
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBlue light-promoted photolysis of aryldiazoacetates†
Igor D.
Jurberg
 *ab and
Huw M. L.
Davies
*a
*ab and
Huw M. L.
Davies
*a
aDepartment of Chemistry, Emory University, 1515 Dickey Drive, Atlanta, Georgia 30322, USA
bInstitute of Chemistry, State University of Campinas, Rua Monteiro Lobato 270, Campinas, São Paulo 13083-970, Brazil. E-mail: idjurberg@iqm.unicamp.br
First published on 22nd May 2018
Abstract
Aryldiazoacetates can undergo photolysis under blue light irradiation (460–490 nm) at room temperature and under air in the presence of numerous trapping agents, such as styrene, carboxylic acids, amines, alkanes and arenes, thus providing a straighforward and general platform for their mild functionalization.
Introduction
The chemistry of diazocompounds has been intensively studied over the past decades.1 Today, a number of catalytic, racemic thermal processes,2 as well as highly stereoselective methods describing cyclopropanations,3 O–H,4 N–H5 and C–H6 insertions, (formal) cycloadditions7 and rearrangements8 have been established, thus allowing the straightforward preparation of a variety of molecules.In contrast to the numerous protocols reported on thermal processes, photochemical studies with diazocompounds have been performed mostly several decades ago,9 and modern applications are relatively scarce.10,11 Among these, Suero and co-workers have recently demonstrated that a hypervalent iodine reagent carrying a diazoacetate moiety could be employed in a photoredox-catalyzed protocol for the C–H functionalization of a variety of arenes.11b In the sequence, the diazocompounds produced could be further engaged in classical reactions, thus representing an overall formal process to access carbyne equivalents. The authors of this communication also noticed the effect of blue light on aryldiazoacetates 1, but did not synthetically exploit this to any significant extent.
In recent years, the chemistry of donor/acceptor metal carbenes has been broadly developed for synthesis.1 These intermediates, typically prepared from aryldiazoacetates or vinyldiazoacetates, have attenuated reactivity compared to metal carbenes lacking a donor group. Donor/acceptor carbenes derived from the thermal decomposition of aryldiazoacetates have been shown to undergo relatively selective cyclopropanations,12 N–H13 and C–H14 insertions.
As opposed to these methods, one might argue that milder, metal-free strategies can be accessible by the use of photochemical conditions, thus being of special importance in scenarios where stereocontrol (typically governed by a metal catalyst) is not a prime concern, such as in chemical-biology applications.15 Based on this background, and motivated by the realization that the use of light could avoid the need for high temperatures and the use of metal catalysts, we have conducted a systematic study on the photochemically-induced reactions of aryldiazoacetates. This approach opens up a new window of chemical reactivity that leads to the mild functionalization of organic molecules, while also being more economical and sustainable.
Results and discussion
Spectroscopic studies
With this mindset, our investigation started by analyzing the UV-Vis absorbance spectra of two aryldiazoacetates 1a and 1b, and comparing them to ethyl 2-diazoacetate 2a and dimethyl 2-diazomalonate 2b. The donor/acceptor diazo compounds 1a and 1b present an almost identical absorbance spectra in the visible region. In contrast to the acceptor-only diazo compounds 2a and 2b, both 1a and 1b display bathochromic shifts in the region of 400–500 nm (violet/blue), which can be presumably attributed to a n–π* transition of the diazo group16 (Fig. 1).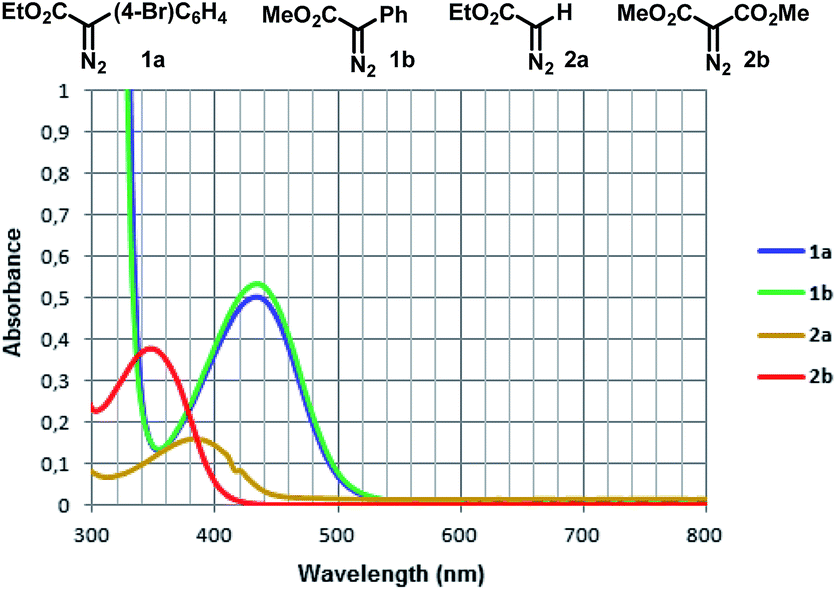 | ||
| Fig. 1 UV-Vis absorbance spectra of diazocompounds 1a, 1b, 2a and 2b in DCM (all in 0.05 mM). | ||
Solvent evaluation studies
In agreement to these observations, preliminary tests promoting the blue-light irradiation of diazocompound 1a in the presence of different solvents, at room temperature and under air led to a variety of compounds in moderate to excellent yields (Scheme 1). These products were derived from [2 + 1] cycloadditions (3a and 3b); O–H insertion events (4a, 4b and 4c. The generation of 4c can be attributed to an ylide formation derived from the O-attack of the THF ring to the free carbene, followed by an elimination event, leading to the formation of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond); and a C–H insertion event (5a). In contrast, when DCM was employed, no appreciable reaction with the solvent was detected. Instead, a complex mixture is observed, presumably derived from dimerization pathways and other self-reactions involving 1a. The compounds formed are the expected products of a carbene, that is being produced as a reactive intermediate derived from the blue light irradiation of aryldiazoacetate 1a
C bond); and a C–H insertion event (5a). In contrast, when DCM was employed, no appreciable reaction with the solvent was detected. Instead, a complex mixture is observed, presumably derived from dimerization pathways and other self-reactions involving 1a. The compounds formed are the expected products of a carbene, that is being produced as a reactive intermediate derived from the blue light irradiation of aryldiazoacetate 1a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 17 (Scheme 1).
17 (Scheme 1).
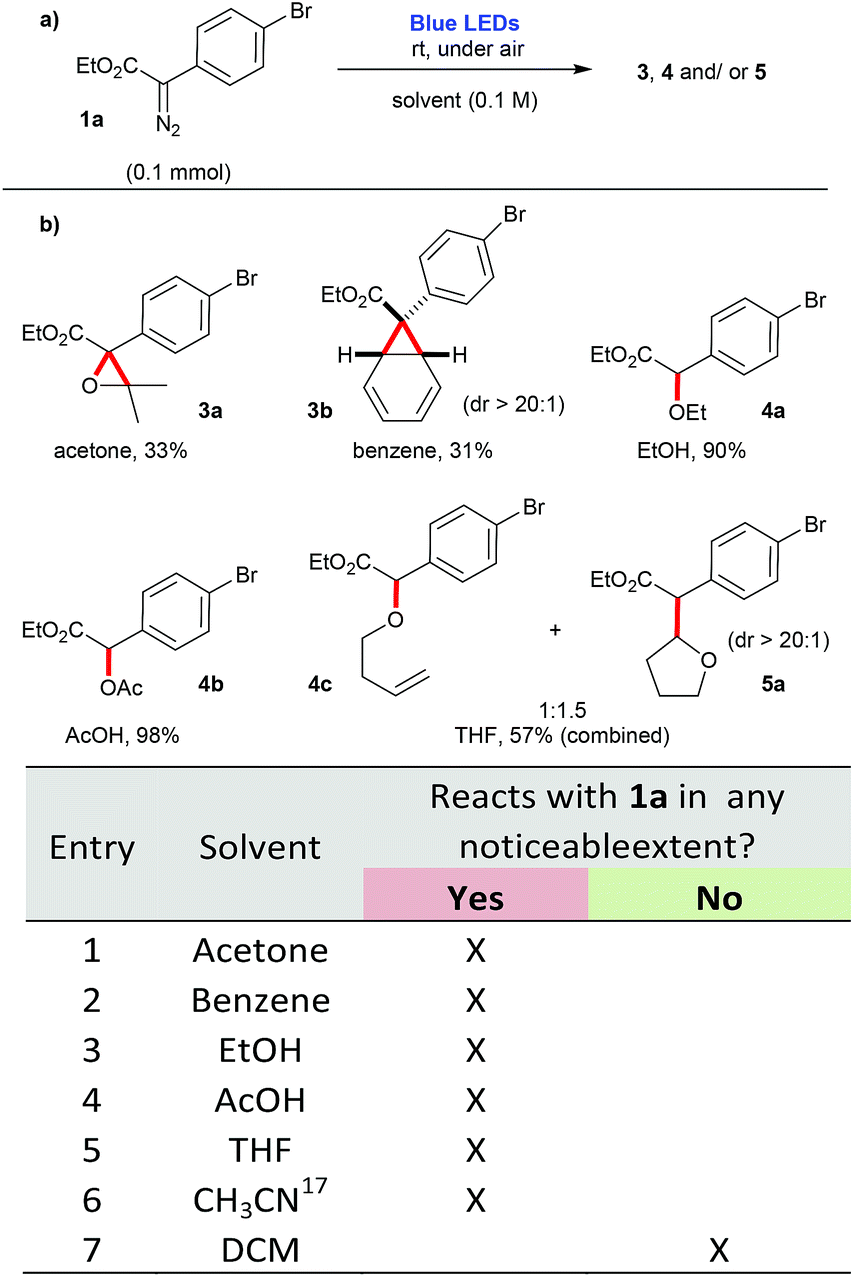 | ||
| Scheme 1 Evaluation of different solvents in the photolysis of 1a using blue light irradiation. | ||
Although this photolysis phenomenon is well known for other diazocompounds using UV-light,9,16,18 one can note here that the presence of a donor group at the aryldiazoacetates 1a and 1b (or the removal of an acceptor group, as one can observe the same trend comparing the UV-Vis absorption spectra of 2a and 2b) is responsible for a significant bathochromic shift in their UV-Vis absorbance spectra, thus allowing the photolysis process to occur in the visible region.10,16
Synthetic applications
Next, the synthetic potential of this new photochemical process involving aryldiazoacetates 1 was evaluated (Scheme 2). As a consequence of the previous investigation, DCM was chosen as the optimal solvent. A range of trapping agents, such as styrene 6 (5 equiv.), carboxylic acids 7 (2 equiv.), amines 8 (3 equiv.), cyclic alkanes 9 (100 equiv.) and arenes 10 (5 equiv.) were examined. In most cases, good yields of the corresponding products were obtained using a moderate excess of the trapping agents, except in the case of cyclic alkanes 9, in which the trapping agent was used as the solvent.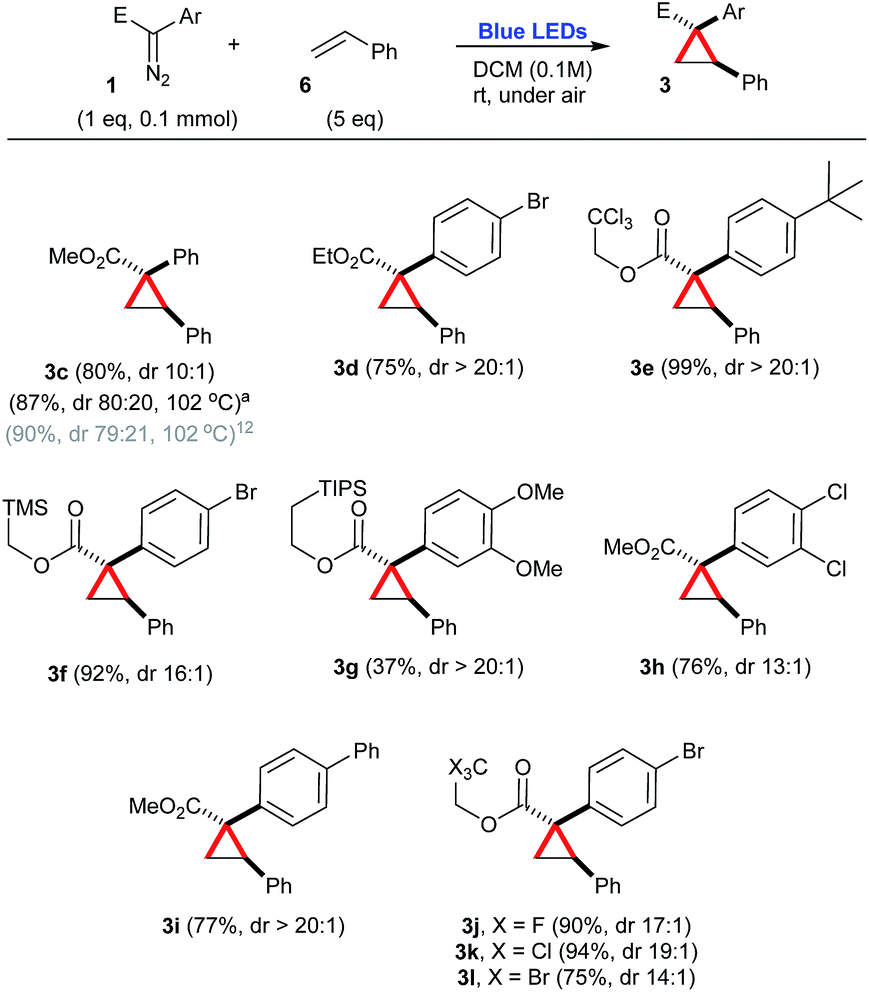 | ||
| Scheme 2 Cyclopropanation of styrene in the presence of a number of aryldiazoacetates, under blue light irradiation. aPrepared under thermal conditions.12 | ||
In the case of styrene 6 as a carbene trap, a number of aryldiazoacetates 1a–1j were employed to produce the corresponding cyclopropanes 3c–3l in good yields, 37–94% and high diastereoselectivities, varying from 10:1 to >20:1 (Scheme 2). Only cyclopropane 3g was produced in a somehow low efficiency (37% yield), due to the competing formation of a diastereoisomeric mixture of β-propiolactones,19 as the result of an intramolecular C–H insertion event (Scheme 2).
Carboxylic acids 7 were found to be excellent carbene traps as they undergo efficient O–H insertions to afford the corresponding esters 4d–4p in generally good yields, 61–99% (Scheme 3a). In this context, control experiments demonstrate that when stronger acids are employed, such as but-2-ynoic acid 7m, 2,3,4,5,6-pentafluorobenzoic acid 7n, 2-oxo-2-phenylacetic acid 7o, and 2,2,3,3,4,4,4-heptafluorobutanoic acid 7p, the corresponding O–H insertions also proceed in the dark (Scheme 3b). Therefore, the preparation of substrates 4m–4p does not proceed via a photochemical process, but it rather follows most likely a classical mechanism of protonation of the starting diazocompound 1a, followed by a SN2 displacement of N2, that is typically found in the esterification mechanism of carboxylic acids using diazomethane.20 For instance, upon addition of 7p to the other components of the reaction mixture, the O–H insertion occurs almost instantly (which is accompanied by the colour change of solution from yellow to transparent and vigorous release of N2).
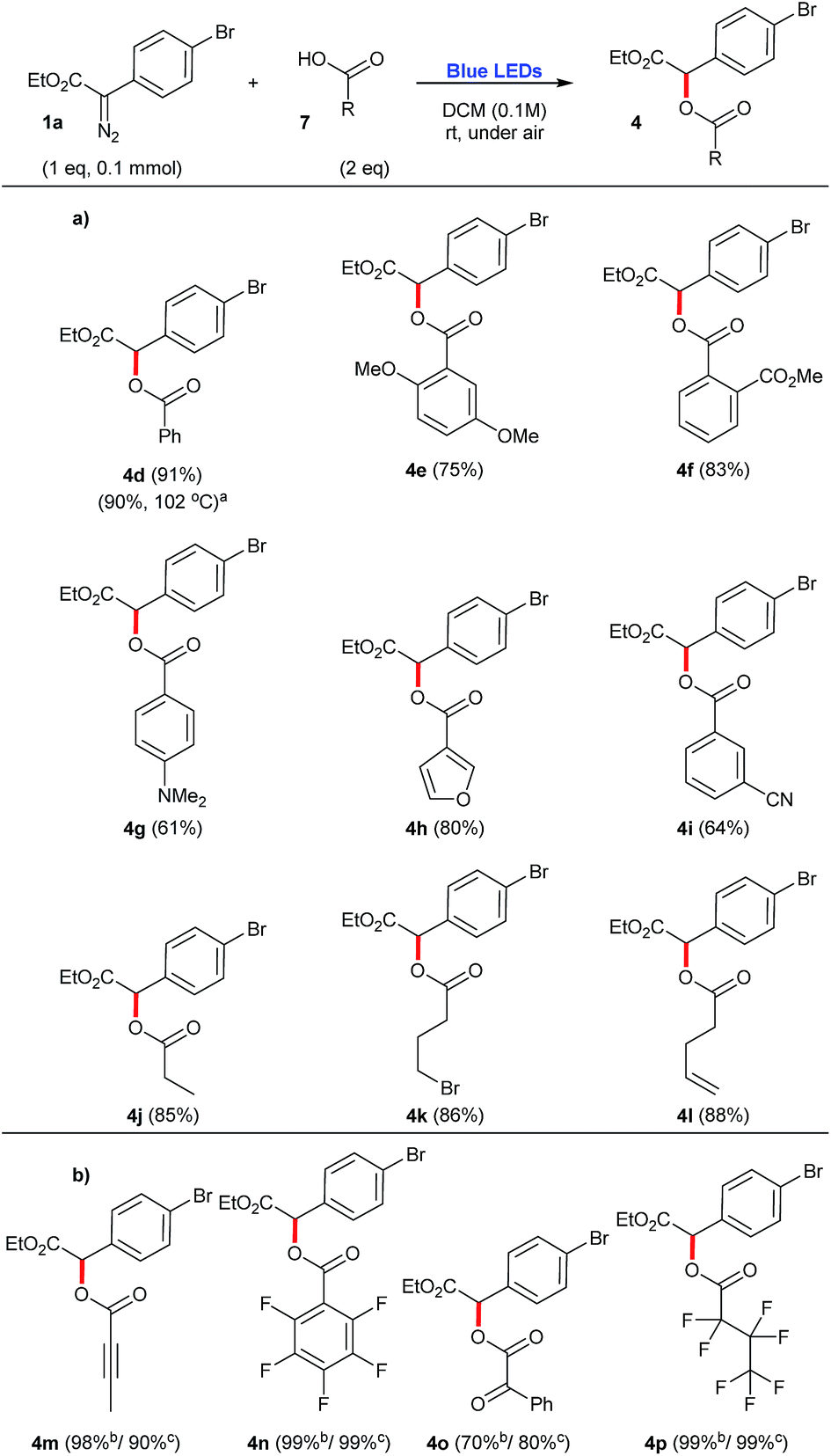 | ||
| Scheme 3 O–H insertion reactions employing aryldiazoacetate 1a and a number of carboxylic acids, under blue light irradiation. (a) Scope of O–H insertions that work exclusively under photochemical conditions. (b) Examples of O–H insertions that work also under thermal conditions (in the absence of blue light). aPrepared under thermal conditions. bYield for the process irradiated with blue light; cyield for the process in the dark. | ||
Concerning the use of primary, secondary and aromatic amines 8 as carbene traps, a variety of N–H insertions can be accomplished in good yields, 58–90%, to produce the corresponding coupled products 11. In this context, it is possible to note that bulky amines produced slightly lower yields, 58–69% (e.g.11e, 11k, 11l), while linear and other unhindered secondary congeners lead to the corresponding coupled products 11 in higher efficiencies, 81–90% (e.g.11a, 11b, 11c, 11i, 11m). Of note, pyrrolidine produced the coupled product 11h in a somehow lower yield, 60% (Scheme 4).
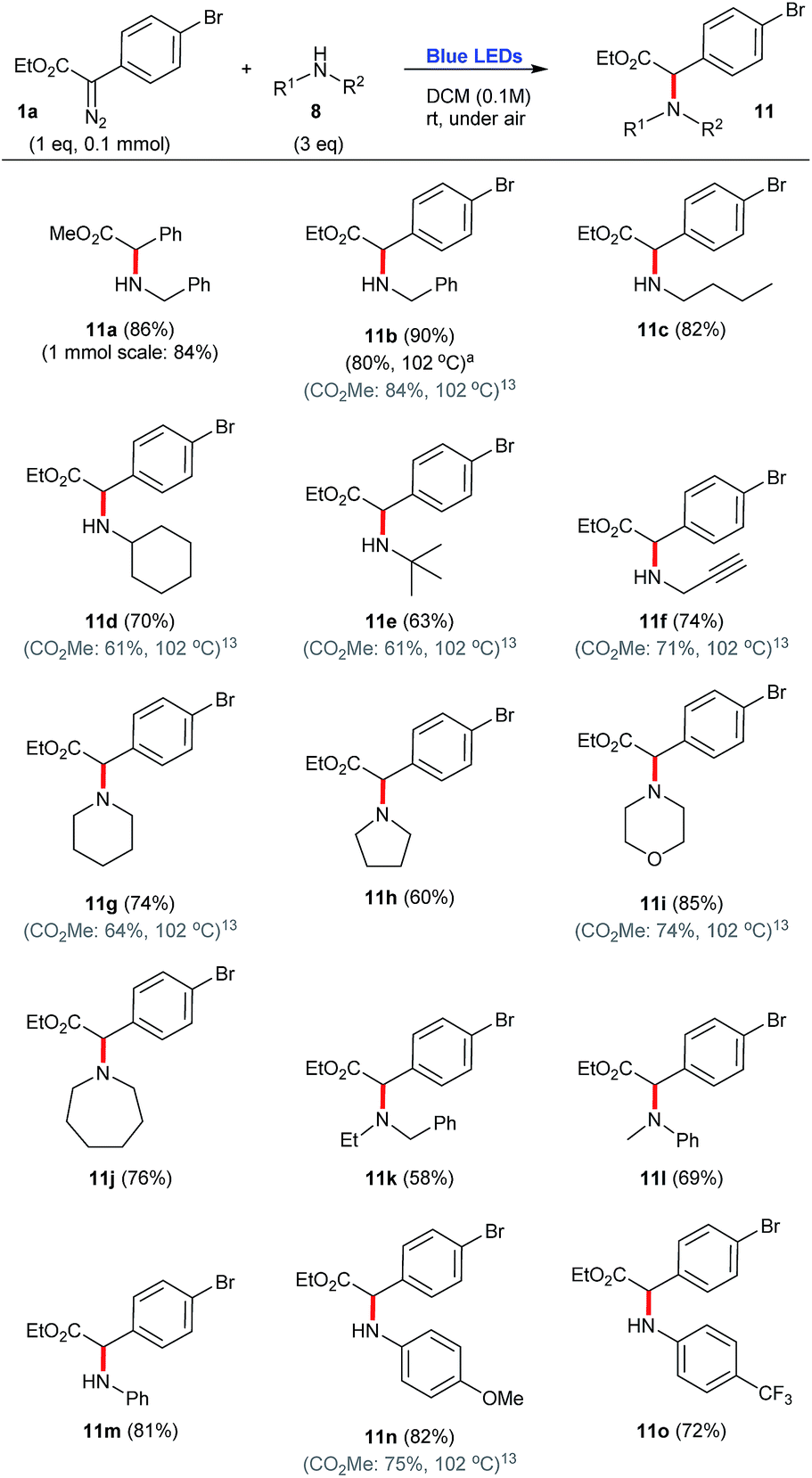 | ||
| Scheme 4 N–H insertion reactions employing aryldiazoacetate 1a and a number of amines, under blue light irradiation. aPrepared under thermal conditions.13 | ||
When cyclic alkanes 9, such as cyclohexane 9a, cyclopentane 9b, and cycloheptane 9c are employed as carbene traps, they need to be used in large excess (100 eq.), in order to afford the corresponding C–H insertion products 5 in a range of moderate to good yields, 55–79% (Scheme 5). In addition, the use of cyclohexadiene 9d as substrate produces an equimolar mixture of allylic C–H insertion (5f) and cyclopropanation (5g).21,22 Furthermore, intramolecular C–H insertions can also be performed. For instance, under these reaction conditions, tert-butyl 2-phenyl-2-diazoacetate 1k and isopropyl 2-phenyl-2-diazoacetate 1l are both converted to the 5-membered lactone 5h, in 91% yield, and the 4-membered lactone 5i, in 57% yield, respectively. In the case of more reactive cyclic alkanes, such as adamantane 9e, it is possible to use lower amounts of trapping alkane (5 eq.) to produce the corresponding C–H inserted product 5j in a respectful 48% yield (Scheme 5).
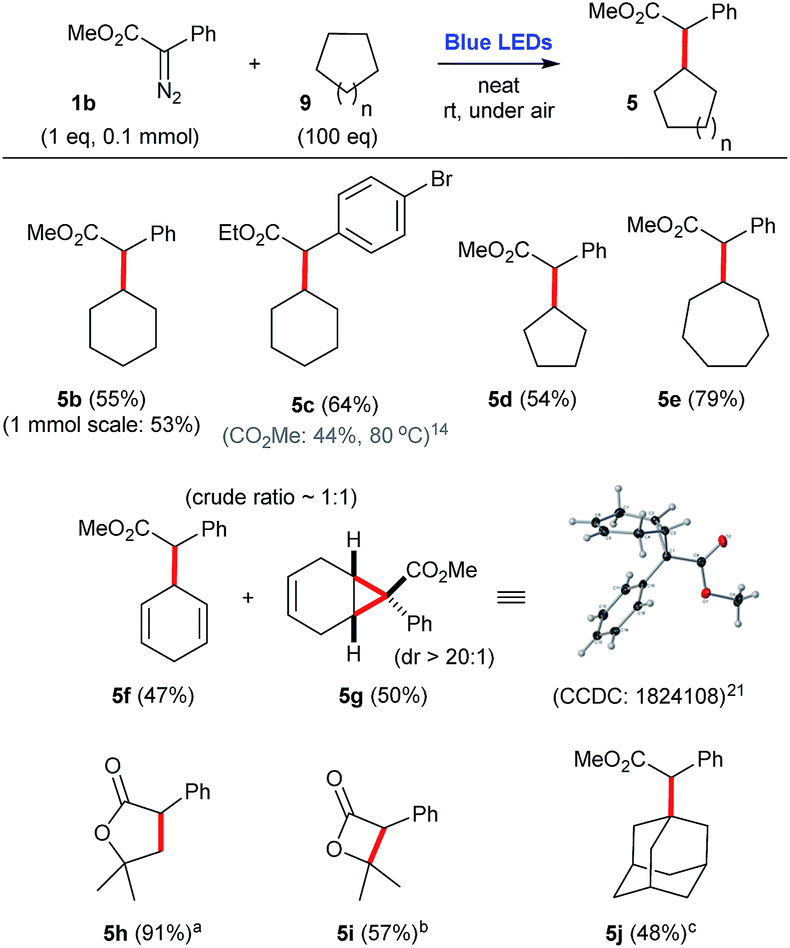 | ||
| Scheme 5 C–H insertion reactions employing aryldiazoacetate 1b and a number of cyclic alkanes and other substrates, under blue light irradiation. aIntramolecular process using tert-butyl 2-phenyl-2-diazoacetate (0.2 mmol). bIntramolecular process using isopropyl 2-phenyl-2-diazoacetate (0.2 mmol). cIntermolecular process employing adamantane (5 equiv.). | ||
The use of arenes 10 as carbene traps can either produce cyclopropanation products or C–H insertion products (Scheme 6).
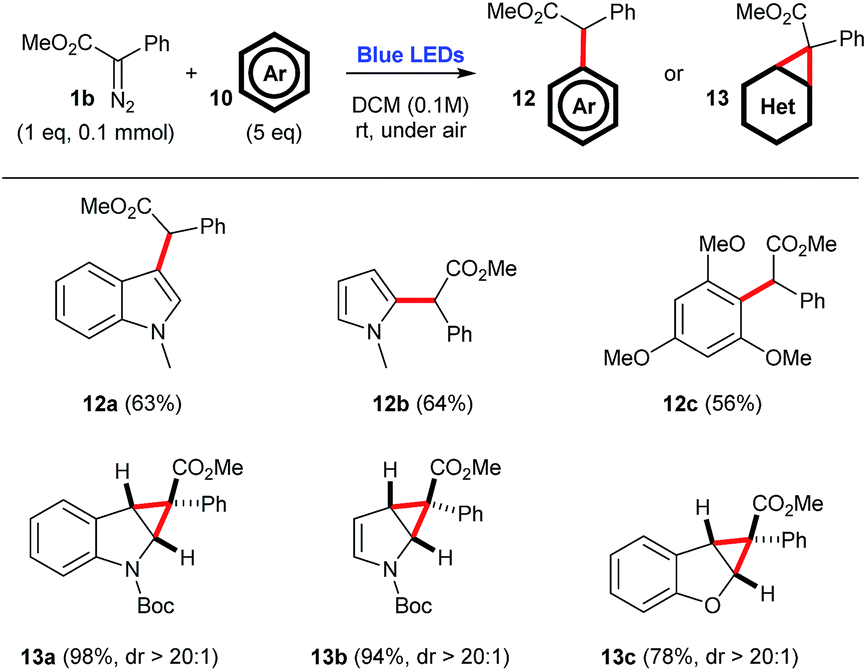 | ||
| Scheme 6 C–H insertion or cyclopropanation reactions employing aryldiazoacetate 1b and a number of arenes, under blue light irradiation. | ||
For instance, when electron-rich N-methyl indole 10a, N-methyl pyrrole 10b, or 1,3,5-trimethoxybenzene 10c are employed, the corresponding C–H inserted products 12a–12c are selectively produced, in 56–64% yield. However, when Boc-protected indole 10d, Boc-protected pyrrole 10e, or benzofuran 10f are employed, one observes the selective formation of the corresponding cyclopropanation products 13a–13c, in high yields 74–98%, and perfect diastereocontrol, dr > 20:1. Of note, both Boc-protected compounds 13a and 13b produce a mixture of rotamers, as evidenced by the coalescence of 1H NMR signals upon heating (see the ESI†).
In order to demonstrate the possibility of scaling up the previously described reactions, substrates 5b and 11a have been also prepared in a 1 mmol-scale, without any significant change in efficiency (Schemes 4 and 5). Importantly, every reaction reported in previous Schemes 2–6 has been also attempted in the dark, and except for the molecules described in Scheme 3b, all failed to provide the corresponding insertion or cyclopropanation products, but rather afforded only recovered starting materials. These control experiments confirm that these processes are indeed promoted photochemically.
Another important consideration is the comparison of the photochemical reactions with the purely thermal reactions. A number of compounds presented in the previous schemes have been also prepared by Davies and co-workers via metal-free thermal processes.12–14 In order to compare the efficiencies of these previously reported protocols with the one reported herein, their yields have been also mentioned, with the corresponding references. In this context, a metal-free thermal cyclopropanation method has been reported for the preparation of 3c, heating the starting aryldiazoacetate 1a and styrene 6 at the reflux of PhCF3, at 102 °C.12 While in the previous report, 3c is obtained in a slightly higher yield (90%), the 4:1 dr reported for this process is considerably inferior to the photochemical transformation reported here, that affords 3c in 80% isolated yield, dr 10:1 [crude dr 9:1] (Scheme 2). Also taking advantage of this thermal protocol for the generation of free carbenes, the O–H insertion of benzoic acid 7a was performed in the presence of diazocompound 1a and produced 4d with the same efficiency as our photochemical process, 90% yield (Scheme 3).
In addition, the preparation of amine 11avia the photochemical process reported herein describes a 90% yield, while the thermal reaction following Davies' conditions (reflux of PhCF3, 102 °C) has been accomplished in our hands in 80% yield; and the corresponding methyl ester derivative has been reported in 84% yield.13 In agreement to this observation, other photochemical processes described here for N–H insertions using ethyl aryldiazoacetate 1a also consistently produced higher yields when compared to methyl analogues employed in thermal processes (Scheme 4).13
Furthermore, an analogous thermal process for the C–H insertion of cyclohexane 9a into methyl 2-phenyl-2-diazoacetate 1b has been described in 44% yield, at 80 °C,14 while the photochemical process reported herein, at room temperature, produces the corresponding ethyl derivative 5c in 64% yield (Scheme 5).
In addition, for comparison purposes, both donor/acceptor diazocompounds 1a and 1b have been compared to acceptor-only diazo compound 2a and acceptor/acceptor diazocompound 2b for the C–H functionalization of cyclohexane. In agreement to the absorbance spectra shown in Fig. 1, both 1a and 1b undergo blue light-promoted photolysis more effectively than 2a and 2b to afford the corresponding C–H insertion products (Scheme 7).
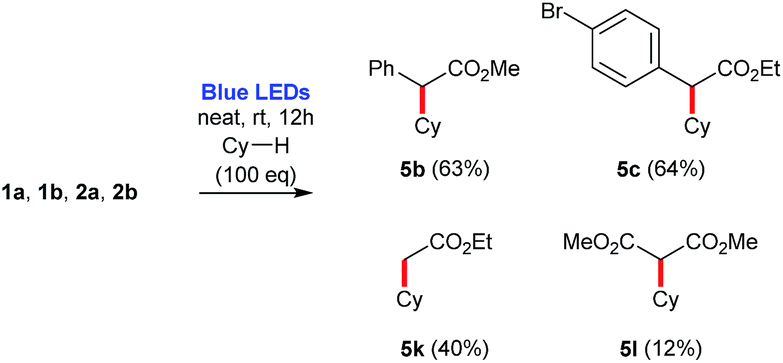 | ||
| Scheme 7 Comparison of C–H insertion reactions involving diazocompounds 1a, 1b, 2a, 2b and cyclohexane. Yields are estimated based on the 1H NMR of the crude reaction mixture using 1,3,5-trimethoxybenzene as internal standard. | ||
Finally, this photochemical protocol was also evaluated for the functionalization of more complex molecules, indometacin 14 and ibopamine 15. In the case of indometacin 14, it is possible to employ a 1:1 mixture of 14:1b to afford the corresponding ester 16 in 77% yield. In the case of ibopamine 15, the optimization of this reaction allowed the reversion of stoichiometry, this time using a 1:3 mixture of 15:1b to produce the corresponding tertiary amine 17 in 57% yield (Scheme 8).
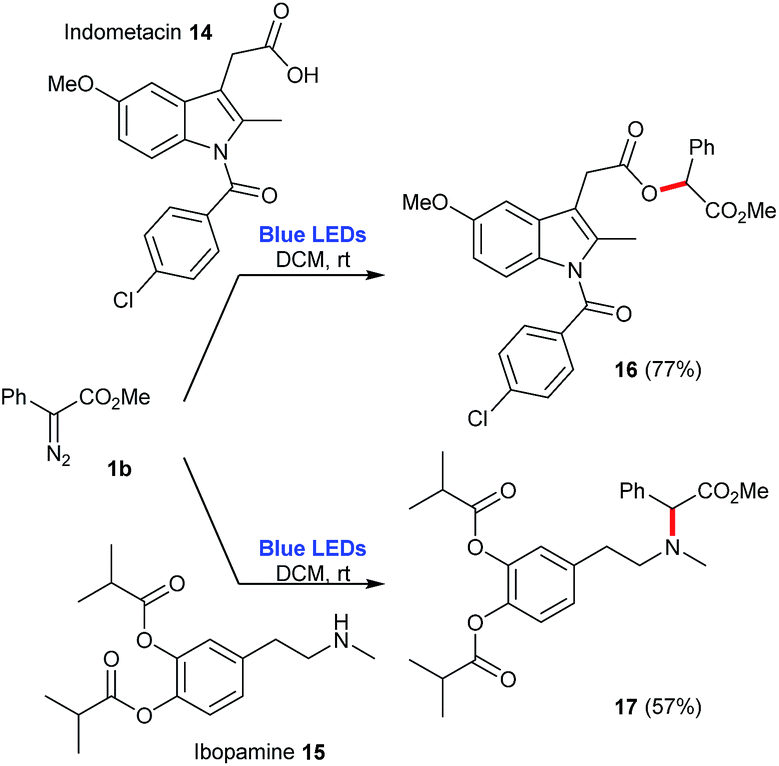 | ||
| Scheme 8 Functionalization of pharmaceuticals employing the blue-light promoted photolysis of methyl phenyldiazoacetate 1b. | ||
Concerning the mechanism of these photochemical reactions and the nature of the carbenes involved, they are likely to proceed via the photoexcitation of the starting diazocompound 1 to a higher energy singlet state 11*, followed by the extrusion of N2, to afford a singlet carbene 118, which can then be trapped by a reacting partner (e.g.6, 7, 8, 9 or 10) to afford the observed final compounds (e.g.3, 4, 5, 11, 12 or 13).13,23 Alternatively, at this point, it is not possible to rule out that the excited diazocompound 11* could react directly with a trapping partner to afford the corresponding final product.24 Although inter system crossing (ISC) converting 118 to 318 is generally a fast process, in this case, experimental evidences suggest that this is a reversible event.25 Subsequent conversion of 318 to the corresponding ketene 19 (via a Wolff-rearrangement) would serve as an additional source for the formation of by-products (Scheme 9).
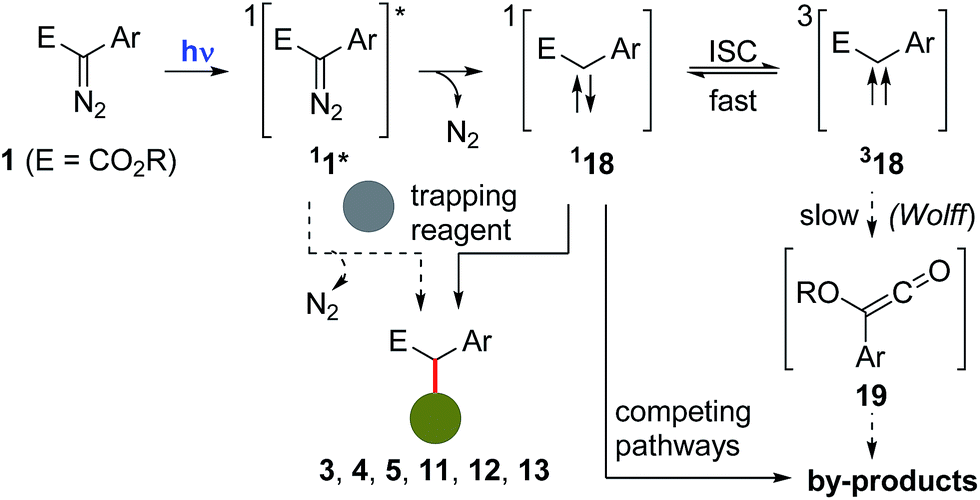 | ||
| Scheme 9 Proposed mechanistic scenario involved in the formation of the products 3, 4, 5, 11, 12 and 13, and plausible explanations for the formation of by-products. | ||
Conclusions
In summary, this study demonstrates that aryldiazoacetates 1 can absorb in the wavelength region of approximately 400–500 nm (visible region), thus being able to undergo photolysis by blue light irradiation in the presence of a variety of partners to afford products of cyclopropanation, O–H, N–H and C–H insertions. Furthermore, this new technology is mild, selective and has been demonstrated to be scalable up to 1 mmol, therefore opening new venues for potential applications in organic synthesis and chemical-biology.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
I. D. J. acknowledges the Fulbright Foundation for a Junior Faculty Award and the support of Fapesp (2015/20809-4) and CNPq (458416/2014-2). H. M. L. D. was supported by the National Science Foundation (CHE 1465189). Instrumentation used in this work was supported by the National Science Foundation (CHE 1531620 and CHE 1626172). We thank Dr John Bacsa (Emory University, U.S.) for the X-ray structure determination of 5g.Notes and references
- For selected recent reviews, see: (a) A. Ford, H. Miel, A. Ring, C. N. Slattery, A. R. Maguire and M. A. McKervey, Chem. Rev., 2015, 115, 9981 CrossRef PubMed; (b) N. Candeias, R. Paterna and P. M. P. Gois, Chem. Rev., 2016, 116, 2937 CrossRef PubMed; (c) Q.-Q. Cheng, Y. Deng, M. Lankelma and M. P. Doyle, Chem. Soc. Rev., 2017, 46, 5425 RSC; (d) H. M. L. Davies and J. S. Alford, Chem. Soc. Rev., 2014, 43, 5151 RSC; (e) H. M. L. Davies and D. Morton, Chem. Soc. Rev., 2011, 40, 1857 RSC; (f) H. M. L. Davies and J. R. Denton, Chem. Soc. Rev., 2009, 38, 3061 RSC . See also:; (g) M. Jia and S. Ma, Angew. Chem., Int. Ed., 2016, 55, 9134 CrossRef PubMed.
- Selected references: Cyclopropanations: (a) A. Prieto, M. R. Fructos, M. Mar Díaz-Requejo, P. J. Pérez, P. Pérez-Galán, N. Delpont and A. M. Echavarren, Tetrahedron, 2009, 65, 1790 CrossRef; (b) J. L. Thompson and H. M. L. Davies, J. Am. Chem. Soc., 2007, 129, 6090 CrossRef PubMed; (c) B. J. Anding, A. Ellern and L. K. Woo, Organometallics, 2012, 31, 3628 CrossRef . OH insertions:; (d) E. D. Couch, T. J. Auvil and A. E. Mattson, Chem.–Eur. J., 2014, 20, 8283 CrossRef PubMed; (e) L. Dumitrescu, K. Azzouzi-Zriba, D. Bonnet-Delpon and B. Crousse, Org. Lett., 2011, 13, 692 CrossRef PubMed; (f) B. Bernardim, E. D. Couch, A. M. Hardman-Baldwin, A. C. B. Burtoloso and A. E. Mattson, Synthesis, 2016, 48, 677 CrossRef; (g) D. P. Hari and J. Waser, J. Am. Chem. Soc., 2016, 138, 2190 CrossRef PubMed . NH insertions:; (h) S. S. So and A. Mattson, J. Am. Chem. Soc., 2012, 134, 8798 CrossRef PubMed; (i) M. E. Morilla, M. M. Díaz-Requejo, T. R. Belderrain, M. C. Nicasio, S. Trofimenko and P. J. Pérez, Chem. Commun., 2002, 2998 RSC; (j) Q.-H. Deng, H.-W. Xu, A. W.-H. Yuen, Z.-J. Xu and C.-M. Che, Org. Lett., 2008, 10, 1529 CrossRef PubMed . CH-insertions:; (k) Z. Yu, B. Ma, M. Chen, H.-H. Wu, L. Liu and J. Zhang, J. Am. Chem. Soc., 2014, 136, 6904 CrossRef PubMed; (l) G. Xu, K. Liu and J. Sun, Org. Lett., 2018, 20, 72 CrossRef PubMed; (m) W.-W. Chan, S.-F. Lo, Z. Zhou and W.-Y. Yu, J. Am. Chem. Soc., 2012, 134, 13565 CrossRef PubMed . Cycloadditions and rearrangements:; (n) T. Shi, X. Guo, S. Teng and W. Hu, Chem. Commun., 2015, 51, 15204 RSC; (o) J. He, L. G. Hamann, H. M. L. Davies and R. E. J. Beckwith, Nat. Commun., 2015, 6, 1 Search PubMed; (p) C. Zhu, G. Xu and J. Sun, Angew. Chem., Int. Ed., 2016, 55, 11867 CrossRef PubMed; (q) A. Sharma, L. Guénée, J.-V. Naubron and J. Lacour, Angew. Chem., Int. Ed., 2011, 50, 3677 CrossRef PubMed; (r) J. Zhu, W. Hu, S. Sun, J.-T. Yu and J. Cheng, Adv. Synth. Catal., 2017, 359, 3725 CrossRef; (s) I. D. Jurberg and H. M. L. Davies, Org. Lett., 2017, 19, 5158 CrossRef PubMed; (t) S. C. Schmid, I. A. Guzei and J. M. Schomaker, Angew. Chem., Int. Ed., 2017, 56, 12229 CrossRef PubMed.
- Selected references: (a) P. Pelphrey, J. Hansen and H. M. L. Davies, Chem. Sci., 2010, 1, 254 RSC; (b) R. Sambasivan and Z. T. Ball, Angew. Chem., Int. Ed., 2012, 51, 8568 CrossRef PubMed; (c) F. G. Adly, M. G. Gardiner and A. Ghanem, Chem.–Eur. J., 2016, 22, 3447 CrossRef PubMed; (d) C. Qin, V. Boyarskikh, J. H. Hansen, K. I. Hardcastle, D. G. Musaev and H. M. L. Davies, J. Am. Chem. Soc., 2011, 133, 19198 CrossRef PubMed; (e) J.-J. Shen, S.-F. Zhu, Y. Cai, H. Xu, X.-L. Xie and Q.-L. Zhou, Angew. Chem., Int. Ed., 2014, 53, 13188 CrossRef PubMed; (f) H. Xu, Y.-P. Li, Y. Cai, G.-P. Wang, S. F. Zhu and Q.-L. Zhou, J. Am. Chem. Soc., 2017, 139, 7697 CrossRef PubMed.
- Selected references: (a) F. Tan, X. Liu, X. Hao, Y. Tang, L. Lin and X. Feng, ACS Catal., 2016, 6, 6930 CrossRef; (b) Y. Zhang, Y. Yao, L. He, Y. Liu and L. Shi, Adv. Synth. Catal., 2017, 359, 2754 CrossRef; (c) S.-F. Zhu, Y. Cai, H.-X. Mao, J.-H. Xie and Q.-L. Zhou, Nat. Chem., 2010, 2, 546 CrossRef PubMed.
- Selected references: (a) S.-F. Zhu, B. Xu, G.-P. Wang and Q.-L. Zhou, J. Am. Chem. Soc., 2012, 134, 436 CrossRef PubMed; (b) Z. Hou, J. Wang, P. He, J. Wang, B. Qin, X. Liu, L. Lin and X. Feng, Angew. Chem., Int. Ed., 2010, 49, 4763 CrossRef PubMed; (c) V. Arredondo, S. C. Hiew, E. S. Gutman, I. D. U. A. Premachandra and D. L. Van Vranken, Angew. Chem., Int. Ed., 2017, 56, 4156 CrossRef PubMed; (d) B. Xu, S.-F. Zhu, X.-L. Xie, J.-J. Shen and Q.-L. Zhou, Angew. Chem., Int. Ed., 2011, 50, 11483 CrossRef PubMed; (e) E. C. Lee and G. C. Fu, J. Am. Chem. Soc., 2007, 129, 12066 CrossRef PubMed.
- Selected references: (a) K. Liao, S. Negretti, D. G. Musaev, J. Bacsa and H. M. L. Davies, Nature, 2016, 533, 230 CrossRef PubMed; (b) K. Liao, T. C. Pickel, V. Boyarskikh, J. Bacsa, D. G. Musaev and H. M. L. Davies, Nature, 2017, 551, 609 Search PubMed; (c) C. Qin and H. M. L. Davies, J. Am. Chem. Soc., 2014, 136, 9792 CrossRef PubMed.
- Selected references: (a) C. Jing, Q.-Q. Cheng, Y. Deng, H. Arman and M. P. Doyle, Org. Lett., 2016, 18, 4550 CrossRef PubMed; (b) C. Qin and H. M. L. Davies, J. Am. Chem. Soc., 2013, 135, 14516 CrossRef PubMed.
- Selected references: (a) D. M. Hodgson, F. Y. T. M. Pierard and P. A. Stupple, Chem. Soc. Rev., 2001, 30, 50 RSC; (b) Z. Zhang, Z. Sheng, W. Yu, G. Wu, R. Zhang, W.-D. Chu, Y. Zhang and J. Wang, Nat. Chem., 2017, 9, 970 CrossRef PubMed.
- For reviews, see: (a) N. R. Candeias and C. A. M. Afonso, Curr. Org. Chem., 2009, 13, 763 CrossRef; (b) O. S. Galkina and L. L. Rodina, Russ. Chem. Rev., 2016, 85, 537 CrossRef.
- Examples of Wolff-rearrangements using diazoketones have been reported under white-light irradiation. See: (a) Y. S. M. Vaske, M. E. Mahoney, J. P. Konopelski, D. L. Rogow and W. J. McDonald, J. Am. Chem. Soc., 2010, 132, 11379 CrossRef PubMed; (b) B. Bernardim, A. M. Hardman-Baldwin and A. C. B. Burtoloso, RSC Adv., 2015, 5, 13311 RSC.
- Using green light, see: (a) T. Xiao, L. Li, G. Lin, Z.-W. Mao and L. Zhou, Org. Lett., 2014, 16, 4232 CrossRef PubMed . Using white light, see:; (b) Z. Wang, A. G. Herraiz, A. M. del Hoyo and M. G. Suero, Nature, 2018, 554, 86 CrossRef PubMed.
- S. R. Ovalles, J. H. Hansen and H. M. L. Davies, Org. Lett., 2011, 13, 4284 CrossRef PubMed.
- S. R. Hansen, J. E. Spangler, J. H. Hansen and H. M. L. Davies, Org. Lett., 2012, 14, 4626 CrossRef PubMed.
- C. Tortoreto, D. Rackl and H. M. L. Davies, Org. Lett., 2017, 19, 770 CrossRef PubMed.
- (a) K. A. Mix, M. R. Aronoff and R. T. Raines, ACS Chem. Biol., 2016, 11, 3233 CrossRef PubMed; (b) N. A. McGrath, K. A. Andersen, A. K. F. Davis, J. E. Lomax and R. T. Raines, Chem. Sci., 2015, 6, 752 RSC; (c) B. M. Ibbeson, L. Laraia, E. Alza, C. J. O'Connor, Y. S. Tan, H. M. L. Davies, G. McKenzie, A. R. Venkitaraman and D. R. Spring, Nat. Commun., 2014, 5, 1 RSC; (d) K. Tishinov, N. Fei and D. Gillingham, Chem. Sci., 2013, 4, 4401 RSC.
- Y.-Z. Li and G. B. Schuster, J. Org. Chem., 1987, 52, 4460 CrossRef.
- Product(s) derived from the use of acetonitrile as solvent, could be noted from the 1H NMR (in CDCl3) of the crude reaction mixture, but were not isolated. Indeed, new protons in the aromatic region and two distinctive methyl groups, with δ = 2.40 and 2.33 ppm could be observed (the chemical shift of the Me group from acetonitrile is 2.10 ppm in CDCl3). Apart from these additional signals, the 1H NMR obtained for the use of CH3CN is the same as the one obtained for DCM, thus suggesting that these common 1H NMR signals are derived from the decomposition of the starting aryldiazoacetate 1a..
- (a) H. Tomioka, M. Itoh, S. Yamakawa and Y. Izawa, J. Chem. Soc., Perkin Trans. 2, 1980, 4, 603 RSC; (b) T. DoMinh, O. P. Strausz and H. E. Gunning, J. Am. Chem. Soc., 1969, 91, 1261 Search PubMed; (c) N. Baumann, Helv. Chim. Acta, 1972, 55, 2716 CrossRef.
- (a) D. M. Guptill, C. M. Cohen and H. M. L. Davies, Org. Lett., 2013, 15, 6120 CrossRef PubMed . See also:; (b) Z. J. Garlets and H. M. L. Davies, Org. Lett., 2018, 20, 2168 CrossRef PubMed.
- For a modern discussion on this mechanism and related transformations, see: E. Kühnel, D. D. P. Laffan, G. C. Lloid-Jones, T. M. del Campo, I. R. Shepperson and J. L. Slaughter, Angew. Chem., Int. Ed., 2007, 46, 7075 CrossRef PubMed.
- CCDC 1824108 contains supplementary crystallographic data for this paper.†.
- See also: H. M. L. Davies, M. G. Coleman and D. L. Ventura, Org. Lett., 2007, 9, 4971 CrossRef PubMed.
- J.-L. Wang, I. Likhotvorik and M. S. Platz, J. Am. Chem. Soc., 1999, 121, 2883 CrossRef.
- H. Tomioka, H. Kitagawa and Y. Izawa, J. Org. Chem., 1979, 44, 3072 CrossRef.
- (a) Z. Zhu, T. Bally, L. L. Stracener and R. J. McMahon, J. Am. Chem. Soc., 1999, 121, 2863 CrossRef . See also:; (b) Y. Wang, T. Yuzawa, H. Hamaguschi and J. P. Toscano, J. Am. Chem. Soc., 1999, 121, 2875 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1824108. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8sc01165f |
| This journal is © The Royal Society of Chemistry 2018 |