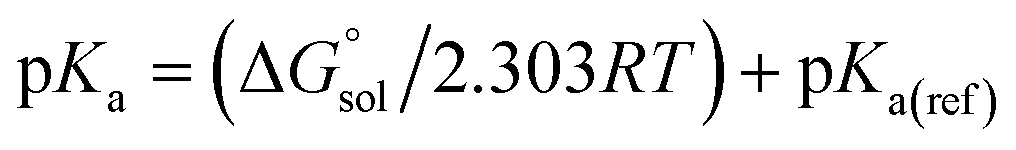DOI:
10.1039/C8SC00093J
(Edge Article)
Chem. Sci., 2018,
9, 4689-4695
Hydrogen gas generation using a metal-free fluorinated porphyrin†
Received
9th January 2018
, Accepted 20th April 2018
First published on 30th April 2018
Abstract
Free-base meso-tetra(pentafluorophenyl)porphyrin, 1, is electrocatalytically active for hydrogen gas generation in the presence of p-toluenesulfonic acid. The electrochemical potential of hydrogen evolution (−1.31 V vs. Fc/Fc+ in THF) is comparable to those of metal containing electrocatalysts such as metallated porphyrins or other metallated macrocycles. Combining experimental observations and DFT computations, we propose the most favorable hydrogen generation mechanism to be a (1) reduction, (2) protonation, (3) reduction, (4) protonation (E–P–E–P) pathway.
Introduction
Production of molecular hydrogen through electrochemical hydrogen evolution reactions (HERs) is crucial for the development of several clean-energy technologies. Such reactions require efficient and robust catalysts that are able to increase the reaction kinetics by lowering the high activation energy of hydrogen generation.1–3 Platinum metal is a highly efficient catalyst which is able to generate high exchange current densities at practically zero overpotential.1,4,5 However, platinum is scarce and expensive.4 Thus, replacement of platinum by other cost-effective alternatives with promisingly high electrocatalytic activity is necessary for the sustainable production of hydrogen gas.6–8 Currently, nearly all HER catalysts developed are based on earth-abundant transition metals such as Mo, Co, Ni, Fe and their complexes.6–15 HER catalytic systems utilizing metal-free compounds are uncommon;16,17 however, organic-based hydrogen generation catalysts may provide superior synthetic flexibility, lower manufacturing costs, and greater chemical stability.
Metalloporphyrins have been well investigated for their ability to generate H2 both electrochemically and photochemically.18–23 In metallated HER electrocatalysts, the metal center is the active site in the hydrogen production process by participating in the formation of metal–hydrogen bonds and is the redox center in the multielectron reduction process.19,24–26 In comparison, free-base porphyrins are known to exhibit rich multielectron redox chemistry.27,28 Two basic imine nitrogen atoms in the porphyrin core can produce a diprotonated porphyrin species.29,30 Thus, free-base porphyrins have the ability to form covalent nitrogen–hydrogen bonds and can also be electrochemically active. In addition, the four-nitrogen porphyrin core is perfectly suited for bringing protons in close proximity to lower the activation energy of dihydrogen production by prearranging the transition state of hydrogen–hydrogen bond formation. However, whether a molecular metal-free porphyrin has the ability to exhibit similar and comparable electrocatalytic HER activity to the existing well-studied metallated macrocyclic complexes has not yet been studied or reported.
In this work we show that free-base meso-tetra(pentafluorophenyl)porphyrin, 1, is a HER electrocatalyst in the presence of p-toluenesulfonic (tosic) acid as the proton source with THF as the solvent. The electrocatalytic activity of 1 was estimated by electrochemical studies and H2 gas analysis in acidic solutions. Spectroscopic measurements including UV-vis spectra and spectroelectrochemical studies reveal the spectral signatures of the intermediates during the catalysis giving insight into the mechanism of H2 generation. In addition, density functional theory (DFT) calculations were performed to provide further support to the mechanistic HER behavior of 1.
Experimental section
Materials
All reagents used for synthesis were purchased from Sigma-Aldrich. Pyrrole was freshly distilled prior to use. Solvents used for electrochemical studies were dried and degassed through a Pure Process Technology solvent purification system. Tetrabutylammonium hexafluorophosphate (TBAPF6) and tosic acid were purchased from Acros Organics. Meso-tetra(pentafluorophenyl)porphyrin and meso-tetraphenylporphyrin were synthesized according to Lindsey's method.31
Synthesis of meso-tetra(pentafluorophenyl)porphyrin, 1
Pentafluorobenzaldehyde (3.00 g, 0.0153 mol) was dissolved in 500 mL of DCM, followed by addition of pyrrole (1.04 g, 0.0153 mol) dropwise. The mixture was stirred and bubbled with N2 for 15 minutes. Next, 0.500 mL of BF3·Et2O was added with a glass syringe without exposing to air. After 2 hours, the resulting porphyrinogen was oxidized by adding 2,3-dichloro-5,6-dicyanobenzoquinone (DDQ) (5.00 g, 0.0220 mol) and letting it react for 30 minutes. Compound 1 (1.80 g, 24.3% yield) was obtained after recrystallization and column chromatography purification on silica gel eluted with a mixture of hexane and dichloromethane (2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). 1H NMR (C6D6): 8.71 ppm (s, 8H), −2.10 ppm (s, 2H); 19F NMR (C6D6): −161.3 ppm (t, 2F), −150.5 ppm (t, 1F), −137.2 ppm (d, 2F); ESI-MS: m/z: 975.5; UV-vis (THF): λ max 408, 503, 543, 584, and 634 nm.
:1). 1H NMR (C6D6): 8.71 ppm (s, 8H), −2.10 ppm (s, 2H); 19F NMR (C6D6): −161.3 ppm (t, 2F), −150.5 ppm (t, 1F), −137.2 ppm (d, 2F); ESI-MS: m/z: 975.5; UV-vis (THF): λ max 408, 503, 543, 584, and 634 nm.
Cyclic voltammetry
All electrochemical measurements were obtained by using a CHI760D potentiostat, with 0.1 M TBAPF6 as the supporting electrolyte. The electrolyte and tosic acid were placed under vacuum and oven dried prior to use. All cyclic voltammograms were obtained in a dry N2-filled glovebox, using a 4 mm diameter glassy carbon working electrode, a Pt mesh auxiliary counter electrode, and an Ag/Ag+ reference electrode. Ferrocene (Fc) was added after each measurement as an internal standard.
Controlled-potential electrolysis and H2 detection
Controlled-potential electrolysis was done in a custom-built two-compartment gas-tight electrochemical cell under an argon atmosphere. One part of the cell contains: (I) a carbon rod working electrode (3 mm diameter and 0.95 mm length); (II) an Ag/Ag+ reference electrode; (III) a gas inlet and gas outlet. The other part of the cell contains a Pt auxiliary wire counter electrode and a gas outlet. The working and counter electrodes are separated through a glass frit. Electrolysis was carried out at −1.7 V vs. Fc/Fc+ in THF, containing 0.1 M TBAPF6 and 10 mM tosic acid with and without adding the catalyst.
Spectroelectrochemistry
Spectroelectrochemistry was performed using an optical transparent 1 mm thin-layer spectroelectrochemical quartz cell containing an Au gauze working electrode, a non-aqueous Ag/Ag+ reference electrode and a Pt wire counter electrode. The absorption spectra were recorded using a UV-vis spectrophotometer while the bulk electrolysis was performed using the CHI760D potentiostat. Solutions of 1 containing 0.1 M TBAPF6 with and without acid were prepared and degassed with N2 before each measurement. The changes of the UV-vis spectra were recorded at one second intervals for 10 minutes at different applied potentials.
Other physical methods
1H NMR and 19F NMR spectra were recorded on a JEOL 600 MHz NMR spectrometer. The proton NMR spectrum was referenced to the residual deuterated solvent signal as an internal calibration (C6D6 = 7.16 ppm). The UV-vis spectra were recorded on a SEC2000 spectra system equipped with VISUAL SPECTRA 2.1 software.
Computational details
The molecular geometries of the free base porphyrin and porphyrin derivatives (Scheme 1) were optimized using density functional theory (DFT). The DFT method employed was Becke's32 three parameter hybrid exchange functional, coupled with the Lee–Yang–Parr33 nonlocal correlation functional (B3LYP) for all calculations. The 6-31+G34,35 Pople basis set, as implemented in Gaussian 09, was used for all atoms.36 Zero point energies and thermodynamic data were calculated at 298.15 K and 1 atm by performing frequency calculations. Calculations were performed on the optimized gas-phase geometries using the solvation model based on density (SMD) as implemented in the Gaussian 09 suite.37–39
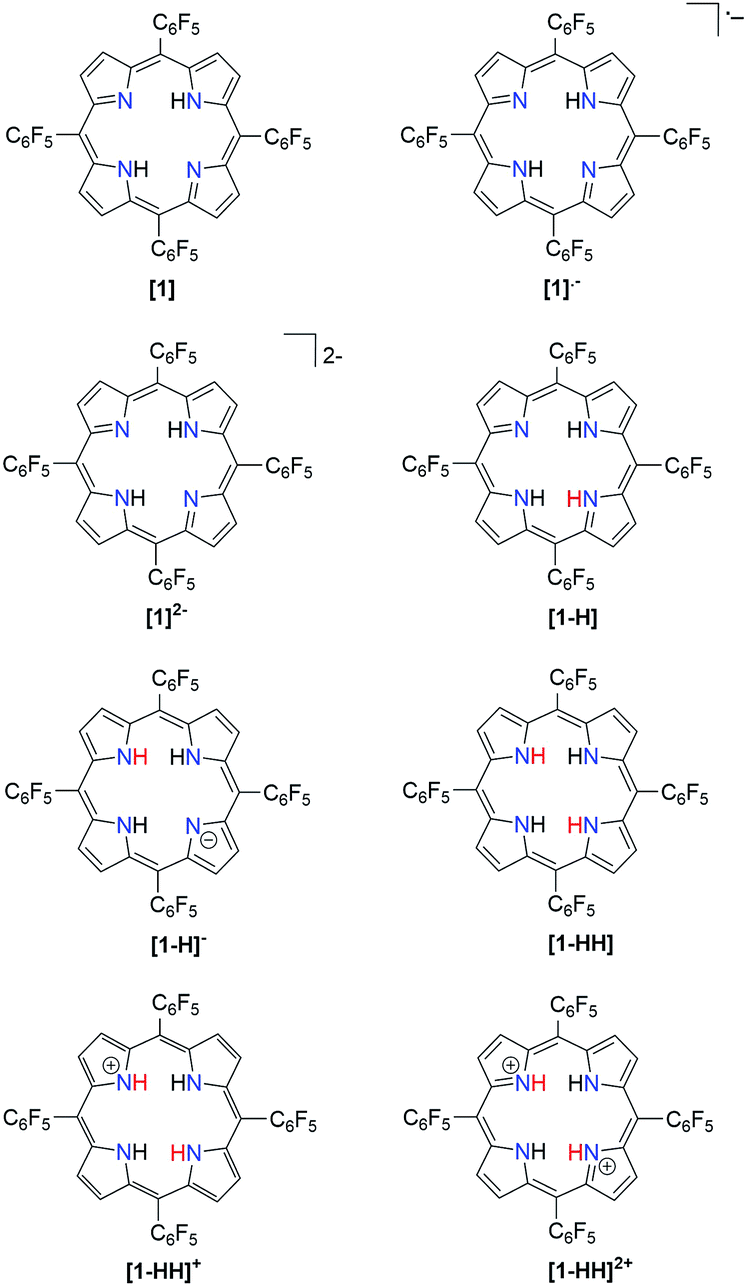
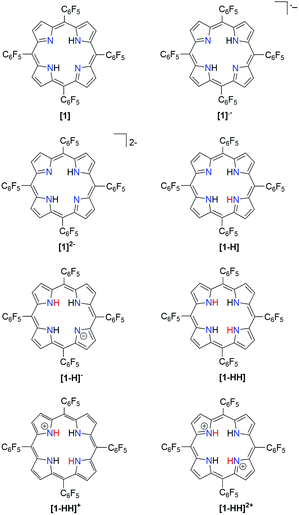 |
| | Scheme 1 Porphyrin derivatives involved in the proposed mechanistic pathways for hydrogen generation. | |
In order to ease the computational expense, a model in which the pentafluorophenyl groups are replaced with chlorine atoms was employed. This substitution is reasonable due to the similar electron-withdrawing ability of Cl compared to that of –C6F5 as supported by their Hammett substitution constants (Table S2†). We followed several theoretical models22,40–49 for the determination of thermodynamic quantities such as reduction potentials and pKas so as to give insight into the hydrogen evolution mechanism.
Calculation of reduction free energies
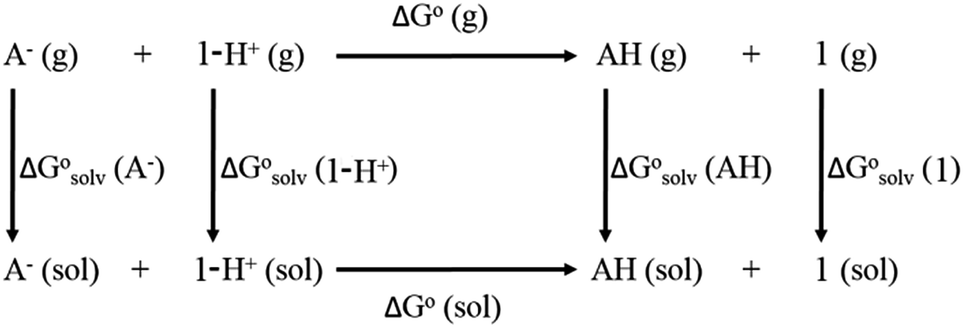
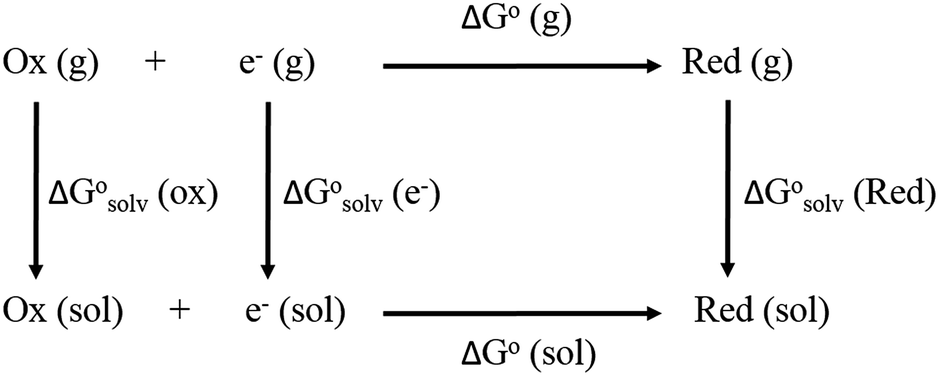
From a Born–Haber thermodynamic cycle (Scheme 2), we can associate the reaction free energies with the gas-phase and solvation energies using eqn (1):| | 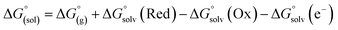 | (1) |
where  and
and  represent the free energies upon solvation for both the reduced and the oxidized species,
represent the free energies upon solvation for both the reduced and the oxidized species, 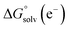 the contribution of the electron to the solvation free energy, and
the contribution of the electron to the solvation free energy, and 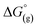 the free energy of the reaction in the gas phase. We can calculate the last value with the expression
the free energy of the reaction in the gas phase. We can calculate the last value with the expression 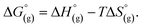
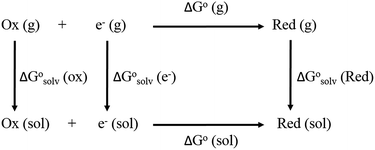 |
| | Scheme 2 Born–Haber thermodynamic cycle for the calculation of the free energy of reduction. | |
Calculation of 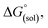 allows the determination of redox potentials using eqn (2):
allows the determination of redox potentials using eqn (2):
| | 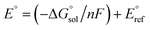 | (2) |
where
E° is the standard reduction potential,
F is the Faraday constant and
n is the number of electrons involved in the redox reaction. In order to allow a direct comparison of calculated
vs. experimental data, the redox potentials reported are referenced to the ferrocene/ferrocenium (Fc/Fc
+) couple.
Calculation of proton dissociation free energies
The direct calculation of reaction free energies in solution cannot be performed because the free energy of the solvated proton is difficult to calculate with accuracy. In order to calculate the thermodynamic value, we need to consider a proton-exchange reaction between a reference compound (AH) and the free base porphyrin (1):Herein, we define the free energy change of reaction (3) as 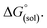 To calculate
To calculate 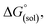 we must relate it to the free energy in the gas phase using the thermodynamic cycle depicted in Scheme 3, obtaining the following equation:
we must relate it to the free energy in the gas phase using the thermodynamic cycle depicted in Scheme 3, obtaining the following equation:| | 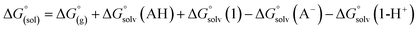 | (4) |
where 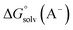 ,
, 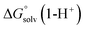 ,
, 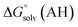 and
and 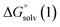 are the free energies upon solvation of the referenced conjugated base, the protonated porphyrin, the referenced acid and the porphyrin, respectively. We can calculate
are the free energies upon solvation of the referenced conjugated base, the protonated porphyrin, the referenced acid and the porphyrin, respectively. We can calculate 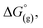 using the expression
using the expression 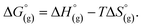 The pKa can be calculated from
The pKa can be calculated from 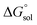 using:
using:| | 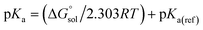 | (5) |
where R is the gas constant, T is the temperature (298.15 K), and pKa(ref) is the experimentally reported pKa of the reference reaction. Since the deprotonation of acetic acid has been studied in THF and the pKa value is available, we chose this reaction as our reference.50
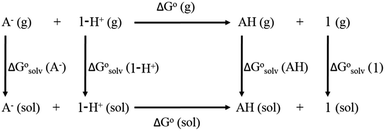 |
| | Scheme 3 Born–Haber thermodynamic cycle for the calculation of the free energy of proton transfer. | |
Results and discussion
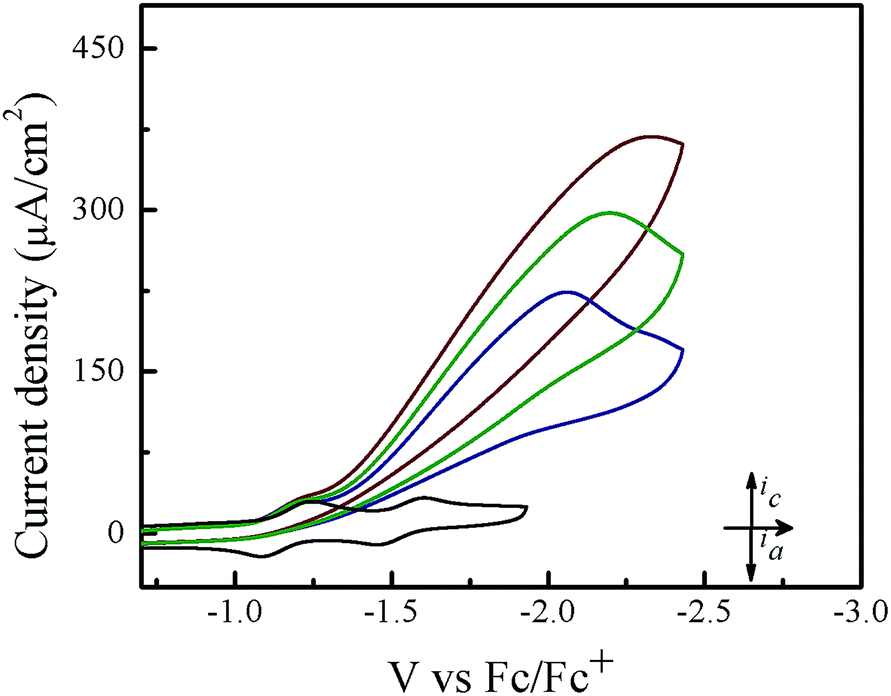
Cyclic voltammetry experiments were conducted in THF in order to assess the electrocatalytic activity of 1. In the absence of acid, 1 features two reversible one-electron reductions at E1/2 = −1.14 V and E1/2 = −1.54 V vs. Fc/Fc+ that yield the porphyrin radical anion [1]˙− and the dianion species [1]2−, respectively (Fig. 1). Upon successive addition of tosic acid, the first reduction wave of 1 remains unchanged, while a catalytic wave appears at a potential near −1.31 V vs. Fc/Fc+, which occurs before the second reduction wave of 1 at −1.54 V. This indicates that protonation of this porphyrin is not possible under these conditions prior to the one-electron reduction. The reduction potential of H+/H2 (EH+) with tosic acid in THF is −0.605 V vs. Fc/Fc+, which corresponds to an overpotential of 1.02 V.51,52 Notably, addition of acid without the presence of 1 shows a negligible current increase (Fig. S1†). As a control, a second metal-free porphyrin, meso-tetraphenylporphyrin was also evaluated as a HER electrocatalyst using tosic acid as the proton source. The electrocatalytic current achieved using this porphyrin was very low (onset potential: −1.89 V vs. Fc/Fc+; peak current: 18 μA. see Fig. S3†).
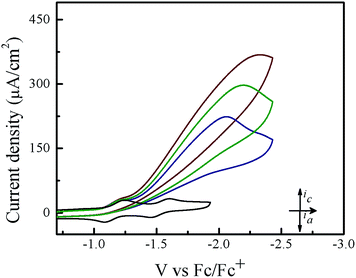 |
| | Fig. 1 Cyclic voltammograms of 0.1 mM 1 in a solution containing 0.1 M TBAPF6 with and without tosic acid: (from bottom to top): 0 equiv. acid, 4 equiv. acid, 9 equiv. acid, and 12 equiv. acid. Scan rate: 100 mV s−1; a glassy carbon working electrode. | |
Hydrogen gas production was confirmed by gas chromatography through the evaluation of the gas product obtained from controlled potential electrolysis. Fig. 2 depicts the dependence of accumulated charge over 40 minutes, resulting in 3.2 C of charge corresponding to 90% faradaic efficiency (see Experimental section for full experimental details).
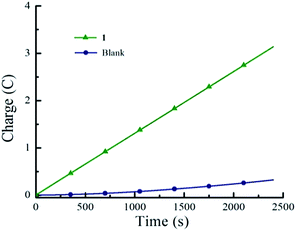 |
| | Fig. 2 Controlled-potential electrolysis experiments containing 1 mM 1, 0.1 M TBAPF6 and 10 mM tosic acid on a carbon rod electrode: (▲) 10 mM tosic acid with 1 mM 1, and (●) 10 mM tosic acid without 1. Potential: −1.7 V vs. Fc/Fc+. | |
In order to investigate whether there is a formation of a heterogeneous metal-free porphyrin thin film on the working electrode, which can be responsible for HER activity, cyclic voltammograms were obtained using a glassy carbon electrode after performing controlled-potential electrolysis. Two glassy carbon electrodes were subjected to 1 h of electrolysis in the presence of 1 with and without tosic acid. After the experiment, the electrodes were rinsed with THF and dried in air. Then they were exposed to fresh acidic solutions (1.5 mM) without the addition of 1. In neither case the working electrode shows any increase of catalytic current (Fig. S6 and S7†), implying that 1 does not adsorb on the working electrode surface during our electrochemical studies.
HER kinetics were measured by foot-of-the-wave analysis (FOWA). The observed rate constant (kobs) was calculated from the slope of the linear region near the foot of the wave (see the ESI† for full experimental details). The calculated kobs are displayed in Table S3,† showing values of 0.528, 0.742 and 0.891 s−1 at acid concentrations ([H+]) of 0.4, 0.9 and 1.2 mM respectively. Plotting kobsversus [H+] gives a linear relationship (Fig. S9†), suggesting a first order dependence of [H+]. A kinetic isotope effect (KIE) of 1.16 was observed when using deuterated tosic acid (C7H7SO3D).
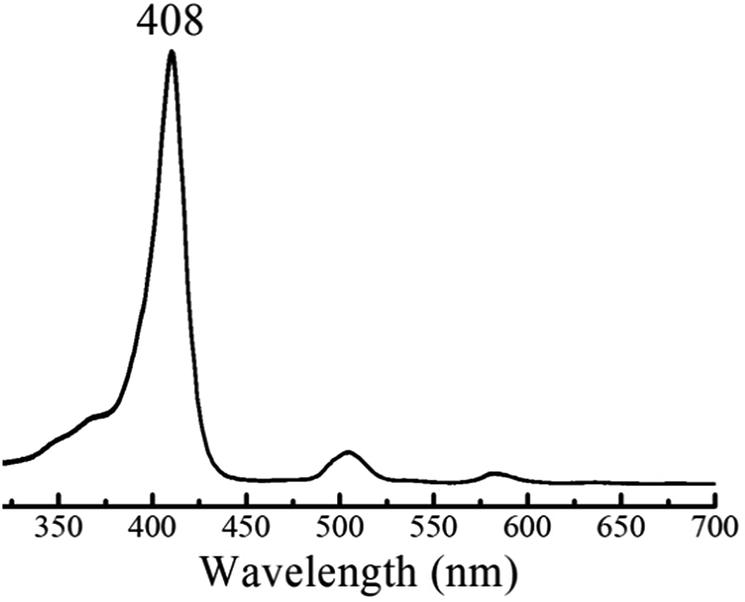
The HER mechanism of 1 was probed through spectroscopic techniques. When two protons and two electrons are involved, several different pathways can be followed in order to generate hydrogen. The following possible sequential mechanistic steps were considered: E–E–P–P, E–P–E–P, E–P–P–E, P–E–E–P, P–E–P–E, and P–P–E–E, where E stands for reduction and P stands for protonation. In THF, 1 exhibits a narrow and intense absorbance in the typical Soret (375–425 nm) region and four small Q-bands between 470 and 650 nm. Upon titration of tosic acid, the electronic spectrum remains constant (Fig. 3). This indicates that in THF, tosic acid is not strong enough to protonate 1, suggesting that the first step of hydrogen generation is a reduction.
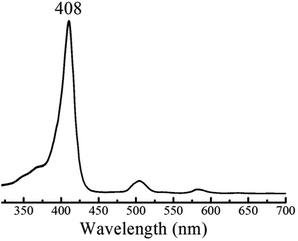 |
| | Fig. 3 UV-Vis spectrum of 1 in THF containing 0.1 M TBAPF6 before and after titrating with tosic acid. | |
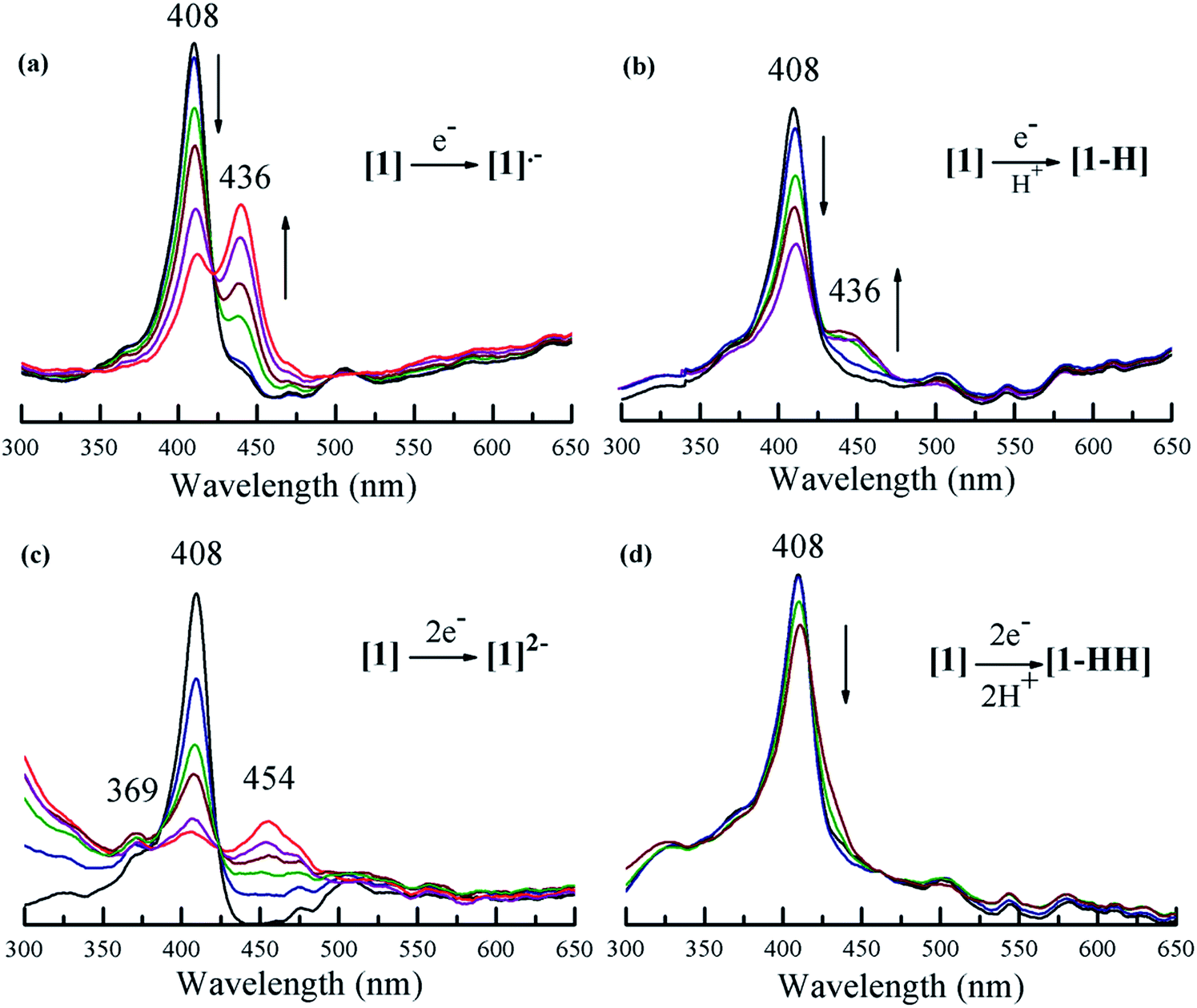
Fig. 4 shows the spectral changes of 1 under different electrolysis conditions. The UV-vis spectrum of 1 upon controlled-potential bulk electrolysis at the potential of the first one-electron reduction of the porphyrin (−1.35 V vs. Fc/Fc+), shows a decrease of the Soret (408 nm) and Q-bands (503, 530, 584, and 634 nm); and the appearance of an absorption band at 436 nm along with an isosbestic point at 428 nm. This new band can be attributed to the generation of the radical anion [1]˙− (Fig. 4a). Under the same controlled potential of −1.35 V vs. Fc/Fc+ but in the presence of tosic acid, the band at 436 nm corresponding to the radical anion [1]˙− is also seen but the isosbestic point at 428 nm disappears as a function of time (Fig. 4b). This implies that in addition to the generation of the radical anion [1]˙−, other chemical species are also produced. Since the only condition changed between Fig. 4a and b is the addition of acid, the second step of the catalytic cycle should be assigned to protonation after the one-electron reduction.
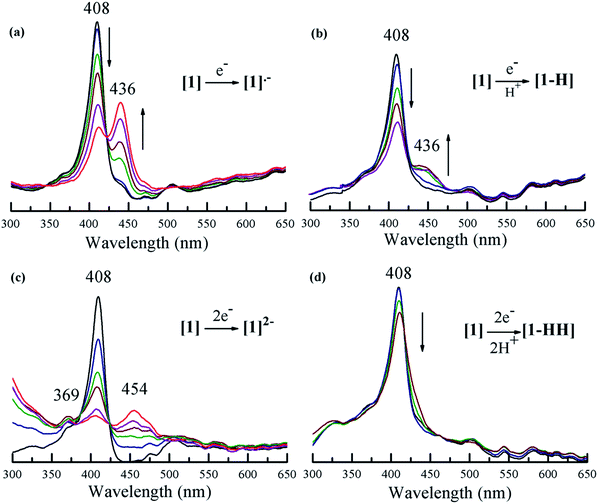 |
| | Fig. 4 Uv-vis spectroelectrochemistry of 1 in the absence and in the presence of tosic acid containing 0.1 M TBAPF6 in THF: (a) 1 at −1.35 V; (b) 1 containing tosic acid at −1.35 V; (c) 1 at −1.7 V; (d) 1 containing tosic acid at −1.7 V (potentials are referred to Fc/Fc+). | |
When performing bulk electrolysis at the potential above the second electron reduction wave (−1.7 V vs. Fc/Fc+) without the presence of acid, the recorded spectrum (Fig. 4c) shows an immediate decay of the Soret absorbance coupled to an increase of a broad absorbance at 454 nm, a new band at 369 nm and another new band in the ultraviolet region (300 nm), along with an isosbestic point at 425 nm. This features the transformation from the free-base porphyrin [1] into the dianion species [1]2−. When bulk electrolysis was conducted at the same potential (−1.7 V vs. Fc/Fc+) in the presence of tosic acid, the absorbance belonging to the dianion [1]2− is not observed and only a decrease of the Soret absorbance occurs (Fig. 4d). However, the rate of decay of the Soret band in the presence of acid is slower compared to that without acid (Fig. S12†). This suggests that 1 is regenerated upon H2 production. Overall these spectroscopy studies show that the first and second events of the mechanism of hydrogen generation with 1 in the presence of tosic acid are reduction and protonation, respectively.
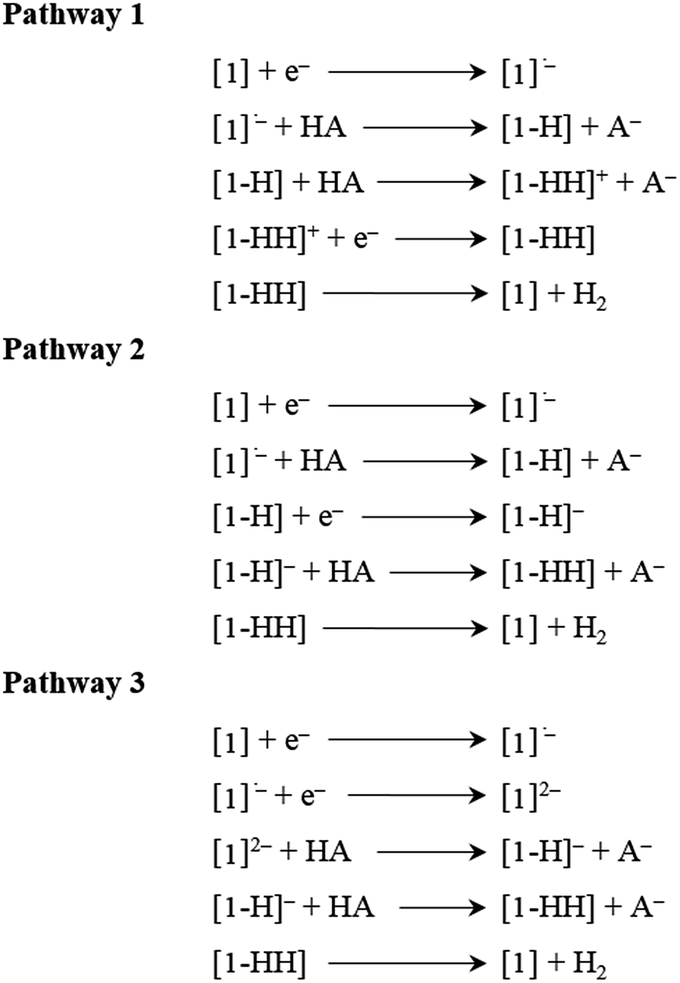
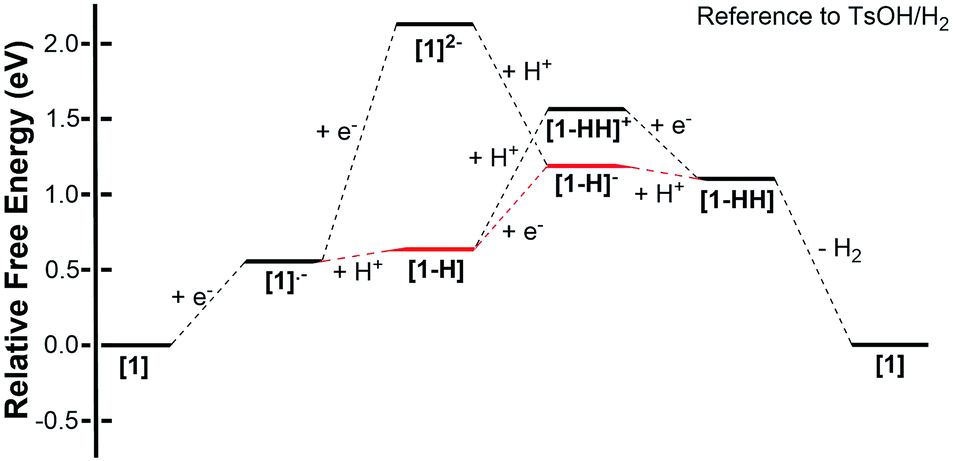
Thermodynamic theoretical calculations were also performed to provide further insight into the next steps of hydrogen generation. Scheme 4 shows the three different pathways for hydrogen generation in THF after the first reduction, and Fig. 5 shows the calculated relative free energies corresponding to each of these pathways. The calculated relative free energies in Fig. 5 are plotted relative to the tosic acid/dihydrogen (TsOH/H2) couple.
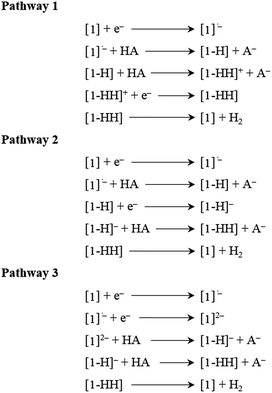 |
| | Scheme 4 Mechanistic pathways proposed for hydrogen generation in THF, with tosic acid as the proton source. | |
 |
| | Fig. 5 Free energy diagram of H2 evolution catalyzed using 1 in THF with tosic acid. The free energies were calculated using the Born–Haber cycles shown in Schemes 2 and 3, and these are plotted relative to the tosic acid/dihydrogen (TsOH/H2) couple. | |
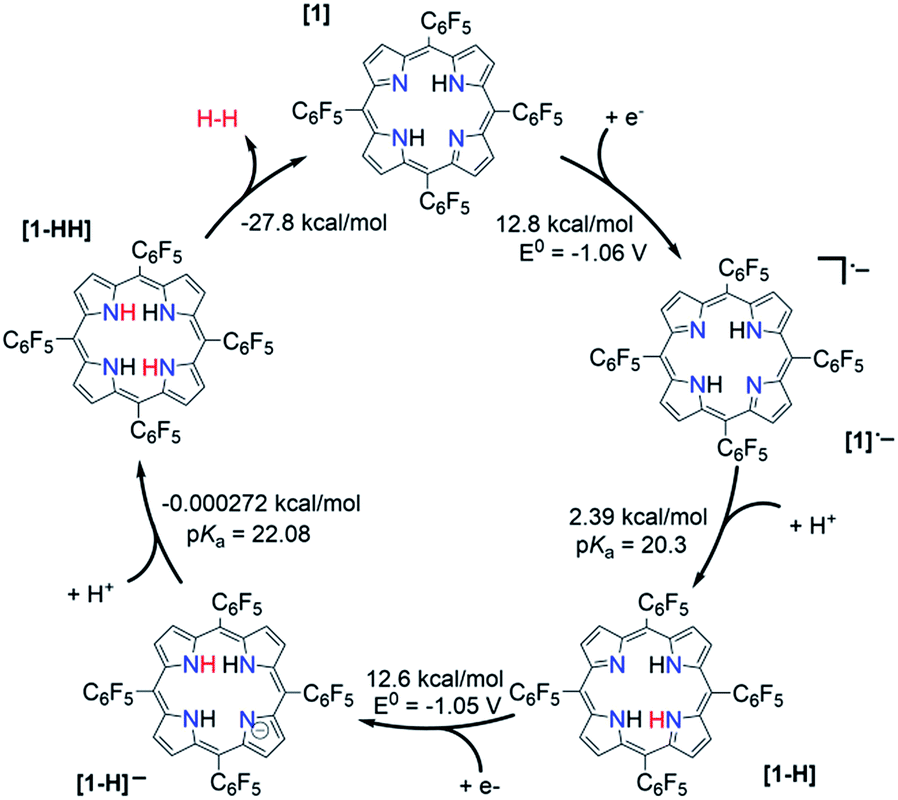
The relative free energy difference between [1] and [1]˙− is calculated to be 12.8 kcal mol−1 (+0.556 eV) which corresponds to a potential of −1.06 V vs. Fc/Fc+. Upon the first reduction, [1]˙− can follow two possible paths: it can be either further reduced to [1]2− or protonated at the N core using tosic acid to yield [1-H]. Protonation to generate [1-H] is thermodynamically favored with a relative free energy of +2.39 kcal mol−1 (+0.104 eV), compared to going uphill +36.3 kcal mol−1 (+1.57 eV) to form [1]2−.
The calculated relative pKa for the deprotonation of [1-H] is 20.3. Since the pKa of tosic acid in THF is predicted to be 11.8, the reduced porphyrin is likely to get protonated. The first two steps of the proposed mechanism (E–P) are in agreement with our experimental observations. The next step can be either electron transfer or protonation, to yield [1-H]− or [1-HH]+, respectively. While we cannot experimentally discern between these two pathways, [1-H]− is calculated to be thermodynamically favored by 8.91 kcal mol−1 when compared to generating [1-HH]+. The following step yields [1-HH], which putatively produces H2, and closes the cycle. The overall proposed mechanism is described in Scheme 5. Calculated relative pKas and redox potentials for all possible mechanistic pathways for H2 generation are presented in Table S3.†
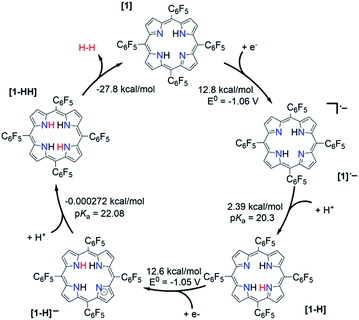 |
| | Scheme 5 Proposed catalytic cycle for H2 evolution, following an E–P–E–P mechanism, with THF as solvent. | |
Conclusion
In conclusion, we have studied the electrocatalytic generation of H2 using a metal-free perfluorinated porphyrin. The catalytic activity was studied through cyclic voltammetry and controlled-potential electrolysis. Hydrogen is produced electrochemically at −1.31 V vs. Fc/Fc+ in THF using tosic acid at 90% faradaic yield. Electronic spectra and spectroelectrochemical experiments combined with thermodynamic calculations using density functional theory computations suggest that the most favorable mechanistic process is an E–P–E–P sequence. This promising finding may contribute to open a new area for replacing noble metals with much more abundant organic compounds for the catalytic generation of hydrogen gas. It should be noted that this observed activity is limited to the meso-tetrapentafluorophenyl porphyrin with tosic acid and in THF as the solvent. We are currently working on the catalytic study of other substitution patterns (and other organic macrocycles) for the electrocatalytic production of hydrogen.
Conflicts of interest
The authors declare no conflicts of interest.
Acknowledgements
NRL acknowledges financial support from CONACYT (Mexico). We thank Prof. Daniel G. Nocera, Dr Dilek Dogutan, and Dr Maher for helpful discussions. This work was supported in part by NSF under award number CHE-1305124.
Notes and references
- N. S. Lewis and D. G. Nocera, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15729–15735 CrossRef CAS PubMed.
- J. A. Turner, Science, 2004, 305, 972–974 CrossRef CAS PubMed.
- M. G. Walter, E. L. Warren, J. R. McKone, S. W. Boettcher, Q. Mi, E. A. Santori and N. S. Lewis, Chem. Rev., 2010, 110, 6446–6473 CrossRef CAS PubMed.
- H. B. Gray, Nat. Chem., 2009, 1, 7 CrossRef CAS PubMed.
- D. Merki and X. Hu, Energy Environ. Sci., 2011, 4, 3878–3888 CAS.
- P. Du and R. Eisenberg, Energy Environ. Sci., 2012, 5, 6012–6021 CAS.
- W. T. Eckenhoff, W. R. McNamara, P. Du and R. Eisenberg, Biochim. Biophys. Acta Bioenerg., 2013, 1827, 958–973 CrossRef CAS PubMed.
- M. Wang, L. Chen and L. Sun, Energy Environ. Sci., 2012, 5, 6763–6778 CAS.
- C. Baffert, V. Artero and M. Fontecave, Inorg. Chem., 2007, 46, 1817–1824 CrossRef CAS PubMed.
- J. F. Callejas, J. M. McEnaney, C. G. Read, J. C. Crompton, A. J. Biacchi, E. J. Popczun, T. R. Gordon, N. S. Lewis and R. E. Schaak, ACS Nano, 2014, 8, 11101–11107 CrossRef CAS PubMed.
- M. R. DuBois and D. L. DuBois, Chem. Soc. Rev., 2008, 38, 62–72 RSC.
- L. L. Efros, H. H. Thorp, G. W. Brudvig and R. H. Crabtree, Inorg. Chem., 1992, 31, 1722–1724 CrossRef CAS.
- T. F. Jaramillo, K. P. Jørgensen, J. Bonde, J. H. Nielsen, S. Horch and I. Chorkendorff, Science, 2007, 317, 100–102 CrossRef CAS PubMed.
- E. J. Popczun, C. G. Read, C. W. Roske, N. S. Lewis and R. E. Schaak, Angew. Chem., Int. Ed., 2014, 53, 5427–5430 CrossRef CAS PubMed.
- A. D. Wilson, R. H. Newell, M. J. McNevin, J. T. Muckerman, M. Rakowski DuBois and D. L. DuBois, J. Am. Chem. Soc., 2006, 128, 358–366 CrossRef CAS PubMed.
- Y. Zheng, Y. Jiao, Y. Zhu, L. H. Li, Y. Han, Y. Chen, A. Du, M. Jaroniec and S. Z. Qiao, Nat. Commun., 2014, 5, ncomms4783 CrossRef PubMed.
- B. C. Patra, S. Khilari, R. N. Manna, S. Mondal, D. Pradhan, A. Pradhan and A. Bhaumik, ACS Catal., 2017, 7, 6120–6127 CrossRef CAS.
- T. Abe, F. Taguchi, H. Imaya, F. Zhao, J. Zhang and M. Kaneko, Polym. Adv. Technol., 1998, 9, 559–562 CrossRef CAS.
- I. Bhugun, D. Lexa and J.-M. Savéant, J. Am. Chem. Soc., 1996, 118, 3982–3983 CrossRef CAS.
- V. Grass, D. Lexa and J.-M. Savéant, J. Am. Chem. Soc., 1997, 119, 7526–7532 CrossRef CAS.
- C. H. Lee, D. K. Dogutan and D. G. Nocera, J. Am. Chem. Soc., 2011, 133, 8775–8777 CrossRef CAS PubMed.
- D. K. Bediako, B. H. Solis, D. K. Dogutan, M. M. Roubelakis, A. G. Maher, C. H. Lee, M. B. Chambers, S. Hammes-Schiffer and D. G. Nocera, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 15001–15006 CrossRef CAS PubMed.
- B. H. Solis and S. Hammes-Schiffer, Inorg. Chem., 2014, 53, 6427–6443 CrossRef CAS PubMed.
- J. P. Collman, P. S. Wagenknecht and N. S. Lewis, J. Am. Chem. Soc., 1992, 114, 5665–5673 CrossRef CAS.
- X. Hu, B. S. Brunschwig and J. C. Peters, J. Am. Chem. Soc., 2007, 129, 8988–8998 CrossRef CAS PubMed.
- S. C. Marinescu, J. R. Winkler and H. B. Gray, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 15127–15131 CrossRef CAS PubMed.
- H. N. Fonda, J. V. Gilbert, R. A. Cormier, J. R. Sprague, K. Kamioka and J. S. Connolly, J. Phys. Chem., 1993, 97, 7024–7033 CrossRef CAS.
- Handbook of Porphyrin Science (Volumes 31–35), http://www.worldscientific.com/worldscibooks/10.1142/8560, accessed January 24, 2017.
- S. Aronoff, J. Phys. Chem., 1958, 62, 428–431 CrossRef CAS.
- F. Hibbert and K. P. P. Hunte, J. Chem. Soc., Perkin Trans. 2, 1977, 1624–1628 RSC.
- J. S. Lindsey, H. C. Hsu and I. C. Schreiman, Tetrahedron Lett., 1986, 27, 4969–4970 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS.
- M. J. Frisch, J. A. Pople and J. S. Binkley, J. Chem. Phys., 1984, 80, 3265–3269 CrossRef CAS.
- P. C. Hariharan and J. A. Pople, Theor. Chim. Acta, 1973, 28, 213–222 CrossRef CAS.
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision C.01, Gaussian, Inc., Wallingford, CT, 2010 Search PubMed.
- R. Cammi, B. Mennucci and J. Tomasi, J. Phys. Chem. A, 2000, 104, 4690–4698 CrossRef CAS.
- R. Cammi, B. Mennucci and J. Tomasi, J. Phys. Chem. A, 1998, 102, 870–875 CrossRef CAS.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS PubMed.
- B. H. Solis, A. G. Maher, T. Honda, D. C. Powers, D. G. Nocera and S. Hammes-Schiffer, ACS Catal., 2014, 4, 4516–4526 CrossRef CAS.
- B. H. Solis and S. Hammes-Schiffer, Inorg. Chem., 2014, 53, 6427–6443 CrossRef CAS PubMed.
- B. H. Solis and S. Hammes-Schiffer, J. Am. Chem. Soc., 2011, 133, 19036–19039 CrossRef CAS PubMed.
- H. Tang and M. B. Hall, J. Am. Chem. Soc., 2017, 139, 18065–18070 CrossRef CAS PubMed.
- B. H. Solis, A. G. Maher, D. K. Dogutan, D. G. Nocera and S. Hammes-Schiffer, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 485–492 CrossRef CAS PubMed.
- K. Aidas, K. Lanevskij, R. Kubilius, L. Juška, D. Petkevičius and P. Japertas, J. Comput. Chem., 2015, 36, 2158–2167 CrossRef CAS PubMed.
- B. H. Solis, A. G. Maher, T. Honda, D. C. Powers, D. G. Nocera and S. Hammes-Schiffer, ACS Catal., 2014, 4, 4516–4526 CrossRef CAS.
- L. E. Fernandez, S. Horvath and S. Hammes-Schiffer, J. Phys. Chem. C, 2012, 116, 3171–3180 CAS.
- B. H. Solis and S. Hammes-Schiffer, Inorg. Chem., 2011, 50, 11252–11262 CrossRef CAS PubMed.
- J. Ho and M. L. Coote, Theor. Chem. Acc., 2010, 125, 3 CrossRef CAS.
- F. Ding, J. M. Smith and H. Wang, J. Org. Chem., 2009, 74, 2679–2691 CrossRef CAS PubMed.
- J. A. S. Roberts and R. M. Bullock, Inorg. Chem., 2013, 52, 3823–3835 CrossRef CAS PubMed.
- A. M. Appel and M. L. Helm, ACS Catal., 2014, 4, 630–633 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Cyclic voltammograms for p-toluenesulfonic acid in THF without the presence of catalysts. Details of the bulk electrolysis experiments. Calculated thermodynamic quantities and additional benchmark details. Cartesian coordinates of the DFT optimized geometries in the gas phase and in solution. See DOI: 10.1039/c8sc00093j |
|
| This journal is © The Royal Society of Chemistry 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *
*
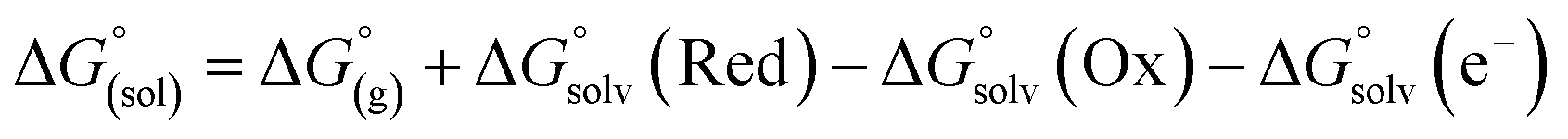
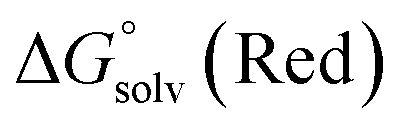 and
and 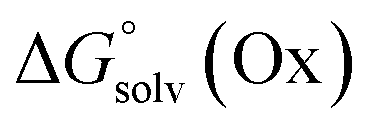 represent the free energies upon solvation for both the reduced and the oxidized species,
represent the free energies upon solvation for both the reduced and the oxidized species, 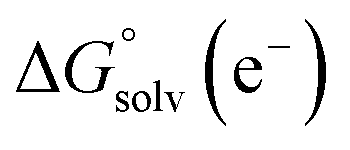 the contribution of the electron to the solvation free energy, and
the contribution of the electron to the solvation free energy, and 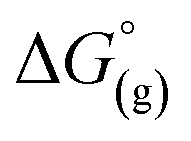 the free energy of the reaction in the gas phase. We can calculate the last value with the expression
the free energy of the reaction in the gas phase. We can calculate the last value with the expression 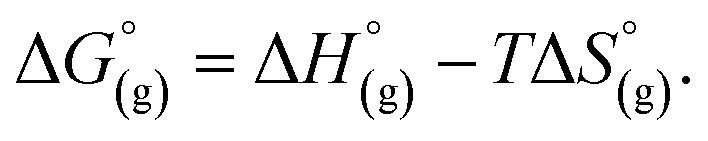
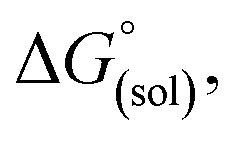 allows the determination of redox potentials using eqn (2):
allows the determination of redox potentials using eqn (2):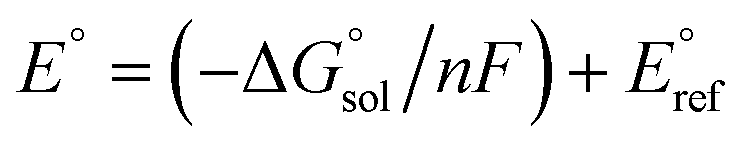
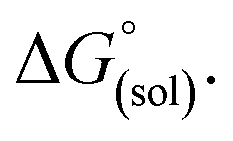 To calculate
To calculate 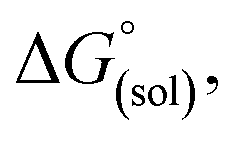 we must relate it to the free energy in the gas phase using the thermodynamic cycle depicted in Scheme 3, obtaining the following equation:
we must relate it to the free energy in the gas phase using the thermodynamic cycle depicted in Scheme 3, obtaining the following equation: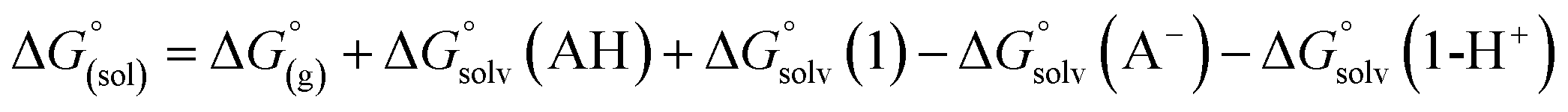
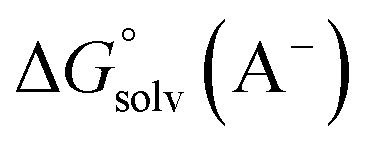 ,
, 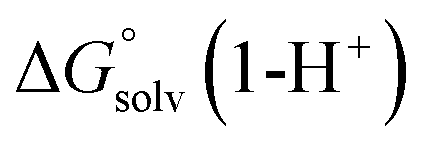 ,
, 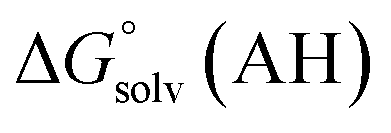 and
and 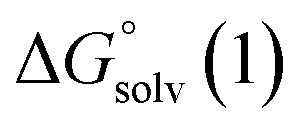 are the free energies upon solvation of the referenced conjugated base, the protonated porphyrin, the referenced acid and the porphyrin, respectively. We can calculate
are the free energies upon solvation of the referenced conjugated base, the protonated porphyrin, the referenced acid and the porphyrin, respectively. We can calculate 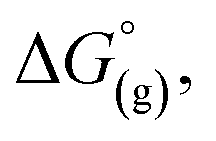 using the expression
using the expression 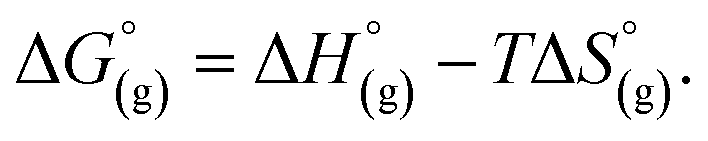 The pKa can be calculated from
The pKa can be calculated from 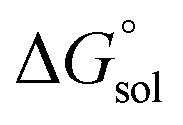 using:
using: