate of decay of 1 was not accelerated by external substrates, with the exception of phosphines. Furthermore, the decay of 1 results in a complicated mixture of paramagnetic products and some degree of ligand oxidation in the mixture can be inferred from the ESI-MS data. Finally, the large calculated KIE for 1 is comparable to other reported metal–oxo complexes.55–57 These data taken together are consistent with the involvement of a Co–oxo intermediate in the case of 1, but are certainly not conclusive on their own. At minimum the data support a distinct reactivity pathway for 1 compared with complexes 2 and 3.
For 2, there is an observed substrate dependence on the rate of decay, suggesting I–O bond scission is not rate-limiting; this observation argues against pathway A. Furthermore, the reactivity of 2 is not inhibited by the addition of excess sPhI, which argues against a reversible oxo formation mechanism as would be observed in pathway B. These combined data support the direct agency of adduct 2 in the observed oxidative activity rather than decomposition into a Co–oxo complex (Fig. 3, pathway C). The observed large KIE for 2 also suggests that this adduct is directly involved in reactivity, as opposed to a dissociative pathway where free sPhIO acts as the active oxidant. Given that the electrochemistry for both the Ad and the tBu systems shows similar electronic properties in both cases, the observed differences in reactivity between 1 and 2 are likely due to the differing steric profiles of these two systems.
The self-decay and observed substrate reactivity for 3 are similar to that of 2, suggesting that a related mechanism may be operative. However, because formation of 2 is frequently observed in the reactivity of 3, it is difficult to deconvolute relative contributions from these two species in certain experiments such as the KIE analysis and the inhibition of reactivity by added sPhI. These factors make it difficult to distinguish between pathways B and C, but the observed rate dependence on substrate concentration for 3 suggests that pathway A can be excluded.
Conclusions
Complexes 1 and 2 represent the first examples of isolable Co iodosylarene complexes and 3 represents the only example of a transition metal iodoxyarene complex to be thoroughly characterized. The studies herein demonstrate that these adducts display O-atom transfer reactivity and C–H bond activation with appropriate substrates. While the data are consistent with complex 1 reacting via transient oxo formation, complexes 2 and 3 appear to react directly as adducts. This series of complexes represents an unusual family of transition metal hypervalent iodine adducts and the detailed reactivity studies reported here further support the diverse reactivity that these species can exhibit. Notably, it must be underscored that these adduct species – which are frequently invoked only as intermediates – can be competent oxidants themselves in oxidative reactions.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
ChemMatCARS Sector 15 is principally supported by the Divisions of Chemistry (CHE) and Materials Research (DMR), National Science Foundation (NSF), under Grant No. NSF/CHE-1346572. Use of the PILATUS3 X CdTe 1M detector is supported by the National Science Foundation under the grant number NSF/DMR-1531283. Use of the Advanced Photon Source, an Office of Science User Facility operated for the U.S. Department of Energy (DOE) Office of Science by Argonne National Laboratory, was supported by the U.S. DOE under Contract No. DE-AC02-06CH11357. We also thank Dr Yu-Sheng Chen for assistance with single crystal X-ray diffraction studies at beamline 15-ID-B. Work presented here was funded by the following sources: NSF CAREER award Grant No. 1654144, M. L. K. supported by the NSF Graduate Student Fellowship Grant No. DGE-1144082 and DGE-1746045, and the University of Chicago.Notes and references
- P. J. Stang and V. V. Zhdankin, Chem. Rev., 1996, 96, 1123–1178 CrossRef CAS PubMed.
- V. V. Zhdankin and P. J. Stang, Chem. Rev., 2002, 102, 2523–2584 CrossRef CAS PubMed.
- V. V. Zhdankin and P. J. Stang, Chem. Rev., 2008, 108, 5299–5358 CrossRef CAS PubMed.
- A. Yoshimura and V. V. Zhdankin, Chem. Rev., 2016, 116, 3328–3435 CrossRef CAS PubMed.
- J. T. Groves, T. E. Nemo and R. S. Myers, J. Am. Chem. Soc., 1979, 101, 1032–1033 CrossRef CAS.
- J. T. Groves, W. J. Kruper and R. C. Haushalter, J. Am. Chem. Soc., 1980, 102, 6375–6377 CrossRef CAS.
- W. J. Song, M. S. Seo, S. D. George, T. Ohta, R. Song, M.-J. Kang, T. Tosha, T. Kitagawa, E. I. Solomon and W. Nam, J. Am. Chem. Soc., 2007, 129, 1268–1277 CrossRef CAS PubMed.
- J. England, M. Martinho, E. R. Farquhar, J. R. Frisch, E. L. Bominaar, E. Münck and L. Que, Angew. Chem., Int. Ed., 2009, 48, 3622–3626 CrossRef CAS PubMed.
- A. I. Nguyen, R. G. Hadt, E. I. Solomon and T. D. Tilley, Chem. Sci., 2014, 5, 2874–2878 RSC.
- J. D. Koola and J. K. Kochi, J. Org. Chem., 1987, 52, 4545–4553 CrossRef CAS.
- A. R. McDonald and L. Que, Coord. Chem. Rev., 2013, 257, 414–428 CrossRef CAS.
- J. P. Collman, A. S. Chien, T. A. Eberspacher and J. I. Brauman, J. Am. Chem. Soc., 2000, 122, 11098–11100 CrossRef CAS.
- W. Nam, S. K. Choi, M. H. Lim, J.-U. Rohde, I. Kim, J. Kim, C. Kim and L. Que, Angew. Chem., Int. Ed., 2003, 42, 109–111 CrossRef CAS PubMed.
- J. P. Collman, A. L. Zeng and J. I. Brauman, Inorg. Chem., 2004, 43, 2672–2679 CrossRef CAS PubMed.
- S. H. Wang, B. S. Mandimutsira, R. Todd, B. Ramdhanie, J. P. Fox and D. P. Goldberg, J. Am. Chem. Soc., 2004, 126, 18–19 CrossRef CAS PubMed.
- K. P. Bryliakov and E. P. Talsi, Angew. Chem., Int. Ed., 2004, 43, 5228–5230 CrossRef CAS PubMed.
- W. J. Song, Y. J. Sun, S. K. Choi and W. Nam, Chem.–Eur. J., 2006, 12, 130–137 CrossRef CAS PubMed.
- P. Leeladee and D. P. Goldberg, Inorg. Chem., 2010, 49, 3083–3085 CrossRef CAS PubMed.
- S. Hong, B. Wang, M. S. Seo, Y.-M. Lee, M. J. Kim, H. R. Kim, T. Ogura, R. Garcia-Serres, M. Clémancey, J.-M. Latour and W. Nam, Angew. Chem., Int. Ed., 2014, 53, 6388–6392 CrossRef CAS PubMed.
- B. Wang, Y.-M. Lee, M. S. Seo and W. Nam, Angew. Chem., Int. Ed., 2015, 54, 11740–11744 CrossRef CAS PubMed.
- J. D. Protasiewicz, in Hypervalent Iodine Chemistry, ed. T. Wirth, Springer, Cham, 2015, pp. 263–288 Search PubMed.
- Y. Kang, X.-X. Li, K.-B. Cho, W. Sun, C. Xia, W. Nam and Y. Wang, J. Am. Chem. Soc., 2017, 139, 7444–7447 CrossRef CAS PubMed.
- M. J. Zdilla and M. M. Abu-Omar, J. Am. Chem. Soc., 2006, 128, 16971–16979 CrossRef CAS PubMed.
- M. M. Abu-Omar, Dalton Trans., 2011, 40, 3435–34444 RSC.
- A. Lennartson and C. J. McKenzie, Angew. Chem., Int. Ed., 2012, 51, 6767–6770 CrossRef CAS PubMed.
- D. P. de Sousa, C. Wegeberg, M. S. Vad, S. Mørup, C. Frandsen, W. A. Donald and C. J. McKenzie, Chem.–Eur. J., 2016, 22, 3810–3820 CrossRef CAS PubMed.
- C. Wang, T. Kurahashi and H. Fujii, Angew. Chem., Int. Ed., 2012, 51, 7809–7811 CrossRef CAS PubMed.
- C. Wang, T. Kurahashi, K. Inomata, M. Hada and H. Fujii, Inorg. Chem., 2013, 52, 9557–9566 CrossRef CAS PubMed.
- C. R. Turlington, J. Morris, P. S. White, W. W. Brennessel, W. D. Jones, M. Brookhart and J. L. Templeton, Organometallics, 2014, 33, 4442–4448 CrossRef CAS.
- K.-C. Au-Yeung, Y.-M. So, G.-C. Wang, H. H.-Y. Sung, I. D. Williams and W.-H. Leung, Dalton Trans., 2016, 45, 5434–5438 RSC.
- G. de Ruiter, K. M. Carsch, S. Gul, R. Chatterjee, N. B. Thompson, M. K. Takase, J. Yano and T. Agapie, Angew. Chem., Int. Ed., 2017, 56, 4772–4776 CrossRef CAS PubMed.
- B. Wang, Y.-M. Lee, W.-Y. Tcho, S. Tussupbayev, S.-T. Kim, Y. Kim, M. S. Seo, K.-B. Cho, Y. Dede, B. C. Keegan, T. Ogura, S. H. Kim, T. Ohta, M.-H. Baik, K. Ray, J. Shearer and W. Nam, Nat. Commun., 2017, 8, 14839 CrossRef CAS PubMed.
- S. Hong, F. F. Pfaff, E. Kwon, Y. Wang, M.-S. Seo, E. Bill, K. Ray and W. Nam, Angew. Chem., Int. Ed., 2014, 53, 10403–10407 CrossRef CAS PubMed.
- F. F. Pfaff, S. Kundu, M. Risch, S. Pandian, F. Heims, I. Pryjomska-Ray, P. Haack, R. Metzinger, E. Bill, H. Dau, P. Comba and K. Ray, Angew. Chem., Int. Ed., 2011, 50, 1711–1715 CrossRef CAS PubMed.
- E. F. Caldin, Chem. Rev., 1969, 69, 135–156 CrossRef CAS.
- Z. Cong, H. Kinemuchi, T. Kurahashi and H. Fujii, Inorg. Chem., 2014, 53, 10632–10641 CrossRef CAS PubMed.
- D. Mandal, D. Mallick and S. Shaik, Acc. Chem. Res., 2018, 51, 107–117 CrossRef CAS PubMed.
- J. M. Mayer, Acc. Chem. Res., 2011, 44, 36–46 CrossRef CAS PubMed.
- D. Macikenas, E. Skrzypczak-Jankun and J. D. Protasiewicz, J. Am. Chem. Soc., 1999, 121, 7164–7165 CrossRef CAS.
- F. Song, C. Wang, J. M. Falkowski, L. Ma and W. Lin, J. Am. Chem. Soc., 2010, 132, 15390–15398 CrossRef CAS PubMed.
- D. Macikenas, E. Skrzypczak-Jankun and J. D. Protasiewicz, Angew. Chem., Int. Ed., 2000, 39, 2007–2010 CrossRef CAS PubMed.
- A. McSkimming and W. H. Harman, J. Am. Chem. Soc., 2015, 137, 8940–8943 CrossRef CAS PubMed.
- Inorganic Syntheses, ed. R. J. Angelici, John Wiley & Sons, Inc., Hoboken, NJ, USA, 1990, vol. 28 Search PubMed.
- C. Goldsmith, R. Jonas and T. Stack, J. Am. Chem. Soc., 2002, 124, 83–96 CrossRef CAS PubMed.
- N. Chakrabarti, W. Sattler and G. Parkin, Polyhedron, 2013, 58, 235–246 CrossRef CAS.
- I. B. Gorrell and G. Parkin, Inorg. Chem., 1990, 29, 2452–2456 CrossRef CAS.
- S. Thyagarajan, C. D. Incarvito, A. L. Rheingold and K. H. Theopold, Inorg. Chim. Acta, 2003, 345, 333–339 CrossRef CAS.
- J. D. Jewson, L. M. Liable-Sands, G. P. A. Yap, A. L. Rheingold and K. H. Theopold, Organometallics, 1999, 18, 300–305 CrossRef CAS.
- J. L. Detrich, R. Konečný, W. M. Vetter, D. Doren, A. L. Rheingold and K. H. Theopold, J. Am. Chem. Soc., 1996, 118, 1703–1712 CrossRef CAS.
- S. Thyagarajan, C. D. Incarvito, A. L. Rheingold and K. H. Theopold, Chem. Commun., 2001, 2198–2199 RSC.
- J. W. Egan, B. S. Haggerty, A. L. Rheingold, S. C. Sendlinger and K. H. Theopold, J. Am. Chem. Soc., 1990, 112, 2445–2446 CrossRef CAS.
- C. R. Groom, I. J. Bruno, M. P. Lightfoot and S. C. Ward, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2016, 72, 171–179 CrossRef CAS PubMed.
- S. J. Kim, R. Latifi, H. Y. Kang, W. Nam and S. P. de Visser, Chem. Commun., 2009, 13, 1562 RSC.
- D. C. Lacy, Y. J. Park, J. W. Ziller, J. Yano and A. S. Borovik, J. Am. Chem. Soc., 2012, 134, 17526–17535 CrossRef CAS PubMed.
- A. Gunay and K. H. Theopold, Chem. Rev., 2010, 110, 1060–1081 CrossRef CAS PubMed.
- L. Q. Shen, S. Kundu, T. J. Collins and E. L. Bominaar, Inorg. Chem., 2017, 56, 4347–4356 CrossRef CAS PubMed.
- S. N. Dhuri, K.-B. Cho, Y.-M. Lee, S. Y. Shin, J. H. Kim, D. Mandal, S. Shaik and W. Nam, J. Am. Chem. Soc., 2015, 137, 8623–8632 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1818929–1818931 and 1818933–1818935. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8sc01167b |
| This journal is © The Royal Society of Chemistry 2018 |