
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceReactivity enhancement of a diphosphene by reversible N-heterocyclic carbene coordination†
Debabrata
Dhara
 a,
Pankaj
Kalita
b,
Subhadip
Mondal
a,
Ramakirushnan Suriya
Narayanan
a,
Kaustubh R.
Mote
a,
Volker
Huch
c,
Michael
Zimmer
c,
Cem B.
Yildiz
*d,
David
Scheschkewitz
*c,
Vadapalli
Chandrasekhar
*ae and
Anukul
Jana
*a
a,
Pankaj
Kalita
b,
Subhadip
Mondal
a,
Ramakirushnan Suriya
Narayanan
a,
Kaustubh R.
Mote
a,
Volker
Huch
c,
Michael
Zimmer
c,
Cem B.
Yildiz
*d,
David
Scheschkewitz
*c,
Vadapalli
Chandrasekhar
*ae and
Anukul
Jana
*a
aTata Institute of Fundamental Research Hyderabad, Gopanpally, Hyderabad-500107, Telangana, India. E-mail: ajana@tifrh.res.in
bSchool of Chemical Sciences, National Institute of Science Education and Research, Bhimpur-Padanpur, Jatni, Khurda, Bhubaneswar-752050, Odisha, India
cKrupp-Chair of General and Inorganic Chemistry, Saarland University, 66123 Saarbrücken, Germany. E-mail: scheschkewitz@mx.uni-saarland.de
dDepartment of Medicinal and Aromatic Plants, University of Aksaray, Aksaray, Turkey. E-mail: cemburakyildiz@aksaray.edu.tr
eDepartment of Chemistry, Indian Institute of Technology Kanpur, Kanpur-208016, India. E-mail: vc@iitk.ac.in
First published on 27th March 2018
Abstract
Diphosphene TerMesP = PTerMes (1; TerMes = 2,6-Mes2C6H3; Mes = 2,4,6-Me3C6H2) and NHCMe42 (NHCMe4 = 1,3,4,5-tetramethylimidazol-2-ylidene) exist in an equilibrium mixture with the NHCMe4-coordinated diphosphene 3. While uncoordinated 1 is inert to hydrolysis, the NHC adduct 3 readily undergoes hydrolysis to afford a phosphino-substituted phosphine oxide with the liberation of NHCMe4. On this basis, conditions suitable for the catalytic use of NHCMe4 were identified. Similarly, while the hydrogenation of free diphosphene 1 with H3N·BH3 is very slow, 3 reacts instantaneously with H3N·BH3 at room temperature to afford a dihydrodiphosphane.
Introduction
Reversible coordination/binding between the substrate and catalyst is pivotal to catalytic reactions. This paradigm has been the raison d'être for the use of a vast number of transition metal complexes in catalysis as exemplified by many celebrated compounds such as Wilkinson's catalyst.1 Another crucial feature of many catalytic reactions is the role of auxiliary Lewis bases/donors in the activation of the catalyst to facilitate product formation.2 While transition metal complexes are a strong mainstay in this field, there have been efforts to examine if systems based on main-group elements can also be useful in catalytic reactions. An important challenge in this endeavor is the design of compounds that, like their transition metal counter parts, are involved in reversible reactions with substrates and auxiliary activators such as bases.3 An important breakthrough in this area has been the seminal report of Stephan and co-workers on the reversible activation of dihydrogen by the frustrated Lewis pair effect of a compound containing a P–B motif.4 Numerous other reports followed including the reversible coordination/binding of ethylene with distannynes,5 phosphines with CO2,6 N-heterocyclic carbenes with cyclotrisilenes,7 and reversible oxidative addition of organoboronate esters at the carbon(II) center of cyclic alkyl amino carbene.8 Main-group compounds can indeed display a transition metal-like behavior as exemplified by the stoichiometric activation of small molecules9 as well as ligand exchange at the reactive low-valent centers.10In the above context, recently Bertrand et al. reported a singlet (phosphino)phosphinidene, I, which is electrophilic despite its P–P multiple bond character and the presence of a formal negative charge at the phosphinidene center.11 It was also shown that ligand exchange could occur at the phosphinidene centre of I (Scheme 1).12 On the other hand, Robinson's NHCDip-stabilized P2, II (Scheme 1)13 which can be considered as an interconnected bis-phosphinidene dimer does not undergo ligand exchange with more nucleophilic NHCs such as NHCMe4 and NHCiPr2Me2.14
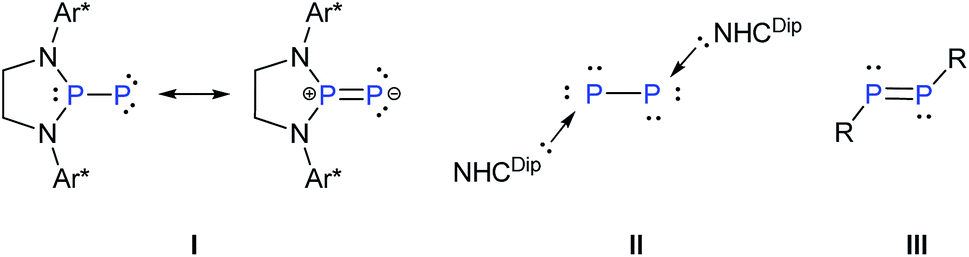 | ||
| Scheme 1 Chemical structures of phosphino phosphinidene I, donor stabilized diphosphorus(0) II, and diphosphene III (Ar* = 2,6-CHPh2-4-tBuC6H2, NHCDip = 1,3-(2,6-iPr2C6H3)2-imidazole-2-ylidene, R = general monoanionic ligand). | ||
The intriguing difference of reactivity between I and II and most importantly the fact that reversible coordination of Lewis bases to multiply bonded main group species is limited7 prompted us to investigate the coordination behaviour of an NHC as an auxiliary base towards a diphosphene III as well as the influence on its reactivity. In this context, it may be mentioned that Matsuo and co-workers recently reported the cleavage of the P–P double bond of Rind-substituted diphosphene (Rind = 1,1,3,3,5,5,7,7-octa-R-substituted s-hydrindacen-4-yl) by an NHC to yield two NHC-coordinated phosphinidene fragments. The mechanism was proposed to proceed via the formation of an NHC-coordinated, highly polarized diphosphene as a transient intermediate.15 The coordination of NHC to the heteronuclear and thus naturally polarised P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond of phosphaalkenes has been studied by Gates et al.16
C bond of phosphaalkenes has been studied by Gates et al.16
Results and discussion
Since the isolation of the sterically protected diphosphene, Mes*PPMes* (Mes* = 2,4,6-tBu3-C6H2), by Yoshifuji and co-workers17 several other diphosphenes have been isolated and characterized.18 We deliberately chose the relatively unreactive diphosphene TerMesPPTerMes, 119 and probed its binding with NHCMe4, 2.20 The treatment of 1 with a stoichiometric amount of 2 at room temperature resulted in an immediate colour change from yellow to deep red (Scheme 2). The 31P{1H} NMR of the reaction mixture showed two upfield doublets of equal intensity at δ = −95.36 ppm and −0.78 ppm (1J(P,P) = 423 Hz) along with the signal of the free diphosphene 1 at δ = 492 ppm in about 70![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :30 ratio. As this ratio remained unaltered even after 2 h, we suspected that the coordination of NHC might indeed be reversible. The remarkable difference in the chemical shifts accompanied by a large coupling constant proves the presence of two non-equivalent phosphorus nuclei in different electronic environments, which is consistent with the formation of the NHC-diphosphene adduct 3 (Scheme 2).
:30 ratio. As this ratio remained unaltered even after 2 h, we suspected that the coordination of NHC might indeed be reversible. The remarkable difference in the chemical shifts accompanied by a large coupling constant proves the presence of two non-equivalent phosphorus nuclei in different electronic environments, which is consistent with the formation of the NHC-diphosphene adduct 3 (Scheme 2).
 | ||
| Scheme 2 Reaction of 1 and 2 under the reversible formation of 3 (TerMes = 2,6-Mes2C6H3, Mes = 2,4,6-Me3C6H2, LA = Ph3B and ZnCl2). | ||
The single crystal X-ray diffraction analysis of 3 reveals that NHCMe4 is coordinated to one of the P-centres of the former diphosphene moiety (Fig. 1). The P–P bond length in 3 is 2.134(2) Å and thus longer than that observed in 1 (2.029(1) Å),21 but still substantially shorter than the P–P single bond of diphosphanes, e.g. (6-Me-2-pyridyl)(SiMe3)2CPH]2 (2.222(3) Å).22 The calculated Wiberg Bond Order (WBO) (1:1.812; 3:1.116) and the partial NBO charges (1: P1 = 0.280, P2 = 0.326; 3: P1 = 0.494, P2 = −0.116) at the B3LYP/6-311G(d,p) level of theory clearly indicate elongation and polarization of the PP bond upon coordination of NHC.23
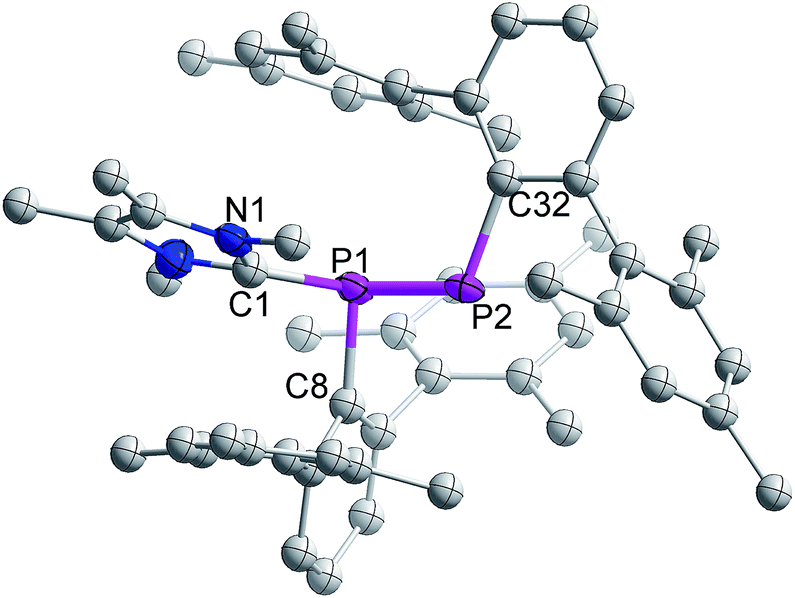 | ||
| Fig. 1 Molecular structure of 3 (thermal ellipsoids at 50% probability; hydrogen atoms omitted for clarity). | ||
The two nonbonding electron pairs at P2 implied by the ylidic nature of 3 correspond to the HOMO and HOMO−1 (Fig. 2). While the HOMO almost exclusively consists of a p-orbital centered at the formally negatively charged P center, the HOMO−1 and the HOMO−2 are delocalized across the P–P unit and therefore correspond to the s-type lone pairs at the phosphorus centers (Fig. 2). The LUMO is mainly delocalized over the P–C bond between the carbenic carbon and P1.
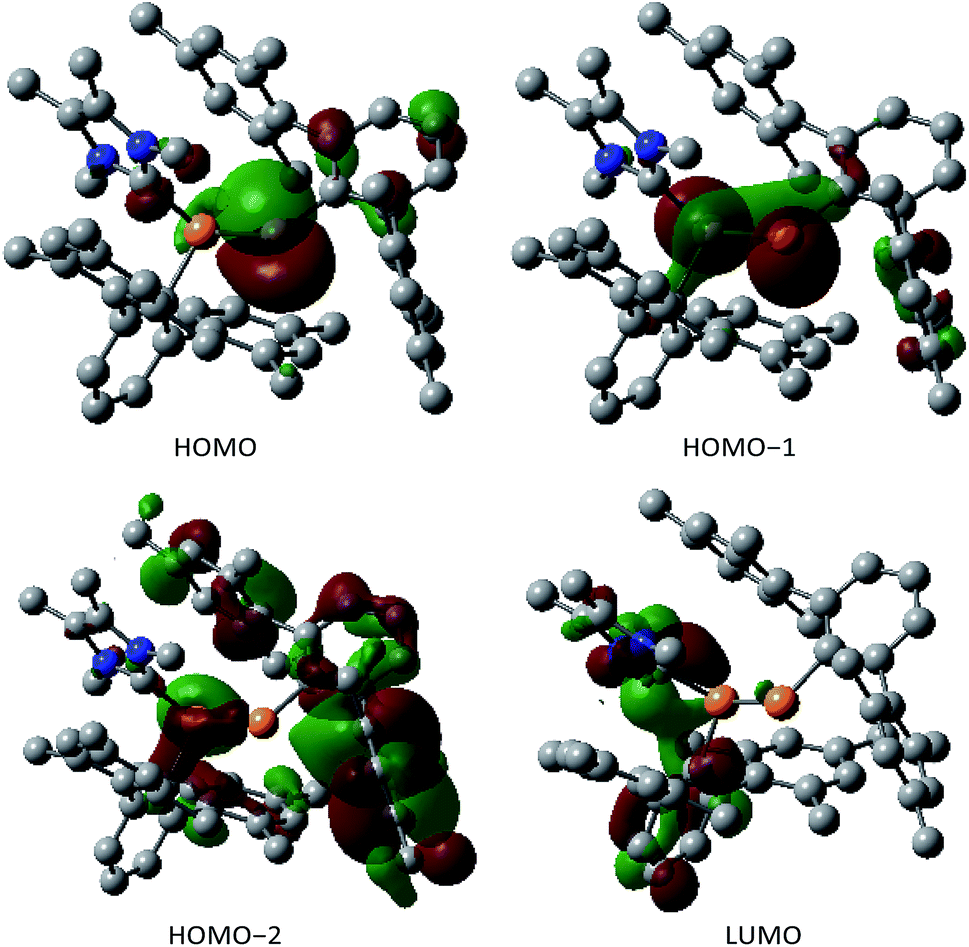 | ||
| Fig. 2 Frontier molecular orbitals of the compound 3 at 0.04 atomic units. | ||
The carbenic carbon C1 is connected to the P1 center with an angle of 113.26(18)° with respect to the P1–P2 bond vector. The two terphenyl ligands adopt a trans-arrangement with a torsion angle of C8–P1–P2–C32 = 166.8(2)°. The bond distance between the carbenic carbon and coordinated phosphorus of 3 is 1.876(2) Å and hence slightly larger than the corresponding distance in NHCMe4-coordinated phosphaalkene, MesPCPh2 (1.8512 Å).16b
To substantiate our assertion of reversible coordination of NHC to the PP moiety, we recorded the 31P{1H} NMR of a 1:1 mixture of 1 and 2 (0.0805 M solution in toluene-d8) at variable temperatures (253 to 293 K). Indeed, the proportion of adduct 3 increases at lower temperature as expected on grounds of entropic arguments. We calculated the Gibbs enthalpy of formation of 3 to be ΔG298 = −11.2 ± 1.1 kJ mol−1.23 In order to check the solvent dependency of the equilibrium, we recorded the solution NMR of a 1:1 mixture of 1 and 2 in C6D6 and THF-d8 at room temperature. While diphosphene 1 and NHC-adduct 3 are observed in an almost 3:7 ratio along with free NHCMe4 in the comparatively apolar C6D6, the more polar THF-d8 leads to a shift of equilibrium in favour of the polarized adduct 3 (90%). As expected on grounds of the law of mass action, the addition of 2 equivalents of NHCMe4 to 1 shifts the equilibrium almost completely towards the adduct 3. On the other hand, when equivalent amounts of Lewis acids such as Ph3B or ZnCl2 are added, NHCMe4 is effectively scavenged and free diphosphene 1 regenerated (Scheme 2). In order to confirm the exclusive presence of 3 in the solid state, we also performed CP-MAS 31P{1H} NMR.23 This reversible addition of NHCMe4, 2, to diphosphene, 1, has similarities with the reactivity of transition metal complexes in terms of ligand exchange.3
In order to further substantiate the reversibility of NHC coordination, we monitored the changes in the absorption of 1 (in THF) with increasing concentrations of 2 by UV/vis spectroscopy. A THF solution of free diphosphene 1 absorbs at λmax = 448, 372, and 317 nm. Upon gradual addition of NHCMe4, 2, the absorbance at 448 nm increases while the absorbance at 372 nm decreases in intensity as the equilibrium is shifted towards 3. The absorbance at λ = 317 nm is shifted to λ = 324 nm when the NHCMe4 concentration exceeds more than 1.11 equivalents (1.33, 2.22, and 4.44 equivalents). A clear isosbestic point is revealed at λ = 392 nm (Fig. 3). From the UV/vis studies the Gibbs enthalpy of formation of 3 is estimated to be ΔG298 = −13.30 kJ mol−1, which is close to the value obtained from the VT-NMR study (ΔG298 = −11.2 ± 1.1 kJ mol−1).23
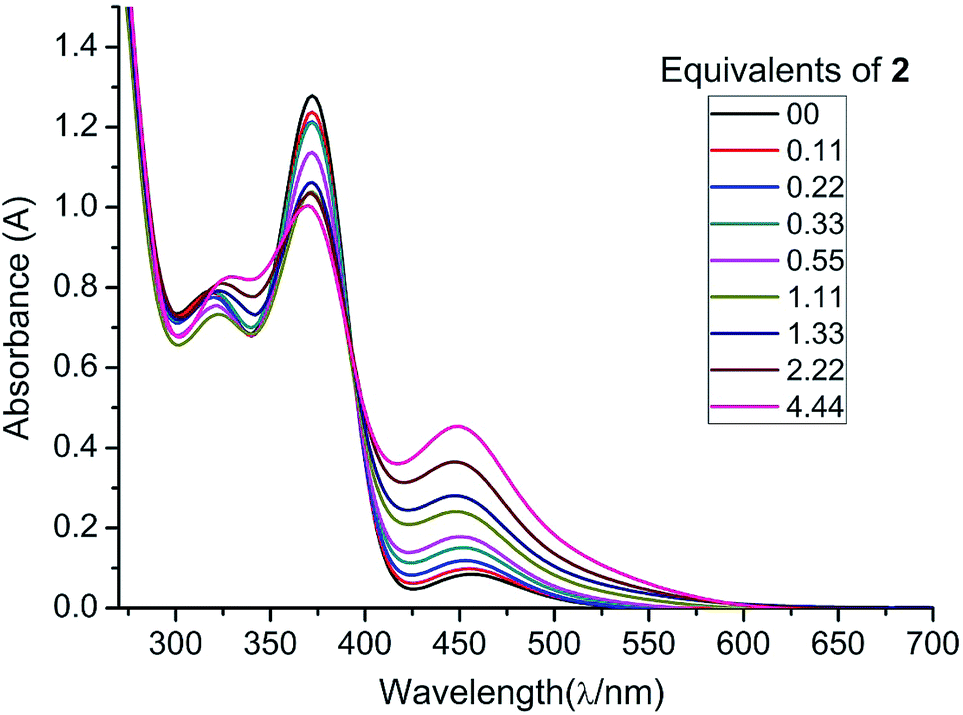 | ||
| Fig. 3 UV/vis spectra of 1 (black line) in THF with increasing concentrations of 2. | ||
Due to the pronounced polarization of the PP moiety upon coordination of NHCMe4, an enhanced reactivity was anticipated. We probed the addition reactions of water and dihydrogen in the presence of NHCMe4 as benchmarks as free diphosphene 1 is inert towards hydrolysis or hydrogenation (Scheme 3). Thus far, only the hydrolysis of Lewis acid-coordinated diphosphene and heteroleptic diphosphenes has been reported.24
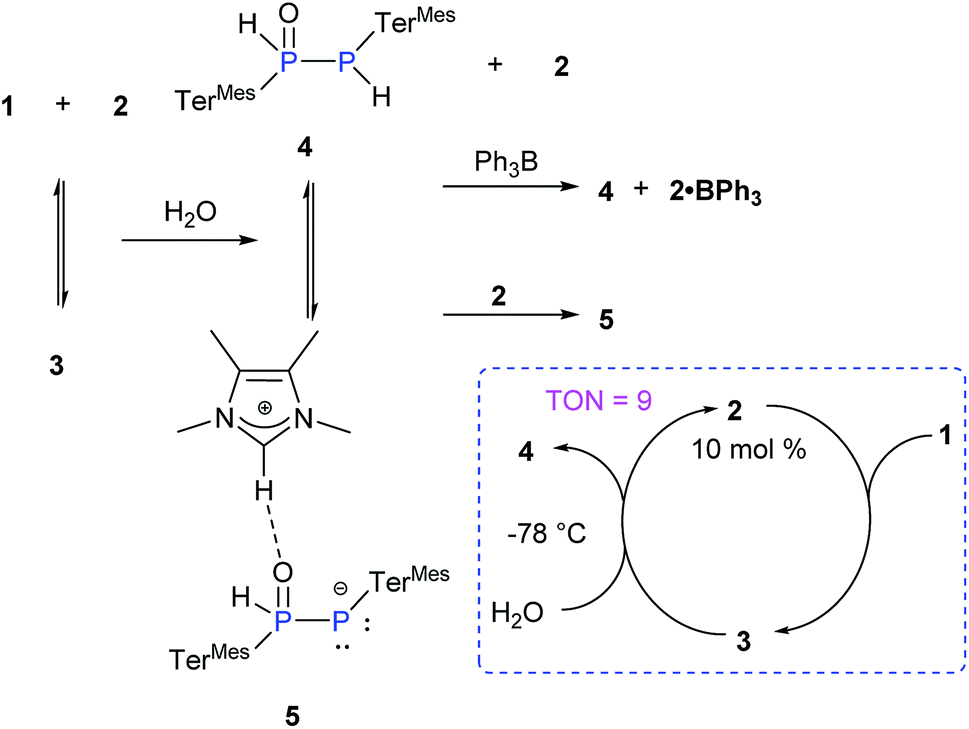 | ||
| Scheme 3 Reaction of 3 with H2O (TerMes = 2,6-Mes2C6H3, Mes = 2,4,6-Me3C6H2). | ||
Addition of one equivalent of H2O to a THF solution of 3 (1:1 mixture of 1 and 2) at RT led to an immediate colour change from red to yellow (Scheme 3). The presence of four new multiplets in the 31P NMR spectrum with each two of equal intensity at δ = 9.55 and −77.36 ppm and δ = 28.50 and −46.76 ppm indicates complete conversion to two new species.
Indeed, the molecular structure determination reveals the formation of phosphino substituted phosphine oxide 4 (Fig. 4) and phosphido phosphine oxide 5 (Fig. 5) in the course of the reaction (Scheme 3). The solid state structures of these two compounds match well with that of solution state NMR data. The P–P distances involved are 2.1946(8) Å for 4 and 2.1099(11) Å for 5. These distances are longer than those in 1 or 3 (1:1.985(2), 3:2.134 Å). The P–O bond length found in 4 is 1.478(2) Å, which is shorter compared with that of 5 (1.492(2) Å) presumably because of a H-bonding interaction with an imidazolium cation.25 The P–P distance in 4 (2.1946(8) Å) is slightly longer than that observed in the metal carbonyl (Lewis acid) free phospha-Wittig–Horner reagent, (Mes*PH–P(O)(OEt)2) (2.1854 Å).26
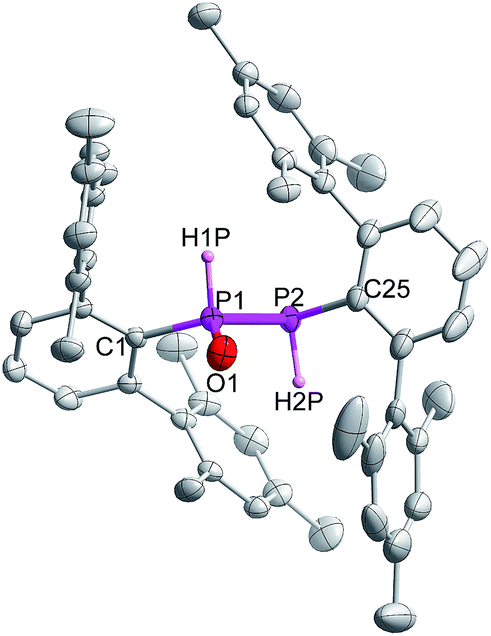 | ||
| Fig. 4 Molecular structure of 4 at thermal ellipsoids of 50% probability level; all the hydrogen atoms are omitted for clarity except P–H. | ||
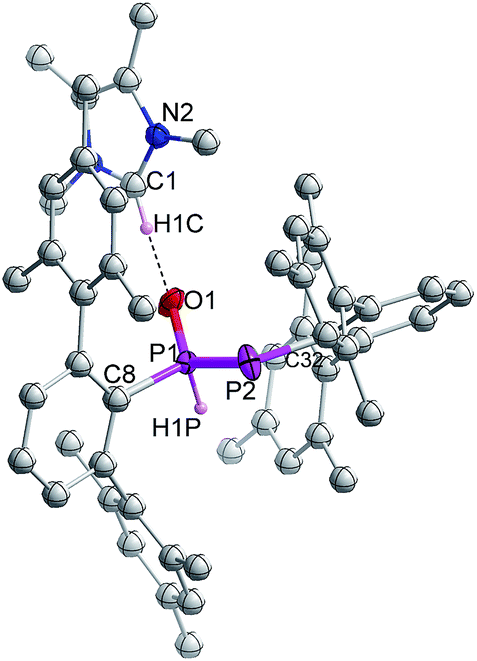 | ||
| Fig. 5 Molecular structures of 5 at thermal ellipsoids of 50% probability level; all the hydrogen atoms are omitted for clarity except P–H and imidazolium C–H. | ||
The Lewis acid free phospha-Wittig–Horner reagent, (Mes*PH-P(O)(OEt)2), is known to be readily deprotonated to the lithiated phospha–Wittig–Horner reagent, Mes*PP(OLi)(OEt)2.26 Similarly, the phosphino substituted phosphine oxide 4 is transformed to the corresponding ion pair 5 through deprotonation by NHCMe4, 2 (Scheme 3). The complete formation of 5 from 4 is observed when 2 equivalents of 2 are added. The 31P NMR spectrum of the phosphino substituted phosphine oxide 4 exhibits two doublets at δ = 9.55 and −77.36 ppm with a 1JP–P of 248 Hz. In 5 the corresponding resonances are observed at δ = 28.50 and −46.76 ppm with a 1JP–P of 464 Hz. These trends are similar to those observed in Mes*PH–P(O)(OEt)2 (1JP–P = 222 Hz) and Mes*PP(OLi)(OEt)2 (1JP–P = 615 Hz).26 Despite the imidazolium character of the NHC moiety in 5, the addition of BPh3 as a NHC scavenger results in the liberation of free 4 as previously observed for standard NHC-coordinated main group species (Scheme 3).27 The exclusive formation of 4 has also been achieved by the addition of Et3N·HCl to the reaction mixture. In view of the apparent equilibrium, we expected the hydrolysis of 1 to be catalytic in NHCMe4. In addition, the calculated relative free energy for the formation of 4 from 3 (3 + H2O → 4 + 2) is determined to be ΔG298 = −12.1 kcal mol−1 at the B3LYP-D3/6311-G(d,p) level of theory.23 Indeed, treatment of 1 at −78 °C with one equivalent of H2O in the presence of 10 mol% of 2 leads to >90% of 4 (TON = 9, Scheme 3).23 At higher temperatures, the hydrolysis of NHCMe4 becomes competitive significantly reducing the yield of 4 and thus the TON.
The reaction of 3 with H2 does not proceed under ambient conditions. On the other hand, 3 reacts instantaneously with NH3·BH3 as a dihydrogen source affording two diastereomers (d/l-6 and meso-6) of dihydrodiphosphane, (Scheme 4). The parent diphosphene 1 only reacts sluggishly with NH3·BH3 taking more than 7 days to afford the product in modest yields.
 | ||
| Scheme 4 Reaction of 3 with NH3·BH3 (TerMes = 2,6-Mes2C6H3, Mes = 2,4,6-Me3C6H2). | ||
The 31P NMR spectra of d/l-6 and meso-6 exhibit an AA′XX′ pattern with the peaks centered at δ = −109.9 and −101.4 ppm, respectively (Fig. 6). To determine the magnitudes of the coupling constants of the AA′XX′ spin system, a simulation of the 1H and 31P NMR spectra was performed (Fig. 6).28 Previously, Erickson et al. reported the formation of one of the diastereomers of compound 6 by the tin-catalyzed dehydrocoupling of TerMesPH2.29 Based on the current study it can be concluded that the previously reported diastereomer had the meso configuration. The initially formed racemic mixture of d/l-6 is slowly converted to the meso-isomer, meso-6, reaching equilibrium within 12 h at room temperature. This has been confirmed by performing the reaction and measuring 31P{1H} NMR at −50 °C. DFT calculations at the B3LYP-D3/6-311G(d,p) level of theory indeed show that the meso-6 isomer is lower in free energy than d/l-6, by ΔG298 = 2.3 kcal mol−1.23
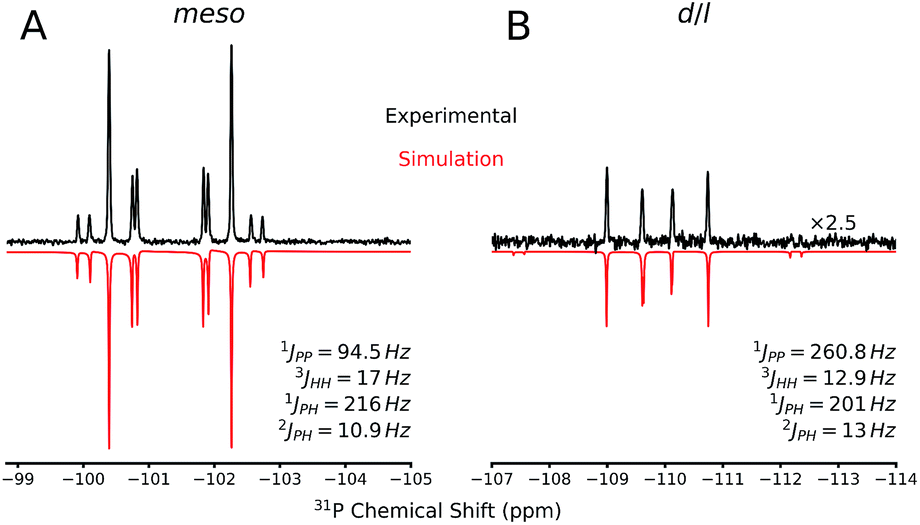 | ||
| Fig. 6 Comparison between the experimental (black, upper curve) and simulated (red, lower curve) 31P NMR spectrum of (A) meso-6 and (B) d/l-6 compounds. The scalar couplings used for the simulations are shown on the lower right side. | ||
Single crystal X-ray diffraction on crystalline samples of 6 exclusively resulted in a structural model in line with meso-6 (Fig. 7). As this model depends on the somewhat ambiguous determination of the hydrogen positions H1P and H1P*, we cannot exclude the presence of a mixture of isomers in the solid state. Indeed, the CP-MAS 31P{1H} NMR of a crystalline sample shows the presence of both diastereomers.
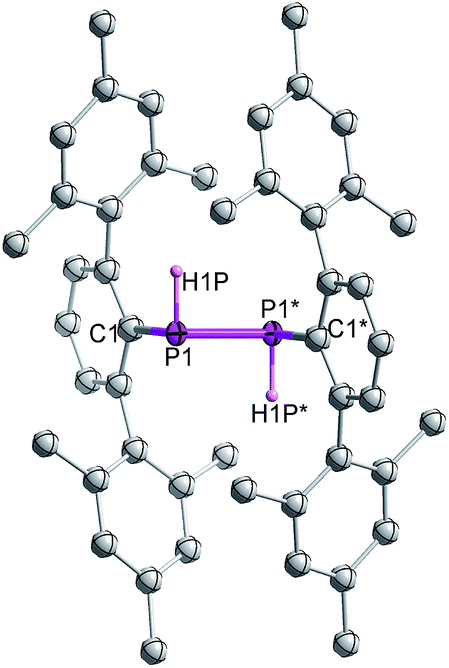 | ||
| Fig. 7 Molecular structure of meso-6 (thermal ellipsoids of 50% probability level; hydrogen atoms are omitted for clarity). | ||
Finally, to address the complete cleavage of the P–P bond present in 3, we monitored the 1:2 reaction between 1 and 2. After four days a colour change from deep red to yellow occurred and we observed the appearance of a new 31P resonance at δ = −77 ppm, which is in the range for a carbene-stabilized phosphinidene.30 Subsequently we heated the reaction mixture at 105 °C for 30 h to ensure complete dissociation resulting in the formation of 7 (Scheme 5).
 | ||
| Scheme 5 Reaction of 3 with NHCMe4 (TerMes = 2,6-Mes2C6H3, Mes = 2,4,6-Me3C6H2). | ||
The distance between phosphorus and the carbenic carbon is 1.786(2) Å (Fig. 8), which is comparable with that reported for a NHC-stabilized phosphinidene.31 However it is shorter than the distance between the phosphorus and carbenic carbon of 3 (1.8306(19) Å).
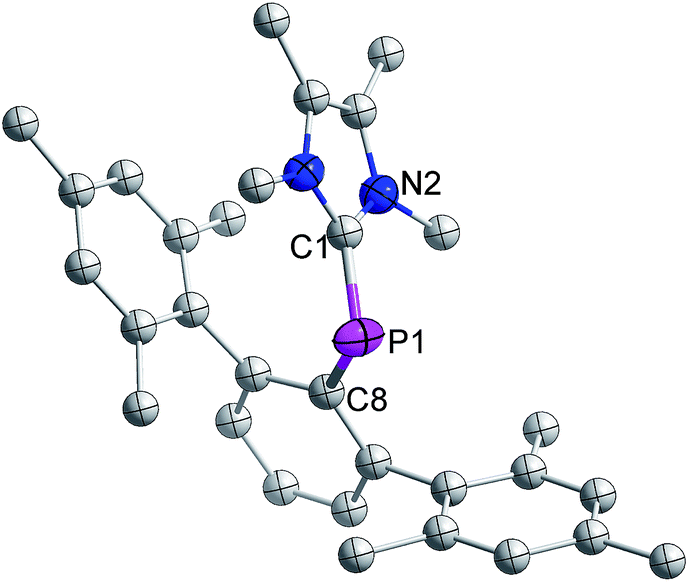 | ||
| Fig. 8 Molecular structure of 7 at thermal ellipsoids of 50% probability level; all hydrogen atoms are omitted for clarity. | ||
The formation of 7 from 3 is strong experimental corroboration for the mechanism proposed by Matsuo and co-workers for the cleavage of a PP bond in a diphosphene.15
Conclusions
In summary, we have demonstrated for the first time that the coordination of an N-heterocyclic carbene to a heavier double bond results in a striking reactivity enhancement. The reversible coordination of the NHC to a diphosphene significantly enhances the reaction rate of hydrolysis and hydrogenation by the ammonia–borane complex. In the case of hydrolysis, we have demonstrated that the reaction is catalytic in NHC.Experimental
General
All experiments were carried out under an argon atmosphere using standard Schlenk techniques or in a PL-HE-2GB Innovative Technology GloveBox. n-Hexane, diethyl ether, THF, and toluene were dried using a PS-MD-5 Innovative Technology solvent purification system. Benzene was refluxed over sodium/benzophenone, and then distilled and stored under argon. Starting compounds TerMes2P2, 119 and NHCMe4, 220 were prepared according to known literature procedures. Benzene-d6 was dried and distilled over potassium under argon. NMR spectra were recorded with a Bruker NanoBay 300 MHz NMR machine. 1H and 13C{1H} NMR spectra were referenced to the peaks of residual protons of the deuterated solvent (1H) or the deuterated solvent itself (13C{1H}). Solid-state 31P{1H} NMR spectra were obtained on a Bruker Avance 400 MHz spectrometer with a wide-bore magnet and operating at 400.13 MHz (1H) and 163.32 MHz (31P). Powdered samples were packed in a 4 mm o.d. zirconia rotor. The diameter of the Probe is 89 mm with a spinning speed of 13 KHz. The 31P{1H} CP MAS experiment was performed using a 1H 90° pulse of 3.3 μs, with a contact time of 5 ms, CPD Spinal64 as the decoupling scheme, and a recycle delay of 3 s. UV/vis spectra were acquired using a Jasco V-670 spectrometer using quartz cells with a path length of 0.1 cm. Elemental analyses were performed on a Leco CHN-900 analyzer. Melting points were determined in closed NMR tubes under an argon atmosphere and are uncorrected.Synthesis of 3
10 mL of THF is added to a Schlenk flask containing 1 (0.357 g, 0.518 mmol) and 2 (0.084 g, 0.673 mmol) at room temperature. After stirring for about 5 minutes, all volatiles are removed under vacuum. The resulting residue is extracted with 10 mL of n-pentane (2 times) and the combined filtrate is kept at −30 °C overnight resulting in the formation of bright red crystals, yield: 0.211 g (51.9%). In solution compound 3 is in equilibrium with 1 and 2. NMR data from a 1:1 stoichiometric ratio of 1 and 2 in C6D6 at RT (almost 30% of 1 and 2 and 70% of 3): mp: 185 °C (decomposed). 1H NMR (300 MHz, C6D6, 298 K): δ = 1.25 (s, 3H, C–CH3 of NHCMe4), 1.30 (s, 3H, C–CH3 of NHCMe4), 1.57 (s, 2H, C–CH3 of 2), 1.89 (br, 14H, 6H from CH3 of Mes and remaining 8H from CH3 of Mes of 1), 2.17 (s, 18H, CH3 of Mes), 2.21 (s, 16H, 12H from CH3 of Mes and remaining 4H from CH3 of Mes of 1), 2.66 (s, 3H, N–CH3 of NHCMe4), 3.35 (br, 5H, 3H from N–CH3 and 2H from N–CH3 of 2), 6.53 (s, br, 2H, Ar–H), 6.62–6.67 (m, 4H, Ar–H), and 6.78–7.04 (m 13H, 8H from Ar–H and 5H from Ar–H of 1) ppm. NMR data from a 1:2 stoichiometric ratio of 1 and 2 in C6D6 at RT to get unambiguous 1H NMR data of 3. 1H NMR (300 MHz, C6D6, 298 K): δ = 1.28 (s, 3H, C–CH3 of NHCMe4), 1.33 (s, 3H, C–CH3 of NHCMe4), 1.82 (br, 6H, CH3 of Mes), 2.16 (s, 18H, CH3 of Mes), 2.21 (s, 12H, CH3 of Mes), 2.66 (s, 3H, N–CH3 of NHCMe4), 3.36 (s, br, 3H, N–CH3 of NHCMe4 and 6H, N–CH3 of 2), 6.52 (s, br, 2H, Ar–H), 6.62–6.66 (m, 4H, Ar–H), 6.78 (s, br, 4H, Ar–H), and 6.86–7.04 (m, 4H, Ar–H) ppm. 13C{1H} NMR (75.43 MHz, C6D6, 298 K): δ = 7.73 (1C, C–CH3 of NHCMe4), 8.29 (1C, C–CH3 of NHCMe4), 21.07 (3C, CH3 of Mes), 21.37 (4C, CH3 of Mes), 21.92 (3C, CH3 of Mes), 22.66 (1C, CH3 of Mes), 22.88 (1C, CH3 of Mes), 30.46 (1C, d, 3J(P,C) = 26.85 Hz, N–CH3 of NHCMe4), 32.83 (1C, d, 3J(P,C) = 19.39 Hz, N–CH3 of NHCMe4), 120.00 (1C, Ar–CH), 123.04 (1C, CH3–C of NHCMe4), 125.82 (1C, CH3–C of NHCMe4), 126.89 (1C, Ar–CH), 127.31 (2C, Ar–CH), 127.63 (2C, Ar–CH), 127.98 (2C, Ar–CH), 128.29 (1C, Ar–CH), 128.58 (3C, Ar–CH), 130.46 (1C, Ar–CH), 130.51 (1C, Ar–CH), 133.77 (2C, Ar–Cquat), 135.55 (2C, Ar–Cquat), 135.70 (3C, Ar–Cquat), 137.46 (2C, Ar–Cquat), 138.22 (1C, Ar–Cquat), 138.40 (1C, Ar–Cquat), 138.78 (2C, Ar–Cquat), 140.95 (2C, Ar–Cquat), 142.78 (1C, Ar–Cquat), 142.84 (2C, Ar–Cquat), 146.93 (1C, Ar–Cquat), 147.18 (1C, Ar–Cquat), 150.17 (1C, Ar–Cquat), 150.28 (1C, NCN), 151.83 (1C, d, 1J(C,P) = 9.50 Hz, Ar–Cquat), and 152.33 (1C, Ar–Cquat) ppm. 31P NMR (121.5 MHz, C6D6, 298 K): δ = −95.36 (d, 1J(P,P) = 423 Hz) and −0.78 (d, 1J(P,P) = 423 Hz) ppm. CP-MAS 31P{1H} NMR (163.32 MHz, 298 K): δ = −99.07 (d, 1J(p,p) = 430 Hz, 1P) and 3.99 (d, 1J(p,p) = 430 Hz, 1P) ppm. UV/vis (n-hexane): IR (KBr, cm−1): ![[small nu, Greek, macron]](https://www.rsc.org/images/entities/i_char_e0ce.gif) = 405 (s), 414 (vs), 422 (s), 435 (w), 449 (vs), 474 (vw), 489 (vw), 501 (vw), 530 (m), 548 (vw), 576 (m), 588 (m), 657 (vw), 669 (m), 712 (s) 738 (br, s), 768 (w), 788 (s), 802 (s), 846 (vs), 872 (vw), 907 (br, vw), 998 (vw), 1030 (s), 1080 (w), 1107 (w), 1178 (m), 1239 (w), 1363 (m), 1439 (m), 1569 (w), 1607 (w), 1652 (w), and 2356 (w).
= 405 (s), 414 (vs), 422 (s), 435 (w), 449 (vs), 474 (vw), 489 (vw), 501 (vw), 530 (m), 548 (vw), 576 (m), 588 (m), 657 (vw), 669 (m), 712 (s) 738 (br, s), 768 (w), 788 (s), 802 (s), 846 (vs), 872 (vw), 907 (br, vw), 998 (vw), 1030 (s), 1080 (w), 1107 (w), 1178 (m), 1239 (w), 1363 (m), 1439 (m), 1569 (w), 1607 (w), 1652 (w), and 2356 (w).
Reaction of 3 with BPh3
In a glovebox, 1 (0.020 g, 0.029 mmol) and 2 (0.0047 g, 0.037 mmol) were dissolved in THF (0.2 mL) in an NMR tube. After about 5 minutes BPh3 (0.0091 g, 0.037 mmol, in 0.2 mL THF) was added resulting in an instant colour change from red to yellow. Subsequent addition of 0.1 mL C6D6 allowed for the recording of a 31P NMR spectrum, which revealed the complete conversion of 2 into 1.Synthesis of 2·BPh3
Inside the glovebox, 0.019 g (0.08 mmol) of BPh3 was weighed into an NMR tube. A benzene-d6 solution of 0.010 g (0.08 mmol, in 0.5 mL C6D6) of 1 was added at room temperature. 1H NMR measurements confirm the formation of 2·BPh3. 1H NMR (300 MHz, C6D6, 298 K): δ = 1.11 (s, 6H, CH3C–CCH3), 2.63 (s, 6H, NCH3), 7.18–7.27 (m, 3H, Ar–H), 7.34 (t, 3J(H,H) = 7.40 Hz, 6H, ArH), and 7.68 (d, 3J(H,H) = 6.95 Hz, 6H, ArH) ppm.Reaction of 3 with ZnCl2
Inside the glovebox, 1 (0.024 g, 0.035 mmol) and 2 (0.0056 g, 0.045 mmol) were dissolved in THF (0.2 mL) in an NMR tube. After about 5 minutes ZnCl2 (0.0061 g, 0.045 mmol, in 0.2 mL THF) was added leading to an instant colour change from red to yellow. After addition of 0.1 mL C6D6, a 31P NMR spectrum was recorded showing complete conversion of 2 into 1.Synthesis of 4
In a 50 mL Schlenk flask, 0.392 g (0.57 mmol) of compound 1 and 0.080 g (0.64 mmol) of 2 were dissolved in 15 mL of THF. The reaction mixture was stirred for 10 minutes. Subsequently, degassed water (12 μL, 0.65 mmol) was added at room temperature and allowed to stir for 1.5 h. The red colour of the solution slowly transformed into orange. This reaction mixture was then added into a THF (5 mL) suspension of Et3N·HCl (0.094 g, 0.68 mmol) and allowed to stir for 30 minutes resulting in the gradual fading of the colour. All volatiles were then evaporated and extracted with hot n-hexane (15 mL). Incipient crystallization happened at room temperature. Isolated yield 0.310 g (78%). Mp: >190 °C. 1H NMR (300 MHz, C6D6, 298 K): δ = 1.79 (s, 6H, CH3 of Mes), 1.93 (s, 6H, CH3 of Mes), 2.05 (s, 6H, CH3 of Mes), 2.28 (s, 6H, CH3 of Mes), 2.31 (s, 12H, CH3 of Mes), 3.21 (ddd, 1J(P,H) = 248.70 Hz, 2J(H,P) = 13.34 Hz, 3J(H,H) = 8.10 Hz, 1H, HP(O)–P–H), 7.19 (ddd, 1J(P,H) = 473.31 Hz, 2J(H,P) = 32.35 Hz, 3J(H,H) = 8.10 Hz, 1H, H–P(O)–PH), 6.70–6.74 (m, 4H, Ar–H), 6.77 (br, 1H, Ar–H), 6.79–6.81 (m, 3H, Ar–H), 6.87 (s, 4H, Ar–H), and 6.96–7.07 (m, 2H, Ar–H) ppm. 13C{1H} NMR (75.43 MHz, C6D6, 298 K): δ = 21.03 (2C, CH3 of Mes), 21.37 (2C, CH3 of Mes), 21.63 (1C, CH3 of Mes), 21.72 (6C, CH3 of Mes), 21.79 (1C, CH3 of Mes), 129.01 (2C, Ar–CH), 129.04 (2C, Ar–CH), 129.14 (2C, Ar–CH), 129.36 (2C, Ar–CH), 129.47 (1C, Ar–CH), 129.87 (1C, Ar–CH), 129.98 (3C, Ar–CH), 132.58 (d, 4J(C,P) = 2.27Hz, 1C, Ar–CH), 133.42 (d, 1J(P,C) = 4.52 Hz, 1C, Ar–Cquat), 134.51 (d, 1J(P,C) = 4.52 Hz, 1C, Ar–Cquat), 136.74 (2C, Ar–Cquat), 136.75 (2C, Ar–Cquat), 136.77 (2C, Ar–Cquat), 137.93 (1C, Ar–Cquat), 139.50 (2C, Ar–Cquat), 145.70 (1C, Ar–Cquat), 145.83 (1C, Ar–Cquat), 147.58 (d, 1C, J(C,P) = 5.28 Hz), and 147.73 (d, 1C, J(C,P) = 4.52 Hz) ppm. 31P NMR (121.5 MHz, C6D6, 298 K): δ = 9.55 (ddd, 1J(P,P) = 248.70 Hz, 1J(P,H) = 473.31 Hz, 2J(H,P) = 13.34 Hz, HP(O)–PH) and −77.36 (dt, 1J(P,P) = 1J(P,H) = 248.70 Hz, 2J(H,P) = 32.35 Hz, Ter-P-H) ppm. 31P{1H} NMR (121.5 MHz, C6D6, 298 K): δ = 9.55 (d, 1J(P,P) = 248.70 Hz, HP(O)–PH) and −77.36 (d, 1J(P,P) = 248.70 Hz, Ter-P-H) ppm. CP-MAS 31P{1H} NMR (163.32 MHz, 298 K): δ = 9.64 (d, 1J(p,p) = 230 Hz, HP(O)–PH) and −78.48 (d, 1J(p,p) = 230 Hz, Ter-P-H) ppm. IR (KBr, cm−1): = 1131 (s), 1187 (s), 1232 (s), 1307 (w), 1370 (m), 1412 (w), 1431 (m), 1450 (m), 1466 (w), 1482 (w), 1501 (m), 1513 (w), 1534 (s), 1553 (s), 1564 (m), 1605 (m), 1633 (w), 1677 (m), 1691 (m), 1712 (m), 1726 (m), 1764 (w), 1785 (w), 1820 (w), 1841 (w), 1862 (w), 1886 (w), 1500 (w), 1555 (s), 1570 (m), 1610 (m), 1641 (m), 1676 (m) 1711 (m), 1727 (m), 2313 (m, P–H), 2352 (s, P–H), 2727 (m), and 2849 (w). Elemental analysis: calcd for C48H52OP2 (706.87): C, 81.56; H, 7.41. Found: C, 80.81; H, 7.17.
Synthesis of 5
In a 50 mL Schlenk flask, 0.510 g (0.740 mmol) of compound 1 and 0.120 g (0.960 mmol) of 2 were dissolved in 15 mL of THF. Water (17.4 μL, 0.960 mmol) was added at room temperature and allowed to stir for 1.5 h. The red colour of the solution slowly transformed into orange. The reaction mixture was then evaporated and extracted with hot toluene (20 mL). Crystallization occurred upon cooling at room temperature. Isolated yield 0.530 g (86.13%). Mp: >190 °C (decomposed) and at 175 °C the color changed slowly towards white. 1H NMR (300 MHz, THF-d8, 298 K): δ = 1.80 (s, 6H, 6H, CH3 of Mes), 1.81 (s, 6H, CH3 of Mes), 1.92 (s, 6H, C–CH3 of NHCMe4), 2.00 (s, 6H, CH3 of Mes), 2.18 (s, 12H, CH3 of Mes), 2.22 (s, 6H, CH3 of Mes), 3.51 (s, 6H, N–CH3 of NHCMe4), 6.32 (s, 1H, Ar–H), 6.34 (s, 1H, Ar–H), 6.44 (s, 2H, Ar–H), 6.47 (s, 2H, Ar–H), 6.50–6.54 (m, 3H, Ar–H), 6.57 (s, 3H, Ar–H), 6.74 (s, 2H, Ar–H), 7.08 (t, 1H, 3J(H,H) = 7.60 Hz, Ar–H), 6.96 (d, 1H, 1J(P,H) = 464.15 Hz, P–H), and 10.65 (s, br, 1H, N–CH–N of NHCMe4) ppm. 13C{1H} NMR (75.43 MHz, C6D6, 298 K): δ = 7.66 (2C, C–CH3 of NHCMe4), 21.02 (1C, CH3 of Mes), 21.05 (1C, CH3 of Mes), 21.34 (2C, CH3 of Mes), 21.44 (2C, CH3 of Mes), 21.58 (1C, CH3 of Mes), 21.61 (1C, CH3 of Mes), 22.14 (1C, CH3 of Mes), 22.32 (1C, CH3 of Mes), 22.47 (1C, CH3 of Mes), 22.55 (1C, CH3 of Mes), 33.42 (2C, CH3–C of NHCMe4), 119.82 (2C, Ar–CH), 126.31 (2C, 4,5-C of NHCMe4), 127.56 (3C, Ar–CH), 128.04 (2C, Ar–CH), 128.18 (2C, Ar–CH), 128.37 (2C, Ar–CH), 128.52 (1C, Ar–CH), 129.63 (1C, Ar–CH), 129.72 (1C, Ar–CH), 134.42 (2C, Ar–Cquat), 134.90 (2C, Ar–Cquat), 136.66 (2C, Ar–Cquat), 137.26 (2C, Ar–Cquat), 137.56 (2C, Ar–Cquat), 137.87 (2C, Ar–Cquat), 141.51 (1C, Ar–Cquat), 141.55 (1C, Ar–Cquat), 142.90 (1C, Ar–Cquat), 142.95 (1C, Ar–Cquat), 143.04 (1C, Ar–Cquat), 143.08 (1C, Ar–Cquat), 143.25 (1C, Ar–Cquat), 144.63 (1C, Ar–Cquat), 144.65 (1C, Ar–Cquat), 144.72 (1C, Ar–Cquat), and 144.76 (1C, NCN) ppm. 31P NMR (121.5 MHz, C6D6, 298 K): δ = 28.50 (t, 1P, 1J(P,P) = 1J(P,H) 464.15Hz, P–P–O) and −46.76 (d, 1P, 1J(P,P) = 464.15 Hz, P–P–O) ppm. 31P{1H} NMR (121.5 MHz, C6D6, 298 K): δ = 28.50 (d, 1J(P,P) = 464.15 Hz, P–P–O) and −46.76 (d, 1J(P,P) = 464.15 Hz, P–P–O) ppm. CP-MAS 31P{1H} NMR (163.32 MHz, 298 K): δ = 27.04 (d, 1J(p,p) = 465 Hz, P–P–O) and −47.00 (d, 1J(P,P) = 465 Hz, P–P–O) ppm. UV/vis (THF): λmax(ε) = 330 (5244), 390 (9754), and 445 (4410) nm (L mol−1 cm−1). IR (KBr, cm−1): = 750 (s), 844 (s), 907 (s), 988 (vs), 1157 (w), 1216 (w), 1270 (w), 1369 (m), 1426 (vw), 1444 (w), 1466 (m), 1485 (m), 1548 (m), 1563 (w), 1582 (w), 1642 (s), 1657 (s), 1688 (s), 1707 (m), 1723 (s), 1754 (m), 1779 (m), 1809 (vw), 1838 (s), 1857 (s), 1879 (m), 1900 (m), 1926 (w), 1951 (w), 1979 (w), 2352 (m P–H), 2380 (m, P–H), and 2921 (br). Elemental analysis: calcd for C55H64N2OP2 (831.06): C, 79.49; H, 7.76; N, 3.37. Found: C, 79.60; H, 7.89; N, 3.73.
Catalytic hydrolysis of 1
0.277 g (0.402 mmol) of 1 and 0.005 g (0.0402 mmol) of 2 were dissolved in about 10 mL of THF (10 mL) and cooled down to −78 °C. Subsequently, 8 μL (0.45 mmol) of water was added and the reaction mixture was allowed to warm to room temperature. Subsequent 31P{1H} NMR measurements show the formation of 4 in about 95% spectroscopic yield.Synthesis of 6
In a 50 mL Schlenk flask, 0.334 g (0.49 mmol) of compound 1 and 0.080 g (0.64 mmol) of 2 were dissolved in 15 mL of THF. The reaction mixture was stirred for 10 minutes. Afterwards, 10 mL of a THF solution of NH3·BH3 (0.020 g, 0.64 mmol) was added at room temperature. The reaction mixture was allowed to stir for 30 minutes during which the red colour of the solution slowly faded to colourless. 31P NMR showed complete formation of compound 6 as d/l and meso isomers. All volatiles were evaporated and the residue was extracted with hot n-hexane (15 mL). Crystallization occurred at room temperature, and a second crop was obtained at −20 °C. Isolated yield 0.300 g (89%). Mp: >190 °C. 1H NMR of the meso isomer (300 MHz, C6D6, 298 K): δ = 1.90 (s, 12H, CH3 of Mes), 2.05 (s, 12H, CH3 of Mes), 2.28 (s, 12H, CH3 of Mes), 3.02 (centre of the AA′XX′ multiplet pattern, 2H, PH–PH, fitted with 1J(31P,31P) = 94.5 Hz, 3J(1H,1H) = 17.0 Hz, 1J(31P,1H) = 216 Hz, and 2J(31P,1H) = 10.9 Hz), 6.74 (s, 2H, Ar–H), 6.76 (s, 2H, Ar–H), 6.81 (s, 4H, Ar–H), 6.83 (s, 4H, Ar–H), and 6.98 (t, 3J(H,H) = 7.41, 2H, Ar–H) ppm. Selected 1H NMR of the d/l isomer (300 MHz, C6D6, 298 K): δ = 1.91 (s, 12H), 1.96 (s, 12H), 2.30 (s, 12H), and 4.01 (centre of the AA′XX′ multiplet pattern, 2H, PH–PH, fitted with 1J(31P,31P) = 260.8 Hz, 3J(1H,1H) = 12.9 Hz, 1J(31P,1H) = 201 Hz, and 2J(31P,1H) = 13 Hz) ppm. 13C{1H} NMR (75.43 MHz, C6D6, 298 K): δ = 21.35 (1C, CH3 of Mes), 21.38 (2C, CH3 of Mes), 21.41 (1C, CH3 of Mes), 21.48 (1C, CH3 of Mes), 21.55 (2C, CH3 of Mes), 21.61 (1C, CH3 of Mes), 21.71 (4C, CH3 of Mes), 128.69 (2C, Ar–CH), 128.79 (2C, Ar–CH), 128.92 (2C, Ar–CH), 129.08 (2C, Ar–CH), 129.02 (2C, Ar–CH), 129.12 (2C, Ar–CH), 129.37 (2C, Ar–CH), 134.14 (1C, Ar–Cquat), 134.27 (1C, Ar–Cquat), 134.41 (1C, Ar–Cquat), 136.12 (1C, Ar–Cquat), 136.36 (3C, Ar–Cquat), 136.54 (1C, Ar–Cquat), 136.65 (3C, Ar–Cquat), 136.76 (3C, Ar–Cquat), 139.88 (3C, Ar–Cquat), 140.05 (1C, Ar–Cquat), 140.70 (1C, Ar–Cquat), 146.78 (2C, Ar–Cquat), and 146.87 (1C, Ar–Cquat) ppm. 31P NMR (121.5 MHz, C6D6, 298 K): δ = −101.4 (meso isomer, centre of the AA′XX′ pattern as seen in the 1H decoupled spectrum; non-decoupled spectrum fit with 1J(31P,31P) = 94.5 Hz, 3J(1H,1H) = 17.0 Hz, 1J(31P,1H) = 216 Hz, and 2J(31P,1H) = 10.9 Hz (the same as that for the corresponding 1H spectrum)) and −109.9 (d/l isomer, centre of the AA′XX′ pattern as seen in the 1H decoupled spectrum; non-decoupled spectrum fit using 1J(31P,31P) = 260.8 Hz, 3J(1H,31H) = 12.9 Hz, 1J(31P,1H) = 201 Hz, and 2J(31P,1H) = 13 Hz (the same as that for the corresponding 1H NMR spectrum)) ppm. 31P{1H} NMR (121.5 MHz, C6D6, 298 K): δ = −101.38 (s, meso isomer) and −109.9 (s, d/l Isomer) ppm. CP-MAS 31P{1H} NMR (163.32 MHz, 298 K)): δ = −101.38 (s, meso isomer) and − 104.13 (s, d/l isomer) ppm. IR (KBr, cm−1): =: 845 (s), 1398 (w), 1432 (w), 1457 (w), 1485 (w), 1567 (s), 1610 (s), 1647 (vw), 1726 (m), 1757 (vw), 1804 (vw), 1866 (m), 1932 (m), 2325 (s, P–H), 2403 (vw, P–H), 2727 (s), 2855 (w), and 2907 (w). Elemental analysis: calcd for C48H52P2 (690.87): C, 83.45; H, 7.59. Found: C, 82.56; H, 7.27.
Synthesis of 7
In a 25 mL Schlenk flask 0.202 g (0.293 mmol) of compound 1 and 0.072 g (0.586 mmol) of 2 were dissolved in 10 mL of toluene. The red reaction mixture was heated at 105 °C for 30 h. During the course of the reaction, the color of the reaction mixture changed from red to yellow. After 12 h at 0 °C yellow crystals of 7 were obtained. The resulting mother liquor was concentrated to 5 mL and kept at −20 °C. After one day a second crop of crystals was collected. Total yield: 0.225 g (81.8%). Single crystals suitable for X-ray diffraction analysis were obtained from a saturated solution of toluene at 0 °C after one day. Mp: >200 °C. 1H NMR (300 MHz, C6D6, 298 K): δ = 1.25 (s, 6H, C–CH3 of NHCMe4), 2.17 (6H, p-CH3 of Mes), 2.40 (12H, o-CH3 of Mes), 2.84 (s, 6H, N–CH3 of NHCMe4), 6.78 (4H, Ar–H), and 6.99–7.08 (m, 3H Ar–H) ppm. 13C{1H} NMR (75.43 MHz, C6D6, 298 K): δ = 8.83 (2C, C–CH3 of NHCMe4), 21.51 (4C, CH3 of Mes), 21.56 (2C, CH3 of Mes), 34.12 (N–CH3 of NHCMe4), 34.24 (N–CH3 of NHCMe4), 122.50 (1C, Ar–CH), 122.55 (2C, CH3–C of NHCMe4), 128.53 (4C, Ar–CH), 128.98 (2C, d, J(C,P) = 1.38, Ar–CH), 134.37 (1C, Ar–Cquat), 136.26 (1C, Ar–Cquat), 142.78 (1C, Ar–Cquat), 142.82 (1C, Ar–Cquat), 144.68 (1C, Ar–Cquat), 144.86 (1C, Ar–Cquat), 150.67 (1C, Ar–Cquat), 151.42 (1C, Ar–Cquat), 167.40 (1C, Ar–Cquat), and 168.70 (1C, Ar–Cquat) ppm. 31P NMR (121.5 MHz, C6D6, 298 K): δ = −77 ppm. CP-MAS 31P{1H} NMR (163.32 MHz, 298 K): δ = −77.05 ppm. UV/vis (n-hexane): λmax(ε) = 446 (11244), 382 (5375), and 310 (2553) nm (L mol−1 cm−1). IR (KBr, cm−1): = 408 (s), 420 (vs), 432 (s), 465 (vs), 552 (s), 583 (m), 593 (vs), 658 (m), 677 (s), 720 (m), 745 (s), 775 (m), 797 (s), 856 (vs), 879 (s), 907 (s), 1042 (br, vs), 1085 (m), 1107 (m), 1185 (w), 1234 (w), 1660 (m), 1705 (m), 1856 (m), 1876 (w), 2346 (br, w), and 2372 (w). Elemental analysis: calcd for C31H37N2P (468.62): C, 79.45; H, 7.96; N, 5.98. Found: C, 79.24; H, 8.02; N, 6.06.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by the Tata Institute of Fundamental Research Hyderabad, Gopanpally, Hyderabad-500107, Telangana, India, SERB-DST (EMR/2014/001237), India, Research Group Linkage Programme of Alexander von Humboldt Foundation, Germany, and Saarland University, Germany. VC is thankful to the Department of Science and Technology, New Delhi, India, for a National J. C. Bose fellowship. We thank the reviewers for their comments to improve the manuscript.Notes and references
- (a) J. A. Osborn, F. H. Jardine, J. F. Young and G. Wilkinson, J. Chem. Soc. A, 1966, 1711–1732 RSC; (b) P. Meakin, J. P. Jesson and C. A. Tolman, J. Am. Chem. Soc., 1972, 94, 3240–3242 CrossRef CAS; (c) S. K. Tanielyan, R. L. Augustine, N. Marin and G. Alvez, ACS Catal., 2011, 1, 159–169 CrossRef CAS.
- (a) N. H. Park, E. V. Vinogradova, D. S. Surry and S. L. Buchwald, Angew. Chem., Int. Ed., 2015, 54, 8259–8262 CrossRef CAS PubMed; (b) P. L. Arrechea and S. L. Buchwald, J. Am. Chem. Soc., 2016, 138, 12486–12493 CrossRef CAS PubMed; (c) D. A. Culkin and J. F. Hartwig, Organometallics, 2004, 23, 3398–3416 CrossRef CAS; (d) B. Su, T.-G. Zhou, X. W. Li, X. R. Shao, P. L. Xu, W. L. Wu, J. F. Hartwig and Z. J. Shi, Angew. Chem., Int. Ed., 2017, 56, 1092–1096 CrossRef CAS PubMed; (e) X. Jiang, J. J. Beiger and J. F. Hartwig, J. Am. Chem. Soc., 2017, 139, 87–90 CrossRef CAS PubMed.
- P. P. Power, Nature, 2010, 463, 171–177 CrossRef CAS PubMed.
- G. C. Welch, R. R. S. Juan, J. D. Masuda and D. W. Stephan, Science, 2006, 314, 1124–1126 CrossRef CAS PubMed.
- Y. Peng, B. D. Ellis, X. Wang, J. C. Fettinger and P. P. Power, Science, 2009, 325, 1668–1670 CrossRef CAS PubMed.
- F. Buß, P. Mehlmann, C. Mück-Lichtenfeld, K. Bergander and F. Dielmann, J. Am. Chem. Soc., 2016, 138, 1840–1843 CrossRef PubMed.
- (a) K. Leszczyńska, K. Abersfelder, A. Mix, B. Neumann, H. G. Stammler, M. J. Cowley, P. Jutzi and D. Scheschkewitz, Angew. Chem. Int. Ed., 2012, 51, 6785–6788 CrossRef PubMed; (b) M. J. Cowley, V. Huch, H. Rzepa and D. Scheschkewitz, Nature Chem., 2013, 5, 876–879 CrossRef CAS PubMed.
- A. F. Eichhorn, S. Fuchs, M. Flock, T. B. Marder and U. Radius, Angew. Chem., Int. Ed., 2017, 56, 10209–10213 CrossRef CAS PubMed.
- (a) G. H. Spikes, J. C. Fettinger and P. P. Power, J. Am. Chem. Soc., 2005, 127, 12232 CrossRef CAS PubMed; (b) G. D. Frey, V. Lavallo, B. Donnadieu, W. W. Schoeller and G. Bertrand, Science, 2007, 316, 439–441 CrossRef CAS PubMed.
- (a) A. Jana, M. Majumdar, V. Huch, M. Zimmer and D. Scheschkewitz, Dalton Trans., 2014, 43, 5175–5181 RSC; (b) A. Jana, I. Omlor, V. Huch, H. S. Rzepa and D. Scheschkewitz, Angew. Chem., Int. Ed., 2014, 53, 9953–9956 CrossRef CAS PubMed.
- (a) L. Liu, D. A. Ruiz, D. Munz and G. Bertrand, Chem, 2016, 1, 147–153 CrossRef CAS; (b) M. M. Hansmann, R. Jazzar and G. Bertrand, J. Am. Chem. Soc., 2016, 138, 8356–8359 CrossRef CAS PubMed.
- M. M. Hansmann and G. Bertrand, J. Am. Chem. Soc., 2016, 138, 15885–15888 CrossRef CAS PubMed.
- Y. Wang, Y. Xie, P. Wei, R. B. King, H. F. Schaefer III, P. v. R. Schleyer and G. H. Robinson, J. Am. Chem. Soc., 2008, 130, 14970–14971 CrossRef CAS PubMed.
- D. Dhara, D. Mandal, A. Maiti, C. B. Yildiz, P. Kalita, N. Chrysochos, C. Schulzke, V. Chandrasekhar and A. Jana, Dalton Trans., 2016, 45, 19290–19298 RSC.
- N. Hayakawa, K. Sadamori, S. Tsujimoto, M. Hatanaka, T. Wakabayashi and T. Matsuo, Angew. Chem., Int. Ed., 2017, 56, 5765–5769 CrossRef CAS PubMed.
- (a) J. I. Bates, P. Kennepohl and D. P. Gates, Angew. Chem. Int. Ed., 2009, 48, 9844–9847 CrossRef CAS PubMed; (b) P. K. Majhi, K. C. F. Chow, T. H. H. Hsieh, E. G. Bowes, G. Schnakenburg, P. Kennepohl, R. Streubel and Derek P. Gates, Chem. Commun., 2016, 52, 998–1001 RSC.
- M. Yoshifuji, I. Shima, N. Inamoto, K. Hirotsu and T. Higuchi, J. Am. Chem. Soc., 1981, 103, 4587–4589 CrossRef CAS.
- M. Yoshifuji, Eur. J. Inorg. Chem., 2016, 607–615 CrossRef CAS.
- E. Urnéžius and J. D. Protasiewicz, Main Group Chem., 1996, 1, 369–372 CrossRef.
- N. Kuhn and T. Kratz, Synthesis, 1993, 561–562 CrossRef CAS.
- J. D. Protasiewicz, M. P. Washington, V. B. Gudimetla, J. L. Payton and M. C. Simpson, Inorg. Chim. Acta, 2010, 364, 39–45 CrossRef CAS.
- P. C. Andrews, S. J. King, C. L. Raston and B. A. Roberts, Chem. Commun., 1998, 547–548 RSC.
- See the ESI† for the details.
- (a) J. Borm, G. Huttner, O. Orama and L. Zsolnai, J. Organomet. Chem., 1985, 282, 53–67 CrossRef CAS; (b) N. Nagahora, T. Sasamori, N. Takeda and N. Tokitoh, Chem.–Eur. J., 2004, 10, 6146–6151 CrossRef CAS PubMed; (c) C. Moser, F. Belaj and R. Pietschnig, Chem.–Eur. J., 2009, 15, 12589–12591 CrossRef CAS PubMed; (d) L. Weber, S. Buchwald, A. Rühlicke, H. G. Stammler and B. Neumann, Z. Anorg. Allg. Chem., 1993, 619, 934–942 CrossRef CAS.
- D. Mandal, B. Santra, P. Kalita, N. Chrysochos, A. Malakar, R. S. Narayanan, S. Biswas, C. Schulzke, V. Chandrasekhar and A. Jana, ChemistrySelect, 2017, 2, 8898–8910 CrossRef CAS.
- K. Esfandiarfard, A. I. Arkhypchuk, A. Orthaber and S. Ott, Dalton Trans., 2016, 45, 2201–2207 RSC.
- (a) A. Jana, V. Huch, H. S. Rzepa and D. Scheschkewitz, Angew. Chem., Int. Ed., 2015, 54, 289–292 CrossRef CAS PubMed; (b) H. Cui, J. Zhang and C. Cui, Organometallics, 2013, 32, 1–4 CrossRef CAS.
- Z. Tošner, R. Andersen, B. Stevensson, M. Edén, N. C. Nielsena and T. Vosegaard, J. Magn. Reson., 2014, 246, 79–93 CrossRef PubMed.
- K. A. Erickson, L. S. H. Dixon, D. S. Wright and R. Waterman, Inorg. Chim. Acta, 2014, 422, 141–145 CrossRef CAS.
- O. Back, M. H. Ellinger, C. D. Martin, D. Martin and G. Bertrand, Angew. Chem., Int. Ed., 2013, 52, 2939–2943 CrossRef CAS PubMed.
- T. Krachko, M. Bispinghoff, A. M. Tondreau, D. Stein, M. Baker, A. Ehlers, J. C. Slootweg and H. Grützmacher, Angew. Chem., Int. Ed., 2017, 56, 7948–7951 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Thermodynamic data of equilibrium between 1 and 3, solution and solid state NMR spectra, UV/vis spectra, NMR simulation of compound 6, X-ray crystallographic data and theoretical details. CCDC 1588456 (3), 1588457 (4), 1588458 (5), 1588459 (6) and 1588460 (7). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8sc00348c |
| This journal is © The Royal Society of Chemistry 2018 |