
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Nitric oxide activation facilitated by cooperative multimetallic electron transfer within an iron-functionalized polyoxovanadate–alkoxide cluster†
F.
Li
 a,
R. L.
Meyer
a,
S. H.
Carpenter
a,
L. E.
VanGelder
a,
A. W.
Nichols
b,
C. W.
Machan
b,
M. L.
Neidig
a and
E. M.
Matson
*a
a,
R. L.
Meyer
a,
S. H.
Carpenter
a,
L. E.
VanGelder
a,
A. W.
Nichols
b,
C. W.
Machan
b,
M. L.
Neidig
a and
E. M.
Matson
*a
aDepartment of Chemistry, University of Rochester, Rochester, New York 14627, USA. E-mail: matson@chem.rochester.edu
bDepartment of Chemistry, University of Virginia, Charlottesville, Virginia 22904-4319, USA
First published on 2nd July 2018
Abstract
A series of NO-bound, iron-functionalized polyoxovanadate–alkoxide (FePOV–alkoxide) clusters have been synthesized, providing insight into the role of multimetallic constructs in the coordination and activation of a substrate. Upon exposure of the heterometallic cluster to NO, the vanadium-oxide metalloligand is oxidized by a single electron, shuttling the reducing equivalent to the {FeNO} subunit to form a {FeNO}7 species. Four NO-bound clusters with electronic distributions ranging from [VV3VIV2]{FeNO}7 to [VIV5]{FeNO}7 have been synthesized, and characterized via1H NMR, infrared, and electronic absorption spectroscopies. The ability of the FePOV–alkoxide cluster to store reducing equivalents in the metalloligand for substrate coordination and activation highlights the ultility of the metal-oxide scaffold as a redox reservoir.
Introduction
The chemical reactivity of nitric oxide (NO) has captivated the field of bioinorganic chemistry, due to the participation of this small molecule in vasodilation, mammalian signalling, and immune defence processes.1 Given the established significance of this substrate in biological systems2,3 and the biogeochemical nitrogen cycle,4,5 the interaction of NO with metal centres, specifically iron, has been an area of intense research. Indeed, the prevalence of heme- and non-heme-containing metalloenzymes in NO reductases has driven interest in understanding the electronic structure of the {FeNO} subunit in metalloproteins and model complexes.6–8 A combination of spectroscopic, crystallographic, and theoretical methods have shown that substrate binding and reduction are key steps in NO activation.2,3,9–13 However, despite reports describing the behaviour of NO with ferric and ferrous heme complexes, the chemistry of this substrate with non-heme derivatives remains underdeveloped (Fig. 1). Toward a more complete understanding of the redox chemistry involved during NO activation, the synthesis, characterization, and reactivity of non-heme models, capable of supporting variable oxidation states of the {FeNO} subunit, are of interest.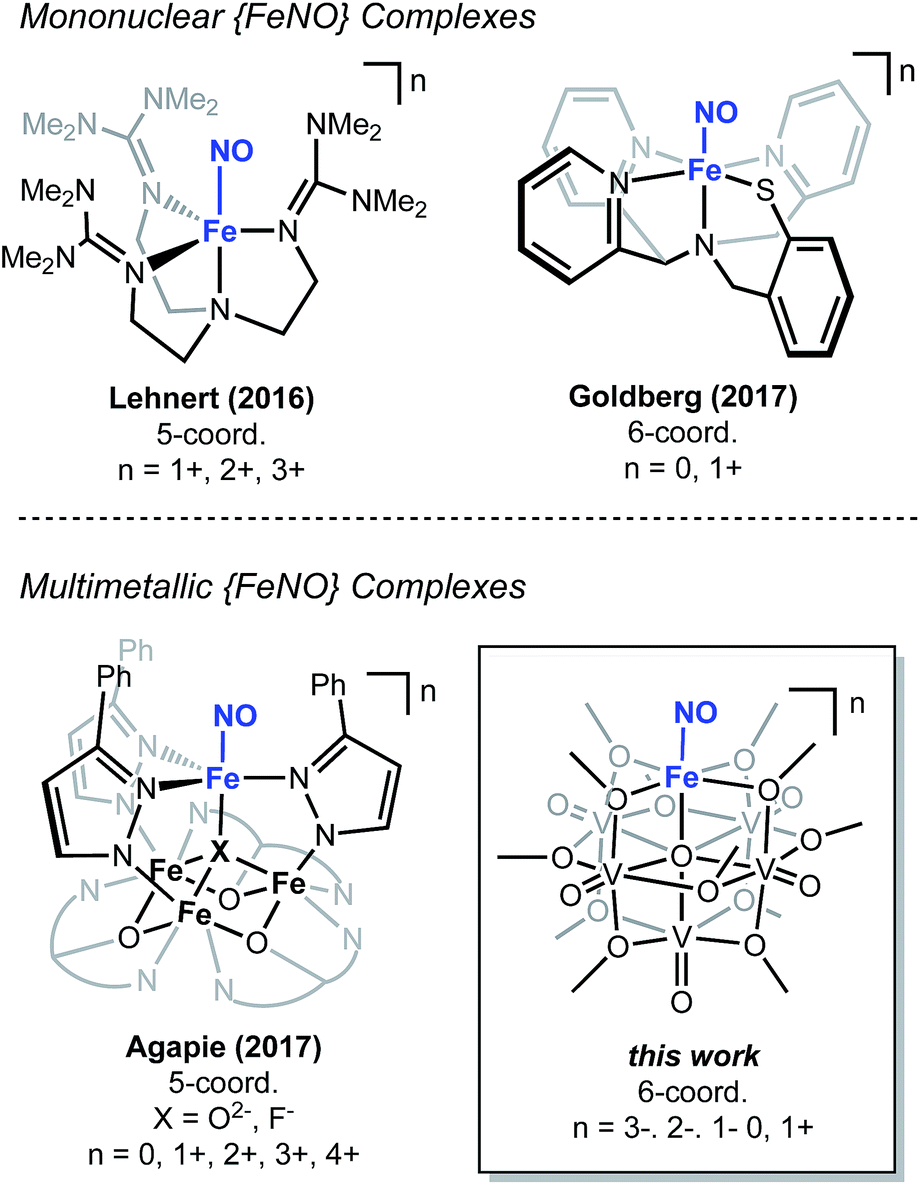 | ||
| Fig. 1 Select examples of non-heme high-spin 5- and 6-coordinate {FeNO} complexes. | ||
The activation of NO requires the simultaneous transfer of multiple electrons and protons to the substrate. In nature, similar chemical transformations of gaseous substrates (e.g. H2, N2, CO2) are often mediated by metalloenzymes that feature multiple, closely associated metal centres within the active site.14–20 Cooperative, multimetallic reactivity has also been noted in heterogeneous systems, where reducible metal-oxide supports have been demonstrated to enhance the activity of metal-catalysts for the activation of small molecules.21–23 In an effort to design homogeneous complexes that mimic the activity of these reactive multimetallic assemblies, our group24,25 and others26–31 have recently reported heterometallic complexes that possess varying degrees of multimetallic electronic communication. Extensive structural and spectroscopic characterization of these clusters has illustrated the participation of the assembly in molecular redox tuning and the storage of electron density. While these studies have expanded our understanding of the static electronic properties of well-defined multinuclear systems, comparatively less is known about the role of electron transfer across a multimetallic assembly during substrate activation.
Recently, our research group has reported the synthesis and characterization of a family of iron-functionalized polyoxovanadate–alkoxide (FePOV–alkoxide) complexes (Fig. 1).24,25,32 Using a suite of analytical techniques, we have demonstrated that the metal-oxide metalloligand functions as a redox reservoir for the ferric centre, storing reducing equivalents across the vanadyl ions in a delocalized cloud of electron density. The established spectroscopic handles, unique to this heterometallic framework, provide the opportunity for in situ analysis of the oxidation state of the metal-oxide scaffold. Therefore, our FePOV–alkoxide complexes are ideal for the investigation of electron transfer within heterometallic assemblies during substrate activation. Given the well-described electronic structures of the {FeNO} subunit, and the large degree of substrate sensitivity to the electronic environment of the bound iron centre,33 we selected NO as an ideal substrate to investigate the reactivity of FePOV–alkoxide clusters. Herein, we report our results that showcase the ability of the POV–alkoxide scaffold to shuttle stored electron density to NO through the ferric centre. This work highlights the role of electron transfer within a multimetallic construct during chemical transformations and substrate activation.
Results and discussion
Reactivity of NO(g) with an FePOV–alkoxide cluster
To investigate the reactivity of NO with high-spin ferric centres embedded within a multimetallic cluster, NO(g) was added to a solution of the neutral, FePOV–alkoxide cluster, [VIV4VVO6(OCH3)12FeIII] (1-V5Fe) (Scheme 1). A gradual colour change from dark-green to brown was observed. Analysis of the reaction mixture via1H NMR spectroscopy revealed formation of a new product, with three paramagnetically shifted and broadened resonances located at 13.86, 20.12, and 127.44 ppm (Fig. S1†). Formation of the desired NO adduct of the FePOV–alkoxide cluster, [V5O6(OCH3)12{FeNO}] (2-V5FeNO), was confirmed by infrared spectroscopy. A new band corresponding to v(NO) was located at 1732 cm−1 (Fig. 2, Table 1), substantially shifted from that of free NO (1900 cm−1).34,35 Due to the coordinative saturation of the vanadyl ions of the FePOV–alkoxide cluster and the shift in v(NO) consistent with formation of an iron–nitrosyl subunit,32,36–38 we concluded that NO most likely binds to the cluster through the vacant site of the 5-coordinate, square pyramidal Fe(III) centre of the multinuclear assembly (Scheme 1).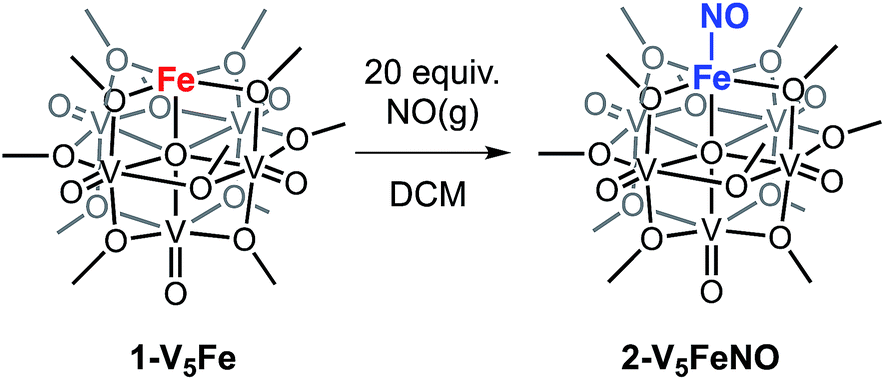 | ||
| Scheme 1 Reactivity of 1-V5Fe with NO(g). | ||
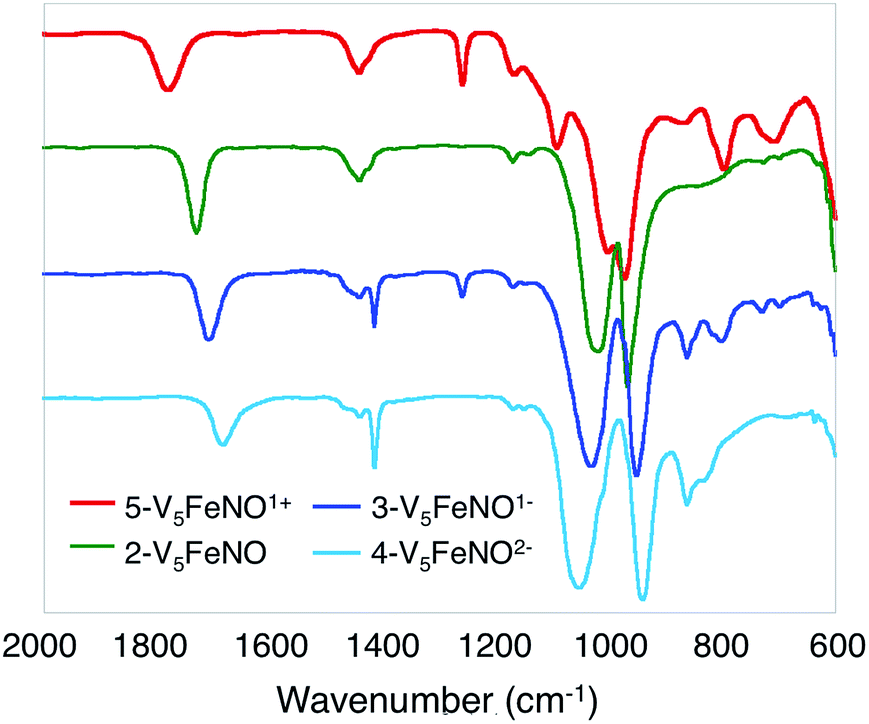 | ||
| Fig. 2 Infrared spectra of complexes 2-V5FeNO, 3-V5FeNO−, 4-V5FeNO2− and 5-V5FeNO+. | ||
| Compound | {FeNO}n | CNa | Fe spin state | v(NO) (cm−1) | v(Ob-CH3) (cm−1) |
ν(V![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Ot) (cm−1) Ot) (cm−1) |
δ (mm s−1)a | ΔEq (mm s−1)a | Ref. |
|---|---|---|---|---|---|---|---|---|---|
a Coordination number.
b Complex readily decomposes. Isolation of this molecule for analysis by Mössbauer spectroscopy was not possible.
c Mössbauer spectrum measured at 80 K.
d Mössbauer spectrum measured at 4.2 K in frozen 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 propionitrile:butyronitrile solution.
e L = 1,3,5-tris(2-di(2′-dipyridyl)hydroxymethylphenyl)benzene.
f Mössbauer spectrum measured at 80 K in solid sample (polycrystalline material mixed with boron nitride). Parameter listed is for the apical iron centre bound to NO. :1 propionitrile:butyronitrile solution.
e L = 1,3,5-tris(2-di(2′-dipyridyl)hydroxymethylphenyl)benzene.
f Mössbauer spectrum measured at 80 K in solid sample (polycrystalline material mixed with boron nitride). Parameter listed is for the apical iron centre bound to NO.
|
|||||||||
| 5-V5FeNO+ | 7 | 6 | 1780 | 1003 | 974 | —b | —b | This work | |
| 2-V5FeNO | 7 | 6 | 1732 | 1022 | 968 | 0.63c | 0.88c | This work | |
| 3-V5FeNO− | 7 | 6 | 1709 | 1032 | 951 | 0.59c | 0.69c | This work | |
| 4-V5FeNO2− | 7 | 6 | 1683 | 1055 | 935 | 0.56c | 0.49c | This work | |
| [(TMG3tren){FeNO}]3+ | 6 | 5 | h.s. | 1879 | — | — | 0.06d | 0.48d | 19 |
| [(TMG3tren){FeNO}]2+ | 7 | 5 | h.s. | 1750 | — | — | 0.48d | 1.42d | 19 |
| [(TMG3tren){FeNO}]1+ | 8 | 5 | h.s. | 1618 | — | — | 0.84d | 2.78d | 19 |
| [(Me3TACN)Fe(NO)(N3)2] | 7 | 6 | l.s. | 1690, 1712 | — | — | 0.62c | 1.28c | 25 |
| cis-[(cyclam)Fe(NO)I]I | 7 | 6 | l.s. | 1726 | — | — | 0.64c | 1.78c | 25 |
| [LFe3F(PhPz)3Fe(NO)]3+e | 7 | 5 | h.s. | 1842 | — | — | 0.62f | 1.51f | 56 |
| [LFe3F(PhPz)3Fe(NO)]2+e | 7 | 5 | h.s. | 1823 | — | — | 0.62f | 1.39f | 56 |
| [LFe3F(PhPz)3Fe(NO)]1+e | 7 | 5 | h.s | 1799 | — | — | 0.63f | 1.67f | 56 |
| [LFe3F(PhPz)3Fe(NO)]e | 8 | 5 | h.s. | 1680 | — | — | 0.95f | 1.63f | 56 |
Given the propensity of NO to disrupt the molecular structure of self-assembled, cluster complexes,39,40 retention of the polynuclear Lindqvist core following exposure of 1-V5Fe to NO was evaluated by IR and electronic absorption spectroscopies. Two characteristic bands of the Lindqvist core were observed in the IR spectrum of 2-V5FeNO, corresponding to v(VOt) and v(Ob–CH3) at 968 and 1022 cm−1, respectively (Ot = terminal vanadyl oxygen atoms, Ob = bridging oxygen atoms of methoxide ligands) (Fig. 2, Table 1). Additionally, the electronic absorption spectrum of 2-V5FeNO reveals two intervalence charge-transfer (IVCT) bands, located at 382 nm (ε = 5.06 × 103 M−1 cm−1) and 982 nm (ε = 5.80 × 102 M−1 cm−1) (Fig. 3). These features correspond to dxy(VIV) → dx2−y2(VV) and dxy(VIV) → dxy(VV) electronic transitions, respectively. Previous reports have established that these absorptions are characteristic of mixed-valent, POV–alkoxide clusters, thus demonstrating retention of the Robin and Day Class II delocalization of electron density across the cluster core upon coordination of NO.25,41 Collectively, IR and electronic absorption spectroscopy confirm retention of the multimetallic assembly following substrate activation.24,25,41,42
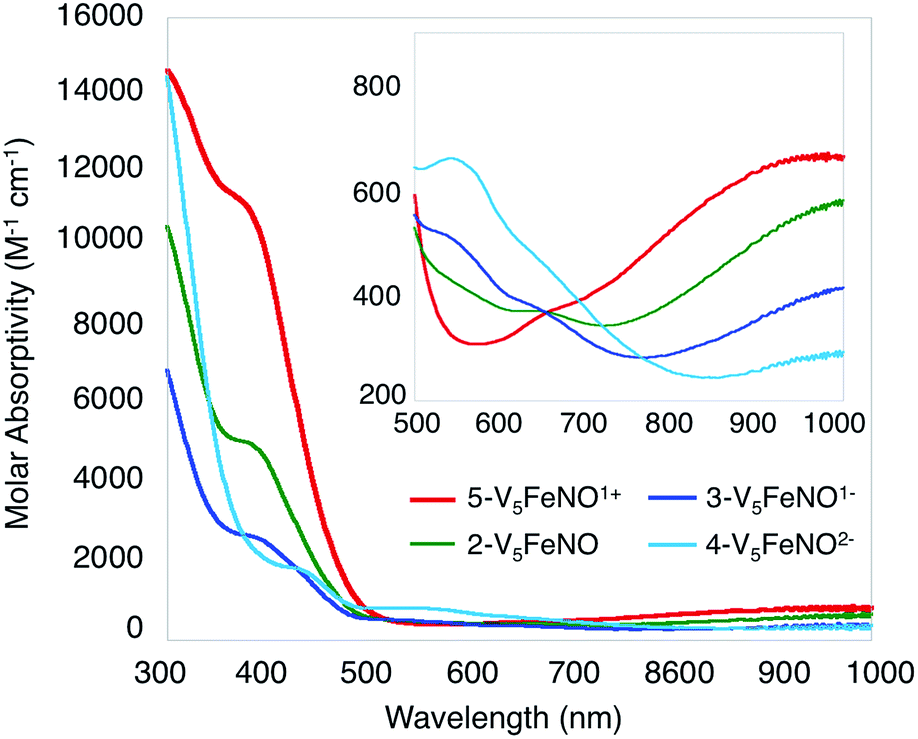 | ||
| Fig. 3 Electronic absorption spectrum of complexes 2–5 collected in CH3CN at 21 °C (the inset shows the low-energy region of the spectrum to more clearly illustrate IVCT bands). | ||
The reactivity of complex 1-V5Fe and NO is surprising, as metalloproteins and model complexes containing high-spin ferric centres have been reported to have a lower affinity for this substrate, in comparison to their ferrous congeners.43,44 This unusual binding of substrate at an FeIII ion prompted the investigation into the participation of the metal-oxide metalloligand in substrate activation. To establish the role of multimetallic, cooperative reactivity, a five coordinate ferric complex, FeIII(salen)Cl (salen = N,N′-bis(salicylidene)ethylenediamine) was reacted with NO(g).45 This monometallic complex was selected as a suitable model for the FePOV–alkoxide clusters, due to their similar iron coordination environments (pseudo-square pyramidal) and the redox non-innocence of the respective organic and inorganic ligands. However, exposure of FeIII(salen)Cl to 20 equiv. of NO(g) yields no reaction. The disparity in the reactivity of the two, 5-coordinate, high-spin ferric complexes (FeIII(salen)Cl and 1-V5Fe) indicates that the POV–alkoxide scaffold is directly involved in NO activation, most likely through the efficient transfer of electron density to the iron centre (vide infra).
Analysis of reduced and oxidized derivatives of 2-V5FeNO
Elucidation of the oxidation state distribution of metal ions in complex 2-V5FeNO is necessary to define the role of the vanadium-oxide metalloligand ligand during NO activation. Previously, our group25 and others41,42,46 have demonstrated that the isolation of the reduced and oxidized derivatives of multimetallic complexes aids in the characterization of the charge distribution of metal ions that compose the cluster core. To assess the accessibility of a redox series of the NO-functionalized FePOV–alkoxide ({FeNO}POV–alkoxide) clusters, the electrochemical profile of 2-V5FeNO was explored via cyclic voltammetry (CV). A voltammogram possessing four redox events located at E1/2 = +0.03, −0.58, −1.16, and −2.68 V vs. Fc/Fc+ was observed (Fig. 4). The reversibility of these events was established by varying the scan rate from 20 to 500 mV s−1 and fitting the data to the Randles–Sevcik equation (Fig. S2†). Linearity of each fully-reversible redox event was confirmed by plotting the current density (jp reported in A cm−2) versus the square root of the scan rate (v1/2 reported in V s−1).25,47 Comparison of the CV of complex 2-V5FeNO to that of the starting material, 1-V5Fe, illustrates the drastic changes in the redox properties of the cluster upon coordination of NO, suggesting substrate coordination has a direct effect on the electronic structure of the multimetallic assembly.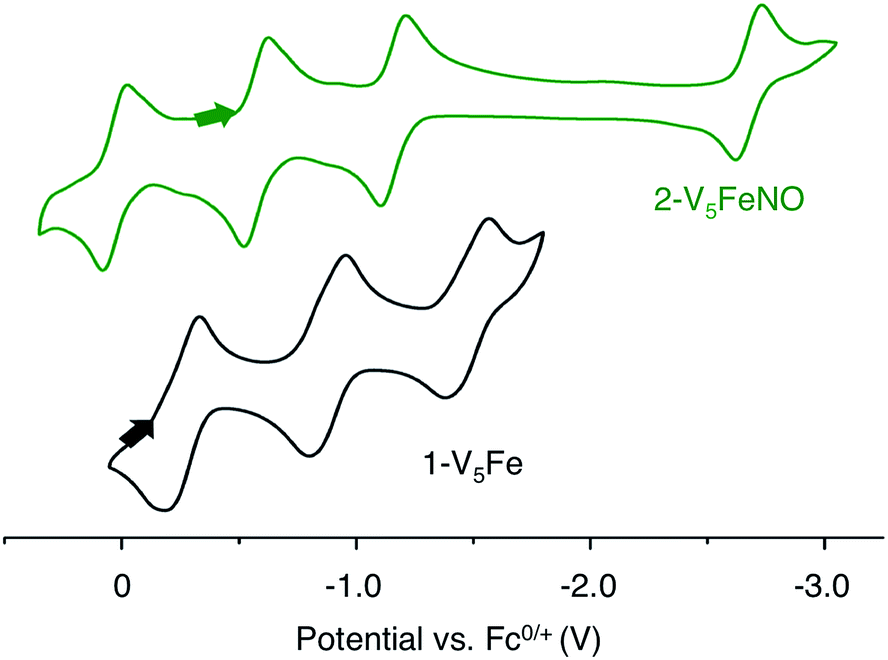 | ||
| Fig. 4 Cyclic Voltammogram of complexes 1-V5Fe and 2-V5FeNO collected in THF with 0.4 M [nBu4N][PF6] as the supporting electrolyte (v = 0.1 V s−1). | ||
The first three redox events in the CV of complex 2-V5FeNO, observed at E1/2 = +0.03, −0.58, and −1.16 V, possess peak-to peak separations (ΔE1/2) of approximately 0.60 V. Similar values of ΔE1/2 have been observed in the case of FePOV–alkoxide clusters25 and their homometallic, hexavanadate congeners,41,42 and are associated with redox events localized to the vanadyl ions. In the case of complex 2-V5FeNO, the ΔE1/2 values for these redox events are similar to those reported for the parent complex, 1-V5Fe, resulting in a similar range of comproportionation constants (Kc) between the two series of FePOV–alkoxide clusters (Kc = 8.8 × 109 to 2.7 × 1010; RTln(Kc) = nF [ΔE01/2]).48 To unambiguously corroborate the hypothesis that the evenly spaced redox events correspond to vanadium-based electrochemical processes, the chemically reduced and oxidized derivatives of 2-V5FeNO were isolated. Exposure of 2-V5FeNO to stoichiometric equivalents of cobaltocene afforded access to the one- and two-electron reduced species, namely 3-V5FeNO− and 4-V5FeNO2− (Scheme 2). Oxidation of 2-V5FeNO with AgClO4 at −40 °C led to the formation of 5-V5FeNO+. The NO adducts (3-V5FeNO− and 5-V5FeNO+) could be independently synthesized by reaction of NO(g) with the appropriate FePOV–alkoxide precursor ([V5O6(OCH3)12Fe]− and [V5O6(OCH3)12Fe]+, respectively) (Scheme 2). In all cases, the products were analysed by 1H NMR spectroscopy (Fig. S3–S5†).
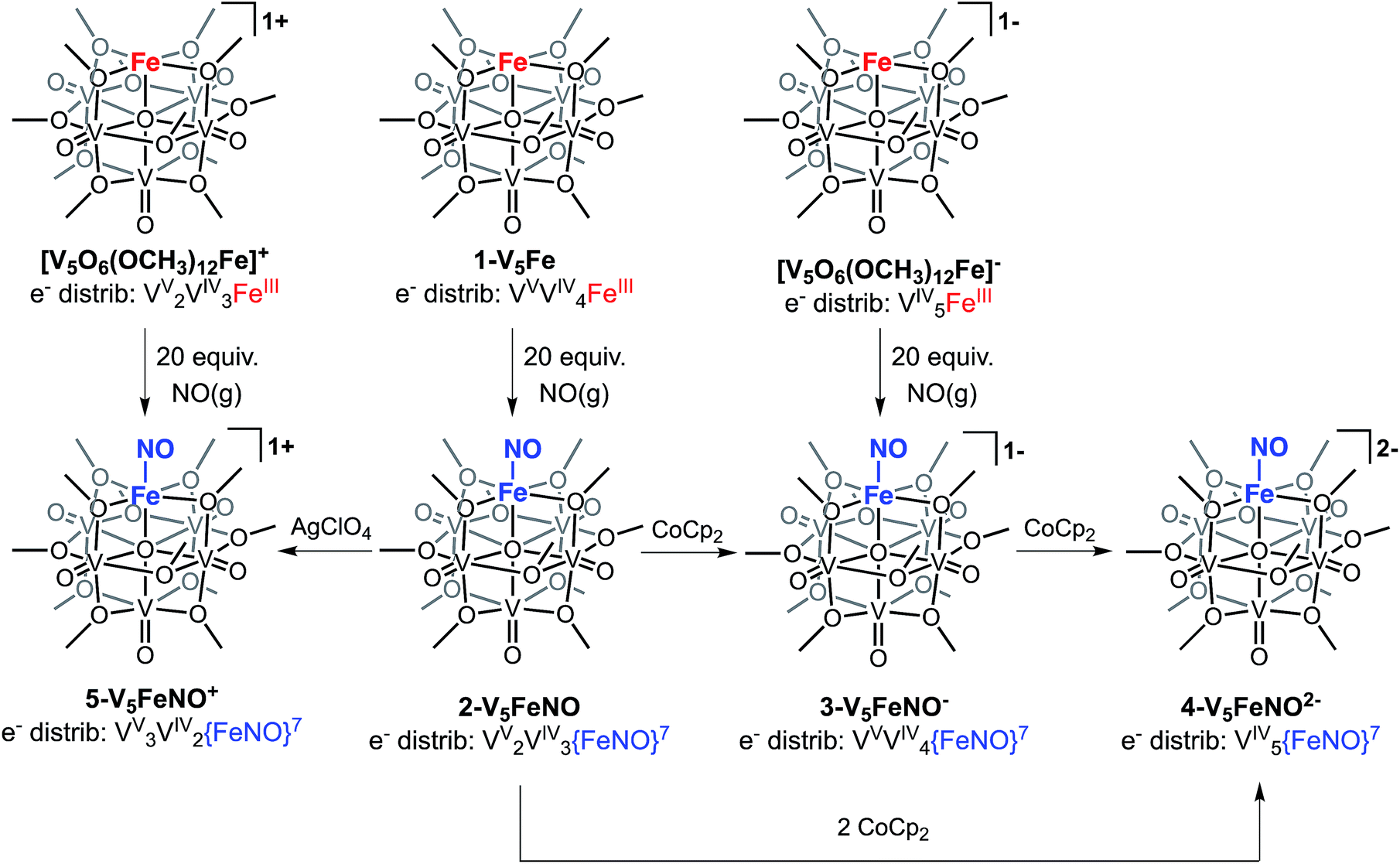 | ||
| Scheme 2 Synthesis of complexes 2-V5FeNO, 3-V5FeNO−, 4-V5FeNO2−, and 5-V5FeNO+. | ||
Upon sequential reduction from 5-V5FeNO+ to 4-V5FeNO2−, the IR spectra reveal that v(VOt) decreases from 974 to 935 cm−1, with corresponding increases in the values of v(Ob–CH3) (1003 to 1055 cm−1) (Fig. 2, Table 1). Similar changes in the v(VOt) and v(Ob–CH3) stretching frequencies have been reported for the redox series of homo- and heterometallic POV–alkoxide complexes.25,41,42 Furthermore, with each electron added to the system, v(NO) shifts ∼30 cm−1 toward lower energies (Fig. 2, Table 1). This small change in stretching frequency of the nitrosyl ligand is inconsistent with the direct reduction of the iron–nitrosyl moiety, as this change in oxidation state of the {FeNO} subunit is typically associated with changes in v(NO) of ∼100 cm−1.37,38,49 Instead, the decrease in stretching frequency of v(NO) compares favourably to changes observed in the {FeNO}6/7 dithiolene systems.50,51 The redox active dithiolene ligands of these complexes are able to store equivalents of electron density upon sequential reduction from the mono-cationic to the mono-anionic species, [Fe(NO)(S2C2R2)2]n (R = p-tolyl; n = +1, 0, −1). As a result of ligand participation in the reduction events of the complex, retention of the {FeNO}6 oxidation state was observed throughout the series. IR spectroscopy of the {FeNO} dithiolate complexes revealed small shifts in v(NO) (∼35 cm−1/e−), consistent with the changes in v(NO) observed in the spectra of the complexes 2–5. These small, yet distinct changes are a consequence of the dependence of the electronic structure of the {FeNO} moiety on the oxidation state of the redox-active ligand.
Likewise, electronic storage across a metal-oxide base has been observed previously, in the case of [LFe3(PhPz)3EFeNO]n+ (L = 1,3,5-triarylbenzene ligand motif, E = O, F).56,57 Agapie and coworkers demonstrate through spectroscopic and crystallographic investigations that changes in oxidation state of the tetra-iron cluster, [LFe3(PhPz)3OFeNO]1+, occur exclusively across the distal iron centres. Mössbauer parameters of complexes [LFe3(PhPz)3OFeNO]n+ (n = 1, 2, 3) reveal little change in the isomer shift of the apical iron centre upon oxidation of the cluster core, indicating the retention of the {FeNO}7 state for the iron–nitrosyl adduct across the redox series. Furthermore, nominal changes in v(NO) (∼30 cm−1/e−) corroborate that no change in the oxidation state of the {FeNO}7 subunit occurs during cluster oxidation. Collectively, these results establish the role of the tri-iron base as a redox-reservoir for the apical iron centre. The POV–alkoxide scaffold acts in analogy to the iron-oxide base of [LFe3(PhPz)3OFeNO]1+ during changes in oxidation state of the cluster, as established via infrared analysis.
The electronic absorption spectra of complexes 2–5 provide additional insight into the electronic structure of the heterometallic Lindqvist cluster. Absorptions diagnostic of IVCT events between V(IV) and V(V) centres are observed in complexes 2-V5FeNO, 3-V5FeNO−, and 5-V5FeNO+, suggesting the presence of a mixed-valent POV–alkoxide core.25,41,42 In previous work, similar features have been considered tell-tale signs of extensive delocalization of electron density across the POV–alkoxide assembly,25,41,42,46,52 In contrast, the electronic absorption spectrum of 4-V5FeNO2− has no IVCT absorptions. Instead, the spectrum contains a weak feature at 542 nm (ε = 661 M−1 cm−1), which has been previously assigned to a forbidden dxy(VIV) → dx2−y2(VIV) excitation of an isovalent POV–alkoxide cluster.25 As such, the charge distribution for complex 4-V5FeNO2− can be described as [VIV5O6(OCH3)12{FeNO}]2−. Taking into consideration that the sequential one-electron oxidations occur across the POV–alkoxide framework, the oxidation state distributions of the vanadium ions in the remaining {FeNO}POV–alkoxide clusters can be assigned as [VVVIV4O6(OCH3)12{FeNO}]− (3-V5FeNO−), [VV2VIV3O6(OCH3)12{FeNO}] (2-V5FeNO), and [VV3VIV2O6(OCH3)12{FeNO}]+ (5-V5FeNO+) (Scheme 2). Thus, following exposure to NO, the POV–alkoxide core is oxidized by one electron, with respect to the parent cluster (Scheme 2).
Electronic structure of the {FeNO} subunit
Following identification of the oxidation states of the vanadium ions within the POV–alkoxide assembly, charge balance reveals, in all cases, an overall 2+ charge on the {FeNO} subunit for complexes 2–5. The required charge is satisfied by multiple resonance structures, namely [FeIINO˙]2+ and [FeIIINO−]2+. Due to the ambiguity in the explicit oxidation state of the iron centre, the electronic structures of the iron–nitrosyl subunits of the FePOV–alkoxide complexes are best described as {FeNO}7 in the Enemark–Feltham notation.53 The v(NO) observed in the IR spectra of clusters 2–5 are consistent with this assignment, with values that resemble previously reported 6-coordinate {FeNO}7 complexes (Table 1).11To more rigorously define the electronic structure of the iron–nitrosyl moiety, zero-field Mössbauer spectroscopy was performed on solid samples of complexes 2–4 (collected at 80 K) (Fig. 5). The broad, slightly asymmetric, quadrupole doublets resemble values reported previously for monodisperse FePOV–alkoxide clusters, indicating a single electronic environment for the iron centre.25 It is worth noting that the isomer shifts obtained from the Mössbauer spectra of the {FeNO}POV–alkoxide clusters resemble previously reported parameters for {FeNO}7 complexes (Table 1). The observed decrease in quadrupole splitting of the {FeNO}POV–alkoxide species as compared to their monometallic, 6-coordinate, {FeNO}7 congeners is likely reflective of the unique structure of the multimetallic cluster complexes.
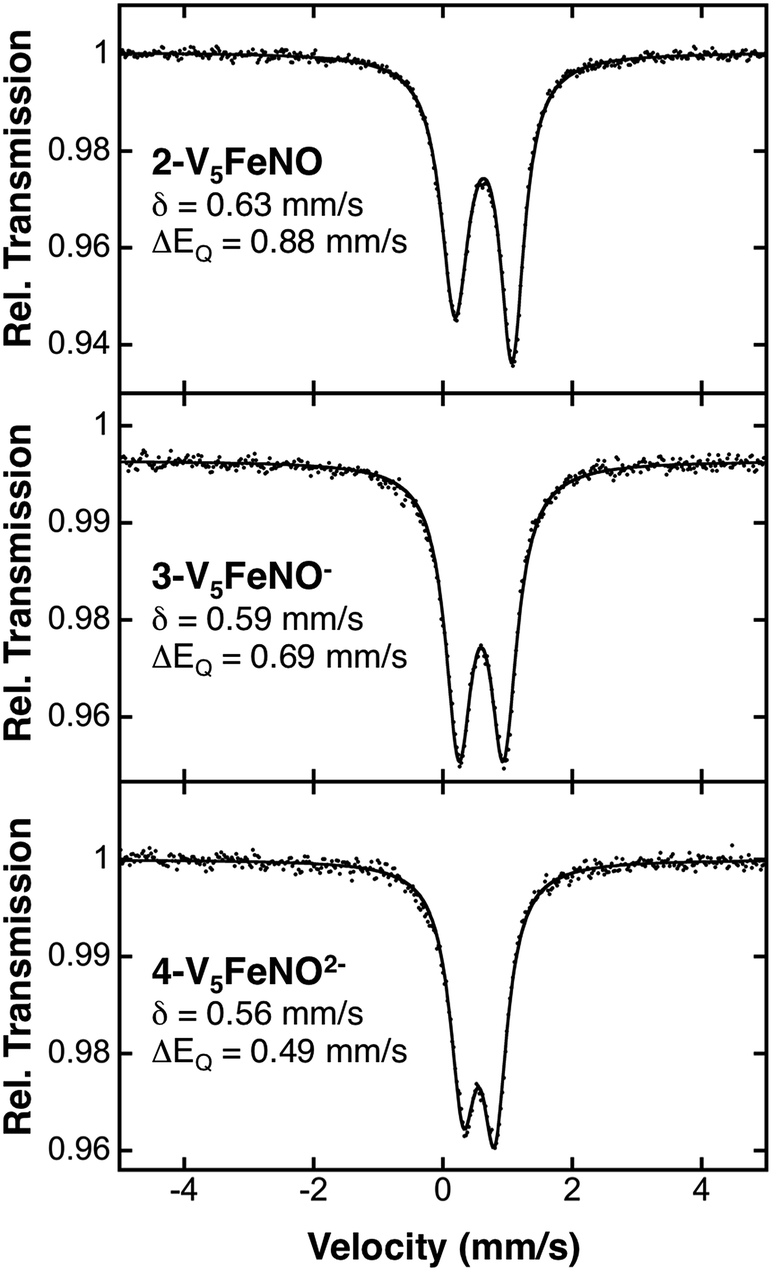 | ||
| Fig. 5 80 K Mössbauer spectra of complexes 2-V5FeNO, 3-V5FeNO−, and 4-V5FeNO2−. For each spectrum, the data (dots) and fit (solid line) are given. The Mössbauer fit parameters for each spectrum are depicted with spectra, and also summarized in Table 1. | ||
Electron paramagnetic resonance (EPR) spectroscopy was also performed on complex 3-V5FeNO− in order to obtain further insight into the spin state of the {FeNO}7 unit of these complexes. In addition to a broad S = 1/2 signal attributed to the POV–alkoxide scaffold,54 significant signal intensity in the g ∼ 4 region is present, consistent with the presence of a high-spin S = 3/2 {FeNO}7 unit (Fig. S6†).
The formation of the {FeNO}7 subunit upon exposure of the FePOV–alkoxide clusters to NO(g) is remarkable, given addition of a nitrosyl ligand to a ferric centre would be expected to result in the formation of an {FeNO}6 subunit. Generation of the {FeNO}7 moiety is consistent with either a metal-centred or substrate-based reduction upon coordination of NO to the multimetallic species. The reduction of the iron nitrosyl moiety occurs simultaneously with the observed oxidation of the POV–alkoxide scaffold, thus we can conclude that the metal-oxide metalloligand transfers electron density to the iron centre to facilitate activation of the substrate. This observed electron transfer between metal-oxide scaffold and iron constitutes a rare example of cooperative multimetallic reactivity for substrate activation. Similar flexibility in site-differentiated multimetallic clusters has recently been reported by Agapie and coworkers.55 This report demonstrates the utility of electron transfer from a mixed-valent iron oxide base to a site-differentiated ferric ion upon coordination of CO, resulting in the formation of [LFe3O(PhIm)Fe(CO)n] (L = 1,3,5-triarylbenzene ligand motif; PhIm = 1-phenyl imidazole) complexes. In both examples, electron transfer clarifies that the metalloligand plays an important part in substrate activation, providing electron density to a ferric centre to facilitate substrate coordination and reduction.
Attempts to access the fully reduced {FeNO}POV–alkoxide
Our attention turned toward identification of the fully reduced species, generated by the one-electron reduction of complex 4-V5FeNO2−. Interest in characterizing the product of this reaction stems from the surprising peak-to-peak separation associated with the redox event centred at E1/2 = −2.68 V vs. Fc/Fc+ (ΔE1/2 = 1.52 V). Similar separations of redox events in heterometal-functionalized polyoxometalates have been attributed to changes in oxidation state of the distinct metal centre incorporated within the cluster.58,59 Consistent with these observations, the reducing nature of this electrochemical process suggests the reduction event is localized to the site-differentiated, iron–nitrosyl moiety. Accordingly, we hypothesized that reduction of the {FeNO}7 subunit would give rise to the formation of a rare, non-heme {FeNO}8 species.36–38,60–63Reduction of 2-V5FeNO in THF was attempted at low-temperatures with a freshly prepared solution of sodium naphthalenide. Stirring the solution for 1 hour results in apparent formation of a single product, as observed by 1H NMR spectroscopy (Fig. S7,†δ = 90.33, 34.48, 16.03 ppm in CD3CN). This three-resonance pattern in the product is similar to complexes 2–5, suggesting clean formation of the desired fully-reduced {FeNO}POV–alkoxide (Scheme 3). However, multiple attempts to characterize the reduced cluster via Mössbauer spectroscopy resulted in identification of three distinct electronic environments for iron within the sample (Fig. S9, Table S1†). Despite the chemical reversibility observed for the most-reducing event in the CV of 2-V5FeNO, and the seemingly straightforward 1H NMR spectrum of the product, the complicated Mössbauer spectrum suggests that substantial decomposition occurs upon attempts to chemically generate [V5O6(OCH3)12{FeNO}]3− (6-V5FeNO3−), to paramagnetic, 1H NMR-silent byproducts.
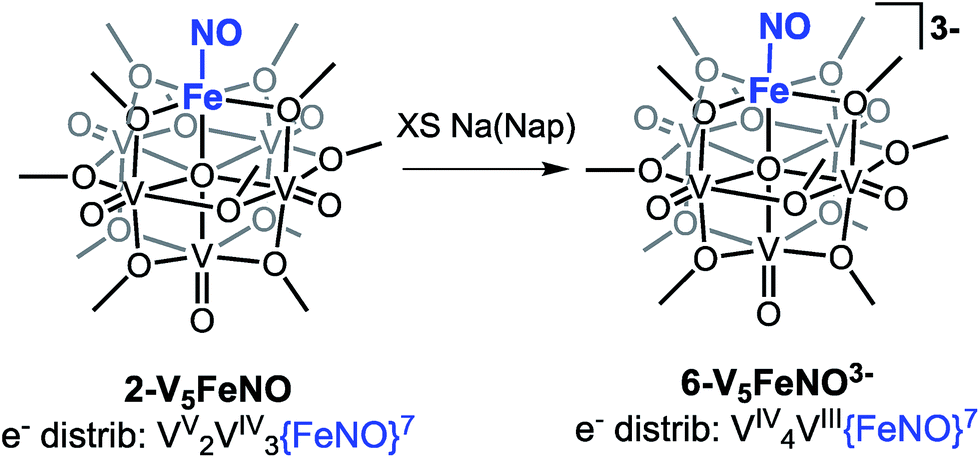 | ||
| Scheme 3 Attempted synthesis of complex 6-V5FeNO3−via the triple reduction of 2-V5FeNO. | ||
Given the apparent thermal instability of the fully reduced cluster, in situ electroanalytical techniques were employed for characterization of the {FeNO}POV–alkoxide clusters (Fig. 6, Table 2). Infrared-spectroelectrochemistry (IR-SEC) allows IR active modes in a compound of interest to be monitored with respect to changes in electrochemical potential as a function of time.64,65 To correlate the electrochemical response directly to the independently synthesized compounds, we started with the electrochemical generation of complexes 2–5. Indeed, a 3 mM solution of 2-V5FeNO in 0.4 M TBAPF6/THF supporting electrolyte at −0.27 V vs. Fc/Fc+ shows an IR absorbance at 1735 cm−1. This value compares well with the IR data from the independently synthesized sample (1732 cm−1, Fig. 3, Table 1). When the applied cell potential was brought to 0.08 V vs. Fc/Fc+, an absorption band at 1785 cm−1 appears and grows in intensity with the concomitant loss of the band at 1735 cm−1, consistent with the electrochemical generation of the one-electron oxidized species 5-V5FeNO1+ (v(NO) (ATR) = 1780 cm−1). Upon returning to −0.27 V, the ν(NO) of the starting material 2-V5FeNO at 1735 cm−1 was regenerated with the loss of the band at 1785 cm−1 (Fig. 6), indicating good reversibility of this electrochemical process. At more negative potentials, −0.68 V vs. Fc/Fc+, an absorption at 1699 cm−1 appears with the disappearance of the band at 1735 cm−1, consistent with the electrochemical generation of the one-electron reduced species 3-V5FeNO1− (v(NO) (ATR) = 1709 cm−1). At −2.0 V vs. Fc/Fc+, a new IR absorbance band at 1664 cm−1 is observed to grow in intensity with the loss of the band at 1699 cm−1, indicating formation of 4-V5FeNO2− (v(NO) (ATR) = 1683 cm−1).
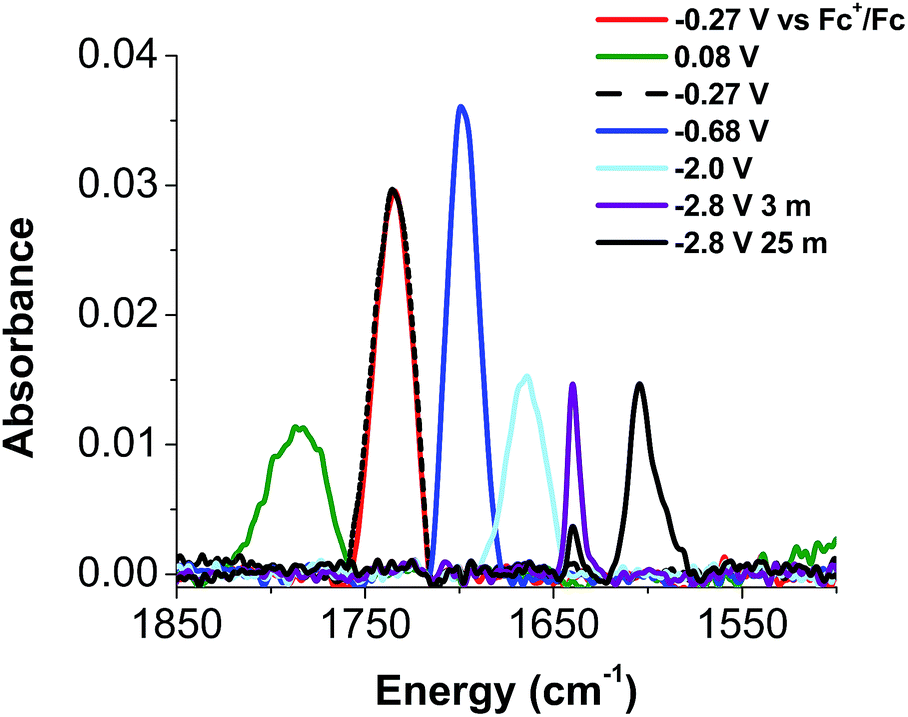 | ||
| Fig. 6 IR-SEC data of complexes 2-V5FeNO measured at −0.27 V (2-V5FeNO, red), 0.08 V (5-V5FeNO+, green), −0.68 V (3-V5FeNO−, blue), −2.0 V (4-V5FeNO2−, light blue), −2.8 V at 3 min (6-V5FeNO3−, purple), −2.8 V at 25 min (black). | ||
| Complex | Chemical reduction/oxidation v(NO) (cm−1) | IR-SEC v(NO) (cm−1) |
|---|---|---|
| a We were unable to isolate complex 6-V5FeNO3− chemically, therefore the IR spectrum of this compound was not obtained. b v(NO) absorbance corresponds to an irreversible chemical transformation that occurs following formation of 6-V5FeNO3−. | ||
| 5-V5FeNO+ | 1780 | 1785 |
| 2-V5FeNO | 1732 | 1735 |
| 3-V5FeNO− | 1709 | 1699 |
| 4-V5FeNO2− | 1683 | 1664 |
| 6-V5FeNO3− | —a | 1640 (3 min) 1604 (25 min)b |
To probe the formation of the fully reduced cluster, [V5O6(OCH3)12FeNO]3− (6-V5FeNO3−), via IR-SEC, the cell potential was held at −2.8 V vs. Fc/Fc+. An IR absorption band appears at 1640 cm−1 and increases in intensity with loss of the band at 1664 cm−1. After holding a potential of −2.8 V for 25 min, the band noted at 1640 cm−1 corresponding to tri-reduced complex, 6-V5FeNO3−, was nearly consumed, with concomitant formation of a new complex with a v(NO) located at 1604 cm−1. This new species is indicative of a subsequent chemical reorganization of the {FeNO}POV–alkoxide cluster, as attempts to return the potential toward oxidizing regimes did not result in regeneration of 2-V5FeNO.
The instability of 6-V5FeNO3−, as confirmed by IR-SEC, is consistent with our experimental data. In the chemical reduction of 2-V5FeNO with excess sodium naphthalenide, the reaction mixture is stirred for one hour, well beyond the lifetime of the reduced cluster as suggested by IR SEC (∼25 min). Attempts to chemically re-oxidize the product mixture isolated from chemical reduction were unsuccessful, indicating that over the course of an hour, complex 6-V5FeNO3− disproportionates to new products with vastly different electronic properties.
The small change in v(NO) (∼25 cm−1) observed in the IR-SEC data of the transiently generated tri-reduced cluster (6-V5FeNO3−) suggests that despite the large ΔE1/2 of the most reducing event, the final reduction likely is localized to the POV–alkoxide metalloligand, as opposed to the {FeNO} subunit. An additional reduction across the POV–alkoxide scaffold requires formation of an FePOV–alkoxide cluster with a single V(III) ion. Accessing a V(III) ion within an iron-functionalized POV–alkoxide scaffold has been reported previously by our laboratory,25 however, the significant peak-to-peak separation observed in the CV of complex 2-V5FeNO was not observed in the case of the parent FePOV–alkoxide cluster, 1-V5Fe. In the case of the nitrosyl functionalized cluster, accessing the V(III) ion within the POV–alkoxide scaffold only occurs at extremely reducing potentials, and immediately affords disproportionation. The seemingly straight-forward 1H NMR spectrum of the product of decomposition and the reduced v(NO) observed by IR-SEC suggests that degradation of 6-V5FeNO3− results in formation of an FePOV–alkoxide cluster with a chemically activated nitrosyl unit. Current investigations into the molecular composition of this highly reactive by-product are underway.
Conclusions
In summary, a series of NO-bound FePOV–alkoxide clusters, 5-V5FeNO+, 2-V5FeNO, 3-V5FeNO−, and 4-V5FeNO2−, varying in oxidation state by a single electron have been synthesized and systematically characterized using a variety of spectroscopic techniques. The charge distributions of complexes 2–5 were determined, revealing electron transfer from the POV–alkoxide scaffold to the iron–nitrosyl moiety upon coordination of NO. These results demonstrate that the metal-oxide metalloligand is capable of providing electron density to the iron centre for substrate coordination and activation.The formation of {FeNO}7 motifs in FePOV–alkoxide clusters illustrates our initial foray into utilizing metal-oxide scaffolds as redox reservoirs for first-row transition metal complexes. The diffuse electron density of the POV–alkoxide scaffold, coupled with a suitable vacant site on the heterometal, enables coordination and the reduction of NO at a ferric centre. Future studies will expand upon this seminal work—extending it to other small molecule substrates and uncovering new approaches to mediating multielectron transformations using earth abundant elements.
Experimental
General considerations
All manipulations were carried out in the absence of water and oxygen in a UniLab MBraun inert atmosphere glovebox under a dinitrogen atmosphere. Glassware was oven dried for a minimum of 4 h and cooled in an evacuated antechamber prior to use. Unless otherwise noted, solvents were dried and deoxygenated on a Glass Contour System (Pure Process Technology, LLC) and stored over activated 3 Å molecular sieves (Fisher Scientific). Celite 545 (J. T. Baker) was dried in a Schlenk flask for at least 14 h at 150 °C under vacuum prior to use. Sodium (Na, 99%+), Silver perchlorate (AgClO4, anhydrous, 97%) and bis(cyclopentadienyl)cobalt(II) (CoCp2, 98%) were purchased from Sigma-Aldrich and used as received. Nitric Oxide (NO, Matheson Gas Products, Inc. 99%) was purified according to literature precedent.66 [V5O6(OCH3)12Fe]+, [V5O6(OCH3)12Fe] (1-V5Fe), [V5O6(OCH3)12Fe]−, and [V5O6(OCH3)12Fe]2− were synthesized according to our previous report.24,251H NMR spectra were recorded on a Bruker DPX-500 MHz spectrometer locked on the signal of deuterated solvents. All chemical shifts were reported relative to the peak of a residual 1H signal in deuterated solvents. CD3CN and CDCl3 were purchased from Cambridge Isotope Laboratories, degassed by three freeze–pump–thaw cycles, and stored over activated 3 Å molecular sieves. Infrared (FT-IR, ATR) spectra of complexes were recorded on a Shimadzu IR Affinity-1 Fourier Transform infrared spectrophotometer and are reported in wavenumbers (cm−1). Electronic absorption measurements were recorded at room temperature in anhydrous acetonitrile in a sealed 1 cm quartz cuvette with an Agilent Cary 60 UV-Vis spectrophotometer. Elemental analyses were performed on a PerkinElmer 2400 Series II Analyzer at the CENTC Elemental Analysis Facility (University of Rochester).
Cyclic Voltammetry experiments were recorded with a CH Instruments Inc. 410c time-resolved electrochemical quartz crystal microbalance. All measurements were performed in a three-electrode system cell configuration that consisted of a glassy-carbon (ø = 3.0 mm) working electrode, a Pt wire counter electrode, and an Ag/AgCl wire reference electrode. All electrochemical measurements were performed at room temperature in a N2-filled glovebox. A 0.4 M nBu4NPF6 solution (anhydrous THF) was used as the electrolyte solution. All redox events were referenced against the ferrocene/ferrocenium (Fc/Fc+) redox couple.
All samples for 57Fe Mössbauer spectroscopy were run as isolated solid samples made from natural abundance iron. All samples were prepared in an inert-atmosphere glovebox equipped with a liquid-nitrogen fill port. This enables freezing of the samples to 77 K within the glovebox. Samples were loaded into a Delrin Mössbauer cup for measurements and loaded under liquid nitrogen. 57Fe Mössbauer measurements were performed using a SEE Co. MS4 Mössbauer spectrometer integrated with a Janis SVT-400T He/N2 cryostat for measurements at 80 K. Isomer shifts were determined relative to α-iron at 298 K. All Mössbauer spectra were fit using the program WMoss (SEE Co.).
Samples for Electron Paramagnetic Resonance analysis were prepared in an inert atmosphere glovebox (N2). EPR samples were prepared as solutions in acetonitrile and loaded into 4 mm OD Suprasil quartz EPR tubes from Wilmad Labglass. X-band EPR spectra were collected at 10 K on a Bruker EMXplus spectrometer equipped with a 4119HS cavity and an Oxford ESR-900 helium flow cryostat. Instrumental parameters employed for all samples were as follows: 1 mW power; 80 ms time constant; 8 G modulation amplitude; 9.38 GHz frequency; and 100 kHz modulation frequency.
IR Spectroelectrochemistry (IR-SEC) experiments were conducted using a custom cell based on a previously published design.67–69 The three-electrode set-up consists of an inner glassy carbon working electrode disc (10 mm diameter), a central circular silver bare metal pseudoreference electrode, and an outer circular glassy carbon counter electrode embedded within a PEEK block. All data were referenced to an internal ferrocene standard (ferricenium/ferrocene reduction potential under stated conditions; obtained by taking a CV with the cell prior to injecting analyte for IR-SEC experiments) unless otherwise specified. IR-SEC solutions were prepared inside a glovebox under N2 atmosphere and injected in IR-SEC cell using an SGE airtight syringe. IR spectra were collected using a Bruker Vertex 80 spectrometer with a liquid nitrogen-cooled detector.
Synthesis of {(VO)5O(OCH3)12FeNO} (2-V5FeNO)
In the glovebox, a 250 mL Schlenk flask was charged with 1-V5Fe (0.119 g, 0.152 mmol) and 40 mL CH2Cl2. The nitrogen atmosphere was removed with two freeze–pump–thaw cycles, and one atmosphere of purified NO gas (normal temperature and pressure) was added via to the evacuated flask. The solution was vigorously stirred for three days, resulting in a colour change from dark-green to brown. Volatiles were removed under reduced pressure, and the flask was taken back into the glovebox. The product was extracted with diethyl ether (Et2O, 10 mL × 3), followed by extraction with n-pentane (10 mL × 3). Removal of solvent under reduced pressure results in isolation of the product, 2-V5FeNO (0.074 g, 0.091 mmol, 60%). 1H NMR (500 MHz, CD3CN): δ = 127.44 (s, J = 283 Hz), 20.12 (s, J = 84 Hz), 13.86 (s, J = 94 Hz). FT-IR (ATR, cm−1): 2920 (C–H), 2814 (C–H), 1730 (NO), 1022 (O–CH3), 968 (VO). UV-Vis [CH3CN; λ, nm (ε, M−1 cm−1)]: 382 (5.06 × 103), 992 (5.80 × 102). Elemental analysis for C12H36NO19V5Fe (MW = 808.96 g mol−1) calcd (%): C, 17.82; H, 4.49; N, 1.73. Found (%): C, 18.16; H, 4.13; N, 1.36.
Synthesis of CoCp2{(VO)5O(OCH3)12FeNO} (3-V5FeNO−)
O), 1032 (O–CH3), 951 (VO). UV-Vis [CH3CN; λ, nm (ε, M−1 cm−1)]: 384 (2.68 × 103), 992 (4.14 × 102). Elemental analysis for C22H46NO19V5FeCo (MW = 998.08 g mol−1) calcd (%): C, 26.47; H, 4.65; N, 1.40. Found (%): C, 26.87; H, 4.43; N, 1.11.
:1) mixture. The flask was capped with a septum, carefully taped, and removed from the glovebox. Purified NO gas (ca. 74 mL at normal temperature and pressure conditions) was added via gas-tight syringe. The solution was vigorously stirred for four days and afforded a colour change from dark-green to brown. The solvent was removed under reduced pressure, and the flask was taken back into the glovebox. Copious amount of CH2Cl2 was added to extract the solid residue until the extraction turned nearly colourless, followed by removal of the volatiles to yield 3-V5FeNO− as a brown powder (0.050 g, 0.059 mmol, 37%). 1H NMR and FT-IR spectra collected for the product are identical to that observed for method A.
Synthesis of (CoCp2)2{(VO)5O(OCH3)12FeNO} (4-V5FeNO2−)
A 20 mL scintillation vial was charged with 2-V5FeNO (0.124 g, 0.153 mmol) and 16 mL THF. In a separate vial, cobaltocene (CoCp2, 59 mg, 0.311 mmol) was dissolved in 4 mL THF and added dropwise to the cluster solution while stirring. A dark grey-brown precipitate started to form within 5 minutes. The mixture was stirred vigorously for an additional 2 h. The precipitate was collected and washed with THF until the filtrate was nearly colourless. The grey-brown solid was dried under vacuum to yield complex 4-V5FeNO2− in good yield (0.148 g, 0.125 mmol, 81%). 1H NMR (500 MHz, CD3CN): δ = 135.68 (s, J = 436 Hz), 23.54 (s, J = 142 Hz), 22.29 (s, J = 128 Hz), 5.68 (s, J = 38 Hz). FT-IR (ATR, cm−1): 3078 (C–H), 2909 (C–H), 2803 (C–H), 1683 (NO), 1055 (O–CH3), 935 (VO). UV-Vis [CH3CN; λ, nm (ε, M−1 cm−1)]: 435 (1.78 × 103), 542 (6.61 × 102). Elemental analysis for C32H56NO19V5Fe2 (MW = 1187.20 g mol−1) calcd (%): C, 32.37; H, 4.75; N, 1.18. Found (%): C, 32.47; H, 4.51; N, 1.22.
Synthesis of {(VO)5O(OCH3)12FeNO}ClO4 (5-V5FeNO+)
O), 1260 (ClO), 1003 (O–CH3), 975 (VO). UV-Vis [CH3CN; λ, nm (ε, M−1 cm−1)]: 386 (1.08 × 104), 992 (6.68 × 102). Elemental analysis of complex 5-V5FeNO+ was not obtained because the compound is not sufficiently stable at room temperature.
Chemical reduction of 2-V5FeNO with Na(Nap)
A 20 mL Scintillation vial was charged with 0.028 g naphthalene (0.218 mmol) and 6 mL THF, to which pieces of freshly cut sodium metal (0.073 g, 3.167 mmol) were added. The mixture was stirred at room temperature for 3 hours, affording a dark green solution of sodium naphthanelide. This solution was decanted into a fresh 20 mL Scintillation vial and frozen in an 80 K cold well. In a separate vial, 2-V5FeNO (0.054 g, 0.067 mmol) was dissolved in 2 mL of THF, and the solution was frozen at 80 K. The 2-V5FeNO solution was allowed to thaw and was added to the frozen sodium naphthanelide solution along with an additional 2 mL thawed THF to complete the transfer. The reaction was stirred vigorously and allowed to warm to room temperature for 1 hour. The resulting brown solution was filtered over a bed of celite (0.5 cm), the volatiles were removed, and the brown solid was washed with toluene (5 mL × 2).Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors would like to acknowledge Samuel D. Weinstein for assistance in the synthesis of the FePOV–alkoxide clusters. This research was funded by the National Science Foundation through grants CHE-1653195 (F. L, R. L. M., L. E. V., and E. M. M.) and CHE-1454370 (S.·H. C. and M. L. N.). F. L., R. L. M., L. E. V., and E. M. M. also acknowledge financial support from the University of Rochester for this work. L. E. V. is supported by the National Science Foundation Graduate Research Fellowship Program (DGE-1419118). A. W. N. and C. W. M. thank the University of Virginia for generous funding.References
- J. L. Zweier, H. Li, A. Samouilov and X. Liu, Nitric Oxide, 2010, 22, 83–90 CrossRef PubMed.
- I. M. Wasser, S. de Vries, P. Moënne-Loccoz, I. Schröder and K. D. Karlin, Chem. Rev., 2002, 102, 1201–1234 CrossRef PubMed.
- J. K. S. Møller and L. H. Skibsted, Chem. Rev., 2002, 102, 1167–1178 CrossRef.
- G. B. Richter-Addo and P. Legzdins, Metal Nitrosyls, Oxford University Press, New York, NY, 1992 Search PubMed.
- J. N. Galloway, F. J. Dentener, D. G. Capone, E. W. Boyer, R. W. Howarth, S. P. Seitzinger, G. P. Asner, C. C. Cleveland, P. A. Green, E. A. Holland, D. M. Karl, A. F. Michaels, J. H. Porter, A. R. Townsend and C. J. Vöosmarty, Biogeochem., 2004, 70, 153–226 CrossRef.
- J. D. M. Kurts, Dalton Trans., 2007, 4115–4121 RSC.
- N. J. Watmough, S. J. Field, R. J. L. Hughes and D. J. Richardson, Biochem. Soc. Trans., 2009, 37, 392 CrossRef PubMed.
- N. Sato, S. Ishii, H. Sugimoto, T. Hino, Y. Fukumori, Y. Sako, Y. Shiro and T. Tosha, Proteins: Struct., Funct., Bioinf., 2013, 82, 1258–1271 CrossRef PubMed.
- P. Moenne-Loccoz, Nat. Prod. Rep., 2007, 24, 610–620 RSC.
- A. J. Timmons and M. D. Symes, Chem. Soc. Rev., 2015, 44, 6708–6722 RSC.
- T. C. Berto, A. L. Speelman, S. Zheng and N. Lehnert, Coord. Chem. Rev., 2013, 257, 244–259 CrossRef.
- S. Chakraborty, J. Reed, J. T. Sage, N. C. Branagan, I. D. Petrik, K. D. Miner, M. Y. Hu, J. Zhao, E. E. Alp and Y. Lu, Inorg. Chem., 2015, 54, 9317–9329 CrossRef PubMed.
- R. Silaghi-Dumitrescu, D. M. Kurtz, L. G. Ljungdahl and W. N. Lanzilotta, Biochem., 2005, 44, 6492–6501 CrossRef PubMed.
- K. N. Ferreira, T. M. Iverson, K. Maghlaoui, J. Barber and S. Iwata, Science, 2004, 303, 1831–1838 CrossRef PubMed.
- O. Einsle, F. A. Tezcan, S. L. A. Andrade, B. Schmid, M. Yoshida, J. B. Howard and D. C. Rees, Science, 2002, 297, 1696–1700 CrossRef PubMed.
- D. Schilter, J. M. Camara, M. T. Huynh, S. Hammes-Schiffer and T. B. Rauchfuss, Chem. Rev., 2016, 116, 8693–8749 CrossRef PubMed.
- S. E. McGlynn, D. W. Mulder, E. M. Shepard, J. B. Broderick and J. W. Peters, Dalton Trans., 2009, 4274–4285 RSC.
- H. Dobbek, V. Svetlitchnyi, L. Gremer, R. Huber and O. Meyer, Science, 2001, 293, 1281 CrossRef PubMed.
- H. Beinert, R. H. Holm and E. Münck, Science, 1997, 277, 653 CrossRef PubMed.
- U. Brandt, Annu. Rev. Biochem., 2006, 75, 69–92 CrossRef PubMed.
- Y.-G. Wang, Y. Yoon, V.-A. Glezakou, J. Li and R. Rousseau, J. Am. Chem. Soc., 2013, 135, 10673–10683 CrossRef PubMed.
- J. A. Rodriguez, P. Liu, D. J. Stacchiola, S. D. Senanayake, M. G. White and J. G. Chen, ACS Catal., 2015, 5, 6696–6706 CrossRef.
- A. Ruiz Puigdollers, P. Schlexer, S. Tosoni and G. Pacchioni, ACS Catal., 2017, 7, 6493–6513 CrossRef.
- F. Li, L. E. VanGelder, W. W. Brennessel and E. M. Matson, Inorg. Chem., 2016, 55, 7332–7334 CrossRef PubMed.
- F. Li, S. H. Carpenter, R. F. Higgins, M. G. Hitt, W. W. Brennessel, M. G. Ferrier, S. K. Cary, J. S. Lezama-Pacheco, J. T. Wright, B. W. Stein, M. P. Shores, M. L. Neidig, S. A. Kozimor and E. M. Matson, Inorg. Chem., 2017, 56, 7065–7080 CrossRef PubMed.
- K. Kastner, J. T. Margraf, T. Clark and C. Streb, Chem.–Eur. J., 2014, 20, 12269–12273 CrossRef PubMed.
- T. M. Powers, N. X. Gu, A. R. Fout, A. M. Baldwin, R. Hernández Sánchez, D. M. Alfonso, Y.-S. Chen, S.-L. Zheng and T. A. Betley, J. Am. Chem. Soc., 2013, 135, 14448–14458 CrossRef PubMed.
- E. V. Eames, R. Hernández Sánchez and T. A. Betley, Inorg. Chem., 2013, 52, 5006–5012 CrossRef PubMed.
- J. S. Kanady, E. Y. Tsui, M. W. Day and T. Agapie, Science, 2011, 333, 733 CrossRef PubMed.
- E. Y. Tsui, R. Tran, J. Yano and T. Agapie, Nat. Chem., 2013, 5, 293–299 CrossRef PubMed.
- D. E. Herbert, D. Lionetti, J. Rittle and T. Agapie, J. Am. Chem. Soc., 2013, 135, 19075–19078 CrossRef PubMed.
- A. L. Speelman, B. Zhang, C. Krebs and N. Lehnert, Angew. Chem., Int. Ed., 2016, 55, 6685–6688 CrossRef PubMed.
- T. W. Hayton, P. Legzdins and W. B. Sharp, Chem. Rev., 2002, 102, 935–992 CrossRef PubMed.
- R. H. Pierson, A. N. Fletcher and E. S. C. Gantz, Anal. Chem., 1956, 28, 1218–1239 CrossRef.
- V. Spagnolo, A. A. Kosterev, L. Dong, R. Lewicki and F. K. Tittel, Appl. Phys. B, 2010, 100, 125–130 CrossRef.
- A. L. Speelman and N. Lehnert, Angew. Chem., Int. Ed., 2013, 52, 12283–12287 CrossRef PubMed.
- R. G. Serres, C. A. Grapperhaus, E. Bothe, E. Bill, T. Weyhermüller, F. Neese and K. Wieghardt, J. Am. Chem. Soc., 2004, 126, 5138–5153 CrossRef PubMed.
- C. Hauser, T. Glaser, E. Bill, T. Weyhermüller and K. Wieghardt, J. Am. Chem. Soc., 2000, 122, 4352–4365 CrossRef.
- E. Victor and S. J. Lippard, Inorg. Chem., 2014, 53, 5311–5320 CrossRef PubMed.
- T. C. Harrop, Z. J. Tonzetich, E. Reisner and S. J. Lippard, J. Am. Chem. Soc., 2008, 130, 15602–15610 CrossRef PubMed.
- C. Daniel and H. Hartl, J. Am. Chem. Soc., 2009, 131, 5101–5114 CrossRef PubMed.
- C. Daniel and H. Hartl, J. Am. Chem. Soc., 2005, 127, 13978–13987 CrossRef PubMed.
- W. R. Scheidt and M. K. Ellison, Acc. Chem. Res., 1999, 32, 350–359 CrossRef.
- C. E. Cooper, Biochim. Biophys. Acta, 1999, 1411, 290–309 CrossRef.
- G. C. Salomão, M. H. N. Olsen, V. Drago, C. Fernandes, L. Cardozo Filho and O. A. C. Antunes, Catal. Commun., 2007, 8, 69–72 CrossRef.
- J. Spandl, C. Daniel, I. Brüdgam and H. Hartl, Angew. Chem., Int. Ed., 2003, 42, 1163–1166 CrossRef PubMed.
- R. Hernández Sánchez, S.-L. Zheng and T. A. Betley, J. Am. Chem. Soc., 2015, 137, 11126–11143 CrossRef PubMed.
- R. R. Gagne and C. L. Spiro, J. Am. Chem. Soc., 1980, 102, 1443–1444 CrossRef.
- B. Hu and J. Li, Angew. Chem., Int. Ed., 2015, 54, 10579–10582 CrossRef PubMed.
- J. A. McCleverty, N. M. Atherton, J. Locke, E. J. Wharton and C. J. Winscom, J. Am. Chem. Soc., 1967, 89, 6082–6092 CrossRef.
- P. Ghosh, K. Stobie, E. Bill, E. Bothe, T. Weyhermüller, M. D. Ward, J. A. McCleverty and K. Wieghardt, Inorg. Chem., 2007, 46, 522–532 CrossRef PubMed.
- M. B. Robin and P. Day, in Adv. Inorg. Chem. and Radiochem., ed. H. J. Emeléus and A. G. Sharpe, Academic Press, 1968, vol. 10, pp. 247–422 Search PubMed.
- J. H. Enemark and R. D. Feltham, Coord. Chem. Rev., 1974, 13, 339–406 CrossRef.
- M. A. Augustyniak-Jabłokow, C. Daniel, H. Hartl, J. Spandl and Y. V. Yablokov, Inorg. Chem., 2008, 47, 322–332 CrossRef PubMed.
- C. H. Arnett, M. J. Chalkley and T. Agapie, J. Am. Chem. Soc., 2018, 140, 5569–5578 CrossRef PubMed.
- G. de Ruiter, N. B. Thompson, D. Lionetti and T. Agapie, J. Am. Chem. Soc., 2015, 137, 14094–14106 CrossRef PubMed.
- C. J. Reed and T. Agapie, Inorg. Chem., 2017, 56, 13360–13367 CrossRef PubMed.
- J. E. Toth and F. C. Anson, J. Am. Chem. Soc., 1989, 111, 2444–2451 CrossRef.
- H. D. Pratt, W. R. Pratt, X. Fang, N. S. Hudak and T. M. Anderson, Electrochim. Acta, 2014, 138, 210–214 CrossRef.
- T. C. Berto, M. B. Hoffman, Y. Murata, K. B. Landenberger, E. E. Alp, J. Zhao and N. Lehnert, J. Am. Chem. Soc., 2011, 133, 16714–16717 CrossRef PubMed.
- C. Kupper, J. A. Rees, S. Dechert, S. DeBeer and F. Meyer, J. Am. Chem. Soc., 2016, 138, 7888–7898 CrossRef PubMed.
- A. M. Confer, A. C. McQuilken, H. Matsumura, P. Moënne-Loccoz and D. P. Goldberg, J. Am. Chem. Soc., 2017, 139, 10621–10624 CrossRef PubMed.
- A. C. Montenegro, V. T. Amorebieta, L. D. Slep, D. F. Martín, F. Roncaroli, D. H. Murgida, S. E. Bari and J. A. Olabe, Angew. Chem., Int. Ed., 2009, 48, 4213–4216 CrossRef PubMed.
- K. Ashley and S. Pons, Chem. Rev., 1988, 88, 673–695 CrossRef.
- W. Kaim and J. Fiedler, Chem. Soc. Rev., 2009, 38, 3373–3382 RSC.
- A. L. Speelman, B. Zhang, A. Silakov, K. M. Skodje, E. E. Alp, J. Zhao, M. Y. Hu, E. Kim, C. Krebs and N. Lehnert, Inorg. Chem., 2016, 55, 5485–5501 CrossRef PubMed.
- I. S. Zavarine and C. P. Kubiak, J. Electroanal. Chem., 2001, 495, 106–109 CrossRef.
- C. W. Machan, M. D. Sampson, S. A. Chabolla, T. Dang and C. P. Kubiak, Organometallics, 2014, 33, 4550–4559 CrossRef.
- A. W. Nichols, S. Chatterjee, M. Sabat and C. W. Machan, Inorg. Chem., 2018, 57, 2111–2121 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: 1H NMR and 57Fe Mössbauer spectra of 2-V5FeNO, 3-V5FeNO−, 4-V5FeNO2−, 5-V5FeNO+. Spectroscopic characterization for complex 6-V5FeNO3− (1H NMR, solution IR). See DOI: 10.1039/c8sc00987b |
| This journal is © The Royal Society of Chemistry 2018 |