
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA bi-terminal protein ligation strategy to probe chromatin structure during DNA damage†
Sinan
Kilic‡§
 ,
Iuliia
Boichenko‡
,
Carolin C.
Lechner¶
and
Beat
Fierz
*
,
Iuliia
Boichenko‡
,
Carolin C.
Lechner¶
and
Beat
Fierz
*
Laboratory of Biophysical Chemistry of Macromolecules, Institute of Chemical Sciences and Engineering, Ecole Polytechnique Fédérale de Lausanne (EPFL), 1015 Lausanne, Switzerland. E-mail: beat.fierz@epfl.ch
First published on 21st March 2018
Abstract
The cellular response to DNA damage results in a signaling cascade that primes chromatin for repair. Combinatorial post-translational modifications (PTMs) play an important role in this process by altering the physical properties of chromatin and recruiting downstream factors. One key signal integrator is the histone variant H2A.X, which is phosphorylated at a C-terminal serine (S139ph), and ubiquitylated within its N-terminal tail at lysines 13 and 15 (K13/15ub). How these PTMs directly impact chromatin structure and thereby facilitate DNA repair is not well understood. Detailed studies require synthetic access to such N- and C-terminally modified proteins. This is complicated by the requirement for protecting groups allowing multi-fragment assembly. Here, we report a semi-synthetic route to generate simultaneously N- and C-terminally modified proteins using genetically encoded orthogonal masking groups. Applied to H2A.X, expression of a central protein fragment, containing a protected N-terminal cysteine and a C-terminal thioester masked as a split intein, enables sequential C- and N-terminal protein modification and results in the convergent production of H2A.X carrying K15ub and S139ph. Using single-molecule FRET between defined nucleosomes in synthetic chromatin fibers, we then show that K15 ubiquitylation (but not S139ph) impairs nucleosome stacking in tetranucleosome units, opening chromatin during DNA repair.
Introduction
DNA stability is continuously compromised by light exposure, chemical reactants and the molecular machineries involved in transcription and replication.1 DNA lesions including DNA double-strand breaks (DSBs) trigger a signaling cascade, the DNA damage response (DDR), which results either in DNA repair or apoptosis.2 In eukaryotes, repair processes are complicated by the presence of chromatin. Nucleosomes, the basic units of chromatin, organize ∼147 bp of DNA around an octamer of the histone proteins H2A, H2B, H3 and H4. Strings of nucleosomes can form compact higher order structures, including segments of chromatin fibers.3–5 A fundamental ordering principle of chromatin fibers are tetranucleosome units, where four consecutive nucleosomes form two stacks connected by DNA in a zig-zag (two-start) pattern4–8 (Scheme 1a). Such chromatin organization restricts access to DNA, and thus hinders repair processes.9–11 DDR processes relieve this inhibition and prime the damaged chromatin region for repair through structural remodeling and chromatin expansion early in the cascade,12,13e.g. as observed by fluorescence imaging in live cells.14 The control of local chromatin structure involves histone post-translational modifications (PTMs).15–18 An early hallmark of DDR signaling is the ataxia-telangiectasia mutated (ATM) kinase-mediated phosphorylation of the histone variant H2A.X on serine 139 (H2A.X S139ph, also known as γH2A.X). This PTM is followed by ubiquitylation, i.e. the attachment of the ∼8 kDa protein ubiquitin to the ε-amino group of lysines 13 or 15 (K13/15ub) of γH2A.X or canonical H2A (Scheme 1a). Nucleosomes carrying such combinatorial PTMs, e.g. phosphorylated and ubiquitylated γH2A.X K15ub, function as recruitment cues for downstream DDR effectors,19 such as 53BP1 and BRCA1.20–23 Changes in chromatin structure, e.g. caused by PTMs on H2A.X,24 contribute to the recruitment mechanism of downstream effectors,25 further determining the subsequent repair pathway. As dysregulation of PTMs on H2A.X is implicated in radiosensitivity, immunodeficiency and cancer,26,27 it is thus important to understand how chromatin structure is remodeled by DDR-associated PTMs.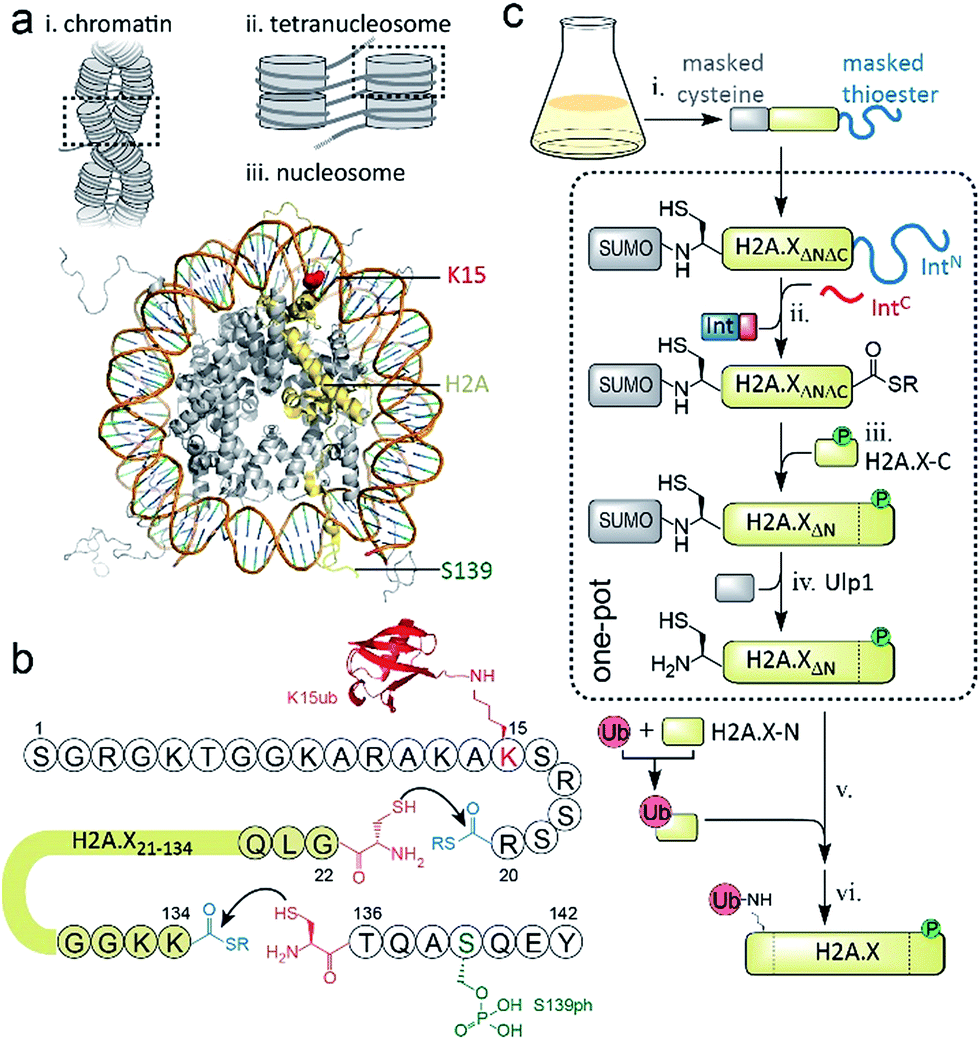 | ||
| Scheme 1 Semisynthetic strategy to produce γH2A.X K15ub. (a) Hierarchical chromatin structure: (i) chromatin fiber. (ii) An individual tetranucleosome unit. (iii) Nucleosome structure (PDB code: 1KX5). H2A is shown in yellow with indicated positions of K15 (red) and S139 (green). (b) Amino acid sequence of human H2A.X, showing the disconnections and EPL reactions to produce γH2A.X K15ub. (c) Scheme of the semisynthesis of γH2A.X K15ub: (i) recombinant expression of H2A.X with truncated N- and C-termini (H2A.XΔNΔC), N-terminally fused to SUMO and C-terminally fused to the N-terminal part of a split intein (IntN). (ii) Split-intein mediated thioester conversion (in the presence of the C-terminal intein fragment, IntC and small molecule thiols). (iii) Ligation to the phosphorylated H2A.X C-terminal octapeptide. (iv) Enzymatic N-terminal deprotection by SUMO protease Ulp1. (v) Ligation to semisynthetic, ubiquitylated H2A.X N-terminal fragment. (vi) Final desulfurization to yield γH2A.X K15ub. Steps (ii–iv), as well as (v and vi) were performed in one-pot. | ||
Chemically modified histones, e.g. prepared by expressed protein ligation (EPL),28 enable direct testing of the individual effects of each PTM on chromatin structure and dynamics.29 To study γH2A.X K15ub function, H2A.X has to be modified on both N- and C-termini. Multistep total synthesis approaches yield access to combinatorially modified proteins,30 using several protecting groups or kinetically controlled activation of thioesters.31–36 Semisynthetic methods can be more convenient, due to a reduced synthetic load. However, they can be difficult to implement, as orthogonal protection schemes in recombinant fragments are required. Here we report the facile convergent semisynthesis of γH2A.X K15ub, employing a recombinantly produced central H2A.X fragment, containing an N-terminal protected cysteine and a C-terminal crypto-thioester. Semisynthetic dual-modified γH2A.X K15ub (and singly-modified variants) are subsequently incorporated into synthetic chromatin fibers, which are further engineered to carry a FRET donor and acceptor dye pair in the DNA of neighboring nucleosomes at precise positions. Employing a single-molecule FRET approach, we demonstrate that S139ph does not alter chromatin structure, whereas K15ub disrupts inter-nucleosomal stacking and opens tetranucleosome units. Together, this shows that K15ub in H2A.X directly opens chromatin structure, providing chromatin access for repair proteins.
Results and discussion
EPL involves the reaction between a C-terminal thioester and an N-terminal cysteine (or related thiol-containing amino acid), resulting in the formation of a native peptide bond.28,37 To synthesize γH2A.X K15ub, we decided to disconnect H2A.X at two alanine residues at positions 21 and 135 (Scheme 1b). After ligation of both the ubiquitylated N-terminal peptide and the phosphorylated C-terminal fragment to the H2A.X core, the non-native cysteines C21 and C135 required for ligation are desulfurized to alanine, restoring the native histone sequence.38,39 In our semisynthetic strategy (Scheme 1c), we decided to produce the core of H2A.X, residues 21–134, recombinantly, thus simplifying the synthetic endeavor. We envisioned using both a genetically encoded N-terminal protection strategy as well as a recombinant intein-based crypto-thioester in this fragment. At the C-terminus of the H2A.X fragment, we decided to add the N-terminal half of the split intein from Nostoc punctiforme (NpuN). Upon addition of the C-terminal intein half (NpuC) and in the presence of suitable thiols this allows the installation of a thioester.40 For protection of the N-terminal cysteine residue, we settled upon a genetic fusion of the H2A.X fragment to small ubiquitin like modifier (SUMO). After a first ligation introducing the phosphorylated C-terminus, the SUMO protecting group can efficiently be removed by the highly specific ubiquitin-like protease 1 (Ulp1). This is then followed by the ligation of a convergently assembled, ubiquitylated N-terminus.41,42 We further envisioned that most steps of γH2A.X K15ub production, including thioester activation and N-terminal deprotection by Ulp1 can be performed in a one-pot reaction (Scheme 1c).To establish the synthetic strategy, we first synthesized the singly-modified γH2A.X. We thus expressed and purified the fragment H2A.X (1–134)-NpuN (1a) (Fig. S1a–c†). We further synthesized a peptide encompassing the C-terminus of H2A.X (residues 135–142, S139ph, 2) by Fmoc SPPS (47% isolated yield) (Fig. S1d–f†). We then proceeded to produce full-length γH2A.X by one-pot thioester conversion, ligation and desulfurization. Due to the poor solubility of 1a, all reactions were performed in 2 M urea, which prevented aggregation while still allowing intein function. Upon addition of the C-terminal intein half NpuC (mutated to prevent trans-splicing, Fig. S1g–i†) and mercapto-ethanesulfonate (MESNa), thioester conversion of 1a was initiated.40,43 We then directly added peptide 2 in the presence of the non-aromatic thiol catalyst methyl thioglycolate (MTG),44 which resulted in simultaneous thioester conversion and ligation (Fig. 1b). In the same mixture, cysteine 135 at the ligation site was finally desulfurized to the native alanine residue, employing radical-based chemistry.39,44 In summary, γH2A.X (3) was synthesized in a three-step one-pot reaction (∼42% isolated yield) (Fig. 1b and c).
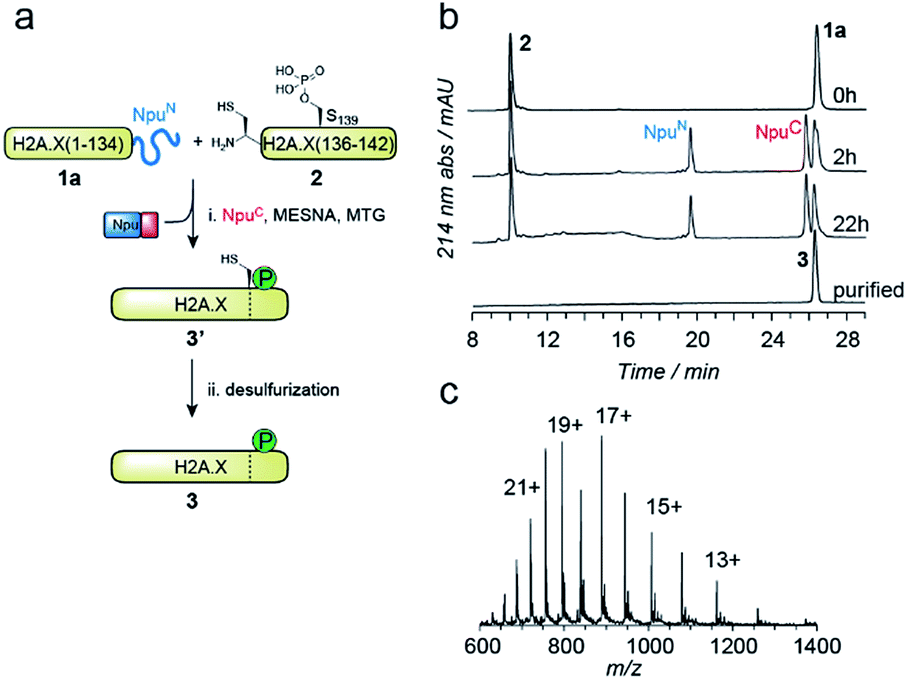 | ||
Fig. 1 One-pot semisynthesis of γH2A.X. (a) Synthetic scheme for the production of γH2A.X. (b) HPLC analysis of reaction progress for the ligation between 1a and 2 in the presence of MESNa, MTG and NpuC peptide. After 6 h of metal-free desulfurization in the same pot, γH2A.X 3 is purified. (c) Mass spectrometry analysis of purified 3. Observed mass is 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 095.0 Da. Expected mass is 15093.3 Da. 095.0 Da. Expected mass is 15093.3 Da. | ||
We thus turned to the task of establishing a convergent route to synthesize the N- and C-terminally modified histone γH2A.X K15ub. Following our envisioned route (Scheme 1b and c) the H2A.X core (21–134) A21C was expressed and purified as an N-terminal fusion to SUMO and as a C-terminal fusion with NpuN (1b, Fig. S2a–c†), enabling selective thioester conversion and N-terminal deprotection. We then initiated thioester formation and protein ligation by addition of the NpuC peptide, MESNa, MTG and phosphorylated H2A.X C-terminal peptide 2, in the presence of 2 M urea (Fig. 2a–b). After 16 h, the reaction was deemed complete by RP-HPLC and MS analysis, yielding ligation product 4a (Fig. S2d–f†). We then continued with the deprotection of the N-terminal cysteine using Ulp1, which proceeded to yield H2A.X (21–42) A21C S139ph (4b) in 90 minutes (Fig. 2a and b).
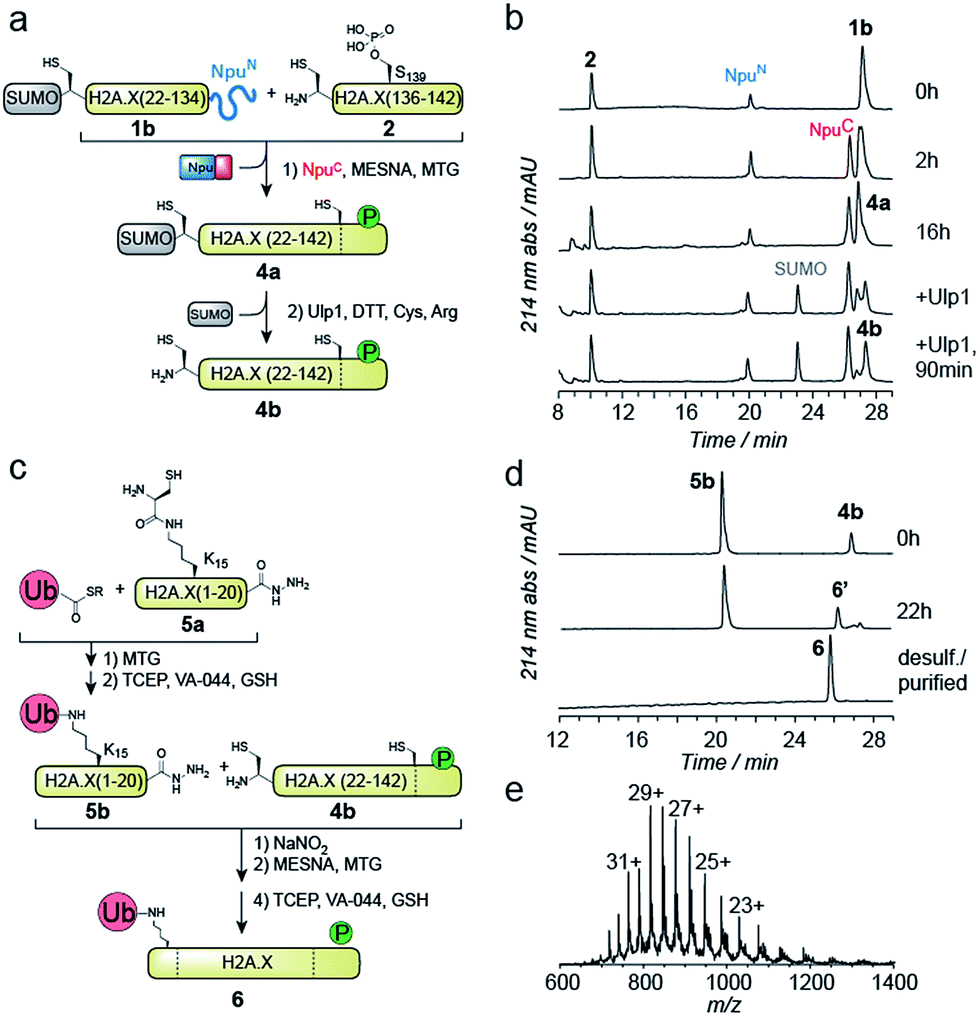 | ||
| Fig. 2 Convergent semisynthesis of γH2A.X K15ub using SUMO and NpuN as recombinant orthogonal protection groups. (a) Scheme for one-pot C-terminal thioester formation of 1b and ligation with 2 to form 4a, followed by N-terminal enzymatic deprotection of SUMO by Ulp1, resulting in 4b. (b) RP-HPLC analysis of the progress of the reactions shown in (a), at the given time points. (c) Scheme of preparation of ubiquitylated peptide 5b by one-pot ligation and desulfurization, followed by one-pot ligation and desulfurization of 5b to H2A.X (21–142) A21C S139ph, 4b, yielding γH2A.X K15ub, 6. (d) RP-HPLC analysis of the reactions shown in (c). (e) ESI-MS analysis of 6 (observed mass: 23658.2 Da, calculated mass: 23654.2 Da). | ||
At this point, we purified 4b by semipreparative RP-HPLC for subsequent ligation. In our convergent strategy, the ubiquitylated N-terminal peptide H2A.X (1–20) K15ub (5b) is prepared by ligation of ubiquitin thioester to H2A.X (1–20), 5a, carrying a cysteine residue attached to the ε-amine of K15.41 To allow subsequent attachment of 5b to the H2A.X core, we incorporated a C-terminal hydrazide, which can be converted into a thioester by oxidation with NaNO2 followed by the addition of thiols45 (Fig. 2c). We thus synthesized the hydrazide 5a, which was ligated to recombinant Ub (1–75) thioester in the presence of MTG (Fig. S2g–p†). Test reactions (using H2A instead of H2A.X) of hydrazide thioester conversion and subsequent ligation did however not result in reaction progress (Fig. S3a–d†). Instead we observed a loss of −32 Da (Fig. S3e–f†). This likely resulted from an intramolecular cyclization reaction between the C-terminal thioester and the cysteine residue remaining at the ubiquitin-H2A.X peptide ligation junction, a side reaction, which, to our surprise, irreversibly blocked further reaction progress (Fig. S3g†). Therefore, we decided to convert the offending cysteine at the ubiquitin ligation junction to alanine by desulfurization before purification of the intermediate, resulting in 5b (Fig. 2c).
With both 4b and 5b in hand, we proceeded to carry out the final ligation. We thus converted the hydrazide in 5b into a thioester and performed the ligation to 4b in the presence of MTG (Fig. 2d). After 22 h, the ligation was complete and the product was desulfurized, converting the two remaining non-native cysteines at positions 21 and 135 into the native alanine residues. The final product, γH2A.X K15ub (6), was subsequently purified by semi-preparative RP-HPLC (Fig. 2d and e, isolated yield 24%). Finally, we prepared H2A.X K15ub, following the same general strategy (Fig. S4†). Together, we thus produced dual modified γH2A.X K15ub (as well as the single-modified variants) in a convergent synthesis from four fragments, employing a central recombinant precursor containing genetically encoded orthogonal protection groups.
We then set out to determine the effects of H2A.X modifications on the modulation of chromatin organization. Some histone PTMs, including ubiquitylation of H2B at K120,16,46 have been shown to result in a loss of chromatin compaction. Recent structural studies identified the tetranucleosome as the fundamental structural unit of chromatin fibers.4–6 We thus decided to use a single-molecule FRET approach47 to measure the ability of nucleosomes containing modified H2A.X to form stacked tetranucleosome units within a chromatin fiber context (Fig. 3a). To this end, we employed a DNA template composed of a 12-mer tandem repeat of the 177 bp 601 nucleosome positioning sequence.48 To introduce FRET dyes into the ∼2 kilobase long chromatin template, we resorted to a multistep DNA ligation approach. Connecting recombinantly produced DNA fragments to PCR generated fluorescently labeled fragments, the two FRET dyes Alexa568 and Alexa647 (resulting in a Förster radius of 82 Å) were positioned within the DNA of nucleosomes 5 and 7 in the center of the 12-nucleosome containing chromatin fiber (Fig. S5†). Based on the crystal-structure of a tetranucleosome unit,4 dye positions were chosen such that they are separated by an interdye distance RDA of 46–64 Å within a compact tetranucleosome unit.47,49 We then proceeded to assemble histone octamers containing either unmodified H2A.X, γH2A.X, H2A.X K15ub or dual-modified γH2A.X K15ub (Fig. S6†). These histone octamers were used to reconstitute chromatin fibers (Fig. 3b).
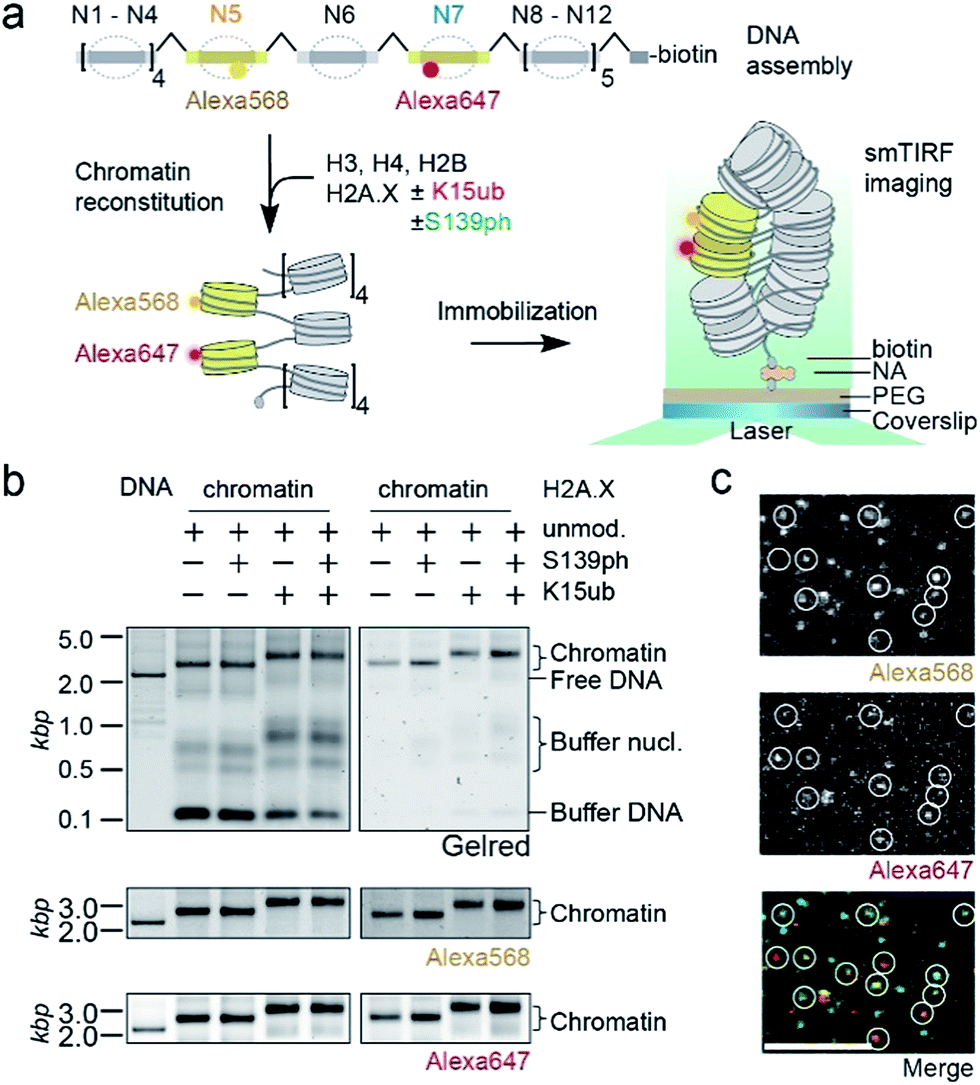 | ||
| Fig. 3 Single-molecule investigation of chromatin structure dependent on γH2A.X K15ub. (a) Scheme of the smFRET assay: DNA is assembled by DNA fragments, two of which (N5 and N7) contain the indicated FRET dyes. Chromatin fibers are reconstituted using the indicated histones, followed by smFRET measurements in a TIRF format. (b) Left panel: crude chromatin assembly, right panel: chromatin fibers purified by Mg2+ precipitation. (c) TIRF microscopy images showing single chromatin fibers containing unmodified H2A.X at 4 mM Mg2+, scale bar: 5 μm. | ||
Tetranucleosome stacking, and thus chromatin folding, can be initiated by addition of bivalent cations (e.g. Mg2+). Initial ensemble FRET experiments demonstrated energy transfer in the assembled chromatin fibers upon Mg2+ addition, and indicated a reduction of nucleosome packing in chromatin fibers containing K15ub, independent of the presence of S139ph (Fig. S7†). However, due to sample heterogeneity, photobleaching and ensemble averaging, exact FRET efficiency values (EFRET) are not accessible from those ensemble experiments. We thus used single-molecule imaging to measure chromatin fiber conformation on the single-fiber level. Using total internal reflection fluorescence (TIRF) imaging (Fig. 3c), we recorded EFRET time-traces from single chromatin fibers with a temporal resolution of 100 ms and Mg2+ concentrations from 0 to 4 mM (Fig. 4a–f). No dynamic structural transitions were observed in the EFRET time-traces, independent on the chromatin modification state, indicating that chromatin dynamics were faster than our time-resolution. Using traces from donor and acceptor dye-containing chromatin fibers, we then constructed EFRET histograms (Fig. 4d–f) for each chromatin state. After inducing tetranucleosome stacking with 4 mM Mg2+, both H2A.X and γH2A.X containing chromatin fibers exhibited a high FRET state with a EFRET distribution centered at 0.6 ± 0.001 (for H2A.X) and 0.59 ± 0.01 (for γH2A.X) (Fig. 4d and e). This indicates that these chromatin fibers form stacked tetranucleosome states, similarly to canonical H2A.47 A second, low EFRET state further reported on fibers in an unstacked conformation. The broad observed EFRET distributions indicate rapid dynamic processes beyond the time-resolution of our TIRF approach.47 γH2A.X K15ub however resulted in a significant reduction in EFRET both in the absence of Mg2+ and at 4 mM Mg2+, where the distribution was centered at EFRET = 0.41 ± 0.07 (Fig. 4f). K15 ubiquitylation (but not S139ph) thus directly disrupts tetranucleosome stacking. For chromatin fibers containing canonical H2A, we could previously identify the dynamically exchanging, underlying structural states that contribute to the observed EFRET value of ∼0.6 at 4 mM Mg2+.47 These states include stacking contacts between nucleosomes within a tetranucleosome unit (RDA = 64 Å) or between neighboring tetranucleosome units (RDA = 46 Å), populated to 35% and 23%, respectively. Assuming that ubiquitylation at K15 in γH2A.X reduces the molecular populations exhibiting close contacts, an overall EFRET value of ∼0.4 corresponds to a more than 50% reduction in compact states. Importantly, attachment of ubiquitin at H2A.X K15 places the ubiquitin moiety close to H2B K120. Ubiquitylation at this site has been shown to induce chromatin opening.16 Within tetranucleosomes, the N-terminal helix of H2A and the C-terminus of H2B of two neighboring nucleosomes form a four-helix bundle, that is susceptible to disruption by ubiquitylation. This region on the nucleosomal surface is thus a hotspot for controlling chromatin structure by PTMs.
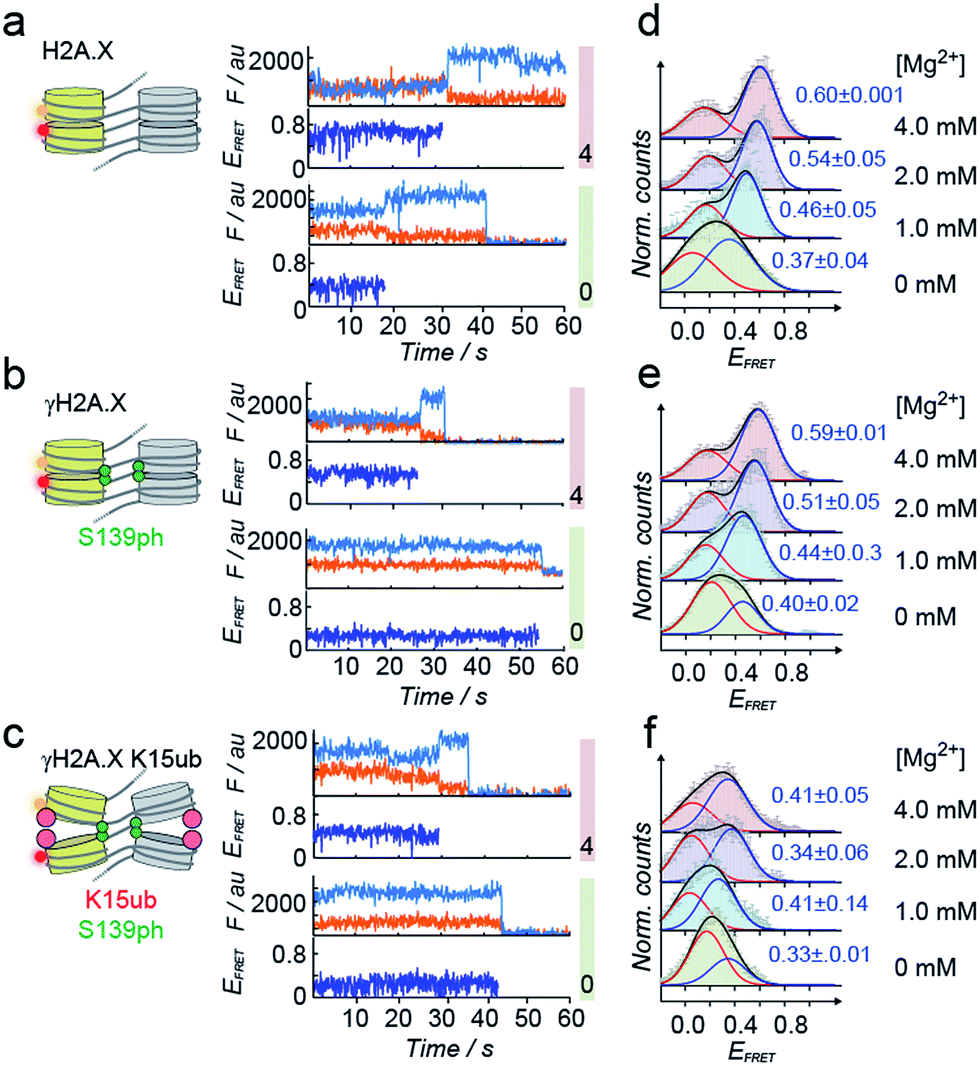 | ||
| Fig. 4 H2A.X K15ub inhibits tetranucleosome compaction. (a) Single-molecule traces (donor: orange, acceptor: red, FRET: blue) for H2A.X at 0 mM Mg2+ (bottom), 4 mM Mg2+ (top) until donor or acceptor photobleaching. (b) FRET traces for γH2A.X, same conditions as in (a). In the scheme, the green sphere indicates S139 phosphorylation. (c) FRET traces for γH2A.X K15ub, same conditions as in (a). The red sphere indicates H2AK15 ubiquitylation. (d) FRET populations observed for H2A.X at the indicated Mg2+ concentrations. The number indicates the center of the high-FRET population (±s.d. from 2 to 3 independent experiments). For the number of replicates and Gaussian fit parameters, see Table S2.† (e) FRET populations observed for γH2A.X at the indicated Mg2+ concentrations. (f) FRET populations observed for γH2A.X K15ub at the indicated Mg2+ concentrations. | ||
Conclusions
In summary, we have developed a general, genetically encoded protection scheme, by utilizing both SUMO as an N-terminal protection group and NpuN as a C-terminal crypto-thioester. This allowed a convergent assembly of γH2A.X K15ub from four starting polypeptides. Importantly, both orthogonal masking groups can individually be removed or converted in a single-pot reaction, enabling high-yield and traceless synthetic reactions. This is showcased in the synthesis of γH2A.X K15ub by the sequential activation of the thioester in the core fragment of H2A.X, followed by a C-terminal protein ligation reaction and a final by N-terminal SUMO deprotection without intermediate purification.Synthetic single- or dual-modified H2A.X proteins were then incorporated into reconstituted chromatin fibers, which carried precisely positioned FRET pairs. This allowed us to demonstrate that S139 phosphorylation, the defining mark of DNA damage and a key recruitment signal for downstream effector enzymes,2 does not directly alter chromatin structure. In contrast, K15 ubiquitylation impairs tetranucleosome stacking and thus chromatin higher-order organization. In addition to functioning as an important recruitment signal (together with S139ph) to coordinate effectors such as 53BP1,23,50,51 or RNF168,52 K15 ubiquitylation shapes chromatin towards a more open structure, thereby increasing accessibility of the nucleosomes and underlying DNA.
Materials and methods
All protein and peptide sequences are given in the ESI.† All reaction yields are tabulated in ESI Table S1.† Analytical data for all reactions (HPLC analyses, electrospray mass spectrometry) are given in ESI Fig. S1–4.† Data on DNA assembly is given in ESI Fig. S5.† Analytical data on histone octamer formation is reported in ESI Fig. S6.† Ensemble FRET spectra are given in ESI Fig. S7.† All Gaussian fit parameters for the analysis of the single-molecule data are given in ESI Table S2.† Detailed descriptions of experimental procedures and reagents are provide in the ESI.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Maeva Tobler and Jade A. Chevalley for materials, and Ruud Hovius for comments on the manuscript. This work was supported by the Sandoz Family Foundation, the Swiss National Science Foundation (grant 31003A_173169), the European Research Council through the 2017 Consolidator Grant chromo-SUMMIT (724022) and EPFL (B. F.). S. K. gratefully acknowledges a Boehringer Ingelheim Fonds PhD fellowship.Notes and references
- S. P. Jackson and J. Bartek, Nature, 2009, 461, 1071–1078 CrossRef CAS PubMed.
- M. Altmeyer and J. Lukas, Cell, 2013, 153, 1431–1434 CrossRef CAS PubMed.
- K. Luger, A. W. Mader, R. K. Richmond, D. F. Sargent and T. J. Richmond, Nature, 1997, 389, 251–260 CrossRef CAS PubMed.
- T. Schalch, S. Duda, D. F. Sargent and T. J. Richmond, Nature, 2005, 436, 138–141 CrossRef CAS PubMed.
- F. Song, P. Chen, D. Sun, M. Wang, L. Dong, D. Liang, R. M. Xu, P. Zhu and G. Li, Science, 2014, 344, 376–380 CrossRef CAS PubMed.
- W. Li, P. Chen, J. Yu, L. Dong, D. Liang, J. Feng, J. Yan, P. Y. Wang, Q. Li, Z. Zhang, M. Li and G. Li, Mol. Cell, 2016, 64, 120–133 CrossRef CAS PubMed.
- T. H. Hsieh, A. Weiner, B. Lajoie, J. Dekker, N. Friedman and O. J. Rando, Cell, 2015, 162, 108–119 CrossRef CAS PubMed.
- V. I. Risca, S. K. Denny, A. F. Straight and W. J. Greenleaf, Nature, 2017, 541, 237–241 CrossRef CAS PubMed.
- K. Luger, M. L. Dechassa and D. J. Tremethick, Nat. Rev. Mol. Cell Biol., 2012, 13, 436–447 CrossRef CAS PubMed.
- J. Widom, Proc. Natl. Acad. Sci. U. S. A., 1992, 89, 1095–1099 CrossRef CAS.
- M. H. Hauer, A. Seeber, V. Singh, R. Thierry, R. Sack, A. Amitai, M. Kryzhanovska, J. Eglinger, D. Holcman, T. Owen-Hughes and S. M. Gasser, Nat. Struct. Mol. Biol., 2017, 24, 99–107 CAS.
- G. Soria, S. E. Polo and G. Almouzni, Mol. Cell, 2012, 46, 722–734 CrossRef CAS PubMed.
- M. L. Li, G. Yuan and R. A. Greenberg, Trends Cell Biol., 2014, 24, 616–618 CrossRef CAS PubMed.
- M. J. Kruhlak, A. Celeste, G. Dellaire, O. Fernandez-Capetillo, W. G. Muller, J. G. McNally, D. P. Bazett-Jones and A. Nussenzweig, J. Cell Biol., 2006, 172, 823–834 CrossRef CAS PubMed.
- M. Shogren-Knaak, H. Ishii, J. M. Sun, M. J. Pazin, J. R. Davie and C. L. Peterson, Science, 2006, 311, 844–847 CrossRef CAS PubMed.
- B. Fierz, C. Chatterjee, R. K. McGinty, M. Bar-Dagan, D. P. Raleigh and T. W. Muir, Nat. Chem. Biol., 2011, 7, 113–119 CrossRef CAS PubMed.
- M. Simon, J. A. North, J. C. Shimko, R. A. Forties, M. B. Ferdinand, M. Manohar, M. Zhang, R. Fishel, J. J. Ottesen and M. G. Poirier, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 12711–12716 CrossRef CAS PubMed.
- A. Dhall, S. Wei, B. Fierz, C. L. Woodcock, T. H. Lee and C. Chatterjee, J. Biol. Chem., 2014, 289, 33827–33837 CrossRef CAS PubMed.
- A. Ciccia and S. J. Elledge, Mol. Cell, 2010, 40, 179–204 CrossRef CAS PubMed.
- S. Burma, B. P. Chen, M. Murphy, A. Kurimasa and D. J. Chen, J. Biol. Chem., 2001, 276, 42462–42467 CrossRef CAS PubMed.
- F. Mattiroli, J. H. Vissers, W. J. van Dijk, P. Ikpa, E. Citterio, W. Vermeulen, J. A. Marteijn and T. K. Sixma, Cell, 2012, 150, 1182–1195 CrossRef CAS PubMed.
- M. Gatti, S. Pinato, E. Maspero, P. Soffientini, S. Polo and L. Penengo, Cell Cycle, 2012, 11, 2538–2544 CrossRef CAS PubMed.
- A. Fradet-Turcotte, M. Canny, C. Escribano-Díaz, A. Orthwein, C. Leung, H. Huang, M.-C. Landry, J. Kitevski-LeBlanc, S. Noordermeer, F. Sicheri and D. Durocher, Nature, 2013, 499, 50–54 CrossRef CAS PubMed.
- M. K. Kocylowski, A. J. Rey, G. S. Stewart and T. D. Halazonetis, Cell Cycle, 2015, 14, 1748–1758 CrossRef CAS PubMed.
- Y. Huyen, O. Zgheib, R. A. Ditullio Jr, V. G. Gorgoulis, P. Zacharatos, T. J. Petty, E. A. Sheston, H. S. Mellert, E. S. Stavridi and T. D. Halazonetis, Nature, 2004, 432, 406–411 CrossRef CAS PubMed.
- G. S. Stewart, S. Panier, K. Townsend, A. K. Al-Hakim, N. K. Kolas, E. S. Miller, S. Nakada, J. Ylanko, S. Olivarius, M. Mendez, C. Oldreive, J. Wildenhain, A. Tagliaferro, L. Pelletier, N. Taubenheim, A. Durandy, P. J. Byrd, T. Stankovic, A. M. Taylor and D. Durocher, Cell, 2009, 136, 420–434 CrossRef CAS PubMed.
- T. Gudjonsson, M. Altmeyer, V. Savic, L. Toledo, C. Dinant, M. Grofte, J. Bartkova, M. Poulsen, Y. Oka, S. Bekker-Jensen, N. Mailand, B. Neumann, J. K. Heriche, R. Shearer, D. Saunders, J. Bartek, J. Lukas and C. Lukas, Cell, 2012, 150, 697–709 CrossRef CAS PubMed.
- T. W. Muir, D. Sondhi and P. A. Cole, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 6705–6710 CrossRef CAS.
- B. Fierz and T. W. Muir, Nat. Chem. Biol., 2012, 8, 417–427 CrossRef CAS PubMed.
- M. Seenaiah, M. Jbara, S. M. Mali and A. Brik, Angew. Chem., Int. Ed., 2015, 54, 12374–12378 CrossRef CAS PubMed.
- G. M. Fang, Y. M. Li, F. Shen, Y. C. Huang, J. B. Li, Y. Lin, H. K. Cui and L. Liu, Angew. Chem., Int. Ed., 2011, 50, 7645–7649 CrossRef CAS PubMed.
- D. Bang and S. B. Kent, Angew. Chem., Int. Ed., 2004, 43, 2534–2538 CrossRef CAS PubMed.
- R. E. Thompson, X. Liu, N. Alonso-Garcia, P. J. Pereira, K. A. Jolliffe and R. J. Payne, J. Am. Chem. Soc., 2014, 136, 8161–8164 CrossRef CAS PubMed.
- D. Bang, B. L. Pentelute and S. B. Kent, Angew. Chem., Int. Ed., 2006, 45, 3985–3988 CrossRef CAS PubMed.
- G. G. Kochendoerfer, S. Y. Chen, F. Mao, S. Cressman, S. Traviglia, H. Shao, C. L. Hunter, D. W. Low, E. N. Cagle, M. Carnevali, V. Gueriguian, P. J. Keogh, H. Porter, S. M. Stratton, M. C. Wiedeke, J. Wilken, J. Tang, J. J. Levy, L. P. Miranda, M. M. Crnogorac, S. Kalbag, P. Botti, J. Schindler-Horvat, L. Savatski, J. W. Adamson, A. Kung, S. B. Kent and J. A. Bradburne, Science, 2003, 299, 884–887 CrossRef CAS PubMed.
- J. B. Li, Y. K. Qi, Q. Q. He, H. S. Ai, S. L. Liu, J. X. Wang, J. S. Zheng, L. Liu and C. Tian, Cell Res., 2017 DOI:10.1038/cr.2017.157.
- P. E. Dawson, T. W. Muir, I. Clark-Lewis and S. B. H. Kent, Science, 1994, 266, 776–779 CAS.
- L. Z. Yan and P. E. Dawson, J. Am. Chem. Soc., 2001, 123, 526–533 CrossRef CAS PubMed.
- Q. Wan and S. J. Danishefsky, Angew. Chem., Int. Ed., 2007, 46, 9248–9252 CrossRef CAS PubMed.
- M. Vila-Perello, Z. Liu, N. H. Shah, J. A. Willis, J. Idoyaga and T. W. Muir, J. Am. Chem. Soc., 2013, 135, 286–292 CrossRef CAS PubMed.
- R. K. McGinty, M. Kohn, C. Chatterjee, K. P. Chiang, M. R. Pratt and T. W. Muir, ACS Chem. Biol., 2009, 4, 958–968 CrossRef CAS PubMed.
- B. Fierz, S. Kilic, A. R. Hieb, K. Luger and T. W. Muir, J. Am. Chem. Soc., 2012, 134, 19548–19551 CrossRef CAS PubMed.
- S. Frutos, J. B. Jordan, M. M. Bio, T. W. Muir, O. R. Thiel and M. Vila-Perello, Org. Biomol. Chem., 2016, 14, 9549–9553 CAS.
- Y. C. Huang, C. C. Chen, S. Gao, Y. H. Wang, H. Xiao, F. Wang, C. L. Tian and Y. M. Li, Chemistry, 2016, 22, 7623–7628 CrossRef CAS PubMed.
- G. M. Fang, J. X. Wang and L. Liu, Angew. Chem., Int. Ed., 2012, 51, 10347–10350 CrossRef CAS PubMed.
- G. T. Debelouchina, K. Gerecht and T. W. Muir, Nat. Chem. Biol., 2017, 13, 105–110 CrossRef CAS PubMed.
- S. Kilic, S. Felekyan, O. Doroshenko, I. Boichenko, M. Dimura, H. Vardanyan, L. C. Bryan, G. Arya, C. A. M. Seidel and B. Fierz, Nat. Commun., 2018, 9, 235 CrossRef PubMed.
- A. Thastrom, P. T. Lowary, H. R. Widlund, H. Cao, M. Kubista and J. Widom, J. Mol. Biol., 1999, 288, 213–229 CrossRef CAS PubMed.
- M. G. Poirier, E. Oh, H. S. Tims and J. Widom, Nat. Struct. Mol. Biol., 2009, 16, 938–944 CAS.
- R. E. Kleiner, P. Verma, K. R. Molloy, B. T. Chait and T. M. Kapoor, Nat. Chem. Biol., 2015, 11, 807–814 CrossRef CAS PubMed.
- Q. Hu, M. V. Botuyan, G. Cui, D. Zhao and G. Mer, Mol. Cell, 2017, 66, 473–487 CrossRef CAS PubMed.
- T. Thorslund, A. Ripplinger, S. Hoffmann, T. Wild, M. Uckelmann, B. Villumsen, T. Narita, T. K. Sixma, C. Choudhary, S. Bekker-Jensen and N. Mailand, Nature, 2015, 527, 389–393 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8sc00681d |
| ‡ These authors contributed equally to this work. |
| § Current address: Department of Molecular Mechanisms of Disease, University of Zürich, 8057 Zürich, Switzerland. |
| ¶ Current address: Bachem AG, 4416 Bubendorf, Switzerland. |
| This journal is © The Royal Society of Chemistry 2018 |