
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAryl bis-sulfonamides bind to the active site of a homotrimeric isoprenoid biosynthesis enzyme IspF and extract the essential divalent metal cation cofactor†
Katharina
Root‡
a,
Konstantin
Barylyuk‡§
a,
Anatol
Schwab
a,
Jonas
Thelemann
a,
Boris
Illarionov
b,
Julie G.
Geist
a,
Tobias
Gräwert
b,
Adelbert
Bacher
c,
Markus
Fischer
b,
François
Diederich
a and
Renato
Zenobi
 *a
*a
aDepartment of Chemistry and Applied Biosciences, ETH Zurich, Zurich, Switzerland. E-mail: zenobi@org.chem.ethz.ch
bHamburg School of Food Science, University of Hamburg, Hamburg, Germany
cDepartment of Chemistry, Technical University of Munich, Garching, Germany
First published on 18th June 2018
Abstract
Characterizing the mode of action of non-covalent inhibitors in multisubunit enzymes often presents a great challenge. Most of the conventionally used methods are based on ensemble measurements of protein–ligand binding in bulk solution. They often fail to accurately describe multiple binding processes occurring in such systems. Native electrospray ionization mass spectrometry (ESI-MS) of intact protein complexes is a direct, label-free approach that can render the entire distribution of ligand-bound states in multimeric protein complexes. Here we apply native ESI-MS to comprehensively characterize the isoprenoid biosynthesis enzyme IspF from Arabidopsis thaliana, an example of a homomeric protein complex with multiple binding sites for several types of ligands, including a metal cofactor and a synthetic inhibitor. While standard biophysical techniques failed to reveal the mode of action of recently discovered aryl-sulfonamide-based inhibitors of AtIspF, direct native ESI-MS titrations of the protein with the ligands and ligand competition assays allowed us to accurately capture the solution-phase protein–ligand binding equilibria in full complexity and detail. Based on these combined with computational modeling, we propose a mechanism of AtIspF inhibition by aryl bis-sulfonamides that involves both the competition with the substrate for the ligand-binding pocket and the extraction of Zn2+ from the enzyme active site. This inhibition mode is therefore mixed competitive and non-competitive, the latter exerting a key inhibitory effect on the enzyme activity. The results of our study deliver a profound insight into the mechanisms of AtIspF action and inhibition, open new perspectives for designing inhibitors of this important drug target, and demonstrate the applicability and value of the native ESI-MS approach for deep analysis of complex biomolecular binding equilibria.
Introduction
Characterizing binding equilibria of oligomeric biomolecular complexes with multiple ligands often poses considerable challenges when using conventional methods such as kinetic binding assays, isothermal titration calorimetry, or surface plasmon resonance.1 These “gold-standard” techniques often fail because they do not reveal how the distribution of individual ligand-bound states changes with ligand concentration but instead describe an average property of the ensemble as a function of ligand concentration.2 In such cases, analysis is often carried out using mixed models comprising both competitive and non-competitive binding. While useful for formally describing the measured signal and extracting some quantitative parameters of the system, such models provide very little insight into the mechanisms of ligand binding and recognition and, if chosen and applied improperly, can lead to an erroneous conclusion. Homomeric symmetrically arranged proteins often feature cooperative effects in ligand binding. That is, occupation of a binding site by a ligand enhances or reduces the affinity of the other binding sites in the protein. This phenomenon brings the problem of characterizing the protein–ligand binding in multimeric proteins to the next level of complexity. Apart from purely fundamental importance, characterizing binding equilibria in multimeric proteins is also a critical step in the drug discovery process because it is crucial for accurately evaluating the inhibitor binding mode and efficiency.In order to precisely describe the binding mode involved in oligomeric biomolecular complexes, new methods and approaches are needed. Native electrospray ionization mass spectrometry (ESI-MS) has recently emerged as a powerful technique for direct, label-free analysis of intact protein complexes. Native ESI-MS has been increasingly recognized for its ability to directly probe such properties of intact macromolecular assemblies as the protein–ligand complex stoichiometry, binding affinities, and allosteric effects. When providing appropriate solution conditions, as well as assuring that collisions with the buffer gas to achieve efficient desolvation are of sufficiently low energy, gentle transfer from the solution to the gas phase has enabled the detection of large multi-protein assemblies, up to intact ribosomes and viruses in the MDa range.3–13 Since proteins and their complexes are detected as distinct signals, ESI-MS provides invaluable information on protein complexes with specifically bound small molecules.2
Although native ESI-MS has been successfully employed in the past to study model systems with already known characteristics including complex stoichiometry and the binding mechanism,4,14 examples in which native ESI-MS was implemented to predict properties and binding mechanisms of an unknown protein–ligand system are scarce.2,15–17 Here we describe a native ESI-MS study of the enzyme IspF (2C-methyl-D-erythritol-2,4-cyclodiphosphate synthase) that catalyzes an intermediate step in the non-mevalonate, or DXP, pathway (DXP: 1-deoxy-D-xylulose-5-phosphate, Scheme 1).18–22 The products of the DXP pathway are isoprenoids, which play a critical role in growth, differentiation, and regulation of the metabolism in living cells. The DXP pathway is exclusively present in protozoan parasites from phylum Apicomplexa, such as malaria-causing Plasmodium spp., Toxoplasma gondii, and Cryptosporidium, as well as in plants, and in numerous pathogenic bacteria including Mycobacterium tuberculosis. Multidrug resistance remains a major obstacle to successful treatment of many human life-threatening and prevalent diseases including malaria,18,23,24 new and previously treated cases of tuberculosis25–27 and toxoplasmosis.28–30 Apicomplexan parasites, such as Neospora, Babesia, Theilleria, as well as Toxoplasma, put a tremendous burden on food security through diseases in livestock and poultry. Repeated use of different herbicide mixtures contributed to the emergence of highly resilient weeds with multiple resistance to almost all commercial herbicides, which puts the future world food production at risk.31–34 Emerging resistance problems of many pathogens as well as weeds urgently call for new affordable therapeutics and herbicides with novel modes of action.18,28,32 Enzymes of the DXP pathway are attractive drug targets, because there are no homologues in mammals.30,32,35 Moreover, these enzymes are clinically validated as drug targets in apicomplexans, pathogenic bacteria, and as targets for herbicides in plants.19–21,23,24,36–39 Any potential inhibitor of the enzymes disrupting the metabolic cascade in the DXP pathway will be devoid of target-related toxicity.40,41
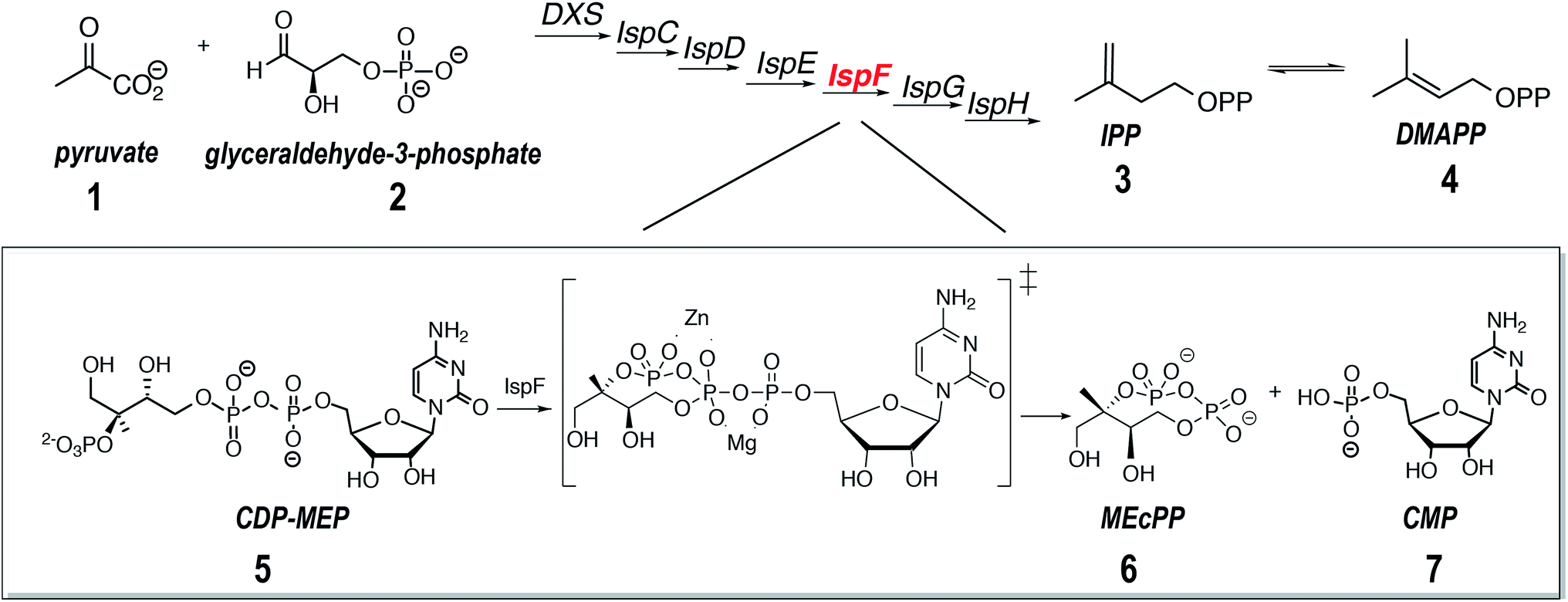 | ||
| Scheme 1 The non-mevalonate (DXP) pathway for the biosynthesis of isoprenoids. The part of the DXP pathway catalyzed by IspF is shown in detail. The transition state is marked by ‡ and shows the charge delocalization by Zn2+. The second metal ion Mg2+ is brought in by the substrate. DXS: 1-deoxy-D-xylulose-5-phosphate-synthase, IspC: 1-deoxy-D-xylulose 5-phosphate reductoisomerase, IspD: 2-C-methyl-D-erythritol 4-phosphate cytidylyltransferase, IspE: 4-diphosphocytidyl-2C-methyl-D-erythritol kinase, IspF: 2C-methyl-D-erythritol-2,4-cyclodiphosphate-synthase, IspG: (E)-4-methyl-D-erythritol-2,4-cyclodiphosphate-reductase, IspH: 1-hydroxy-2-(E)-methylbutenyl-4-diphosphate-reductase, IPP: isopentenyldiphosphate, DMAPP: dimethylallyldiphosphate, CDP-MEP: 4-diphosphocytidyl-2-C-methyl-D-erythritol 2-phosphate, MEcPP: 2-C-methyl-D-erythritol-2,4-cyclodiphosphate, CMP: cytidine monophosphate. | ||
The enzyme IspF catalyses the cyclization of 4-diphosphocytidyl-2C-methyl-D-erythritol 2-phosphate (CDP-MEP, 5, Scheme 1) to 2C-methyl-D-erythritol-2,4-cyclodiphosphate (MEcPP, 6, Scheme 1) with the release of cytidine monophosphate (CMP, 7, Scheme 1).20,29 Published X-ray crystal structures show IspF to be a globular bell-shaped C3-symmetric homotrimer.20 The enzyme's active sites are located at the interfaces between adjacent subunits, which also contain tetrahedrally coordinated zinc ions that is essential for catalysis.19 Depletion of IspF in E. coli and Bacillus subtilis has a significant impact on cell wall biosynthesis leading to cell death and proves, therefore, a legitimate target for the development of new therapeutics.42 While the number of potent inhibitors of IspF available today is limited,43,44 new promising molecules based on the aryl-sulfonamide moiety that inhibit IspF from Arabidopsis thaliana and Plasmodium falciparum with submicromolar IC50in vitro were recently identified.45 No co-crystal structure of AtIspF with any of these inhibitors could, however, be obtained. Rational derivation of the inhibition mechanism was complicated by the availability of several binding sites, as well as binding partners including the metal ion cofactor and the inhibitor. Although some synthetic ligands showed strong inhibition, the interpretation of biochemical assay data was difficult due to a lack of information on the mode of action of the inhibitors.46 Therefore, the stoichiometry of protein–ligand binding remains unknown, as well as the binding site location, inhibition mode (competitive/allosteric), and exact binding constants.
We applied native ESI-MS to study interactions of recombinant IspF from A. thaliana (AtIspF) with various ligands including zinc ions, its natural substrate, and aryl sulfonamide inhibitors. Thanks to the ability of the method to resolve multiple co-existing ligand-bound states and simultaneously trace their abundances in the mixture as a function of concentration, we disentangled the parallel binding events occurring in the system. Based on the MS data, we suggest a model for the binding and inhibition mechanism of aryl sulfonamides that combines a competitive binding to the enzyme active site and removal of the essential Zn2+ ion co-factor.
Experimental section
Mass spectrometry
Native ESI-MS was carried out on a Synapt G2S HDMS (Waters, Manchester, UK). All experiments were carried out in positive ion mode. Au/Pd-coated glass capillary emitters (1 μm outlet inner diameter; Thermo Scientific, Madison, WI, USA) were used to load 3 μL of the protein sample. A commercial nano-ESI source was used at ambient temperature by applying 0.8–1.3 kV to generate the electrospray. A backing pressure of 0.25–0.3 bar was applied to assist the sample flow. Several tuning parameters such as the cone voltage, collision energy, and the gas trap pressure were carefully optimized to preserve noncovalent complexes and at the same time obtain the most efficient ion transfer, and the highest signal intensity and resolution. The parameters were tuned as follows: the cone voltage was set to 80 V. The trap and transfer collision energies were optimized to 50 V and 30 V, respectively. The gas trap pressure was set to 6.0 mL min−1. The quadrupole profile was adjusted for the desired m/z range. Mass spectra were obtained in a window of m/z 50–8000 with a scan time of 2 s and an inter-scan delay of 0.1 s. Typically, fifty scans were combined to produce a mass spectrum.Mass spectra were recorded using the MassLynx 4.0 software (Waters, Manchester, UK). All mass spectra were baseline-corrected, normalized, and smoothed using MATLAB R2017a (MathWorks, Natick, MA, USA). An aqueous CsI solution (40 mg mL−1) was used to perform calibration of the mass spectrometer. After averaging the recorded spectra (50 scans), they were smoothed with a moving average algorithm (span of ±3 steps) and centroid spectra were generated at 80% peak height. The m/z axis was calibrated by fitting a polynomial function.
Inhibitor titration experiments
Direct titration experiments of AtIspF with aryl-sulfonamides were carried out as follows: the ligand stock solution was used to prepare a dilution series in DMSO, which was further diluted 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 (v/v) in 200 mM aqueous ammonium acetate solution (pH 8.0). These solutions were mixed 1:1 (v/v) with the protein solution at a trimer concentration of 8 μM in 200 mM aqueous ammonium acetate (pH = 8.0). Such a mixing procedure ensures constant protein and DMSO concentrations (ctrimer = 4 μM and 1%, respectively) but a variable ligand concentration (Scheme SI1†). The ligand concentration spanned the range of 1–100 μM. Each measurement was replicated 3 times. For the ligand competition test, the samples were prepared in analogous fashion.
:50 (v/v) in 200 mM aqueous ammonium acetate solution (pH 8.0). These solutions were mixed 1:1 (v/v) with the protein solution at a trimer concentration of 8 μM in 200 mM aqueous ammonium acetate (pH = 8.0). Such a mixing procedure ensures constant protein and DMSO concentrations (ctrimer = 4 μM and 1%, respectively) but a variable ligand concentration (Scheme SI1†). The ligand concentration spanned the range of 1–100 μM. Each measurement was replicated 3 times. For the ligand competition test, the samples were prepared in analogous fashion.
Results and discussion
Analysis of AtIspF under denaturing and native-like conditions using native ESI-MS
The crystal structure of IspF in complex with CMP was previously resolved and shows IspF as a globular bell-shaped homotrimer (Fig. 1a).47 We produced a recombinant full-length AtIspF in house (see ESI Methods for details†). In order to confirm the protein's molecular weight, stoichiometry, and purity, nano-ESI mass spectra of recombinant AtIspF were recorded under denaturing and native-like conditions (Fig. SI1,†1b, respectively). The signal detected in the mass spectrum of the denatured protein was attributed to a single species with the measured molecular weight of 19225.0 ± 0.3 Da, which is in perfect agreement with the mass of AtIspF monomer calculated based on its amino acid sequence (19225.1 Da; Fig. SI1, Table SI2†). The acquired native ESI mass spectrum showed dominant trimer signals (T; MWT ≈ 57681 ± 5.4 Da) of AtIspF based on the measured mass (Fig. 1b, Table SI2†). The trimeric stoichiometry of AtIspF was confirmed by collision-induced dissociation (CID) tandem MS of the isolated T14+ ion (m/z = 4120) (Fig. 1c). The parent ion (T14+) asymmetrically dissociated48–50via ejection of relatively highly charged unfolded monomer ions and compact dimer ions carrying the remaining charge. This experiment also confirmed the non-covalent nature of the association of monomers into trimer, because only intact protein chains were found in the fragment ion spectrum.
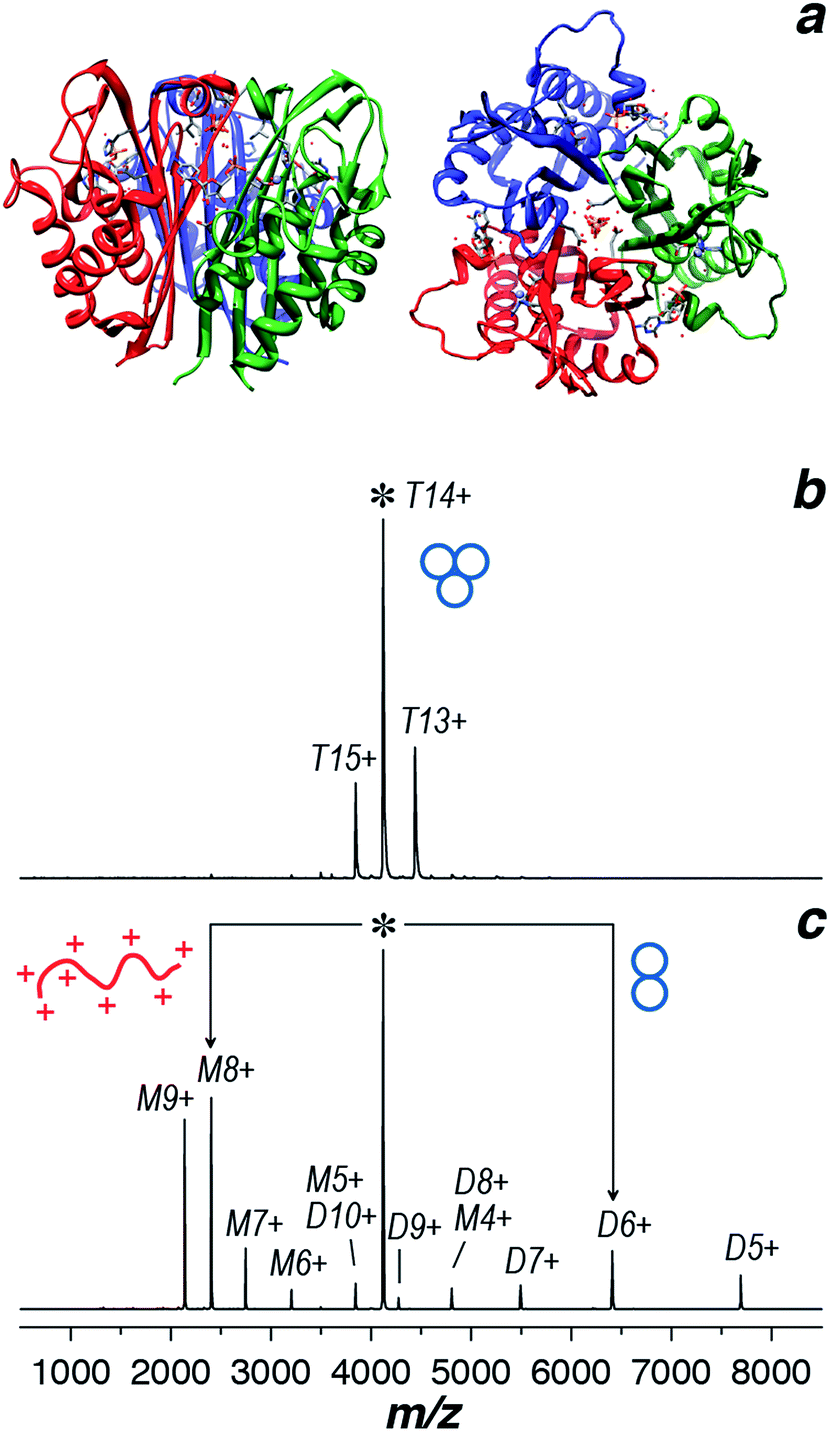 | ||
| Fig. 1 (a) Ribbon diagram representing the X-ray structure of AtIspF in complex with 7 and Zn2+ shown as stick models. Side (left) and top51 views are presented (PDB entry 2PMP).52 Individual protein chains are in different color. (b) ESI mass spectrum of AtIspF recorded under native-like conditions (ammonium acetate 150 mM, pH = 8.0). The dominant peaks were attributed to the trimer ions (T) (+13 to +15). (c) The trimer ion (+14; m/z = 4120) was isolated in the quadrupole and subjected to collisional activation inside the collision cell of the mass spectrometer (CID-MS/MS). The parent ion undergoes asymmetric charge partitioning ejecting highly charged unfolded monomer (M) ions below the parent ion peak and compact dimer (D) ions carrying the remaining charge at m/z above that of the parent ion peak. | ||
Binding of the natural ligands, CDP-MEP and Zn2+ to AtIspF
The zinc cofactor is essential for catalysis in AtIspF: it positions the β-phosphate group of the substrate and favours nucleophilic attack by decreasing the negative charge on the β-phosphate oxygen.29 Upon incubation of AtIspF with zinc, we expected saturation of each active site with one Zn2+ ion. We expected that the enzymatic activity could be influenced depending on depletion or addition of zinc ions in AtIspF. Indeed, the spectrum of Zn2+-saturated AtIspF differs from that of the bare protein (Fig. 2a and b) by a shift in m/z attributed to the binding of three Zn2+ ions to the protein active sites (Fig. 2e and f). Incubation of the Zn2+-depleted enzyme with its substrate, 4-diphosphocytidyl-2-C-methyl-D-erythritol 2-phosphate (5, CDP-MEP, Scheme 1), afforded complexes containing up to three molecules of 5 in addition to the bare protein signal in the mass spectrum (Fig. 2c and d). As expected, no substrate conversion was observed in the mass spectrum suggesting that the specific enzymatic activity was disabled by the absence of Zn2+. Upon incubation of Zn2+-saturated AtIspF with the substrate (Fig. 2g and h), several peaks were mass-shifted relative to the bare protein trimer by approximately n × 322 Da (n = 1, 2, 3), which matches the mass of CMP (7, Scheme 1). The detected complexes were not ESI artefacts but present in solution, because only negligible – if any – signals of PREFL complexes with a non-binding reference protein (PREF: PMSF-inactivated bovine cationic trypsin) were detected in the control measurements (Fig. SI2†). At the same time, no signals corresponding to Zn2+-saturated AtIspF in complex with the substrate 5 or the second product of the reaction, MEcPP 6, were observed (Scheme 1). Interestingly, similar to our observation, only 7 was found bound in the active site when AtIspF was co-crystallized with the substrate.53,54 While 6 is released from the active site, 7 is bound tighter and stays in the cytidyl-binding pocket. Thus, the complex can survive in electrospray and be detected in the mass spectrum. The absence of free 6 in the mass spectrum can be explained by its acidity, making it undetectable in the positive ion mode. We demonstrated that we can monitor and influence the specific enzymatic activity of AtIspF, as well as binding of the metal cation and the natural ligands, 5 and 7, by native ESI-MS. Our results also have implications for the mechanism of the negative-feedback regulation of the enzyme activity via competitive inhibition by the product.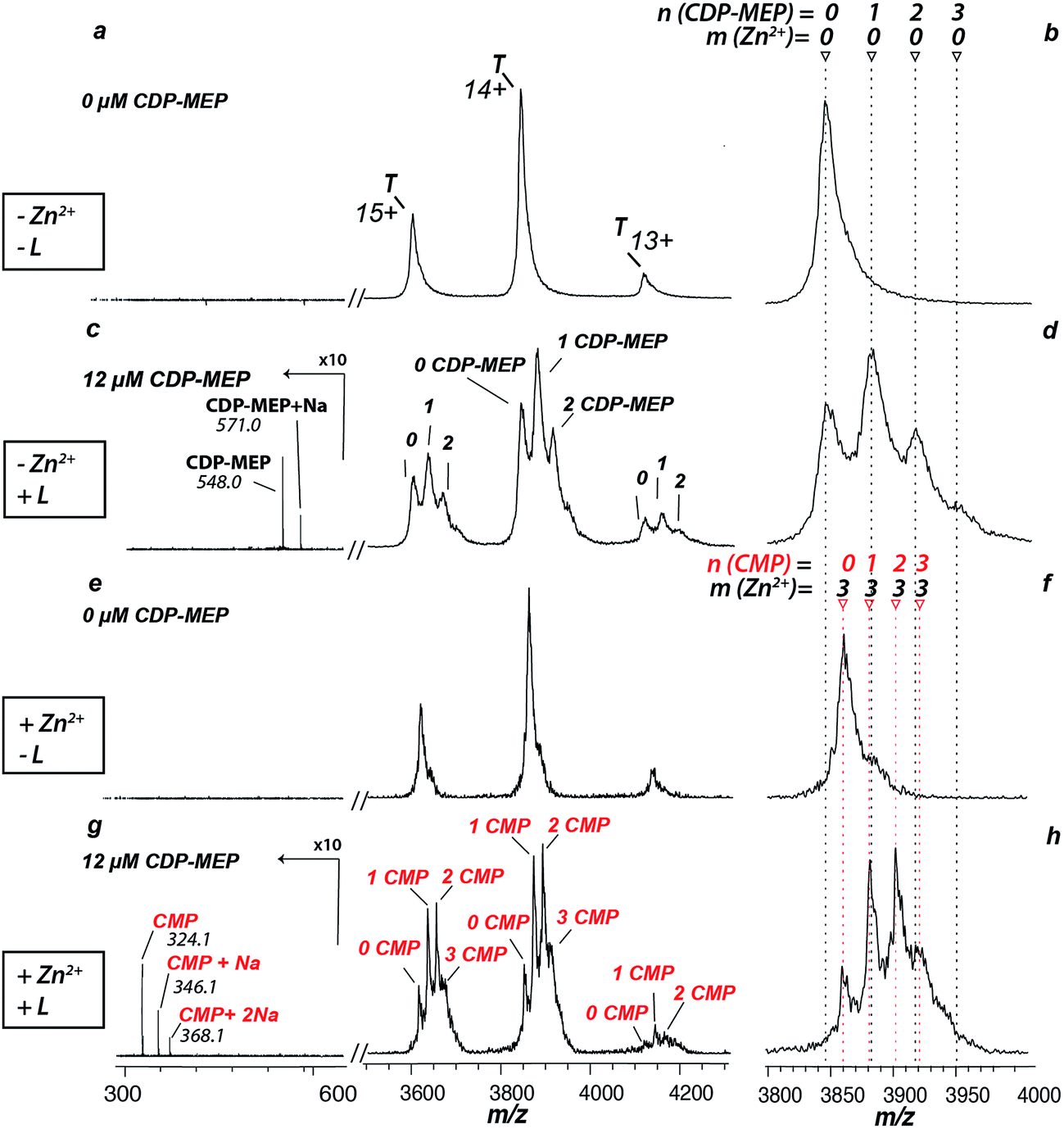 | ||
| Fig. 2 ESI-mass spectra of AtIspF incubated with 5 (T:L ratio 1:0 to 1:2) in the absence and presence of Zn2+ (a, b and e, f, respectively). A zoom-in into the most intense signal (T14+) is shown next to the individual spectra. A native ESI mass spectrum of the bare protein is given for reference (a). AtIspF was incubated with 5 (T:L ratio of 1:2) in the absence (c, d) and presence of Zn2+ (g, h). Red and black markers indicate the number of 7 or 5 molecules bound to AtIspF, respectively. (c, d) Incubation of zinc-deficient AtIspF with the substrate yields complexes containing up to three 5 molecules bound to the protein trimer. (e, f) Saturating AtIspF with Zn2+ results in a mass shift corresponding to three Zn2+ ions bound to the protein. (g, h) Upon incubation of Zn2+-saturated AtIspF with 5, fine-structured complex peaks were observed with a mass shift of approx. 322 Da matching to 7, one of the products of the reaction. Ions corresponding to free 5 (c) or 7 (g), protonated or with sodium cations bound, were found in the mass spectrum. The signal intensity in spectra c and g was magnified 10-fold in the range from 300 to 600 m/z. | ||
Aryl bis-sulfonamide inhibitors bind to AtIspF trimer and extract Zn2+ cation from the active site
Having demonstrated the validity of the native ESI-MS approach for AtIspF with its natural ligands, we explored the binding mechanism of aryl sulfonamide inhibitors. Computational simulations suggested that aryl sulfonamide-based inhibitors bind to the active site of AtIspF,46 but the detailed binding mechanism and the mode of inhibition are unknown. In the absence of Zn2+, we initially anticipated no or weak non-specific binding of aryl sulfonamide based inhibitors (Scheme 2, Table SI1†), but we expected to detect specific TL3 complexes in the presence of Zn2+, as well as a trend in the observed binding affinity that would correspond to the trend in inhibitor potency (IC50). Surprisingly, all bis-sulfonamides pronouncedly bound to the Zn2+-deficient protein trimer regardless of their solution-phase inhibitory activity (Fig. 3a, SI3a–7a†). The signal of every TLn complex increases gradually followed by a gradual decrease when TLn+1 complex takes over (Fig. 3a). Complexes containing up to six inhibitor molecules were observed in the spectrum. The situation, however, changed when we incubated Zn2+-saturated AtIspF with the inhibitors (Fig. 3b, SI3b–7b†). In this case, three key observations were made. First, up to six ligands bound to the protein trimer, similar to the case of Zn2+-free AtIspF. The detected complexes were not ESI artifacts, because only negligible – if any – signals of PREFL complexes with the reference protein (PREF) were detected in the control measurements (Fig. SI9†). Second, in contrast to Zn2+-free AtIspF, the binding pattern was clearly more complex, with a dominant TL3 (in some cases TL4 or TL2) peak. Third, the number of bound Zn2+ ions per trimer got increasingly scrambled as the ligand concentration increased. On the one hand, Zn2+-depleted states were observed for T, TL1 and TL2 ions, while, on the other hand, excess numbers of zinc ions were observed for the complexes containing three ligands and onwards.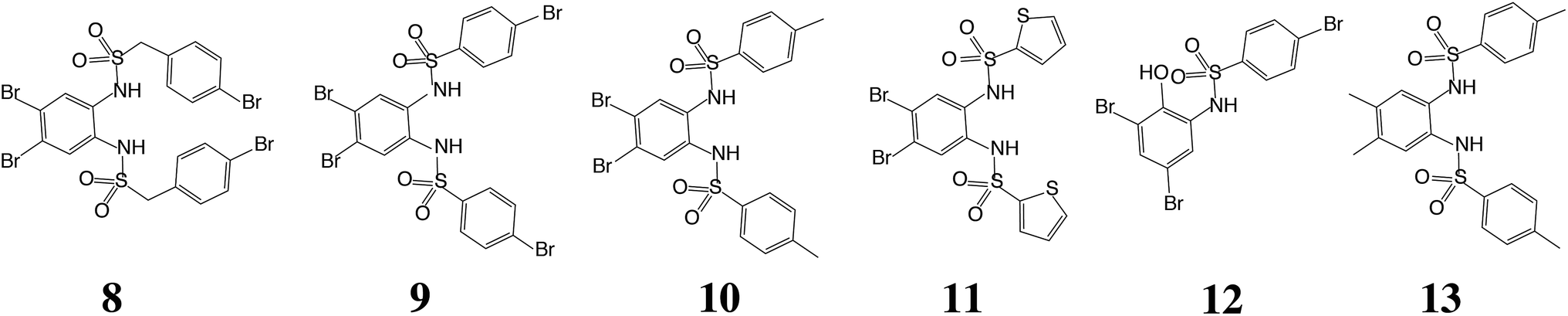 | ||
| Scheme 2 Chemical structures of the aryl bis-sulfonamide inhibitors of AtIspF used in this study. 8: N,N′-(4,5-dibromo-1,2-phenylene)bis[1-((4-bromophenyl)methanesulfonamide)], 9: N,N′-(4,5-dibromo-1,2-phenylene)bis(4-bromobenzenesulfonamide), 10: N,N′-(4,5-dibromo-1,2-phenylene)bis(4-methylbenzenesulfonamide), 11: N,N′-(4,5-dibromo-1,2-phenylene)bis(thiophene-2-sulfonamide), 12: 4-bromo-N-(3,5-dibromo-2-hydroxyphenyl)benzenesulfonamide, 13: N,N′-(4,5-dimethyl-1,2-phenylene)bis(4-methylbenzenesulfonamide). | ||
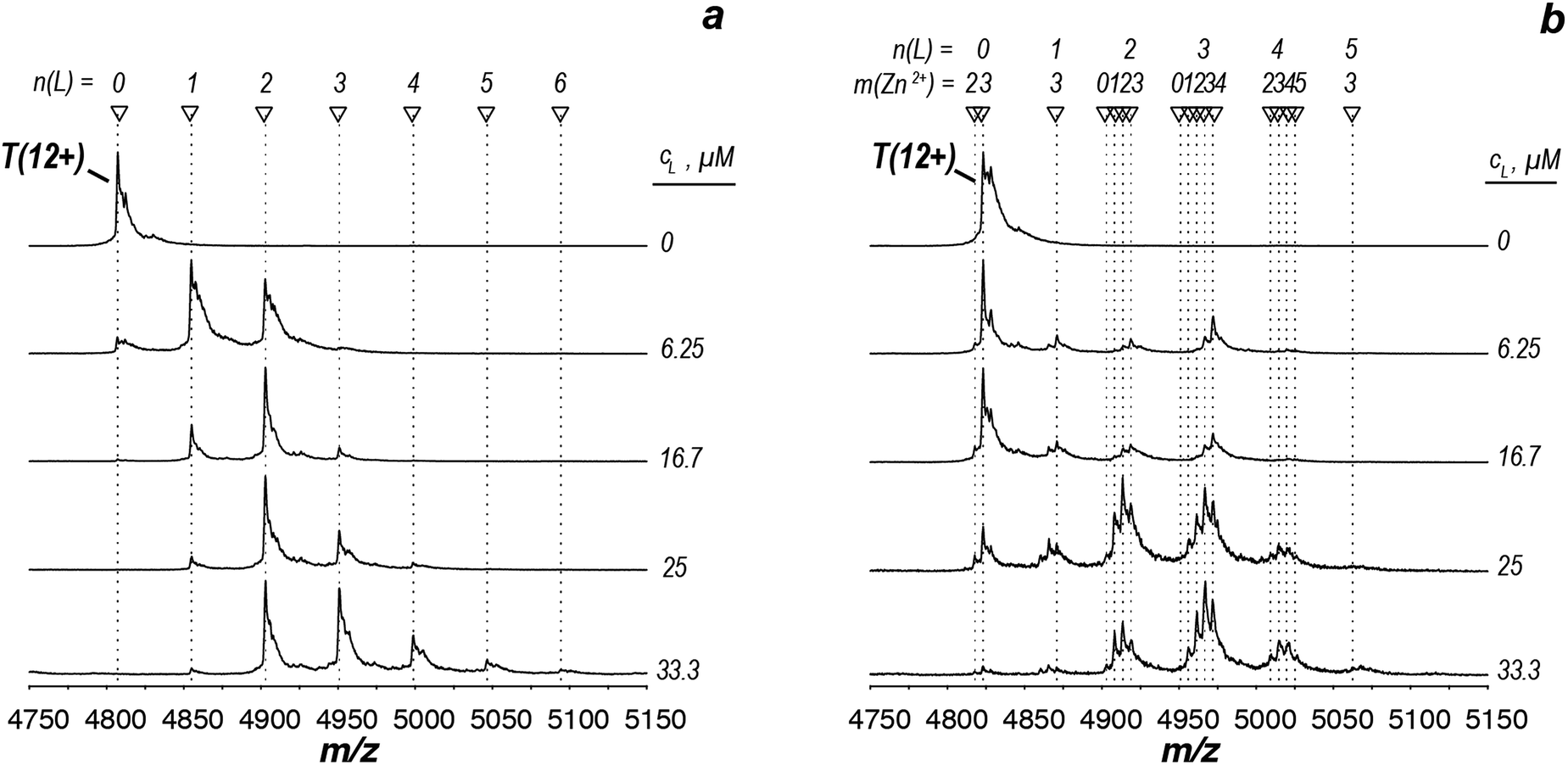 | ||
| Fig. 3 ESI-mass spectra of the titration series of AtIspF (at a monomer concentration of 24 μM) with increasing concentration (cL = 0–50 μM) of bis-sulfonamide inhibitor 10 in the absence (a) and presence (b) of Zn2+. The spectra are recorded in 150 mM ammonium acetate (pH = 8.0) containing 1% DMSO (Fig. SI10†). The spectral region around the 12+ charge state of the protein trimer is shown. The markers indicate the number of ligands (n) and Zn2+ cations (m) bound to the protein trimer. (a) In the absence of Zn2+, sequential ligand binding yields complexes containing up to six ligands. (b) Upon an increase of the 10 concentration, Zn2+-depleted states (m < 3) of T, TL1, and TL2 are observed. In addition, binding of the dimeric ligand chelate complex [L2Zn]2− brings extra Zn2+ ions (m > 3) to the TLn species, where n ≥ 2. | ||
We can conclude that all tested bis-sulfonamides bind to AtIspF in solution in the absence of Zn2+ in a sequential, independent fashion, producing TLn complexes with n ≤ 6, i.e. up to two ligands per AtIspF monomer. Surprisingly, even the inhibitor with the lowest IC50 value, 13, exhibited pronounced binding suggesting that Zn2+-depleted AtIspF lacks selectivity towards aryl bis-sulfonamides. However, the presence of Zn2+ dramatically altered the protein–ligand binding and conferred selectivity on AtIspF. For instance, AtIspF bound 8 with a higher affinity compared to 12, which is in a good qualitative agreement with the trend in IC50 values for the inhibitors. Furthermore, the distribution of peak intensities for various TLn complexes (0 ≤ n ≤ 6) in the mass spectra suggested a preference for certain complex stoichiometries (n = 2–4). This, together with the observed scrambling of the Zn2+ binding stoichiometry, suggests that aryl bis-sulfonamides could extract the cation from the protein active site and associate into a dimeric chelate complex [L2Zn]2− in solution (Fig. SI10†),46 which in turn could also bind to the protein.
We hypothesized that if aryl bis-sulfonamide ligands could indeed remove Zn2+ from the active site of AtIspF, presaturation of the ligand with the cation would prevent Zn2+ extraction from the protein complex. Therefore, no Zn2+-deficient complexes should be observed. We mixed Zn2+-saturated AtIspF with Zn2+-saturated 8 (Fig. 4). The increase of the concentration of 8 (T:L ratio from 1:0 to 1:7) resulted in complexes containing up to six ligands, but no Zn2+-depleted states. An excess of Zn2+ (3 < m ≤ 6) was detected in complexes containing two to six ligands. The peak intensity distribution showed only a small fraction of complexes containing 1, 2, and 3 ligands bound, whereas the signals of the complexes containing four to six ligands (4 ≤ n ≤ 6) were highly pronounced. This strongly indicates positive binding cooperativity. The binding of the 1st chelated dimer complex increases the affinity of the 2nd and 3rd binding site for the binding of the 2nd and 3rd chelated dimer complex. This explains the dominant signal intensities corresponding to complexes containing four and six ligands in the mass spectrum. Additionally, the chelated dimer complex is in equilibrium with its free constituents (with an association constant of 15.6 × 106 M−2).46 Binding of the free ligand to the abundant protein–ligand complex, when it already contains two chelated dimer complexes, explains the pronounced signal intensity of the complex containing five ligands. The inhibitor bound to the protein trimer mainly in the form of [L2Zn]2− complex, which also explains the excess of Zn2+ ions observed in the spectra. Overall, these results are consistent with binding of a dimeric chelate complex of aryl bis-sulfonamide inhibitors with Zn2+ to the active sites of AtIspF trimer.
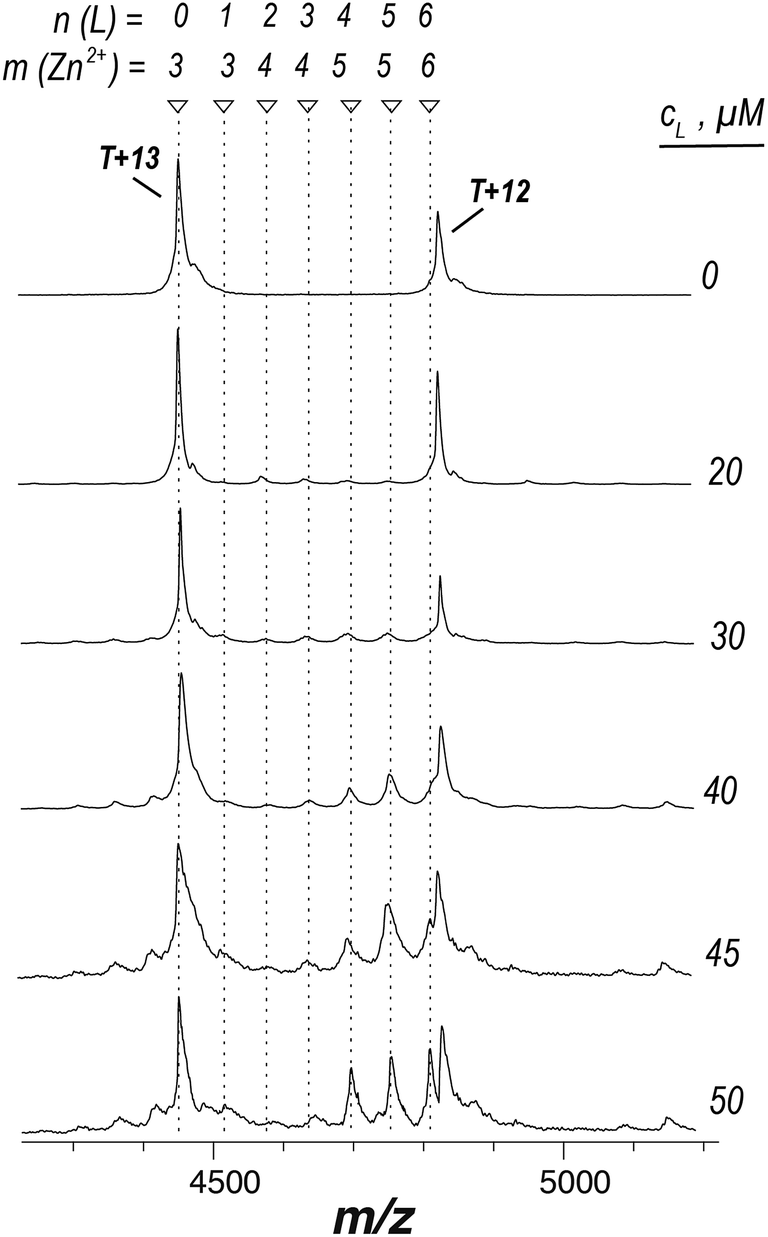 | ||
| Fig. 4 ESI-mass spectra of AtIspF (at a monomer concentration of c = 24 μM) presaturated with Zn2+ and incubated with an increasing concentration (c = 0–50 μM) of Zn2+-presaturated 8. A spectral region around the 13+ charge state is shown. The markers indicate the peak position and the number of 8 and Zn2+ cations bound (n and m, respectively). The ligand concentration in μM is indicated next to the traces. No Zn2+-depleted states were observed for any of the complexes formed. An excess amount of Zn2+ was observed for complexes containing two ligands onwards. | ||
Docking of bis-sulfonamide ligands
To rationalize the observed binding of six ligands per IspF homotrimer, possible binding modes were evaluated computationally. Crystallographic data are available for the trimeric IspF enzyme of Arabidopsis thaliana.47 However, co-crystallization attempts with bis-sulfonamide ligands have remained unsuccessful. The structure-based analysis was therefore performed on the crystal structure of CMP-bound AtIspF (PDB ID: 2PMP, 2.3 Å resolution).20 The trimeric complex has three equivalent active sites, which present the only cavity capable of binding small organic molecules in an enclosed environment. As a representative for other inhibitors, the para-methyl substituted derivative 10 was chosen. As a prerequisite for successful docking of the ligands, the space filling of the active site by two ligands was calculated. The program HOLLOW was used to generate a cast of the active site to the periphery of the enzyme, which amounted to a volume of 2100 Å3 (Fig. 5a).55 The ligand 10 was calculated to have a volume of 350 Å3, resulting in a packing coefficient of 33% if two ligand molecules occupy each active site. This value is consistent with an ideal packing coefficient of 55%, showing that the active site provides sufficient room for two ligands, and further permits that larger derivatives of the series could be bound in a similar fashion.56 Ligands were positioned to be in agreement with the previously reported structure–activity relationship,46 and to account for the conformational preferences of their functional groups (Fig. 5b, for details see ESI Experimental†). Inhibitors bearing bromine substituents on the diamine scaffold constitute the most active derivatives of the ligand class. Superior inhibition and binding of bromide over the corresponding methyl derivatives, such as 10vs.13, suggest favourable halogen bonding interactions of the two halide substituents.57 The modelled binding mode of 10 demonstrates the ability of the active site to favourably accommodate two ligand molecules and could be used for the design of larger inhibitors filling the entire active site.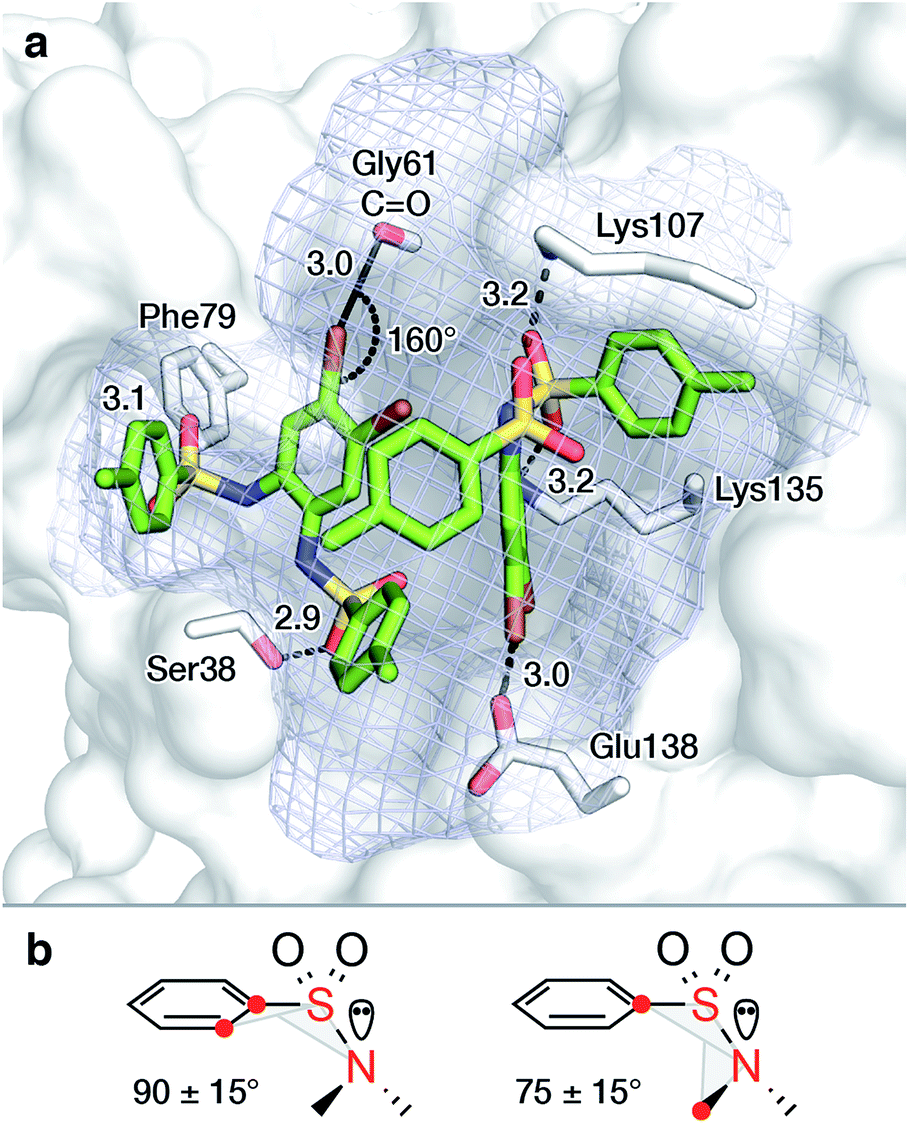 | ||
| Fig. 5 Proposed binding mode of two molecules of 10 in the active site of AtIspF ((a), PDB ID: 2PMP,20 2.3 Å) and conformational constraints for aryl sulfonamides (b). The mesh surface spans the volume of the active site. Atom coloring: Br brown, N blue, O red, S yellow, distances are given in Å. | ||
To complement binding studies in the presence of Zn2+, the [L2Zn]2− complex, previously characterized by small molecule crystallography, was modeled in the active site (ESI Experimental, Fig. SI11†).46 In the obtained binding mode, the zinc ion coordinates with Asp66 in the complex, analogous to the magnesium ion, which coordinates with the diphosphate of CDP-MEP and Asp66 during turnover (Scheme 1).29,58,59 A favorable electrostatic contribution to the anionic ligand complex binding, similar to that of the anionic substrate CDP-MEP, is expected.
CDP-ME and aryl bis-sulfonamide inhibitors compete for the same binding site in AtIspF
In order to determine the binding site as well as the inhibition mode exhibited by aryl sulfonamides a ligand competition experiment was carried out. Fig. 6 shows Zn2+-saturated AtIspF mixed with both 5 and 8 at different ratios. As expected, independent binding of multiple 7 residues was found when the protein was incubated with 5 alone (Fig. 6, 1st row). In the case of 8, signals corresponding to the binding of one, two, three, and four ligands per trimer were detected by ESI-MS (Fig. 6, 1st column). Remarkably, when both ligands were added to the protein together, the resulting native ESI mass spectrum looked like a superposition of those recorded in the presence of each ligand individually (Fig. 6, bottom row). No multiplexing of spectra or appearance of new signals was found. Instead, the peak intensities of protein–ligand complexes were attenuated.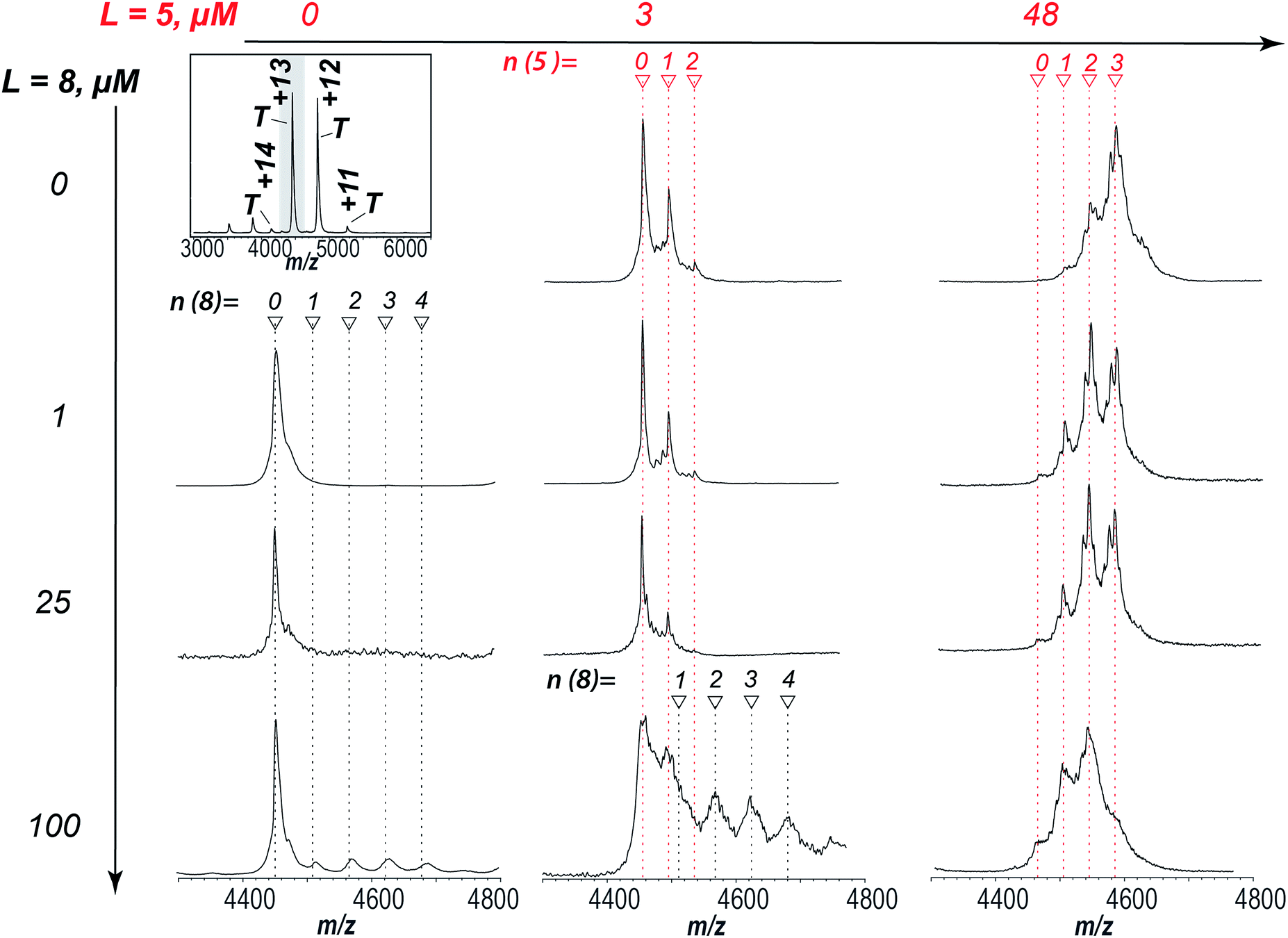 | ||
| Fig. 6 Competitive binding of 8 and 5 to AtIspF. The entire spectrum of the bare protein is given for reference (top left). AtIspF (at a monomer concentration c = 24 μM) was mixed with increasing concentrations of 5 (3–48 μM, middle and right columns), and/or 8 (1–100 μM, second to fourth row). The spectral region around the T13+ signal is shown. Red and black markers indicate the number of 7 and 8 ligands bound, respectively. Upon addition of 5 to the protein, peaks matching to one, two, or three 7 residues bound to the trimer were found in the spectrum (first row). When 8 was added exclusively, signals of the protein complex with up to four ligands were detected (bottom row). No additional peaks were observed when two ligands were added to the protein together (third and fourth row). Increasing the concentration of 5 at a fixed concentration of 8 (100 μM) resulted in a superposition of both signal patterns of the complexes formed. A fine structure of the complex peaks was attributed to scrambling of Zn2+ bound states as already shown for the protein titration with 8 alone (Fig. SI4†). | ||
At higher concentrations of 8, scrambling of Zn2+-bound states was observed, similar to the results of AtIspF titration with 8 only (Fig. SI3†). These results suggest that the natural substrate and the aryl bis-sulfonamide inhibitor compete for the same binding site in AtIspF, or that their binding sites overlap significantly. The nucleotide binds much stronger, but the intensity of the corresponding peaks in the mass spectra is attenuated by the presence of the bis-sulfonamide – even when the signal of the AtIspF–8 complex is hardly seen in the absence of the substrate. Our results suggest that the inhibition mechanism is mixed competitive and non-competitive. The competitive component is relatively weak, but the non-competitive inhibition occurs via extraction of the Zn2+ cation from the active site, which has a much more detrimental effect to the enzyme activity (see Fig. 2 and the associated discussion).
Suggested mechanism of bis-sulfonamide inhibitors binding to AtIspF
The results obtained provide insight into the mechanism of bis-sulfonamide inhibitor binding to AtIspF. Scheme 3 shows two binding scenarios depending on whether or not AtIspF was preincubated with Zn2+. In the absence of Zn2+, the binding sites of AtIspF are sequentially occupied by up to six ligand molecules (Scheme 3a). AtIspF contains three binding pockets that are sequentially filled by up to two inhibitor molecules each. We observed only a minor difference in the apparent affinity of AtIspF towards different aryl bis-sulfonamides in the absence of Zn2+. However, the binding must have occurred in solution, because no complexes were formed between a reference protein and the inhibitors (see Fig. SI9† and associated discussion).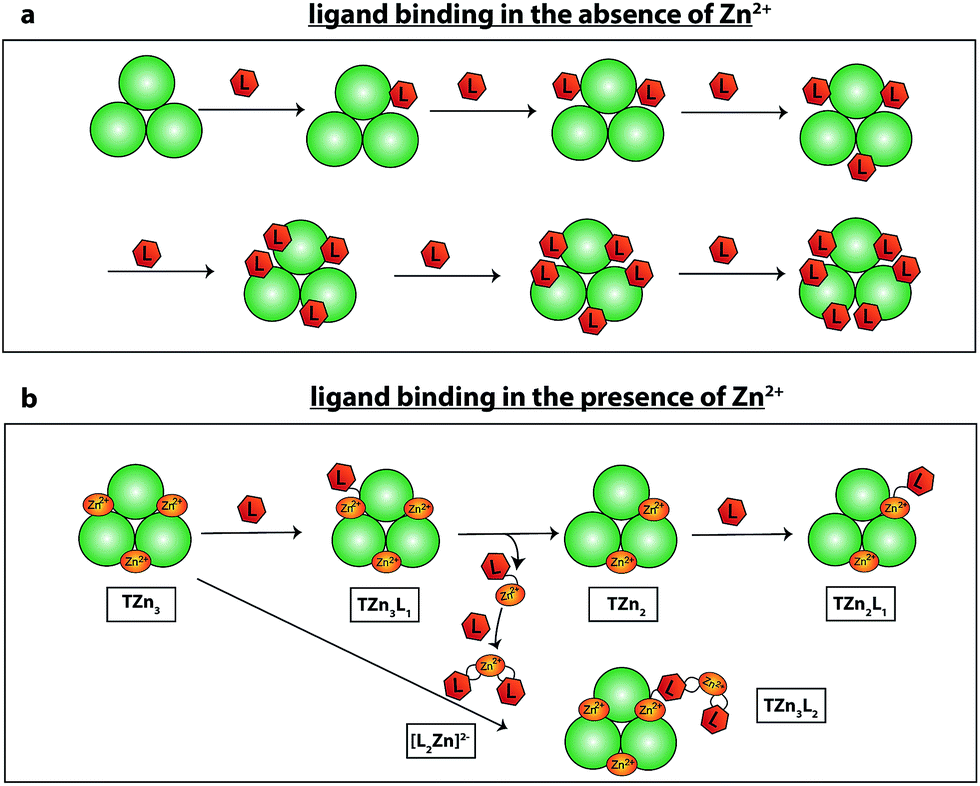 | ||
| Scheme 3 Proposed binding mechanism of bis-sulfonamide inhibitors (L) to AtIspF in the absence (a) and presence (b) of Zn2+. In (b), only a few initial steps are shown. (a) In the absence of Zn2+, a sequential occupation of the three available binding sites of AtIspF takes place. In total, up to six ligands can bind to AtIspF. (b) The ligand binds to Zn2+ saturated AtIspF resulting in the complex containing one ligand (TZn3L1). The bound monomeric ligand can extract Zn2+ from the active site and dissociate into the bulk phase where it can form a dimeric chelate complex [L2Zn]2−. Zn2+ deficient AtIspF (TZn2) emerges in solution. A ligand molecule can bind to the Zn2+ deficient complex producing TZn2L1. In turn, the dimeric chelate complex [L2Zn]2− can bind to Zn2+ saturated AtIspF (TZn3) resulting in TZn4L. Combinations of these basic steps yield all the other complexes detected by ESI-MS. | ||
Scheme 3b shows the scenario for an aryl bis-sulfonamide inhibitor binding to Zn2+-presaturated AtIspF (no free Zn2+ in solution). The ligand competition experiment showed that the natural substrate and the synthetic inhibitor compete for the same binding site in AtIspF. Zinc-depleted complexes are formed as a result of zinc extraction from the protein active sites by the aryl-sulfonamide inhibitor, which forms a dimeric chelate complex with zinc in the condensed phase.46 Enzyme trimers containing more than six inhibitor molecules also carry an excess of zinc due to binding of dimeric chelate Zn2+-inhibitor complex, [L2Zn]2−.
Conclusions
We demonstrated how a thoroughly planned experimental design combining native ESI-MS and computational modelling delivers answers to fundamental questions in a complex biological system, where a more conventional approach involving “gold-standard” biophysical techniques could not be applied. We focused our study on structural and functional aspects of protein–ligand interactions in A. thaliana IspF, an enzyme of the DXP pathway of isoprenoid biosynthesis and an attractive target for the development of new antimalarial and antituberculosis drugs, as well as herbicides with novel mode of action. Using direct native ESI-MS titration, we comprehensively explored the processes of the protein binding with various ligands, including the zinc cation, the natural substrate, and a range of synthetic aryl bis-sulfonamide inhibitors.Our data strongly suggest that the zinc cation in the protein active site plays a key role for the catalytic activity and inhibition of AtIspF. Extraction of Zn2+ from the active site with EDTA fully inhibits the specific enzymatic activity of AtIspF, which was reflected in the native ESI mass spectra by detection of the protein complexes with the intact substrate molecule, 5. Adding Zn2+ back fully restored the catalytic activity of the protein, so that signals of the complexes with one of the reaction products, 7, were present in mass spectra. These results are consistent with the previously published data on AtIspF high-resolution 3D structure and function, hence, demonstrating the validity of the native ESI-MS approach for the analysis of such protein systems.
We analysed the stoichiometry, specificity, selectivity, and relative strength of binding of a range of synthetic aryl bis-sulfonamide inhibitors to AtIspF – both in the presence and absence of Zn2+. In the absence of zinc, up to two inhibitor molecules per active site could bind to the protein in a sequential, independent fashion. We computationally evaluated the binding mode and showed the ability of the spacious active site to favorably accommodate two inhibitor molecules. However, the Zn-deficient AtIspF exhibited a weak selectivity towards different aryl bis-sulfonamides. The binding pattern changed dramatically in the presence of Zn2+. The relative binding affinity for the range of studied ligands followed the previously characterized trend in the corresponding IC50 values. The most potent inhibitor 8 was binding to AtIspF in our direct native ESI-MS titration experiments more readily, indicating a high binding affinity, while incubation of the protein with the inactive 13 resulted in negligibly small amount of complexes.
Aryl bis-sulfonamide binding proceeded with scrambling of the protein trimer to Zn2+ stoichiometry: we detected a range of the protein trimer and TLn complexes containing between zero and six Zn2+ ions. We attributed this observation to two parallel processes taking place in solution: first, the extraction of Zn2+ from the protein active site by the inhibitors, to form a dimeric chelate complex [L2Zn]2− in the bulk phase, and second, the binding of these [L2Zn]2− complexes to the protein. Hence, the former process reduced the number of zinc cations bound, whereas the latter increased the Zn2+:protein stoichiometric ratio. In agreement with our interpretation, zinc-depleted states were not observed in a control experiment when both the protein and the ligand were pre-incubated with Zn2+, with up to six ligands bound to the protein trimer. On the contrary, TLn complexes contained extra Zn2+ in addition to the three cations that were present in the protein originally: a total of four Zn2+ cations were found for n = 2 to 3, five for n = 4 to 5, and six for n = 6. Interestingly, the distribution of TLn peak intensities in mass spectra revealed a preference for the complexes binding four to six ligands (4 ≤ n ≤ 6), strongly indicating a positive cooperativity effect. Overall, these results are consistent with binding of a dimeric chelate complex of aryl bis-sulfonamide inhibitors with Zn2+ to the active sites of AtIspF trimer.
Based on the results of our analysis, we propose a mixed competitive and non-competitive mechanism of AtIspF inhibition by the synthetic aryl bis-sulfonamides. Competition experiments revealed that the natural substrate and bis-sulfonamide inhibitors compete for the same binding site in AtIspF, but the inhibitor binds weaker than 7, which is one of the reaction products and a moiety present in the substrate. However, non-competitive inhibition via zinc extraction from the active site has a more detrimental effect on the enzyme activity. Noteworthy, divalent metal cations are often present in the buffer for spectrophotometric enzyme-coupled assays, rendering the analysis of metal ion cofactor binding to proteins like AtIspF difficult.
While native ESI-MS has been extensively used in a number of proof-of-principle studies of well-characterized model proteins, application of this method to predict the behavior and binding mechanisms of truly unknown systems are rare. The work presented here demonstrates that the native ESI-MS approach can be efficiently used to elucidate unknown binding mechanisms of oligomeric proteins involved into complex, multistage binding equilibria with cofactors, drugs, and other ligands. The native ESI-MS titration approach provides a direct, label-free access to the information on protein complex composition and stoichiometry and quantitative analysis of binding. A careful design of experiment allows one to keep potential artefacts under control and get an insight into the specificity and selectivity of protein–ligand interactions. Binding of several ligands can be monitored simultaneously permitting parallel analysis of multiple binding equilibria and designing more sophisticated binding assays, such as ligand competition. In summary, the native ESI-MS data alone or in combination with complementary solution-phase assays and/or computational modelling deliver an unprecedented insight into the mechanisms of multimeric protein binding of several ligands.
Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
We thank the Swiss National Science Foundation for financial support of this research (grant # 200020_159929). The authors thank the group of Prof. Dr D. Hilvert for sharing the UV-spectrophotometer. We also thank Prof. Dr R. Riek's group for sharing their protein expression and purification facilities and Dr N. Nespovitaya for assistance in designing the method for recombinant AtIspF expression and purification. The data used in this publication are freely accessible in a curated data archive at ETH Zurich (http://https://www.researchcollection.ethz.ch) under the DOI: 10.3929/ethz-b-000270414.References
- P. Wolff, C. Da Veiga, E. Ennifar, G. Bec, G. Guichard, D. Burnouf and P. Dumas, J. Am. Soc. Mass Spectrom., 2017, 28, 347–357 CrossRef PubMed.
- A. Dyachenko, R. Gruber, L. Shimon, A. Horovitz and M. Sharon, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 7235–7239 CrossRef PubMed.
- N. Tahallah, M. Pinkse, C. S. Maier and A. J. Heck, Rapid Commun. Mass Spectrom., 2001, 15, 596–601 CrossRef PubMed.
- D. Cubrilovic, W. Haap, K. Barylyuk, A. Ruf, M. Badertscher, M. Gubler, T. Tetaz, C. Joseph, J. Benz and R. Zenobi, ACS Chem. Biol., 2014, 9, 218–226 CrossRef PubMed.
- J. L. P. Benesch and C. V. Robinson, Nature, 2009, 462, 576–577 CrossRef PubMed.
- P. Lossl, M. van de Waterbeemd and A. J. Heck, EMBO J., 2016, 35, 2634–2657 CrossRef PubMed.
- A. C. Leney and A. J. R. Heck, J. Am. Soc. Mass Spectrom., 2017, 28, 5–13 CrossRef PubMed.
- S. Mehmood, T. M. Allison and C. V. Robinson, Annu. Rev. Phys. Chem., 2015, 66, 453–474 CrossRef PubMed.
- C. Schmidt and C. V. Robinson, FEBS J., 2014, 281, 1950–1964 CrossRef PubMed.
- D. R. Benjamin, C. V. Robinson, J. P. Hendrick, F. U. Hartl and C. M. Dobson, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 7391–7395 CrossRef.
- J. Snijder, R. J. Rose, D. Veesler, J. E. Johnson and A. J. Heck, Angew. Chem., Int. Ed., 2013, 52, 4020–4023 CrossRef PubMed.
- C. Uetrecht, C. Versluis, N. R. Watts, P. T. Wingfield, A. C. Steven and A. J. R. Heck, Angew. Chem., Int. Ed., 2008, 47, 6247–6251 CrossRef PubMed.
- J. T. S. Hopper and C. V. Robinson, Angew. Chem., Int. Ed., 2014, 53, 14002–14015 CrossRef PubMed.
- H. Rogniaux, S. Sanglier, K. Strupat, S. Azza, O. Roitel, V. Ball, D. Tritsch, G. Branlant and A. Van Dorsselaer, Anal. Biochem., 2001, 291, 48–61 CrossRef PubMed.
- M. Goth, V. Badock, J. Weiske, K. Pagel and B. Kuropka, ChemMedChem, 2017, 12, 1201–1211 CrossRef PubMed.
- X. Chen, S. Qin, S. Chen, J. Li, L. Li, Z. Wang, Q. Wang, J. Lin, C. Yang and W. Shui, Sci. Rep., 2015, 5, 8361 CrossRef PubMed.
- S. Mehmood, J. Marcoux, J. Gault, A. Quigley, S. Michaelis, S. G. Young, E. P. Carpenter and C. V. Robinson, Nat. Chem., 2016, 8, 1152–1158 CrossRef PubMed.
- H. Jomaa, J. Wiesner, S. Sanderbrand, B. Altincicek, C. Weidemeyer, M. Hintz, I. Turbachova, M. Eberl, J. Zeidler, H. K. Lichtenthaler, D. Soldati and E. Beck, Science, 1999, 285, 1573–1576 CrossRef PubMed.
- H. K. Lichtenthaler, Annu. Rev. Plant Physiol. Plant Mol. Biol., 1999, 50, 47–65 CrossRef PubMed.
- B. M. Calisto, J. Perez-Gil, M. Bergua, J. Querol-Audi, I. Fita and S. Imperial, Protein Sci., 2007, 16, 2082–2088 CrossRef PubMed.
- C. Obiol-Pardo, J. Rubio-Martinez and S. Imperial, Curr. Med. Chem., 2011, 18, 1325–1338 CrossRef PubMed.
- A. Frank and M. Groll, Chem. Rev., 2017, 117, 5675–5703 CrossRef PubMed.
- S. Konzuch, T. Umeda, J. Held, S. Hahn, K. Brucher, C. Lienau, C. T. Behrendt, T. Grawert, A. Bacher, B. Illarionov, M. Fischer, B. Mordmuller, N. Tanaka and T. Kurz, J. Med. Chem., 2014, 57, 8827–8838 CrossRef PubMed.
- A. Schwab, B. Illarionov, A. Frank, A. Kunfermann, M. Seet, A. Bacher, M. C. Witschel, M. Fischer, M. Groll and F. Diederich, ACS Chem. Biol., 2017, 12, 2132–2138 CrossRef PubMed.
- A. Sandgren, M. Strong, P. Muthukrishnan, B. K. Weiner, G. M. Church and M. B. Murray, PLoS Med., 2009, 6, e2 CrossRef PubMed.
- A. Jain and P. Dixit, J. Biosci., 2008, 33, 605–616 CrossRef PubMed.
- A. Jain and R. Mondal, FEMS Immunol. Med. Microbiol., 2008, 53, 145–150 CrossRef PubMed.
- X. Wang and D. Moazed, Science, 2017, 356, 88–91 CrossRef PubMed.
- S. Steinbacher, J. Kaiser, J. Wungsintaweekul, S. Hecht, W. Eisenreich, S. Gerhardt, A. Bacher and F. Rohdich, J. Mol. Biol., 2002, 316, 79–88 CrossRef PubMed.
- V. Illarionova, J. Kaiser, E. Ostrozhenkova, A. Bacher, M. Fischer, W. Eisenreich and F. Rohdich, J. Org. Chem., 2006, 71, 8824–8834 CrossRef PubMed.
- B. Peters and H. J. Strek, Pest Manage. Sci., 2018 DOI:10.1002/ps.4768.
- M. C. Witschel, H. W. Hoffken, M. Seet, L. Parra, T. Mietzner, F. Thater, R. Niggeweg, F. Rohl, B. Illarionov, F. Rohdich, J. Kaiser, M. Fischer, A. Bacher and F. Diederich, Angew. Chem., Int. Ed., 2011, 50, 7931–7935 CrossRef PubMed.
- J. M. Green and M. D. K. Owen, Abstr. Pap., 2010, 59, 5819–5829 Search PubMed.
- A. Flaus and T. Owen-Hughes, Science, 2017, 355, 245–246 CrossRef PubMed.
- C. M. Crane, J. Kaiser, N. L. Ramsden, S. Lauw, F. Rohdich, W. Eisenreich, W. N. Hunter, A. Bacher and F. Diederich, Angew. Chem., Int. Ed., 2006, 45, 1069–1074 CrossRef PubMed.
- A. R. Odom and W. C. Van Voorhis, Mol. Biochem. Parasitol., 2010, 170, 108–111 CrossRef PubMed.
- T. Haemers, J. Wiesner, S. Van Poecke, J. Goeman, D. Henschker, E. Beck, H. Jomaa and S. Van Calenbergh, Bioorg. Med. Chem. Lett., 2006, 16, 1888–1891 CrossRef PubMed.
- H. K. Lichtenthaler, Biochem. Soc. Trans., 2000, 28, 785–789 CrossRef PubMed.
- F. Rohdich, A. Bacher and W. Eisenreich, Biochem. Soc. Trans., 2005, 33, 785–791 CrossRef PubMed.
- B. M. Lange, T. Rujan, W. Martin and R. Croteau, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 13172–13177 CrossRef PubMed.
- M. Rohmer, M. Seemann, S. Horbach, S. BringerMeyer and H. Sahm, J. Am. Chem. Soc., 1996, 118, 2564–2566 CrossRef.
- T. L. Campbell and E. D. Brown, J. Bacteriol., 2002, 184, 5609–5618 CrossRef PubMed.
- J. G. Kang, J. H. Hur, S. J. Choi, G. J. Choi, K. Y. Cho, L. N. Ten, K. H. Park and K. Y. Kang, Biosci., Biotechnol., Biochem., 2002, 66, 2677–2682 CrossRef PubMed.
- P. Purushottamachar, A. Khandelwal, T. S. Vasaitis, R. D. Bruno, L. K. Gediya and V. C. Njar, Bioorg. Med. Chem., 2008, 16, 3519–3529 CrossRef PubMed.
- J. G. Geist, S. Lauw, V. Illarionova, B. Illarionov, M. Fischer, T. Grawert, F. Rohdich, W. Eisenreich, J. Kaiser, M. Groll, C. Scheurer, S. Wittlin, J. L. Alonso-Gomez, W. B. Schweizer, A. Bacher and F. Diederich, ChemMedChem, 2010, 5, 1092–1101 CrossRef PubMed.
- J. Thelemann, B. Illarionov, K. Barylyuk, J. Geist, J. Kirchmair, P. Schneider, L. Anthore, K. Root, N. Trapp, A. Bacher, M. Witschel, R. Zenobi, M. Fischer, G. Schneider and F. Diederich, ChemMedChem, 2015, 10, 2090–2098 CrossRef PubMed.
- P. E. O'Rourke, J. Kalinowska-Tluscik, P. K. Fyfe, A. Dawson and W. N. Hunter, BMC Struct. Biol., 2014, 14, 1 CrossRef PubMed.
- S. V. Sciuto, J. Liu and L. Konermann, J. Am. Soc. Mass Spectrom., 2011, 22, 1679–1689 CrossRef PubMed.
- J. C. Jurchen and E. R. Williams, J. Am. Chem. Soc., 2003, 125, 2817–2826 CrossRef PubMed.
- R. L. Beardsley, C. M. Jones, A. S. Galhena and V. H. Wysocki, Anal. Chem., 2009, 81, 1347–1356 CrossRef PubMed.
- D. D. He and J. Marles-Wright, New Biotechnol., 2015, 32, 651–657 CrossRef PubMed.
- B. M. Calisto, J. Perez-Gil, M. Bergua, J. Querol-Audi, I. Fita and S. Imperial, Protein Sci., 2007, 16, 2082–2088 CrossRef PubMed.
- F. Rohdich, W. Eisenreich, J. Wungsintaweekul, S. Hecht, C. A. Schuhr and A. Bacher, Eur. J. Biochem., 2001, 268, 3190–3197 CrossRef PubMed.
- S. Herz, J. Wungsintaweekul, C. A. Schuhr, S. Hecht, H. Luttgen, S. Sagner, M. Fellermeier, W. Eisenreich, M. H. Zenk, A. Bacher and F. Rohdich, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 2486–2490 CrossRef PubMed.
- B. K. Ho and F. Gruswitz, BMC Struct. Biol., 2008, 8, 49 CrossRef PubMed.
- S. Mecozzi and J. Rebek, Chem.–Eur. J., 1998, 4, 1016–1022 CrossRef.
- G. Cavallo, P. Metrangolo, R. Milani, T. Pilati, A. Priimagi, G. Resnati and G. Terraneo, Chem. Rev., 2016, 116, 2478–2601 CrossRef PubMed.
- L. E. Kemp, C. S. Bond and W. N. Hunter, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 6591–6596 CrossRef PubMed.
- L. E. Kemp, M. S. Alphey, C. S. Bond, M. A. Ferguson, S. Hecht, A. Bacher, W. Eisenreich, F. Rohdich and W. N. Hunter, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2005, 61, 45–52 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures for the protein expression and purification, protein denaturation, buffer exchange and data analysis (PDF). See DOI: 10.1039/c8sc00814k |
| ‡ K. R. and K. B. contributed equally to this work. |
| § Present address: Department of Biochemistry, University of Cambridge, Hopkins Building, Downing Site, Tennis Court Road, CB2, 1QW, UK. |
| This journal is © The Royal Society of Chemistry 2018 |