
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA porous, electrically conductive hexa-zirconium(IV) metal–organic framework†
Subhadip
Goswami
 a,
Debmalya
Ray
b,
Ken-ichi
Otake
a,
Chung-Wei
Kung
a,
Sergio J.
Garibay
a,
Timur
Islamoglu
a,
Ahmet
Atilgan
a,
Yuexing
Cui
a,
Christopher J.
Cramer
b,
Omar K.
Farha
ac and
Joseph T.
Hupp
*a
a,
Debmalya
Ray
b,
Ken-ichi
Otake
a,
Chung-Wei
Kung
a,
Sergio J.
Garibay
a,
Timur
Islamoglu
a,
Ahmet
Atilgan
a,
Yuexing
Cui
a,
Christopher J.
Cramer
b,
Omar K.
Farha
ac and
Joseph T.
Hupp
*a
aDepartment of Chemistry, Northwestern University, 2145 Sheridan Road, Evanston, IL 60208, USA. E-mail: j-hupp@northwestern.edu
bDepartment of Chemistry, Chemical Theory Center, Minnesota Supercomputing Institute, University of Minnesota, 207 Pleasant Street SE, Minneapolis, MN 55455, USA
cDepartment of Chemistry, King Abdulaziz University, Jeddah 21589, Saudi Arabia
First published on 11th April 2018
Abstract
Engendering electrical conductivity in high-porosity metal–organic frameworks (MOFs) promises to unlock the full potential of MOFs for electrical energy storage, electrocatalysis, or integration of MOFs with conventional electronic materials. Here we report that a porous zirconium-node-containing MOF, NU-901, can be rendered electronically conductive by physically encapsulating C60, an excellent electron acceptor, within a fraction (ca. 60%) of the diamond-shaped cavities of the MOF. The cavities are defined by node-connected tetra-phenyl-carboxylated pyrene linkers, i.e. species that are excellent electron donors. The bulk electrical conductivity of the MOF is shown to increase from immeasurably low to 10−3 S cm−1, following fullerene incorporation. The observed conductivity originates from electron donor–acceptor interactions, i.e. charge-transfer interactions – a conclusion that is supported by density functional theory calculations and by the observation of a charge-transfer-derived band in the electronic absorption spectrum of the hybrid material. Notably, the conductive version of the MOF retains substantial nanoscale porosity and continues to display a sizable internal surface area, suggesting potential future applications that capitalize on the ability of the material to sorb molecular species.
Introduction
Metal–organic frameworks (MOFs) are a large and growing class of porous, or potentially porous, materials. They are characterized by well-ordered (i.e., crystalline) combinations of multitopic organic ligands connected by coordination bonds to one or more metal ions, or metal-ion-containing clusters.1–4 The enormous potential and experimentally realized compositional variety of MOFs,5–8 together with their typically large internal surface areas and their molecular-scale voids, has inspired exploration of myriad candidate applications including, but not limited to, gas storage and release,9,10 chemical separations,11,12 chemical sensing,13 catalysis,14–17 energy transfer,18–20 drug delivery,21 and solar fuel production.22–24 Electrically conductive MOFs, while few in absolute number, constitute an intriguing and increasingly important sub-class of materials, especially in view of the insulating character (conductivity <10−10 S cm−1) of most MOFs. The insulating versions typically make use of redox-resistant d(0) or d(10) metal ions that are energetically poorly matched or otherwise ineffective at interacting with linker-based π–π* systems. Thus, nodes may behave as local insulators between otherwise potentially conductive arrays of linkers (and/or vice versa).Nevertheless, the demonstrated feasibility of deploying electrically conductive MOFs in field-effect transistors (FETs),25 chemiresistive sensors,26 electrochromic devices,27,28 supercapacitors29,30 and batteries,31 has motivated the search for suitably conductive MOFs. The common design rule is to provide a low energy pathway for charge transport, and reported strategies include: (a) π stacking of conjugated organic ligands,32 (b) formulation of MOFs as laterally conjugated 2D coordination compounds,33 (c) templating the growth of conductive polymers within suitably large MOF channels,34,35 (d) intentionally matching energies of frontier orbitals of metal-ion nodes and organic linkers,36 (e) post-synthetic modification or augmentation of MOF nodes, either with metals featuring partially filled d-shells32 or with redox couples such as ferrocenium/ferrocene,37 to yield redox-hopping-type conductivity, and (f) electrochemically oxidizing or reducing the linkers of solution-immersed MOFs, again to enable redox-hopping-type conduction.38,39
The availability of free space within MOFs suggests another route to eliciting electrical conductivity, namely, the incorporation of guest molecules (electron acceptors or donors) that are complementary, in a charge-transfer sense, to MOF nodes or linkers.40,41 The approach has clear parallels, for example, to the formation of conductive, organic charge-transfer salts by combining (co-crystallizing) a strong electron donor (D), such as tetrathiafulvalene (TTF), with a strong electron acceptor (A) such as tetracyanoquinodimethane (TCNQ). Generally, in order to effectively transport charges, long-range ordering of donor and acceptor components is necessary. MOFs, with their inherent crystallinity and their ability to encapsulate guest molecules, offer platforms for achieving the desired long-range ordering of electronically complementary species. In a groundbreaking study, Talin et al. demonstrated that infiltration of the microporous MOF Cu3(btc)2 (also known as HKUST-1; btc = 1,3,5-benzenetricarboxylate) with TCNQ boosts the MOF's electrical conductivity by a remarkable six orders of magnitude (i.e., from 10−8 S cm−1 to 0.07 S cm−1).42 The basis for increase is the ligation of open Cu(II) sites by TCNQ nitrile groups such that a continuous charge-transfer pathway is formed (each TCNQ is thought to connect to four copper ions, with the overall Cu(II)trimesate unit functioning as an electron donor, and TCNQ as an acceptor).43 Among other D–A pairs incorporated within MOFs to study charge transfer are: (a) TTF/naphthalenetetracarboxydiimide (NDI),44,45 (b) oligothiophene/[6,6]-phenyl-C61-butyric acid methyl ester (PCBM),44,46 and (c) N,N′-bis(4-pyridyl)-2,6-dipyrrolidyl napthalenediimide/methylviologen.47 However, only in case (c) has bulk electrical conductivity been investigated (with a 35-fold increase in conductivity accompanying viologen incorporation).
Among the challenges in using guest infiltrated MOFs for device applications are: (a) limited stability of the MOF, as most of the MOFs evaluated to date feature comparatively weakly ligating nodes containing Zn(II), Cu(II) or Ni(II), and (b) substantial loss of porosity upon guest inclusion. For example, after TCNQ infiltration, the BET surface area of HKUST-1 decreases from 1844 ± 4 m2 g−1 to 214 ± 0.5 m2 g−1. Hence it is of particular interest to design stable and porous electrically conductive MOFs. Stability can be defined in many ways. For oxidative heterogeneous catalysis of gas phase reactions, thermal stability is typically paramount, while for solution-phase electrocatalysis processes such as water splitting, aqueous stability may be paramount.
These stability considerations are well satisfied by MOFs featuring Zr(IV)-oxygen(anion) coordination as the basis for node and linker connectivity.48–51 With this in mind, we settled on a high-surface-area MOF, NU-901,28,52 as a guest-accessible, electron-donating scaffold, and the iconic electron acceptor, buckminsterfullerene (C60) as the complementary component for studies of MOF electrical conductivity. Notably, there are previous reports on C60 incorporation within the MOF channels, the earliest being a report from Chae et al.53 To our knowledge, however, this is the first report of host–guest interaction to induce electrical conductivity in an otherwise insulating zirconium based MOF.
NU-901 consists of Zr6(μ3-O)4(μ3-OH)4(H2O)4(OH)4 nodes and tetratopic 1,3,6,8-tetrakis(p-benzoate)pyrene (TBAPy4−) linkers. We hypothesized that NU-901 would be an attractive candidate for encapsulation of C60 and electronic activation due to: (a) the availability of diamond-shaped channels that can withstand removal of synthesis solvent and that are reasonably well size-matched to C60 (van der waals diameter ∼7 Å), (b) the modest onset potential for electrochemical oxidation of NU-901,28 which suggests that its linkers may be strong enough donors to form charge-transfer complexes with easily reducible fullerenes, and (c) the arrangement of the diamond-shaped voids, as defined by the scu topology of the MOF, which should allow for a spatially continuous network of charge-transfer type interactions (and, therefore, electrical conductivity) in the a-b plane.
Results and discussion
NU-901 was synthesized by a slightly modified literature procedure.52 Infiltration with C60 was performed by soaking a microcrystalline powder sample of activated (i.e., solvent-evacuated and fully porous) NU-901 in a saturated o-dichlorobenzene solution of C60 for four days at 60 °C. The powder X-ray diffraction (PXRD) pattern of NU-901-C60 matches well with the experimentally obtained and computationally simulated patterns for pure NU-901, implying that guest uptake does not alter the material's crystallinity (Fig. 2a). Preservation of microcrystal morphology was confirmed by comparing scanning electron microscopy images of NU-901 and NU-901-C60 (Fig. S1†). The presence of fullerene in NU-901-C60 was probed by Raman spectroscopy, based on 785 nm excitation (Fig. 2d). The scattering spectrum of NU-901-C60 displays peaks attributable to both the MOF and the fullerene, including the Ag(1) vibration of C60 at 492 cm−1. Consistent with interactions with the fullerene, the vibrational peaks for NU-901 are slightly shifted in NU-901-C60.The porosity of both NU-901 and NU-901-C60 was confirmed by nitrogen adsorption–desorption measurements (Fig. 2b). Both display approximately type I isotherms, albeit with slight hysteresis presumably arising from inter-crystallite macroporosity. Brunauer–Emmett–Teller (BET) analyses of the isotherms return gravimetric surface areas of 2120 m2 g−1 for guest-free NU-901 and 1550 m2 g−1 for NU-901-C60. DFT pore-size distribution analyses of the isotherms reveal a large peak centered at 12 Å, together with a small peak at 27 Å. The former is consistent with the crystallographically observed diamond-shaped pores of NU-901. The latter we speculatively attribute to pores associated with defects.
After installation of C60, the incremental pore volume corresponding to the diamond shaped channels decreased from 0.54 cm3 g−1 to 0.38 cm3 g−1, whereas the pore volume remains unchanged for pores generated by defect sites, suggesting fullerenes primarily reside within the diamond channels of the MOF (Fig. 2c). From the decrease in gravimetric pore volume, we estimate that the fullerene incorporation to be ∼0.6 per node (∼0.6 per linker-defined diamond void). Notably, DFT calculations (geometry optimized) show siting in diamond pores is overwhelmingly energetically favored (by 0.8 eV) compared to siting in the smaller pores defined by pairs of nodes along the c axis (Fig. S8b†). The representations in the bottom of Fig. 1 are derived from the DFT geometry.54
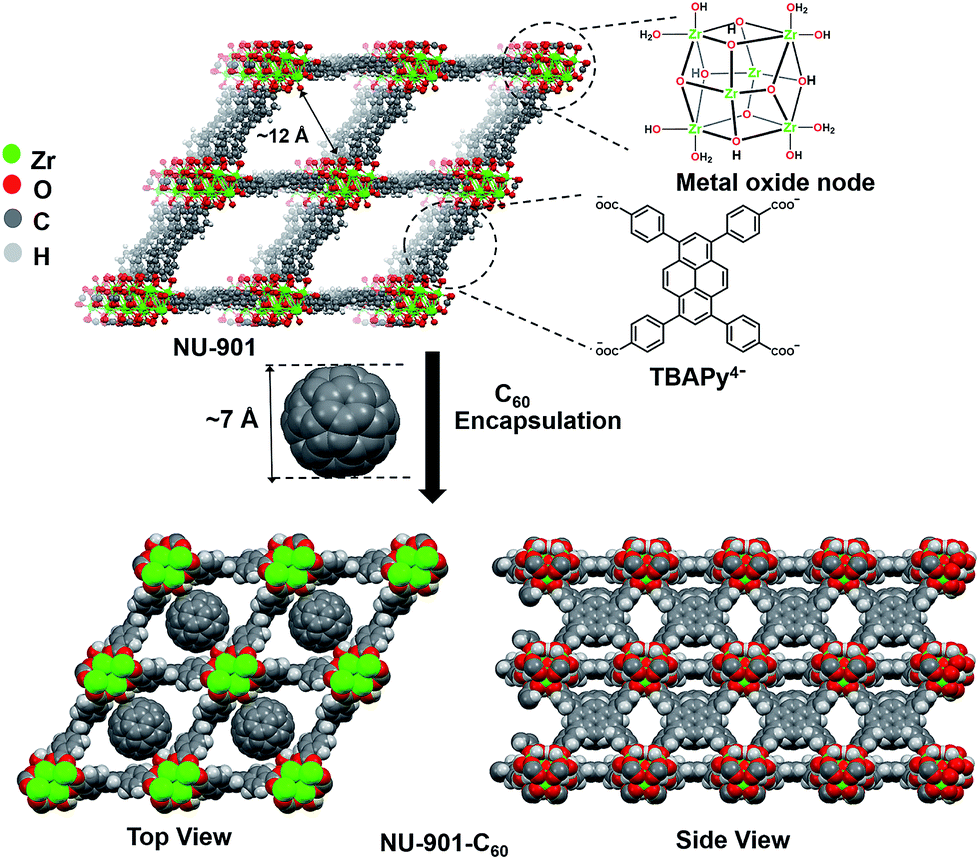 | ||
Fig. 1 Immobilization of C60 within diamond shaped channels of NU-901. The chemical structures of the individual components of NU-901 are shown. The diameter of C60 (∼7 Å) is well suited for encapsulation in NU-901 with pore aperture of 12 Å. The top view and side view of the composite NU-901-C60 are shown. The top and side views are DFT-optimized structures in the limit of full occupancy of the diamond pores by the fullerene guest, i.e. 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 occupancy. :1 occupancy. | ||
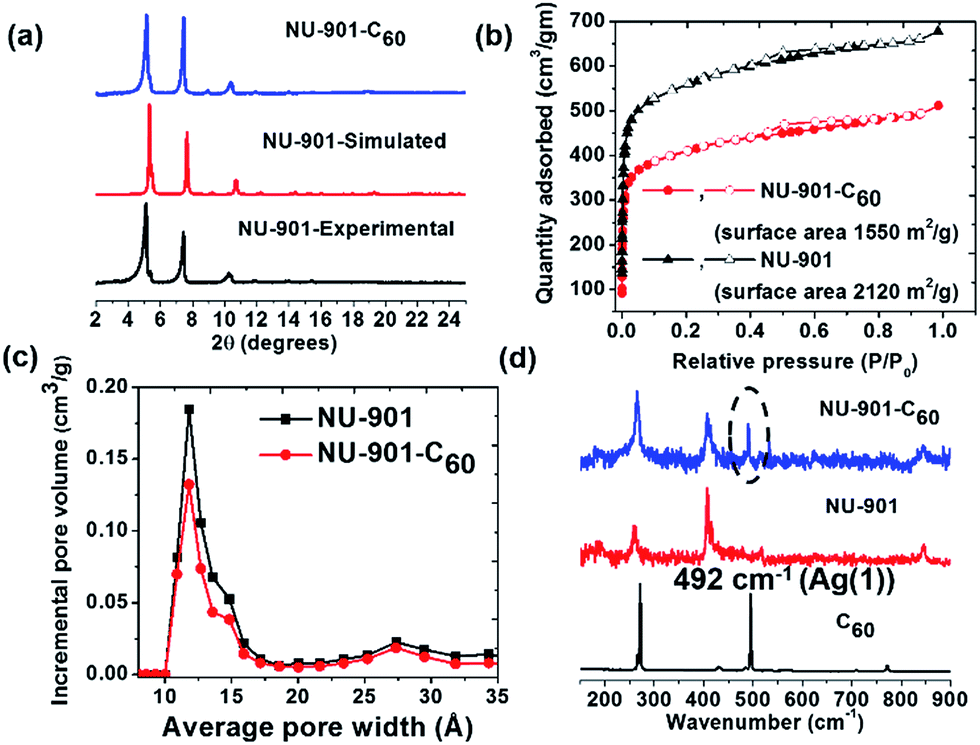 | ||
| Fig. 2 (a) PXRD pattern of simulated NU-901, experimental NU-901, and experimental NU-901-C60, (b) N2 adsorption–desorption isotherms (77 K) (NU-901 and NU-901-C60), (c) density functional theory (DFT) pore size distribution (NU-901-C60 and NU-901) and, (d) Raman spectra (NU-901, NU-901-C60 and C60). | ||
C60 encapsulation by NU-901 is accompanied by a change in color of the MOF from bright yellow to light brown. The visible color change is observable in corresponding diffuse reflectance UV-visible absorption spectra (Fig. S2a†). Guest-free NU-901 shows a broad absorption band beginning at 490 nm (2.53 eV) and maximizing in the UV, with no absorption beyond 490 nm. The spectrum of NU-901-C60 likewise displays the UV-blue band, but also presents a broad band extending from 500 nm to 700 nm (∼1.77 eV). We attribute the new band to a donor/acceptor (pyrene linker/C60) charge-transfer transition.55 Consistent with the introduction of a strong electron acceptor,56,57 the encapsulated fullerene quenches the steady-state luminescence of the tetra-phenyl-pyrene linker by ca. 90%; see Fig. S2b.†
Qualitative assessments of electrical conductivity were made by first dropcasting MOF suspensions on interdigitated array electrodes (IDEs; 5 μm gap) and then recording the current (I) transmitted between the electrodes, via electrode-gap-spanning MOF crystallites, in response to applied biases (V; two-point measurements). NU-901-C60 samples yielded measurable currents whereas guest-free crystallites of NU-901 did not; see Fig. S7.† Unfortunately, we were unable with this approach to quantify the conductivity, σ, of NU-901-C60. We therefore turned to macroscopic, two-point, I–V measurements using pressed pellets. The stability of the MOF pellets before and after C60 encapsulation was verified by PXRD and nitrogen adsorption isotherms (Fig. S3 and S4†). Consistent with IDE measurements, NU-901 alone displayed no measurable conductivity (where the lower limit of detection is estimated, from the magnitude of electrode noise, to be about 10−14 S cm−1). In contrast (Fig. 3), NU-901-C60 shows remarkable electrical conductivity of σ ∼ 10−3 S cm−1,58i.e. eleven orders of magnitude (or more) greater than NU-901 alone, and similar to that of organic semiconductors (σ > 10−6 S cm−1).59 Unlike conventional organic semiconductors, however, the MOF/fullerene material is characterized by substantial internal surface area and considerable molecular-scale porosity.
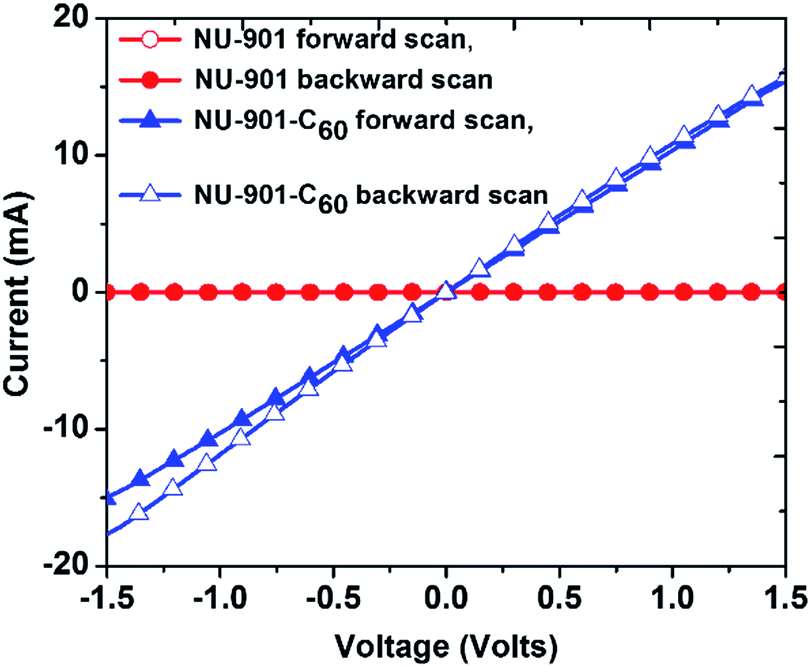 | ||
| Fig. 3 Current (I) vs. voltage (V) plot for pressed pellets of NU-901 and NU-901-C60. The scan was performed from −2 V to 2 V and the scan rate was 50 mV s−1. | ||
We reasoned that if TBAPy4−/C60 donor/acceptor interactions are responsible for the observed electrical conductivity, increasing the strength of the MOF linker as an electron donor would increase the conductivity. We therefore replaced TBAPy4− with a tetra-amino (phenyl-substituted) analogue.60 PXRD (Fig. S6†) and SEM (Fig. S1†) assessments of NU-901-amino-C60 indicated formation of a material that is structurally similar (Fig. S5†) to NU-901-C60 (see (Fig. S5†)). N2 adsorption and desorption measurements established the permanent porosity of the material, and yielded BET surface area of 980 m2 g−1 for NU-901-amino and 370 m2 g−1 for NU-901-amino-C60 (Fig. S6†). IDE based measurements show that C60 incorporation imparts electrical conductivity to NU-901-amino, with the observed currents being larger for NU-901-amino-C60 than for NU-901-C60;61 see Fig. S7.†
Next we turn our attention to density functional theory (the details of the methods used are in the ESI†) to gain further insight into the nature of the observed electron-donor/electron-acceptor interactions and their manifestation as electrical conductivity in the MOF/fullerene assemblies. The results of electronic density of states (DOS) calculations for NU-901, NU-901-amino, NU-901-C60 and NU-901-amino-C60 are summarized in Fig. S9.† Briefly, for the guest-free MOFs, both the valence band maximum (VBM) and conduction band minimum (CBM) are dominated by contributions from linker p orbitals (i.e., those that contribute to the linkers' aromatic systems). For the C60-containing materials, the VBM remains predominantly linker defined, but the CBM consists of fullerene p orbitals, consistent with donor/acceptor complex formation. The calculated CBM – VBM difference, i.e. the nominal bandgap, decreases by 1.2 eV with incorporation of C60, a result that is qualitatively replicated in the visible-region reflectance spectra for fullerene-containing versus fullerene-free versions of the MOF (note that DFT methods often underestimate absolute magnitudes of bandgaps, particularly when local functionals like PBE are used).62,63
Given the spatial and energetic proximity of the putative donors and acceptors and their associated bands, we also calculated charge-transfer integrals for relevant clusters cut from the NU-901-C60 and NU-901-amino-C60 periodic structures (Table S1†). Both the electron-transfer and hole-transfer integrals are larger (by 2- and 4-fold, respectively) for NU-901-amino-C60 compared to NU-901-C60, consistent with larger IDE currents for the amino version.30
Conclusions
In summary, we find that the zirconium-based MOF, NU-901, can be rendered electronically conductive by installing isolated C60 molecules in roughly half the linker-defined, diamond shaped pores presented by the MOF. Indeed, the experimentally evaluated conductivity increases from immeasurably small (<10−14 S cm−1) to ∼10−3 S cm−1. The basis for the conductivity is in electron-donor (pyrene)/electron-acceptor (fullerene) interactions, as evidenced, in part, by the appearance of a visible-region charge-transfer band and by the results of DFT band structure calculations. Notably, the MOF internal surface area (gravimetric surface area) decreases by only about 27% with fullerene incorporation while the N2-accessible void volume (as expressed gravimetrically) decreases by only about a third. DFT computes that appending NH2 substituents (strongly electron-donating groups) to the linker phenyl groups of NU-901 increases hole- and electron-transfer integrals, which is consistent with the increased conductivity measured experimentally. These findings point to the potential for systematic chemical tunability of the observed conductivity.Finally, while electrocatalysis applications are beyond the scope of this initial report, it is worth considering how much conductivity is needed for a MOF to function effectively in that context. A common motif for electrochemical applications of MOFs is a thin film (a fraction of a micron to several microns thick) sited on a conventionally conductive platform – for example, a planar glassy carbon or platinum electrode. A benchmark catalytic current density for solar-driven electrochemical processes is 10 mA cm−2. For a hypothetical 10 micron thick MOF film having the modest conductivity observed here, i.e., ∼10−3 S cm−1, and operating at 10 mA cm−2, the energetic penalty (i-R drop) due to film resistivity would be just 10 mV – a value barely within the reproducibility of kinetic overpotentials for many electrocatalytic systems. We suggest that for these types of applications, conductivities in the range of 10−3 S cm−1, despite falling in the semiconducting regime (phenomenologically),64 will typically be sufficient.
Author contributions
J. T. H., O. K. F. and S. G. conceived the project, design the experiments and wrote the manuscript. S. G. performed the synthesis and experiments. D. R. and C. J. C. contributed to the theoretical studies. K. O. and C. W. C. assist in performing Raman spectroscopy and IDE measurements respectively. S. J. G., T. L. and A. A. contributed in amino linker synthesis. Y. C. assist in performing diffuse reflectance UV measurements.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
J. T. H. and C. J. C. gratefully acknowledge support from the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, Division of Chemical Sciences, Geosciences and Biosciences (grant no. DE-FG02-17ER16362). SEM images were obtained by using the EPIC facility (NUANCE Center, Northwestern University), which has received support from the MRSEC program (NSF DMR-1121262) at the Materials Research Center, the International Institute for Nanotechnology (IIN), and the State of Illinois, through the IIN.Notes and references
- H. Furukawa, K. E. Cordova, M. O'Keeffe and O. M. Yaghi, Science, 2013, 341, 1230444 CrossRef PubMed.
- G. Ferey, Chem. Soc. Rev., 2008, 37, 191–214 RSC.
- O. K. Farha and J. T. Hupp, Acc. Chem. Res., 2010, 43, 1166–1175 CrossRef CAS PubMed.
- S. Horike, S. Shimomura and S. Kitagawa, Nat. Chem., 2009, 1, 695 CrossRef CAS PubMed.
- Y. G. Chung, J. Camp, M. Haranczyk, B. J. Sikora, W. Bury, V. Krungleviciute, T. Yildirim, O. K. Farha, D. S. Sholl and R. Q. Snurr, Chem. Mater., 2014, 26, 6185–6192 CrossRef CAS.
- C. E. Wilmer, M. Leaf, C. Y. Lee, O. K. Farha, B. G. Hauser, J. T. Hupp and R. Q. Snurr, Nat. Chem., 2011, 4, 83 CrossRef PubMed.
- P. G. Boyd, Y. Lee and B. Smit, Nat. Rev. Mater., 2017, 2, 17037 CrossRef CAS.
- D. Nazarian, P. Ganesh and D. S. Sholl, J. Mater. Chem. A, 2015, 3, 22432–22440 CAS.
- M. Dincă and J. R. Long, Angew. Chem., Int. Ed., 2008, 47, 6766–6779 CrossRef PubMed.
- O. K. Farha, A. Özgür Yazaydın, I. Eryazici, C. D. Malliakas, B. G. Hauser, M. G. Kanatzidis, S. T. Nguyen, R. Q. Snurr and J. T. Hupp, Nat. Chem., 2010, 2, 944 CrossRef CAS PubMed.
- J.-R. Li, R. J. Kuppler and H.-C. Zhou, Chem. Soc. Rev., 2009, 38, 1477–1504 RSC.
- Z. Bao, G. Chang, H. Xing, R. Krishna, Q. Ren and B. Chen, Energy Environ. Sci., 2016, 9, 3612–3641 CAS.
- L. E. Kreno, K. Leong, O. K. Farha, M. Allendorf, R. P. Van Duyne and J. T. Hupp, Chem. Rev., 2012, 112, 1105–1125 CrossRef CAS PubMed.
- T. Islamoglu, S. Goswami, Z. Li, A. J. Howarth, O. K. Farha and J. T. Hupp, Acc. Chem. Res., 2017, 50, 805–813 CrossRef CAS PubMed.
- V. Bernales, M. A. Ortuño, D. G. Truhlar, C. J. Cramer and L. Gagliardi, ACS Cent. Sci., 2018, 4, 5–19 CrossRef CAS PubMed.
- J. Lee, O. K. Farha, J. Roberts, K. A. Scheidt, S. T. Nguyen and J. T. Hupp, Chem. Soc. Rev., 2009, 38, 1450–1459 RSC.
- J. Gascon, A. Corma, F. Kapteijn and F. X. Llabrés i Xamena, ACS Catal., 2014, 4, 361–378 CrossRef CAS.
- C. A. Kent, B. P. Mehl, L. Ma, J. M. Papanikolas, T. J. Meyer and W. Lin, J. Am. Chem. Soc., 2010, 132, 12767–12769 CrossRef CAS PubMed.
- W. A. Maza, R. Padilla and A. J. Morris, J. Am. Chem. Soc., 2015, 137, 8161–8168 CrossRef CAS PubMed.
- S. Goswami, L. Ma, A. B. F. Martinson, M. R. Wasielewski, O. K. Farha and J. T. Hupp, ACS Appl. Mater. Interfaces, 2016, 8, 30863–30870 CAS.
- P. Horcajada, T. Chalati, C. Serre, B. Gillet, C. Sebrie, T. Baati, J. F. Eubank, D. Heurtaux, P. Clayette, C. Kreuz, J.-S. Chang, Y. K. Hwang, V. Marsaud, P.-N. Bories, L. Cynober, S. Gil, G. Férey, P. Couvreur and R. Gref, Nat. Mater., 2009, 9, 172 CrossRef PubMed.
- J.-L. Wang, C. Wang and W. Lin, ACS Catal., 2012, 2, 2630–2640 CrossRef CAS.
- I. Hod, M. D. Sampson, P. Deria, C. P. Kubiak, O. K. Farha and J. T. Hupp, ACS Catal., 2015, 5, 6302–6309 CrossRef CAS.
- N. Kornienko, Y. Zhao, C. S. Kley, C. Zhu, D. Kim, S. Lin, C. J. Chang, O. M. Yaghi and P. Yang, J. Am. Chem. Soc., 2015, 137, 14129–14135 CrossRef CAS PubMed.
- G. Wu, J. Huang, Y. Zang, J. He and G. Xu, J. Am. Chem. Soc., 2017, 139, 1360–1363 CrossRef CAS PubMed.
- M. G. Campbell, S. F. Liu, T. M. Swager and M. Dincă, J. Am. Chem. Soc., 2015, 137, 13780–13783 CrossRef CAS PubMed.
- K. AlKaabi, C. R. Wade and M. Dincă, Chem, 2016, 1, 264–272 CAS.
- C.-W. Kung, T. C. Wang, J. E. Mondloch, D. Fairen-Jimenez, D. M. Gardner, W. Bury, J. M. Klingsporn, J. C. Barnes, R. Van Duyne, J. F. Stoddart, M. R. Wasielewski, O. K. Farha and J. T. Hupp, Chem. Mater., 2013, 25, 5012–5017 CrossRef CAS.
- D. Sheberla, J. C. Bachman, J. S. Elias, C.-J. Sun, Y. Shao-Horn and M. Dincă, Nat. Mater., 2016, 16, 220 CrossRef PubMed.
- K. M. Choi, H. M. Jeong, J. H. Park, Y.-B. Zhang, J. K. Kang and O. M. Yaghi, ACS Nano, 2014, 8, 7451–7457 CrossRef CAS PubMed.
- D. Wu, Z. Guo, X. Yin, Q. Pang, B. Tu, L. Zhang, Y.-G. Wang and Q. Li, Adv. Mater., 2014, 26, 3258–3262 CrossRef CAS PubMed.
- S. S. Park, E. R. Hontz, L. Sun, C. H. Hendon, A. Walsh, T. Van Voorhis and M. Dincă, J. Am. Chem. Soc., 2015, 137, 1774–1777 CrossRef CAS PubMed.
- D. Sheberla, L. Sun, M. A. Blood-Forsythe, S. Er, C. R. Wade, C. K. Brozek, A. Aspuru-Guzik and M. Dincă, J. Am. Chem. Soc., 2014, 136, 8859–8862 CrossRef CAS PubMed.
- T. C. Wang, I. Hod, C. O. Audu, N. A. Vermeulen, S. T. Nguyen, O. K. Farha and J. T. Hupp, ACS Appl. Mater. Interfaces, 2017, 9, 12584–12591 CAS.
- B. Le Ouay, M. Boudot, T. Kitao, T. Yanagida, S. Kitagawa and T. Uemura, J. Am. Chem. Soc., 2016, 138, 10088–10091 CrossRef CAS PubMed.
- L. Sun, T. Miyakai, S. Seki and M. Dincă, J. Am. Chem. Soc., 2013, 135, 8185–8188 CrossRef CAS PubMed.
- I. Hod, W. Bury, D. M. Gardner, P. Deria, V. Roznyatovskiy, M. R. Wasielewski, O. K. Farha and J. T. Hupp, J. Phys. Chem. Lett., 2015, 6, 586–591 CrossRef CAS PubMed.
- S. R. Ahrenholtz, C. C. Epley and A. J. Morris, J. Am. Chem. Soc., 2014, 136, 2464–2472 CrossRef CAS PubMed.
- D. M. D'Alessandro, Chem. Commun., 2016, 52, 8957–8971 RSC.
- M. D. Allendorf, M. E. Foster, F. Léonard, V. Stavila, P. L. Feng, F. P. Doty, K. Leong, E. Y. Ma, S. R. Johnston and A. A. Talin, J. Phys. Chem. Lett., 2015, 6, 1182–1195 CrossRef CAS PubMed.
- J. Liu, T. Wächter, A. Irmler, P. G. Weidler, H. Gliemann, F. Pauly, V. Mugnaini, M. Zharnikov and C. Wöll, ACS Appl. Mater. Interfaces, 2015, 7, 9824–9830 CAS.
- A. A. Talin, A. Centrone, A. C. Ford, M. E. Foster, V. Stavila, P. Haney, R. A. Kinney, V. Szalai, F. El Gabaly, H. P. Yoon, F. Léonard and M. D. Allendorf, Science, 2014, 343, 66–69 CrossRef CAS PubMed.
- X. Nie, A. Kulkarni and D. S. Sholl, J. Phys. Chem. Lett., 2015, 6, 1586–1591 CrossRef CAS PubMed.
- C. F. Leong, B. Chan, T. B. Faust and D. M. D'Alessandro, Chem. Sci., 2014, 5, 4724–4728 RSC.
- Z. Guo, D. K. Panda, M. A. Gordillo, A. Khatun, H. Wu, W. Zhou and S. Saha, ACS Appl. Mater. Interfaces, 2017, 9, 32413–32417 CAS.
- K. Leong, M. E. Foster, B. M. Wong, E. D. Spoerke, D. Van Gough, J. C. Deaton and M. D. Allendorf, J. Mater. Chem. A, 2014, 2, 3389–3398 CAS.
- Z. Guo, D. K. Panda, K. Maity, D. Lindsey, T. G. Parker, T. E. Albrecht-Schmitt, J. L. Barreda-Esparza, P. Xiong, W. Zhou and S. Saha, J. Mater. Chem. C, 2016, 4, 894–899 RSC.
- V. Bon, I. Senkovska, I. A. Baburin and S. Kaskel, Cryst. Growth Des., 2013, 13, 1231–1237 CAS.
- M. Kandiah, M. H. Nilsen, S. Usseglio, S. Jakobsen, U. Olsbye, M. Tilset, C. Larabi, E. A. Quadrelli, F. Bonino and K. P. Lillerud, Chem. Mater., 2010, 22, 6632–6640 CrossRef CAS.
- A. J. Howarth, Y. Liu, P. Li, Z. Li, T. C. Wang, J. T. Hupp and O. K. Farha, Nat. Rev. Mater., 2016, 1, 15018 CrossRef CAS.
- Y. Bai, Y. Dou, L.-H. Xie, W. Rutledge, J.-R. Li and H.-C. Zhou, Chem. Soc. Rev., 2016, 45, 2327–2367 RSC.
- P. Deria, J. Yu, T. Smith and R. P. Balaraman, J. Am. Chem. Soc., 2017, 139, 5973–5983 CrossRef CAS PubMed.
- H. K. Chae, D. Y. Siberio-Pérez, J. Kim, Y. Go, M. Eddaoudi, A. J. Matzger, M. O'Keeffe and O. M. Yaghi, Nature, 2004, 427, 523 CrossRef CAS PubMed.
- The DFT calculations also indicate a slight change pore geome-try in response to complete fullerene incorporation (1:1 incorporation). Based on PXRD measurements, the anticipated changes are absent suggesting minor differences in framework coordinates for C60 incorporation at 0.6:1 versus 1:1. As yet, we have been unable to boost the loading beyond 0.6:1.
Nevertheless, it will be inter-esting to see (eventually) if higher loading translates not only into detectable MOF structural changes, but also significant further changes in electronic conductivity (see subsequent results and discussion).
- Based on the (+/0) redox potential of TBAPy linkers in NU-901 (1.4 V vs. Ag/AgCl) and (−/0) redox potential of C60 (−0.33 V vs. Ag/AgCl), the charge transfer (CT) band should appear near 1.8 eV (690 nm). The experimental band maximum is at near 550 nm and the onset is near 700 nm (1.77 eV).
- D. M. Guldi, F. Spänig, D. Kreher, I. F. Perepichka, C. van der Pol, M. R. Bryce, K. Ohkubo and S. Fukuzumi, Chem.–Eur. J., 2008, 14, 250–258 CrossRef CAS PubMed.
- P. Dallas, G. Rogers, B. Reid, R. A. Taylor, H. Shinohara, G. A. D. Briggs and K. Porfyrakis, Chem. Phys., 2016, 465–466, 28–39 CrossRef CAS.
- We recognize that two-point measurements with pelletized samples do exclude effects due to elec-trode/sample contact resistance and do not distinguish between inter-crystallite and intra-crystallite con-tributions to sample resistivity. Thus, the σ value of ∼10−3 S cm−1 should be regarded as a lower-limit estimate for electrical conductivity within a single crystallite. Also obscured by pellet-based measurements is any conductivity anisotropy – specifically, differences in conductivity in the MOF ab plane versus in the c direction (the direction aligned with the diamond-shaped channels).
- D. Hinderberger, in EPR Spectroscopy: Applications in Chemistry and Biology, ed. M. Drescher and G. Jeschke, Springer Berlin Heidelberg, Berlin, Heidelberg, 2012, pp. 67–89, DOI:10.1007/128_2011_236.
- T. Islamoglu, M. A. Ortuño, E. Proussaloglou, A. J. Howarth, N. A. Vermeulen, A. Atilgan, A. M. Asiri, C. J. Cramer and O. K. Farha, Angew. Chem., 2018, 130, 1967–1971 CrossRef.
- Given uncertainties in MOF loading on the IDE platforms, we are unable to say with certainty that the conductivity of NU-901-amino-C60 exceeds that of NU-901-C60. Unfortunately, we lack sufficient NU-901-amino-C60 to extend the study pelletized samples, i.e. samples that could yield quantitative conductivity data.
- A. J. Garza and G. E. Scuseria, J. Phys. Chem. Lett., 2016, 7, 4165–4170 CrossRef CAS PubMed.
- J. P. Perdew, W. Yang, K. Burke, Z. Yang, E. K. U. Gross, M. Scheffler, G. E. Scuseria, T. M. Henderson, I. Y. Zhang, A. Ruzsinszky, H. Peng, J. Sun, E. Trushin and A. Görling, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 2801–2806 CrossRef CAS PubMed.
- Definitive demonstration (or not) of the putative semiconducting behavior will require variable temperature conductivity measurements. We anticipate reporting on these in due course.
Footnote |
| † Electronic supplementary information (ESI) available: Specifics on materials used, synthesis of the MOFs, fabrication of pressed pellets for conductivity measurements, details of characterization, instrumentation, and computational methods are available in the supporting information. See DOI: 10.1039/c8sc00961a |
| This journal is © The Royal Society of Chemistry 2018 |