
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDiffusion across a gel–gel interface – molecular-scale mobility of self-assembled ‘solid-like’ gel nanofibres in multi-component supramolecular organogels†
Jorge
Ruíz-Olles
and
David K.
Smith
 *
*
Department of Chemistry, University of York, Heslington, York, YO10 5DD, UK. E-mail: david.smith@york.ac.uk
First published on 30th May 2018
Abstract
This paper explores macroscopic-scale diffusion of the molecular-scale building blocks of two-component self-assembled organogel nanofibres using a diffusion cell in which two different gels are in contact with one another. Both components of the ‘solid-like’ nanofibres (lysine peptide dendron acids and amines) can diffuse through these gels and across a gel–gel interface, although diffusion is significantly slower than that of a non-interactive additive in the ‘liquid-like’ phase of the gel. Amine diffusion was probed by bringing similar gels with different amines into contact. Dendron acid diffusion was tested by bringing similar gels with enantiomeric dendrons into contact. Surprisingly, dendron and amine diffusion rates were similar, even though the peptide dendron is more intimately hydrogen bonded in the self-assembled nanofibres. It is proposed that thermal disassembly of the acid–amine complex delivers both components into the liquid-like phase, allowing them to diffuse via a decomplexation/recomplexation mechanism. This is a rare observation in which molecules assembled into solid-like gel nanofibres are mobile – in dynamic equilibrium with the liquid-like phase. Gel nanofibre diffusion and reorganisation are vital in understanding dynamic materials processes such as metastability, self-healing and adaptability.
Introduction
Supramolecular gels are colloidal materials constituted by a ‘liquid-like’ phase with a sample-spanning ‘solid-like’ nanoscale network self-assembled from low-molecular-weight gelators (LMWGs).1 They have wide-ranging applications, from current industrial use in greases, personal care products and adhesives, to rapidly developing high-tech uses including tissue engineering and nanoscale electronics.2 One of the most intriguing aspects of such gels is their dynamic nature – they self-assemble in a responsive manner from small molecule building blocks, and typically contain ca. 99% of liquid-like solvent. There has been considerable interest in diffusion within gels – for example, solvent molecule mobility has been studied using NMR methods.3 Tritt-Goc and co-workers characterised solvent diffusion in supramolecular gels, showing that in the absence of interactions with the gelator network on short timescales solvent diffusion is similar to bulk solvent.4 However, interactions between solvent and gel nanofibres, which increase with gelator concentration, decrease diffusion coefficients by several orders of magnitude,4 although the relationship is not always straightforward.5 In the absence of interactions with gel nanofibres, ionic components in the liquid-like phase can also rapidly diffuse,6 leading to conductivity – important for applications such as solar cells, lithium ion batteries and super-capacitors.7 Molecular additives in the liquid-like solvent phase also have interesting diffusion profiles. Influential work from Adams and co-workers demonstrated that network mesh size could control diffusion, with larger molecules becoming physically trapped within the network.8 This has applications, for example immobilising enzymes for biocatalysis.9 Control over diffusion has also been important in forming unique crystal morphologies in self-assembled gels.10 Non-covalent interactions between a nanofibre gel network and molecular additives can also affect diffusion rates,11 of interest for applications such as drug delivery.12 Interestingly, related studies using polymer gels,13 have stimulated considerable theoretical interest in diffusion through gels.14In contrast to polymer gels, supramolecular gels are considerably more dynamic as a result of their ability to disassemble into small molecules. There has been interest in the kinetics/dynamics of gel-assembly,15 and increasing focus on the metastability of gel nanostructures.16 However, although assembly dynamics have been studied, there is less insight into the ability of the molecular-scale components of gel nanofibres to diffuse through gels across macroscopic length scales. In principle, the dynamic nature of supramolecular interactions means supramolecular gels may be able to self-heal when damaged.17 However, although self-healing is increasingly recognised, studies of network reorganisation dynamics are rare.18 In an influential study, Aida and co-workers reported a gel, which they cut into cubes – when the solvated cube surfaces were brought together, there was healing across the interface yielding a physically robust structure, indicative of nanostructure reorganisation.19 More recently, a similar approach was employed using gel blocks either doped or undoped with methyl orange – when healing across the interface occurred, this was accompanied with diffusion of the dye from one block to the next.20 Perhaps surprisingly, for supramolecular gels, however, there remains a lack of studies of gel–gel diffusion. This research therefore aimed to explore the diffusion of molecular-scale gel components across gel–gel interfaces, focussing on the ability of the molecules constituting the self-assembled nanofibres to diffuse and exchange between two self-assembled gels.
A two-component organogel based on a lysine peptide dendron acid combined with an amine (e.g., Fig. 1) was selected for study. This acid–amine gelation system is well-established in our research group and well understood.21 It assembles primarily via intermolecular hydrogen bond interactions between peptides, with the amine complexing the acid, modifying solubility, and promoting assembly and gelation. Advantageously, such gels can form in situ with rapid kinetics on simple mixing of the two components in toluene.22 NMR has demonstrated that adding a different amine, in solution, to these gels leads to amine exchange on the self-assembled nanofibres, to give the thermodynamically most stable system.23 This led us to believe a degree of diffusion of the ‘solid-like’ components may be possible in this system. Furthermore, having two components, this organogel provides the opportunity to probe the diffusion of each constituent part of the solid-like nanofibres.
 | ||
| Fig. 1 Structures of components used to assemble two-component organogels in toluene for diffusion studies and schematic of key diffusion experiments to probe mobility of acid and amine components in two-component organogels. | ||
Results and discussion
We proposed to use different amines and/or acids and monitor diffusion across a gel–gel interface (Fig. 1). Initially, gels were based on a lysine peptide dendron acid (L-Lys-G2-COOH) combined with either (i) hexylamine (non-fluorescent) or (ii) 1-naphthylmethylamine (fluorescent) (referred to as L-Lys-HexGel and L-Lys-NaphGel respectively, Fig. 1 (top)). All gels were formed with 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 acid:amine ratios. We reasoned that if such gels were brought into contact, amine diffusion across the interface, could be visualised with a simple fluorescent output.
:1 acid:amine ratios. We reasoned that if such gels were brought into contact, amine diffusion across the interface, could be visualised with a simple fluorescent output.
Characterisation of gels
Prior to studying diffusion, we characterised these two organogels. At a concentration of 10 mM, L-Lys-HexGel and L-Lys-NaphGel had similar thermal stabilities (Tgel) of ca. 70 °C, determined using simple reproducible tube inversion methodology – suggesting the two gels have similar thermodynamic stability. Rheology was used to follow the gelation process in situ, and it was observed that on mixing the two components in solution under ambient conditions a rapid gelation event took place, with an increase in elastic modulus, followed by a further slower increase in modulus as the network aged and optimised the formation of a sample-spanning network (Fig. S1 and S2, ESI†). This suggests significant dynamic character to these gels. Similar changes were observed for both L-Lys-HexGel and L-Lys-NaphGel suggesting the mechanism of gelation is similar in each case. Atomic Force Microscopy (AFM), scanning electron microscopy (SEM) and transmission electron microscopy (TEM) were used to probe morphology (Fig. S3–S7, ESI†). Both systems assembled into nanoscale fibrillar morphologies. In general, L-Lys-HexGel nanofibres were wider than L-Lys-NaphGel, especially for the gel formed by simple mixing at ambient temperature. It is likely there is greater bundling of L-Lys-HexGel fibrils than for L-Lys-NaphGel, with the latter being more compatible with the toluene solvent as a result of π–π stacking. The difference in diameter was less marked for samples produced using a heat-cool cycle, during which the L-Lys-HexGel nanofibers can rearrange into a more thermodynamically favoured form. In summary, L-Lys-HexGel and L-Lys-NaphGel formed gels with equivalent thermal stabilities underpinned by fibrillar solid-like self-assembled morphologies. There were some differences in fibre diameter dependent on the amine, but the gels were similar – suitable for study in diffusion experiments.Qualitative diffusion experiment
In a qualitative initial experiment to test the hypothesis that components of the solid-like gel nanofibres may diffuse, simple vials were used and two gels formed on top of one another by stepwise room-temperature mixing of components to form each gel layer in turn. The bottom layer was formed by mixing L-Lys-G2-COOH and 1-naphthylmethylamine in toluene, then once it had formed, the upper layer was formed on top of it by mixing L-Lys-G2-COOH and hexylamine in toluene. Diffusion of the fluorescent amine was tracked visually under UV irradiation (Fig. 2). At the start of the experiment, the lower layer of the gel was fluorescent (L-Lys-NaphGel), while the upper layer was not (L-Lys-HexGel). After 5 hours, the fluorescent amine (Naph) had diffused somewhat into the upper gel layer. After 48 hours, diffusion of the fluorescent amine appeared (visually) to be reasonably complete. | ||
| Fig. 2 Qualitative diffusion experiment demonstrating diffusion of the fluorescent amine from a lower layer of L-Lys-NaphGel (10 mM) into an upper L-Lys-HexGel (10 mM) layer. | ||
Mobility of the fluorescent amine component of the nanoscale network clearly appears to be allowed. This was a remarkable result, as such networks are usually considered ‘solid-like’. We anticipated that the non-fluorescent amine may also diffuse in the opposite direction, but it obviously cannot be detected using this simple approach.
Experimental design of gel–gel diffusion cell
To understand the intriguing diffusion event described above in a more quantitative way, we designed an experiment to probe diffusion across a gel–gel interface. This presented considerable difficulties as a result of the volatility of toluene and its ability to dissolve or leach into plastics. Glass and metal were used for the construction of the diffusion cell. The cell design (Fig. 3 and S10–S12†) had length (4 cm), width (3.5 cm) and height (2.5 cm), was made of soda glass, and has the capacity to load two gels, one on each side. The volume of each gel introduced into the cell was 5.44 mL creating an interfacial area of 1.6 cm2 between the two gels. Separation between the two sides of the cell during loading was achieved using a home-made ultra-thin aluminium ‘separator’, which was sufficiently thin that, when removed, the two gels come into direct contact. Given the gels are formed in situ within the cell (see below), it was essential to eliminate any leakage. A silicone seal was used to join the two sides of the diffusion cell, swollen in toluene, and trimmed – the aluminium separator intimately contacted this seal (see ESI†). Loading the gels (5 mM) in the cell was initially attempted by simple room temperature mixing of acid and amine leading to rapid in situ gelation. However, gelation kinetics were faster than mixing kinetics and to obtain homogeneous gels, hot solutions (>Tgel) of the chosen acid–amine complexes were instead poured into either side of the cell, with gels forming on cooling. Gel homogeneity must be high to form an effective gel–gel interface. To avoid toluene evaporation, the diffusion cell was placed in a chamber containing solvent. | ||
| Fig. 3 Diffusion cell under UV illumination with L-Lys-NaphGel (5 mM) loaded in the left-hand side of the cell and L-Lys-HexGel (5 mM) loaded in the right-hand side of the cell – the cell is shown at the start of the experiment (left) and after some time has elapsed – supportive of the view that diffusion and amine exchange is taking place. | ||
Fig. 3 illustrates fluorescent L-Lys-NaphGel loaded into the left-hand-side of the cell and non-fluorescent L-Lys-HexGel loaded into right-hand-side at the start of an experiment. The choice of a fluorescent amine once again allowed us to visually monitor progress. After some time had elapsed, it was clear the fluorescent amine had diffused into the right-hand-side of the cell. No disruption of the gels on either side of the gel–gel interface was observed.
It was necessary to sample from the gel to quantify the diffusion of both fluorescent 1-naphthylmethylamine and non-fluorescent hexylamine. The fluorescent amine could have been quantified by non-invasive fluorescence imaging, but this was not possible for hexylamine – sampling was therefore combined with NMR for quantification. The cuvette was artificially divided into six regions of equivalent size (Fig. 4). Regions 3 and 4 are closest to the gel–gel interface, and regions 1 and 6 are most distant. Gel was sampled from each of the regions, transferred to a numbered vial, evaporated to dryness, and fully dissolved in d4-methanol. The concentration of amine in each region was quantified with reference to the dendron acid (L-Lys-G2-COOH), which has a constant known concentration (5 mM) across the cell and hence acts as an internal standard. The amines had NMR spectra that could be differentiated from one another and L-Lys-G2-COOH, allowing quantification of all components. Specifically, we used the 1H NMR resonances for the four aromatic protons on the unsubstituted naphthalene ring for 1-naphthylmethylamine, the CH2–NH2 protons for hexylamine and the proton at the chiral centre closest to the carboxylic acid for L-Lys-G2-COOH. Sampling from the cell disrupts the diffusion experiment, and to investigate multiple timepoints, a number of cells were used in parallel.
 | ||
| Fig. 4 Division of the diffusion cell into 6 regions for purposes of sampling. | ||
Diffusion of amines across the gel–gel interface
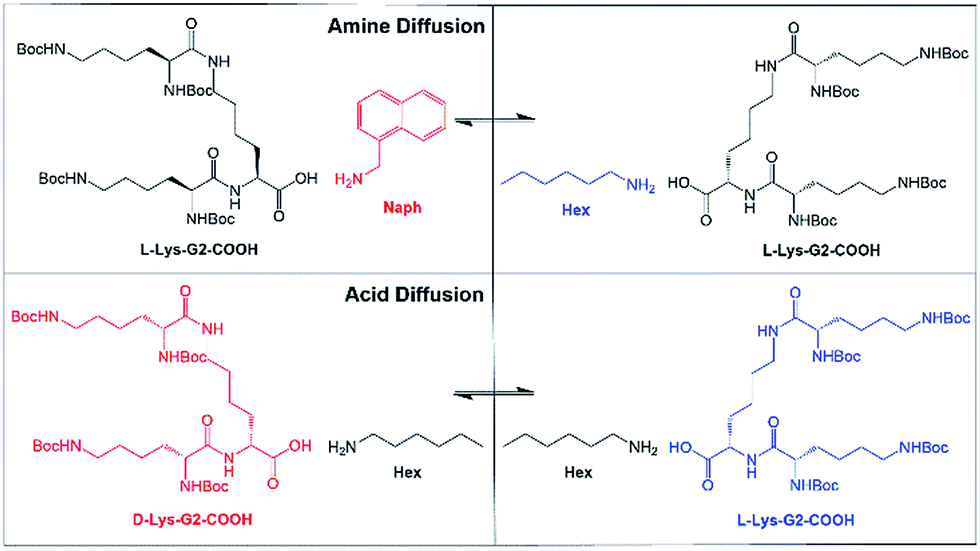
Diffusion cells were prepared, filled with L-Lys-HexGel and L-Lys-NaphGel (5 mM) and diffusion monitored at a controlled temperature of 25 °C. NMR analysis generated profiles representing amine concentration and diffusion distance for given times (Fig. 5). Pleasingly, the concentration graphs are broadly symmetric about the gel–gel interface and show reliable, reproducible trends. As time progresses, the concentration of hexylamine in cell region 3 increases, while that in cell region 4 decreases, suggesting the diffusion of hexylamine from right to left in the diffusion cell. The same trends are seen, but with smaller concentration changes in cell regions 2 and 5, as expected given they are further away from the gel–gel interface. Concentration changes in cell regions 1 and 6, most distant from the gel–gel interface, are small. The concentration plots for 1-naphthylmethylamine indicate it diffuses in the opposite direction (i.e., left to right) – concentration increases in cell region 4 (and to a lesser extent 5) and decreases in cell region 3 (and to a lesser extent 2). This experiment indicates both amines diffuse through these gels, across the gel–gel interface and exchange with one another, even though they are an integral part of the self-assembled fibre network. However, even after 300 hours (12.5 days) diffusion is incomplete and the system has not fully equilibrated.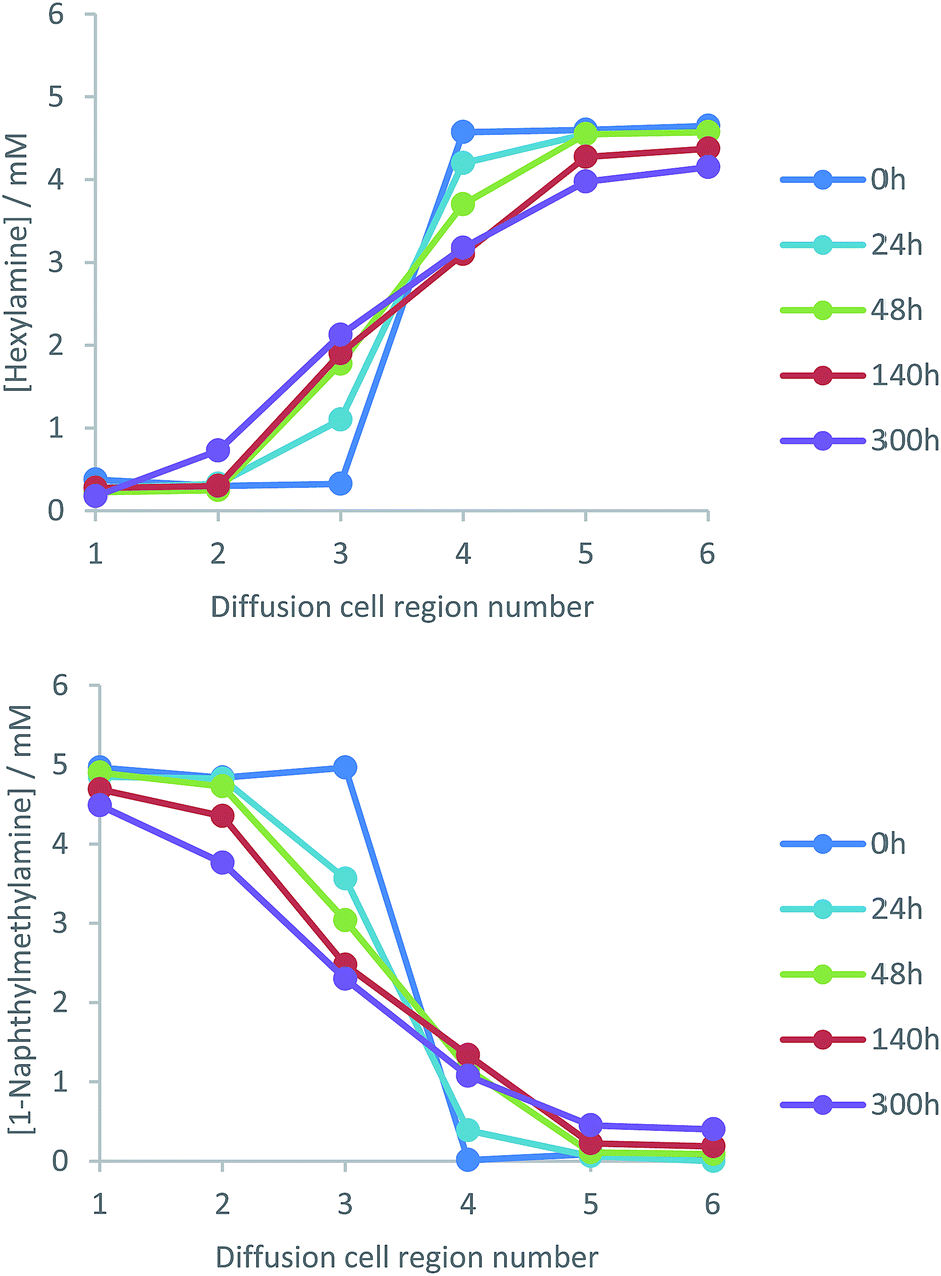 | ||
| Fig. 5 Concentration profiles of (top) hexylamine and (bottom) 1-naphthylmethylamine between regions 1 and 6 of the diffusion cell. At the start of the experiment, hexylamine is loaded in regions 4–6 of the diffusion cell and 1-naphthylmethylamine is loaded in regions 1–3. As time progresses, the concentrations of the amines change as diffusion leads to exchange across the gel–gel interface. The points in these graphs are joined together solely as a guide to the eye. | ||
TEM imaging was performed on each region of the gel after 48 h of diffusion (Fig. S13–S18, ESI†). Regions 1–3 showed narrow nanofibre morphologies typical of L-Lys-NaphGel, and regions 4–6 showed wider nanofibres typical of L-Lys-HexGel. However, in region 4, the nanofibres associated with L-Lys-HexGel had a clear helical bias. We suggest this may result from 1-naphthylmethylamine interacting with fibres that predominantly include hexylamine, leading to a subtle change in morphology. Furthermore, TEM imaging of region 3 suggested the narrow L-Lys-NaphGel fibres were better defined than in regions 1 and 2 – i.e., they could be more clearly imaged. This may suggest an increasing influence of hexylamine diffusing into this side of the cell.
Diffusion of a non-interactive small molecule
The results for amine diffusion were compared with a system in which a non-interactive additive was added to the gel and allowed to diffuse from one side of the cell to the other. In this case, L-Lys-HexGel was loaded into each side of the cell, but on one side (regions 4–6) diphenylmethane (5 mM), a small molecule which does not interact with the gel network,23 was also added. In this case, we sampled from the gel at time points of hours rather than days (Fig. 6). The concentration of diphenylmethane on the right-hand side of the cell rapidly decreased, while that on the left-hand side of the cell increased. After 48 hours, an equal concentration of diphenylmethane was observed across the cell. This is different to amine diffusion, which was only partial, even after 300 hours. This clearly indicates that although the amine gel components can diffuse through the gel, they do so much more slowly than an unrestrained small molecule. This leads us to propose a mechanism in which the amine de-complexes from the nanofibres, and diffuses through the gel between acidic sites of L-Lys-G2-COOH (see below for further evidence). As such, amine diffusion is restrained by its non-covalent interactions. This is in analogy with reports of drug delivery using LMWGs, in which drug release can be controlled by non-covalent interactions with a self-assembled gel network.11 | ||
| Fig. 6 Concentration profiles of diphenylmethane between regions 1 and 6 of the diffusion cell. At the start of the experiment, diphenylmethane is loaded in regions 4–6 of the cell. The points in these graphs are joined together solely as a guide to the eye. | ||
Temperature dependence of amine diffusion
The effect of temperature on amine diffusion was then investigated, with experiments performed at 5 °C and 45 °C. As previously, diffusion from one side of the gel to the other was observed, but progress was significantly less at 5 °C (Fig. 7) than 25 °C (Fig. 5). Indeed, at 5 °C, diffusion had barely progressed at all into sections 2 and 5 of the cell. At 45 °C (importantly still <Tgel), it was more difficult to obtain reliable results as on extended standing, the gels began to dry. Nonetheless, the first four days of diffusion were monitored (Fig. 7) – the rate of concentration change was much greater. After only 96 h, the concentration of hexylamine in cell region 3 had risen to ca. 2 mM and in cell region 1, distant from the gel–gel interface, to 1 mM. After 140 h, amine concentrations were close to the equilibrium value of 2.5 mM across the diffusion cell.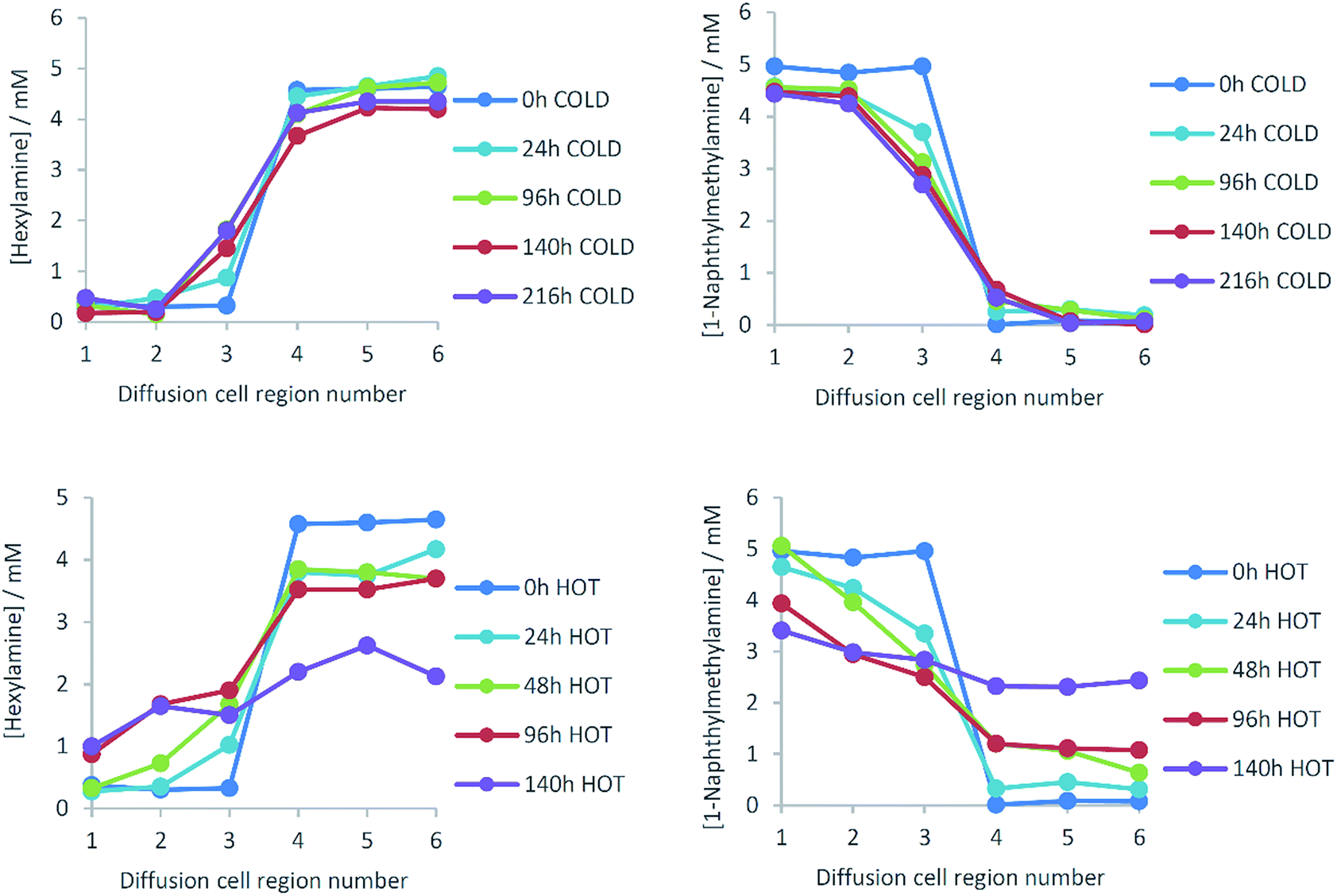 | ||
| Fig. 7 Concentration profiles of (left) hexylamine and (right) 1-naphthylmethylamine between regions 1 and 6 of the diffusion cell at temperatures of (top) 5 °C and (bottom) 45 °C demonstrating the very significant impact of temperature on diffusion kinetics. At the start of the experiment, hexylamine is loaded in regions 4–6 of the diffusion cell and 1-naphthylmethylamine is loaded in regions 1–3. The points in these graphs are joined together solely as a guide to the eye. | ||
There are two possible reason for the increased diffusional mobility of amines at elevated temperatures:
(1) Increase of internal mobility of all liquid-like substances within the gel because of an increase of translational kinetic energy.
(2) Disruption of the acid/amine complex on increasing temperature increases [amine] in the liquid-like phase (and the on–off rate) and may thus have very significant impact on the observed diffusion rate.
To approximate an effective diffusion coefficient to reflect the rate of mass transfer, Fick's second law (eqn (1)), which relates the rate of change of concentration at position x and time t to the rate of change of the gradient of concentration at the same position, was used.24
 | (1) |
Assuming the solute is initially present in half of the container, the concentration profile at any time can then be calculated using eqn (2).25
 | (2) |
This model was compared with the data obtained at each timepoint, and fitting of all data for each diffusion event was performed using least squares error analysis (Fig. S19–S24, ESI†). This allowed us to estimate average diffusion coefficients for each amine at each temperature (Table 1). The resulting D values of ca. 5 × 10−7 cm2 s−1 are below typical diffusion coefficients of liquids (ca. 1 × 10−5 cm2 s−1), but significantly higher than typical diffusion coefficients of solids (<1 × 10−9 cm2 s−1). This would support the view that the overall mobility of the amine within these gels is between the diffusional mobility of a solid and a liquid. The data were also fitted for diphenylmethane diffusion within the gel at 25 °C (Fig. S25†), and a D value of 3.3 × 10−4 cm2 s−1 was obtained – three orders of magnitude faster diffusion than the amines, and in-line with expectations for the diffusion of a liquid/solute.
| 5 °C | 25 °C | 45 °C | |
|---|---|---|---|
| Hexylamine | 1 × 10−7 | 7 × 10−7 | 6 × 10−6 |
| 1-Naphthylmethylamine | 3 × 10−7 | 5 × 10−7 | 6 × 10−6 |
Increasing temperature increased the apparent average diffusion coefficients of the amines (Table 1). It should be noted that the fitting of the data at 45 °C was less robust, for reasons described above, but it was clear from visual inspection that diffusion was significantly faster (ca. one order of magnitude), in agreement with the derived D values. The relationship between diffusion coefficient and temperature depends on which state a material is in. For a liquid this is expressed by the Stokes Einstein equation.26 Assuming viscosity is constant with temperature (as is essentially the case in gels), diffusion coefficients should be proportional to absolute temperature. This is not the case here, as the amine diffusion coefficient increases ca. >10-fold on increasing temperature from 278 K to 318 K, when a 1.14-fold increase would be predicted. Clearly, enhanced diffusion is not just due to mechanism (i) in which greater internal mobility of the liquid-like substances within the gel encourages enhanced mobility. We therefore suggest temperature acts through mechanism (ii), disrupting the acid–amine complex responsible for gelation, and promoting mobility of the amine within the gel.
We have previously studied gels formed by L-Lys-G2-COOH and hexylamine, 1-naphthylmethylamine and mixtures of the two in some detail.23 This helps us understand further the results presented here. We used VT 1H NMR spectroscopy to demonstrate that for these gels, increasing amounts of amine become mobile, and hence visible in the NMR spectrum, on raising temperature. At 25 °C, ca. 10% of the amine (or the dendron, Fig. S8 and S9†) in L-Lys-HexGel was mobile on the NMR timescale.23a This clearly suggests that there is indeed some molecular-scale mobility of the amine component possible in L-Lys-HexGel and that the ‘on–off’ rates of the complexes are relatively fast. Similar amounts of 1-naphthylmethylamine were mobile in L-Lys-NaphGel. Interestingly, however, in our diffusion experiment here (Fig. 5), by the end of the experiment >20% of the total amine has diffused across the gel–gel interface. It is therefore clearly not only the initial ‘mobile amine’ (ca. 10%) that diffuses, while the rest remains bound to gel nanofibres. We propose that diffusion of the mobile amine is coupled with decomplexation of the amine from the acid–amine complex (and from the nanofibres) as a result of Le Chatelier's principle, and in this way, the system equilibrates, with diffusion acting to exchange components. These observations are compatible with a mechanism in which amine diffusion is partially liquid-like, but with reversible interactions being possible with the acid groups on the nanofibre network. We also previously reported23a that hexylamine has a higher pKa than 1-naphthylmethylamine and for this reason the acid–amine complex formed by hexylamine is stronger. This meant that when gels were formed from a mixture of the two amines, hexylamine was preferentially incorporated over 1-naphthylmethylamine (Fig. S9†). This previous work therefore demonstrated that the acid–amine complexation event played a key role in determining the incorporation of amines into the gel-phase nanofibres. Furthermore, we also previously reported23a that if a sample of NaphGel was challenged with the addition of hexylamine, the amine immobilised on the gel fibres changed – clearly demonstrating a reversible exchange mechanism. We thus propose that acid–amine decomplexation is the likely mechanism by which mobility of these components through the gel becomes possible. Ultimately, when coupled with diffusion of the amines, this mechanism gives rise to compositional change of these gels, which impacts on physical properties (e.g. fluorescence and nanofibre morphology).
Diffusion of dendron acids
We then turned our attention to the diffusion of the other component of the solid-like nanofibres. The self-assembly model for these two-component gels relies on intermolecular amide–amide hydrogen bond interactions between peptide dendron acids, and it might thus be anticipated that this component is less able to diffuse through the gel. NMR could not be used to quantify diffusion, as changing the structure of the dendron acid can quite radically affect self-assembly.24 An experiment was therefore proposed taking advantage of dendron chirality. There are various possible stereoisomers for the dendron acid, and we used the L,L,L form as above (L-Lys-G2-COOH) and the D,D,D enantiomer (D-Lys-G2-COOH). Gels formed from these enantiomers are identical in every way, except for properties associated with chirality. When combined with hexylamine the enantiomeric dendrons form mirror image gels with equivalent thermal stabilities and nanoscale morphologies but equal and opposite circular dichroism (CD) spectroscopy signals.27 Diffusion experiments were planned using our diffusion cell, with one side of the cell loaded with L-Lys-HexGel (5 mM) and the other side loaded with D-Lys-HexGel (5 mM) (Fig. 1 (bottom)). Samples would be taken from the six regions of the gel and analysed using CD spectroscopy to determine the enantiomeric excess in each case, to uncover the extent to which the L,L,L dendron acid diffused into the gel based on the D,D,D acid and vice versa.In many cases, mixing enantiomers disrupts gelation28 – this would be highly undesirable in this diffusion experiment as it would potentially lead to destruction of the gels as diffusion progressed. The thermal stability of gels formed from mixtures of enantiomeric dendrons was therefore probed using simple reproducible tube inversion methodology (Fig. 8). Importantly, mixing enantiomers had limited impact on thermal stability (ca. 70 °C), allowing enantiomeric dendrons to be used to test diffusion.
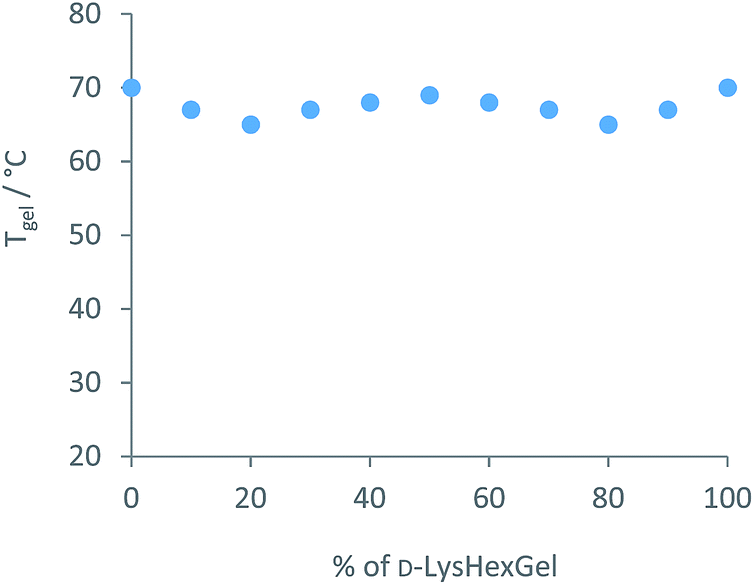 | ||
| Fig. 8 Thermal stability of mixtures of enantiomeric L-Lys-HexGel and D-Lys-HexGel at a total concentration of 10 mM, as determined using tube inversion methodology. | ||
To quantify the extent of diffusion, CD spectroscopy was used. It was essential to check this approach could be used in a quantitative way. CD spectra were measured in methylcyclohexane:dioxane (95:5) as, unlike toluene, this solvent is optically transparent in the relevant part of the spectrum and also supports assembly.23 We prepared samples with mixed enantiomers (total concentration 5 mM) and plotted the ellipticity of the CD signal at 222 nm, associated with the peptide chromophore, against % enantiomer content. A linear relationship was observed at this concentration (Fig. 9), making analysis very simple as CD signal can be directly related to composition.
 | ||
| Fig. 9 (Top) CD spectra of mixtures of L-Lys-HexGel and D-Lys-HexGel at a total concentration of 5 mM in methylcyclohexane/dioxane (95/5), (Bottom) ellipticity at 220 nm extracted from the CD spectra and plotted against the % enantiomer content, demonstrating an approximately linear relationship between CD ellipticity and composition of enantiomers. | ||
In our experimental method, after specific diffusion times, the cells were sampled from their six different regions into six different vials, which were evaporated and weighed on an accurate balance. Solvent (methylcyclohexane/dioxane 95/5) was then added to the weighed samples in order to obtain concentrations of 5 mM in each vial, ensuring that the extent of self-assembly is the same in each case, and the samples were then analysed by CD. All samples were left to stand for 15 min in the quartz cell prior to analysis.
Fig. 10 presents data from a diffusion cell sampled after 240 h (other timepoints can be found in Fig. S28–S30, ESI†). The most intense CD signals (negative and positive) correspond to regions 1 and 6 of the cell. They have the highest absolute values because they are most distant from the gel–gel interface and have not experienced any mixing of chirality from the diffusing enantiomeric dendrons. However, in regions 3 and 4 closest to the gel–gel interface, there is a significant decrease of the expected CD signal. This suggests diffusion of dendrons is occurring, decreasing the enantio-purity of the gels either side of the interface. These results therefore suggest that, like the amines, the dendron acids are also able to move through the gel. This was a very surprising result given we anticipated the dendron acids were more intimately involved in the self-assembled ‘solid-like’ network than the amines, as result of intermolecular peptide–peptide hydrogen bonds.
 | ||
| Fig. 10 CD spectra of regions 1–6 of the diffusion cell after 240 h. | ||
We converted these data into diffusion profile plots (Fig. 11). As expected, the diffusion of L-Lys-COOH mirrors that of D-Lys-COOH. Over time, concentration changes the most in regions 3 and 4 of the cell, but also, over extended periods, to some extent in regions 2 and 5 – suggesting, as for the amines, an ability to diffuse across and away from the gel–gel interface.
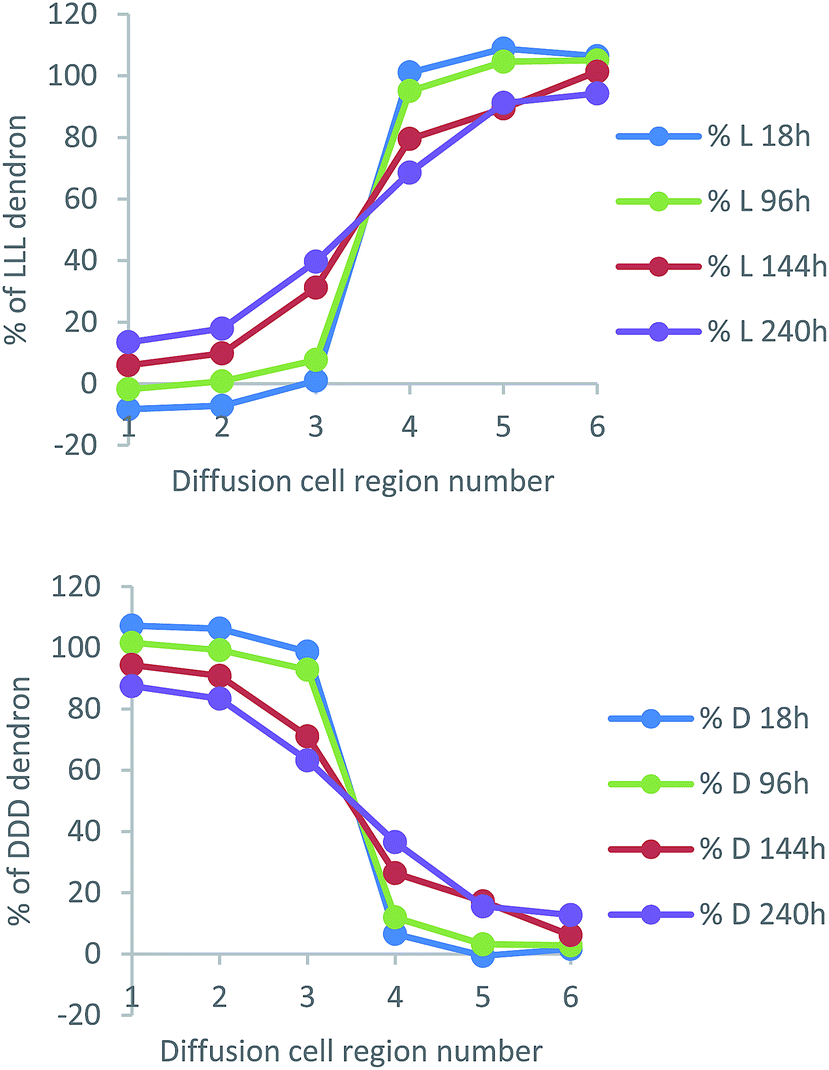 | ||
| Fig. 11 Concentration profiles of (top) L-Lys-CO2H and (bottom) D-Lys-CO2H between regions 1 and 6 of the diffusion cell. At the start of the experiment, L-Lys-CO2H is loaded in regions 4–6 of the diffusion cell and D-Lys-CO2H is loaded in regions 1–3. The points in these graphs are joined together solely as a guide to the eye. | ||
Comparing the results in a simple qualitative way against the diffusion of the amines described above, at ca. 140 hours, the dendron has achieved ca. 30% diffusion into Section 3, and 10% diffusion into Section 2. At the same time, hexylamine had diffused ca. 40% into Section 3 and ca. 10% into Section 2, while 1-naphthylmethylamine had diffused ca. 30% into Section 4 and <10% into Section 5. It would therefore appear that the dendron acid and the amines have similar mobilities. More quantitatively, we estimated average diffusion coefficients for the dendron acids in this experiment, using the same methods outlined above (Fig. S26 and S27, ESI†), and found D values of 3 × 10−7 cm2 s−1 for both L-Lys-COOH and D-Lys-COOH at 25 °C. This is a little lower than the diffusion coefficients reported for the amines, which may suggest slightly slower diffusion rates for peptide acids, but given the limitations of the methodology, is very similar.
The similarity between amine and acid diffusion was surprising to us, and suggests that both components are equally able to move within the gel, i.e., the dendron acid is not additionally immobilised as a result of peptide–peptide hydrogen bonding interactions. This suggests that the acid and amine can each dissociate from the gel nanofibres, probably associated with breakage of the acid–amine complex, and either component can then be delivered into the ‘liquid-like’ phase, where it starts diffusing down the concentration gradient. We propose each component therefore diffuses via a mechanism in which it decomplexes from the gel nanofibres and then recomplexes if it comes into contact with a complementary binding partner in the gel network. We refer to this as a decomplexation/recomplexation mechanism. This is supported by the effect of diffusion on the nanoscale morphology of the gel.
It is interesting, and important, to speculate how general this unexpected phenomenon might be amongst self-assembled gels. We note that these two-component organogels have very rapid gelation kinetics – indeed they form gels simply on mixing the two-components. This suggests a low kinetic barrier to gel assembly. Furthermore, NMR experiments previously indicated that some of the acid and amine are both observed by NMR indicating a degree of liquid-like behaviour, suggesting the molecular components of these self-assembled nanofibres have relatively fast ‘on–off’ kinetics (see NMR spectra in ESI†).23 It therefore seems likely that the highly dynamic nature of this gelation system is one of the reasons that the components of the nanofibres themselves show significant diffusional mobility. Many gels are based on one-component systems, that become fully immobilised on the NMR timescale, and we would suggest that such systems show limited diffusional mobility – as evidenced by the many studies in which a drug is shown to be released from a self-assembled gel, while the components of the nanofibres themselves are not.12 We would therefore suggest that it is important to have dynamic equilibrium between gel-phase nanofibres and the solution-phase in order for this diffusional mechanism to be possible. This is more likely to be the case in multi-component gels in which a reversible interaction between two units is required to form the active gelator. Furthermore, we suggest that simple NMR observations may offer a good predictive approach for finding gels in which components of the solid-like nanofibres are able to diffuse and reorganise on experimentally useful timescales, as in such gels, NMR resonances will be observed even in a standard 1H NMR experiment, whereas in less dynamic gels, all of the NMR resonances will be fully broadened. As such, we suggest NMR as a useful screening tool for gels that are capable of diffusional reorganisation of their nanofibres.
Conclusions
In summary, this paper reports an experimental approach to study diffusion across a gel–gel interface. Diffusion in self-assembled gels is crucial in a variety of settings, such as energy technology and controlled drug release. A non-interactive additive (diphenylmethane) diffused rapidly (hours) across the gel–gel interface consistent with its behaviour as a solution-phase ‘liquid-like’ species. It was then demonstrated that both components of a two-component acid–amine organogel could slowly diffuse (days) across the gel–gel interface in spite of being integrated within the ‘solid-like’ self-assembled nanofibres of the material. The diffusion of amines was probed by bringing similar gels with different amines into contact with one another. Diffusion was highly temperature dependent, which was ascribed to thermally induced disruption, delivering more of the amine into the liquid-like state and hence diffusing through the cell. The diffusion of dendron acids was probed by bringing enantiomeric gels into contact, and using CD spectroscopy to determine the extent of mixing. Surprisingly, the diffusion rate of the dendron acids was similar to that of the amines, even though the lysine dendrons are more directly engaged in the self-assembled nanofibres via intermolecular amide–amide hydrogen bonds. We propose that disassembly delivers both components into the liquid-like phase, allowing them to diffuse via a decomplexation/recomplexation mechanism. As such, this is a rare observation in which the molecular-scale components assembled into solid-like nanofibres are shown to be mobile within a gel, and capable of diffusing across a gel–gel interface.We anticipate that gel nanofibres such as those reported here in which the molecular-scale components are dynamic and have the capacity to diffuse will be particularly suitable for use in adaptive and self-healing materials. Given the importance of this in a wide range of applications, we suggest that this approach to understanding the diffusion potential of solid-like nanofibres in self-assembled gels has considerable general significance and should, in the future, be explored for a wide range of different gelation scaffolds. Simple NMR experiments may provide a useful screening tool to identify gels capable of this type of intriguing and potentially useful type of behaviour.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by European Commission Marie Curie ITN 316656 SMARTNET (JRO). We thank William Edwards for providing thermal stability analysis of the enantiomeric mixtures of Lys-HexGel.Notes and references
- (a) R. G. Weiss, J. Am. Chem. Soc., 2014, 136, 7519–7530 CrossRef PubMed; (b) E. R. Draper and D. J. Adams, Chem, 2017, 3, 390–410 CrossRef; (c) D. J. Amabilino, D. K. Smith and J. W. Steed, Chem. Soc. Rev., 2017, 46, 2404–2420 RSC.
- (a) N. M. Sangeetha and U. Maitra, Chem. Soc. Rev., 2005, 34, 821–836 RSC; (b) A. R. Hirst, B. Escuder, J. F. Miravet and D. K. Smith, Angew. Chem., Int. Ed., 2008, 47, 8002–8018 CrossRef PubMed; (c) S. S. Babu, V. K. Praveen and A. Ajayaghosh, Chem. Rev., 2014, 114, 1973–2129 CrossRef PubMed; (d) X. Du, J. Zhou, J. Shi and B. Xu, Chem. Rev., 2015, 115, 13165–13307 CrossRef PubMed; (e) B. O. Okesola, V. M. P. Vieira, D. J. Cornwell, N. K. Whitelaw and D. K. Smith, Soft Matter, 2015, 11, 4768–4787 RSC.
- Y. E. Shapiro, Prog. Polym. Sci., 2011, 36, 1184–1253 CrossRef.
- (a) J. Kowalczuk, S. Jarosz and J. Tritt-Goc, Tetrahedron, 2009, 65, 9801–9806 CrossRef; (b) J. Bielejewski and J. Tritt-Goc, Langmuir, 2010, 26, 17459–17464 CrossRef PubMed; (c) J. Bielejewski, J. Kowalczuk, J. Kaszynska, A. Lapinski, R. Luboradzki, O. Demchuk and J. Tritt-Goc, Soft Matter, 2013, 9, 7501–7514 RSC.
- (a) J. Kowalczuk, J. Bielejewski, A. Lapinski, R. Luboradzki and J. Tritt-Goc, J. Phys. Chem. B, 2014, 118, 4005–4015 CrossRef PubMed; (b) J. Kowalczuk, A. Rachocki, M. Bielejewsjki and J. Tritt-Goc, J. Colloid Interface Sci., 2016, 472, 60–68 CrossRef PubMed.
- (a) K. Hanabusa, K. Hiratsuka and M. Kimura, Chem. Mater., 1999, 11, 649–655 CrossRef; (b) K. Hanabusa, H. Fukui, M. Suzuki and H. Shirai, Langmuir, 2005, 21, 10383–10390 CrossRef PubMed; (c) B. A. Voss, J. E. Bara, D. L. Gin and R. D. Noble, Chem. Mater., 2009, 21, 3027–3029 CrossRef; (d) A. Maršavelski, V. Smrečki, R. Vianello, M. Žinić, A. Moguš-Milanković and A. Šantić, Chem.–Eur. J., 2015, 21, 12121–12128 CrossRef PubMed.
- (a) N. Mohmeyer, P. Wang, H.-W. Schmidt, S. M. Zakeeruddin and M. Grätzel, J. Mater. Chem., 2004, 14, 1905–1909 RSC; (b) Q. Yu, C. Yu, F. Guo, J. Wang, S. Jiao, S. Gao, H. Li and L. Zhao, Energy Environ. Sci., 2012, 5, 6151–6155 RSC; (c) V. R. Basrur, J. Guo, C. Wang and S. R. Raghavan, ACS Appl. Mater. Interfaces, 2013, 5, 262–267 CrossRef PubMed; (d) J.-D. Decoppet, T. Moehi, S. S. Babkair, R. A. Alzubaydi, A. A. Ansari, S. S. Habib, S. M. Zakeeruddin, H.-W. Schmidt and M. Grätzel, J. Mater. Chem. A, 2014, 2, 15972–15977 RSC.
- (a) S. Sutton, N. L. Campbell, A. I. Cooper, M. Kirkland, W. J. Frith and D. J. Adams, Langmuir, 2009, 25, 10285–10291 CrossRef PubMed; (b) M. Wallace, D. J. Adams and J. A. Iggo, Soft Matter, 2013, 9, 5483–5491 RSC.
- (a) Q. G. Wang, Z. M. Yang, L. Wang, M. L. Ma and B. Xu, Chem. Commun., 2007, 1032–1034 RSC; (b) L. A. Solomon, J. B. Kronenberg and H. C. Fry, J. Am. Chem. Soc., 2017, 139, 8497–8507 CrossRef PubMed.
- (a) R. Daly, O. Kotova, M. Boese, T. Gunnlaugsson and J. J. Boland, ACS Nano, 2013, 7, 4838–4845 CrossRef PubMed; (b) J. A. Foster, K. K. Damodaran, A. Maurin, G. M. Day, H. P. J. Thompson, G. J. Cameron, J. Cuesta Bernal and J. W. Steed, Chem. Sci., 2017, 8, 78–84 RSC.
- (a) E. J. Howe, B. O. Okesola and D. K. Smith, Chem. Commun., 2015, 51, 7451–7454 RSC; (b) S. Gupta, M. Singh, M. A. Reddy, P. S. Yawari, A. Srivastava and A. Bajaj, RSC Adv., 2016, 6, 19751–19757 RSC; (c) P. R. A. Chivers and D. K. Smith, Chem. Sci., 2017, 8, 7218–7227 RSC; (d) V. M. P. Vieira, L. L. Hay and D. K. Smith, Chem. Sci., 2017, 8, 6981–6990 RSC.
- K. J. Skilling, F. Citossi, T. D. Bradshaw, M. Ashford, B. Kellam and M. Marlow, Soft Matter, 2014, 10, 237–256 RSC.
- (a) Polymer Gels: Fundamentals and Applications, ed. H. B. Bohidar, P. Dubin and Y. Osada, American Chemical Society, Washington DC, 2002 Search PubMed; (b) J. Li and D. J. Mooney, Nat. Rev. Mater., 2016, 1, 16071 CrossRef PubMed.
- See for example: (a) P. A. Netz and T. Dorfmuller, J. Chem. Phys., 1997, 107, 9221–9233 CrossRef; (b) S. K. Ghosh, A. G. Cherstvy and R. Metzler, Phys. Chem. Chem. Phys., 2015, 17, 1847–1858 RSC.
- (a) X. Huang, P. Terech, S. R. Raghavan and R. G. Weiss, J. Am. Chem. Soc., 2005, 127, 4336–4344 CrossRef PubMed; (b) P. Jonkheijm, P. van der Schoot, A. P. H. J. Schenning and E. W. Meijer, Science, 2006, 313, 80–83 CrossRef PubMed; (c) X. Huang, S. R. Raghavan, P. Terech and R. G. Weiss, J. Am. Chem. Soc., 2006, 128, 15341–15352 CrossRef PubMed; (d) S. Cantekin, Y. Nakano, J. C. Everts, P. van der Schoot, E. W. Meijer and A. R. A. Palmans, Chem. Commun., 2012, 48, 3803–3805 RSC; (e) Y. Liu, R.-Y. Yao, J.-L. Li, B. Yuan, M. Han, P. Wang and X.-Y. Liu, CrystEngComm, 2014, 16, 5402–5408 RSC; (f) D. J. Cornwell, O. J. Daubney and D. K. Smith, J. Am. Chem. Soc., 2015, 137, 15486–15492 CrossRef PubMed; (g) D. Nonappa, Saman and E. Kolehmainen, Magn. Reson. Chem., 2015, 53, 256–260 CrossRef PubMed; (h) G. M. Peters, L. P. Skala and J. T. Davis, J. Am. Chem. Soc., 2016, 138, 134–139 CrossRef PubMed; (i) S. Onogi, H. Shigemitsu, T. Yoshii, T. Tanida, M. Ikeda, R. Kubota and I. Hamachi, Nat. Chem., 2016, 8, 743–752 CrossRef PubMed; (j) S. M. Ramalhete, K. P. Nartowski, N. Sarathchandra, J. S. Foster, A. N. Round, J. Angulo, G. O. Lloyd and Y. Z. Khimyak, Chem.–Eur. J., 2017, 23, 8014–8024 CrossRef PubMed; (k) E. A. Appel, F. Biedermann, D. Hoogland, J. del Barrio, M. D. Driscoll, S. Hay, D. J. Wales and O. A. Scherman, J. Am. Chem. Soc., 2017, 139, 12985–12993 CrossRef PubMed.
- (a) J. Boekhoven, W. E. Hendriksen, G. J. M. Koper, R. Eelkema and J. H. van Esch, Science, 2015, 349, 1075–1078 CrossRef PubMed; (b) F. Tantakitti, J. Boekhoven, X. Wang, R. V. Kazantsev, T. Yu, J. Li, E. Zhuang, R. Zandi, J. H. Ortony, C. J. Newcomb, L. C. Palmer, G. S. Shekhawat, M. Olvera de la Cruz, G. C. Schatz and S. I. Stupp, Nat. Mater., 2016, 15, 469–476 CrossRef PubMed.
- (a) X. Yu, L. Chen, M. Zhang and T. Yi, Chem. Soc. Rev., 2014, 43, 5346–5371 RSC; (b) D. L. Taylor and M. H. Panhuis, Adv. Mater., 2016, 28, 9060–9093 CrossRef PubMed; (c) J. Li, L. Geng, G. Wang, H. Chu and H. Wei, Chem. Mater., 2017, 29, 8932–8952 CrossRef.
- T. Yan, K. Schroter, F. Herbst, W. H. Binder and T. Thurn-Albrecht, Sci. Rep., 2016, 6, 32356 CrossRef PubMed.
- Q. Wang, J. L. Mynar, M. Yoshida, E. Lee, M. Lee, K. Okuro, K. Kinbara and T. Aida, Nature, 2010, 463, 339–343 CrossRef PubMed.
- S. Bera and D. Haldar, J. Mater. Chem. A, 2016, 4, 6933–6939 Search PubMed.
- (a) A. R. Hirst, D. K. Smith, M. C. Feiters and H. P. M. Geurts, J. Am. Chem. Soc., 2003, 125, 9010–9011 CrossRef PubMed; (b) A. R. Hirst, D. K. Smith, M. C. Feiters and H. P. M. Geurts, Langmuir, 2004, 20, 7070–7077 CrossRef PubMed; (c) A. R. Hirst and D. K. Smith, Org. Biomol. Chem., 2004, 2, 2965–2971 RSC; (d) A. R. Hirst, D. K. Smith, M. C. Feiters and H. P. M. Geurts, Chem.–Eur. J., 2004, 10, 5901–5910 CrossRef PubMed; (e) B. Huang, A. R. Hirst, D. K. Smith, V. Castelletto and I. W. Hamley, J. Am. Chem. Soc., 2005, 127, 7130–7139 CrossRef PubMed; (f) A. R. Hirst, D. K. Smith and J. P. Harrington, Chem.–Eur. J., 2005, 11, 6552–6559 CrossRef PubMed; (g) A. R. Hirst, J. F. Miravet, B. Escuder, L. Noirez, V. Castelletto, I. W. Hamley and D. K. Smith, Chem.–Eur. J., 2009, 15, 372–379 CrossRef PubMed; (h) J. G. Hardy, A. R. Hirst and D. K. Smith, Soft Matter, 2012, 8, 3399–3406 RSC.
- S. S. Rohner, J. R. Olles and D. K. Smith, RSC Adv., 2015, 5, 27190–27196 RSC.
- (a) W. Edwards and D. K. Smith, J. Am. Chem. Soc., 2013, 135, 5911–5920 CrossRef PubMed; (b) W. Edwards and D. K. Smith, J. Am. Chem. Soc., 2014, 136, 1116–1124 CrossRef PubMed.
- It should be noted that gels are often considered as porous materials, although the solid-like network constitutes <1% of the material in this case. It is difficult to experimentally determine the porosity of solvated gels, and for this reason we decided to use a simple Fickian approach to approximate diffusion coefficients, which are very useful for comparative purposes: A. Fick, Ann. Phys., 1855, 170, 59–86 CrossRef.
- (a) J. Crank, Mathematics of Diffusion, Clarendon, Oxford, 1975 Search PubMed; (b) E. L. Cussler, Diffusion: Mass Transfer in Fluid Systems, Cambridge, London, 1997 Search PubMed; (c) Website:http://www.pojman.com/mg_materials/Diffusion/Diffusion.html accessed 06/03/2018.
- A. Einstein, Ann. Phys., 1905, 17, 549–560 CrossRef.
- W. Edwards and D. K. Smith, Gels, 2018, 4, 31 CrossRef.
- See for selected examples: (a) X. Luo, B. Liu and Y. Liang, Chem. Commun., 2001, 1556–1557 RSC; (b) T. Koga, M. Matsuoka and N. Higashi, J. Am. Chem. Soc., 2005, 127, 17596–17597 CrossRef PubMed; (c) J. Puigmartí-Luis, V. Laukhin, A. Pérez del Pino, J. Vidal-Gancedo, C. Rovira, E. Laukhina and D. B. Amabilino, Angew. Chem., Int. Ed., 2007, 46, 238–241 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: AFM, SEM and TEM imaging, fitting of diffusion cell data to yield diffusion coefficients and additional spectroscopic and rheological data. See DOI: 10.1039/c8sc01071d |
| This journal is © The Royal Society of Chemistry 2018 |