
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNear infrared two-photon-excited and -emissive dyes based on a strapped excited-state intramolecular proton-transfer (ESIPT) scaffold†
Naoya
Suzuki
a,
Kayo
Suda
b,
Daisuke
Yokogawa
 *ab,
Hirotaka
Kitoh-Nishioka
a,
Stephan
Irle
ab,
Akihiro
Ando
c,
Luis M. G.
Abegão
cd,
Kenji
Kamada
*c,
Aiko
Fukazawa
a and
Shigehiro
Yamaguchi
*ab
*ab,
Hirotaka
Kitoh-Nishioka
a,
Stephan
Irle
ab,
Akihiro
Ando
c,
Luis M. G.
Abegão
cd,
Kenji
Kamada
*c,
Aiko
Fukazawa
a and
Shigehiro
Yamaguchi
*ab
aDepartment of Chemistry, Graduate School of Science and Integrated Research Consortium on Chemical Sciences (IRCCS), Nagoya University, Japan
bInstitute of Transformative Bio-Molecules (WPI-ITbM), Nagoya University, Furo, Chikusa, Nagoya, 464-8602, Japan
cIFMRI, National Institute of Advanced Industrial Science and Technology (AIST), 1-8-31 Midorigaoka, Ikeda, Osaka, 563-8577, Japan
dDepartamento de Física, Universidade Federal de Sergipe, 49100-000 São Cristovão, SE, Brazil
First published on 1st February 2018
Abstract
Fluorophores that can undergo excited-state intramolecular proton transfer (ESIPT) represent promising scaffolds for the design of compounds that show red-shifted fluorescence. Herein, we disclose new near infrared-emissive materials based on a dialkylamine-strapped 2,5-dithienylpyrrole as an ESIPT scaffold. The introduction of electron-accepting units to the terminal positions of this scaffold generates acceptor–π–donor–π–acceptor (A–π–D–π–A) type π-conjugated compounds. Following the ESIPT, the electron-donating ability of the core scaffold increases, which results in a substantially red-shifted emission in the NIR region, while increasing the oscillator strength. The electron-accepting units play a vital role to achieve intense and red-shifted emission from the ESIPT state. The strapped dialkylamine chain that forms an intramolecular hydrogen bond is also essential to induce the ESIPT. Moreover, an extended A–π–D–π–A skeleton enables two-photon excitation with the NIR light. One of the derivatives that satisfy these features, i.e., borylethenyl-substituted 5, exhibited an intense NIR emission in polar solvents such as acetone (λem = 708 nm, ΦF = 0.55) with a strong two-photon-absorption band in the NIR region.
Introduction
Fluorescent molecules that exhibit emission in the near infrared (NIR) region have attracted increasing attention due to their versatile utility in biological applications1 and optoelectronics.2,3 In particular, the emission of such long-wavelength light reduces the phototoxicity toward cells, avoids interference from the auto-fluorescence background, and enables an in-depth penetration due to the low absorptivity between 650 nm and 900 nm in biological tissue.4 Fluorophores with these features are highly desirable for optical imaging in biological research such as deep-tissue imaging.5 Moreover, the fabrication of organic light-emitting diodes using NIR fluorophores would provide a promising light source for various bio-sensing applications such as pulse oximeters.6 Thus, considerable efforts have recently been devoted to the development of NIR fluorophores based on various design principles, such as construction of donor–acceptor-type skeletons,7–10 modifications of polymethine-based skeletons such as cyanine dyes,11 xanthene dyes,12–15 and BODIPY,16,17 as well as the formation of emissive excimers18 or aggregates.19 One particularly intriguing approach is based on the construction of a skeleton that features excited-state intramolecular proton transfer (ESIPT), i.e., the intramolecular transfer of an acidic proton to a basic moiety in the excited state (ES).20 Consequently, such a fluorophore can exhibit fluorescence with a large Stokes shift, which is the most notable feature of ESIPT-capable fluorophores compared to other types of emitting materials.Various types of the ESIPT systems have been reported, e.g. those containing 2-hydroxyphenyl-substituted benzothiazoles,21 benzoxazoles,21a,22 imidazoles,23 imidazo[1,2-a]pyridines,24,25 or 10-hydroxy benzo[h]quinolines.26 In particular, some ESIPT fluorophores with azole-type heterocycles have a suitable structure to avoid a nonradiative twisted intramolecular charge transfer process,27 and thereby exhibit intense ESIPT emission in the visible region. However, ESIPT fluorophores often suffer from low fluorescence quantum yields when their emission wavelengths reach the NIR region. One of the reasons for the inefficient ESIPT emission is slow radiative decay of the ES following proton transfer. Most conventional ESIPT systems rely on keto–enol tautomerism in the ES. This structural change results in a localization of the HOMO and LUMO in the ESIPT state, which results in a reduced overlap between ground and ES wavefunction and consequently leads to a small oscillator strength associated with the radiative decay.28 In addition, according to the energy-gap law,29 the nonradiative decay rate constants are expected to significantly increase upon bathochromically shifting the electronic transition. Therefore, a new design of an ESIPT-capable skeleton that does not rely on the keto–enol tautomerism is required to attain an intense emission in the NIR region based on the ESIPT mechanism.
Herein, we disclose a design strategy for fluorophores that exhibit a NIR-excited NIR emission based on a unique ESIPT skeleton. We have previously reported a 2,5-dithienylpyrrole that contains a dialkylamine strap as a core skeleton to induce ESIPT.30 The underlying concept for the design of this scaffold is to introduce a basic amine moiety as a proton acceptor to an emissive π-conjugated skeleton with an acidic proton in a shape suitable for formation of an intramolecular hydrogen bond (Fig. 1a). Indeed, strapped 2,5-dithienylpyrrole 1, which contains dimesitylboryl groups at the terminal positions, exhibited an emission band at ∼600 nm that was accompanied by a large Stokes shift in polar solvents, resulting from a zwitterionic ESIPT structure. Notably, a symmetric acceptor–π–donor–π–acceptor structure usually benefits efficient two-photon-absorption (TPA) properties.31 This fact prompted us to aim at a more red-shifted NIR emission upon excitation with NIR light through TPA. To optimize the structure to this end, the key features should be (1) the electron-accepting ability of the terminal groups and (2) the hydrogen-bond-forming ability of the strap moiety. To explore the substituent effects, we synthesized a series of compounds 2–7 that contain various terminal groups and strap moieties (Fig. 1b). Among these, compound 5, which was extended with borylvinyl groups, indeed exhibited the desired bathochromically shifted NIR emission with high intensity.
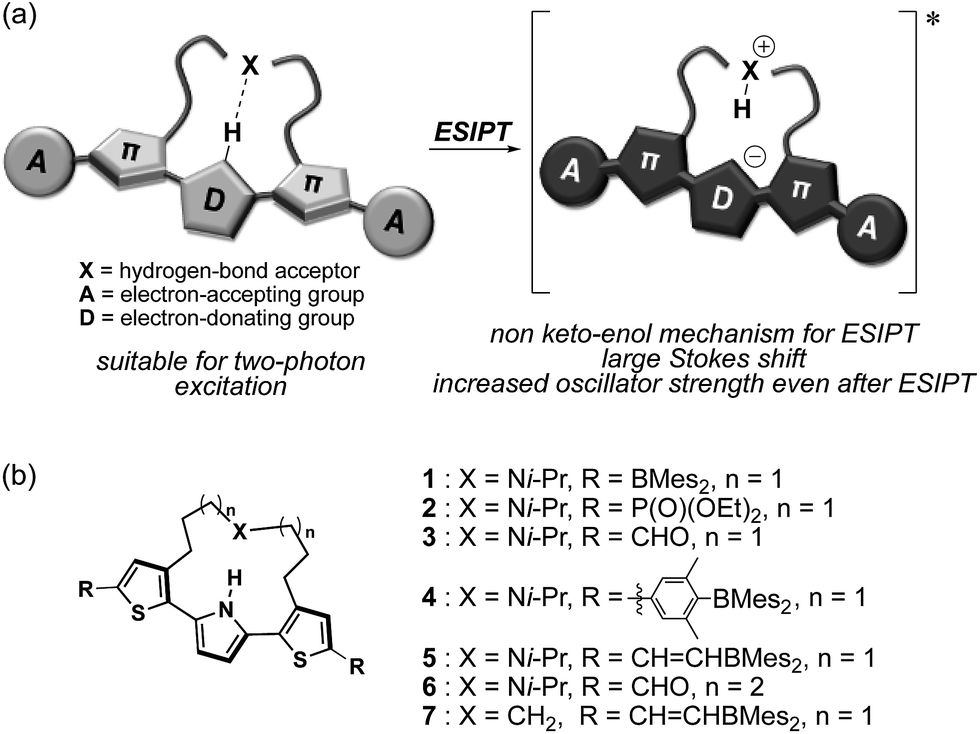 | ||
| Fig. 1 (a) Schematic illustration of NIR two-photon-excited and -emissive strapped ESIPT dyes and (b) chemical structures of 1–7, which were examined in this study. | ||
Results and discussion
Synthesis of a series of strapped dithienylpyrroles
The synthesis of compounds 2–5 is shown in Scheme 1. Compounds 2 and 3 were synthesized in three steps from strapped dithienylpyrrole 8. The pyrrole N–H moiety in 8 was initially protected with a t-butoxycarbonyl (Boc) group to afford 9, which was transformed into 10 and 11via lithiation with n-BuLi followed by treatment with the corresponding electrophiles. A subsequent heat-induced deprotection of the Boc group on the pyrrole afforded 2 and 3 as a colorless oil and a brown solid, respectively. Dimesitylborylxylyl-substituted 4 was synthesized in three steps from 9. Namely, 9 was dilithiated with LDA, followed by treatment with 1,2-dibromotetrachloroethene to furnish 12. After removal of the Boc group by treatment with an excess amount of sodium methoxide, Suzuki–Miyaura coupling reaction with the corresponding boron compound afforded 4 as a yellow powder. Dimesitylborylethenyl-substituted 5 was synthesized in three steps from 12. Sonogashira–Hagihara coupling reaction between 12 and trimethylsilylacetylene afforded 13, from which the trimethylsilyl and Boc groups were removed simultaneously by treatment with an excess amount of sodium methoxide to generate 14. A subsequent hydroborylation with dimesitylborane afforded 5 as an orange powder. Reference compounds 6 and 7 with a longer or non-amine strapped chain, respectively, were synthesized in a similar manner as described for 3 and 5, respectively.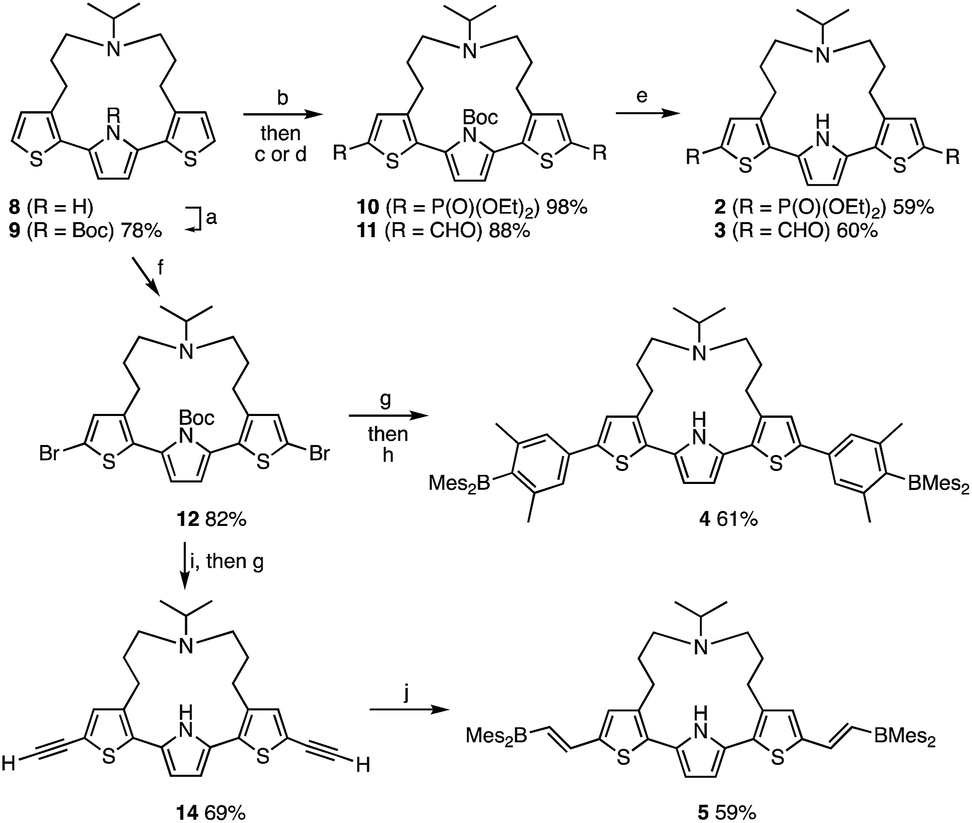 | ||
| Scheme 1 Synthesis of 2–5. Reagents and conditions: (a) Boc2O, DMAP, MeCN, 80 °C; (b) n-BuLi, THF, −78 °C; (c) diethyl chlorophosphate, THF, −78 °C to rt; (d) DMF, THF, −78 °C to rt; (e) neat, 150 °C for 2; 170 °C for 3; (f) LDA, 1,2-dibromotetrachloroethane, THF, −78 °C to rt; (g) NaOMe, THF/MeOH, 80 °C; (h) 4-Mes2B-3,5-Me2C6H2B(pin), Pd(PPh3)4, K2CO3, THF/H2O, 80 °C; (i) trimethylsilylacetylene, Pd(PPh3)4, CuI, i-Pr2NH, 70 °C; (j) HBMes2, THF, rt. | ||
The effect of terminal electron-accepting groups on the ESIPT behavior
Initially, we explored the impact of the terminal groups on the ESIPT behavior. For that purpose, we introduced diethoxy phosphoryl (–P(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)(OEt)2) or formyl groups into 2 and 3, respectively. Importantly, while the phosphoryl group can be considered as an electron-withdrawing group based on the inductive effect, the formyl group serves as an electron-accepting group that shows the mesomeric effect and participates in the delocalization of the LUMO (Fig. S22†). Consequently, 3 exhibits a much lower LUMO compared to that of 2, and this difference is consistent with the photophysical properties of these compounds (Fig. 2a). The absorption spectrum of 3 in THF shows an absorption maximum (λabs) at 423 nm, which is by 61 nm bathochromically shifted compared to that of 2. The fluorescence spectra in the same solvent show an even more pronounced difference. While phosphoryl-substituted 2 exhibits an emission band (λem) at 456 nm with shoulders at 420 nm and 487 nm, formyl-substituted 3 shows two distinct emission bands at 490 nm and 552 nm with Stokes shifts of 3200 cm−1 and 5500 cm−1, respectively. Picosecond time-resolved fluorescence spectroscopy measurements showed that the fluorescence decay curves can be fitted with double-exponential functions for both compounds (2
O)(OEt)2) or formyl groups into 2 and 3, respectively. Importantly, while the phosphoryl group can be considered as an electron-withdrawing group based on the inductive effect, the formyl group serves as an electron-accepting group that shows the mesomeric effect and participates in the delocalization of the LUMO (Fig. S22†). Consequently, 3 exhibits a much lower LUMO compared to that of 2, and this difference is consistent with the photophysical properties of these compounds (Fig. 2a). The absorption spectrum of 3 in THF shows an absorption maximum (λabs) at 423 nm, which is by 61 nm bathochromically shifted compared to that of 2. The fluorescence spectra in the same solvent show an even more pronounced difference. While phosphoryl-substituted 2 exhibits an emission band (λem) at 456 nm with shoulders at 420 nm and 487 nm, formyl-substituted 3 shows two distinct emission bands at 490 nm and 552 nm with Stokes shifts of 3200 cm−1 and 5500 cm−1, respectively. Picosecond time-resolved fluorescence spectroscopy measurements showed that the fluorescence decay curves can be fitted with double-exponential functions for both compounds (2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.74 and 0.97 ns, 3:0.73 and 1.7 ns in THF, respectively. Fig. S12 and S13†), indicative of dual emission from the locally-excited (LE) and ESIPT states for both compounds. However, it is obvious that the ESIPT emission is more enhanced in formyl-substituted 3. This difference implies that the degree of the electron-accepting ability of the terminal group should affect the acidity of the pyrrole N–H group in the ES.
:0.74 and 0.97 ns, 3:0.73 and 1.7 ns in THF, respectively. Fig. S12 and S13†), indicative of dual emission from the locally-excited (LE) and ESIPT states for both compounds. However, it is obvious that the ESIPT emission is more enhanced in formyl-substituted 3. This difference implies that the degree of the electron-accepting ability of the terminal group should affect the acidity of the pyrrole N–H group in the ES.
 | ||
| Fig. 2 UV-vis absorption and emission spectra of (a) 2, 3, and 6 in THF and (b) 3 in various solvents; dotted lines represent absorption, while solid lines show emission. | ||
Interestingly, 3 exhibited a distinct dependence of its dual emission properties on the solvent polarity (Fig. 2b). In nonpolar cyclohexane, 3 exhibited an intense structured emission band at 466 nm (ΦF = 0.37). The single exponential decay of the fluorescence suggests the existence of only LE state in this solvent. As the solvent polarity increases from benzene to THF to acetone, the emission band from the ESIPT state increased in intensity and was slightly red-shifted. Eventually, 3 showed emission only from the ESIPT state in acetone, as was evident from a single component of the fluorescence lifetime. It should be noted that the total fluorescence quantum yield increases with the solvent polarity and ultimately reaches ΦF = 0.60 in acetone. This is opposite to the general trend observed for conventional A–π–D–π–A-type compounds, whose quantum yields tend to decrease with increasing solvent polarity. This trend should result from the higher fluorescence quantum yield for the ESIPT emission compared to that for the LE emission. The radiative (kr) and nonradiative (knr) decay rate constants from both states were determined based on the ΦF and τ values (Table S1†). While the kr and knr values are 0.41 × 109 s−1 and 0.70 × 109 s−1 for the LE state in cyclohexane, the values for the ESIPT state in acetone are 0.27 × 109 s−1 and 0.18 × 109 s−1, respectively. This comparison clearly shows that the nonradiative decay from the ESIPT state is retarded to a larger extent.
The effect of the length of the strap chain on the ESIPT behavior
To induce ESIPT, an appropriate chain length of the strap moiety is also crucial. Compound 6 bears the longer strap chain and shows, in sharp contrast to 3, only a single emission band even in polar solvents (Fig. 2a). Its emission band is located in a similar region to that of the LE band of 3. Time-dependent fluorescence measurements revealed that 6 has only a single component of the fluorescence lifetime. These results demonstrate that 6 is ESIPT inert even though it contains a proton-accepting amine in the strap chain.The behavioral difference between 3 and 6, despite sharing an identical fluorophore skeleton, should be attributed to differences in the ability to form an intramolecular hydrogen bond between the pyrrole N–H bond and the amine moiety of the strap. To obtain clear-cut evidence for such intramolecular hydrogen bonds, the molecular structures of 3 and 6 in the crystalline state were determined (Fig. 3). In the single crystals, the dithienylpyrrole scaffold of 6 adopts an unsymmetrically twisted structure. The dihedral angle between the central pyrrole ring and the thiophene rings are 27.9° and 54.4°. In contrast, 3 exhibits a relatively coplanar structure with dihedral angles of 25.2° and 17.9°. This difference should be responsible for the slightly red-shifted absorption maxima of 3 (λabs,max = 423 nm) compared to that of 6 (λabs,max = 393 nm). More importantly, while the distance between the two nitrogen atoms N1 and N2 in 6 is 5.21 Å, that in 3 is 2.83 Å, which clearly demonstrates the existence of an intramolecular hydrogen bond.32 This comparison shows that the chain length of the strap moiety is critical for the formation of an intramolecular hydrogen bond.
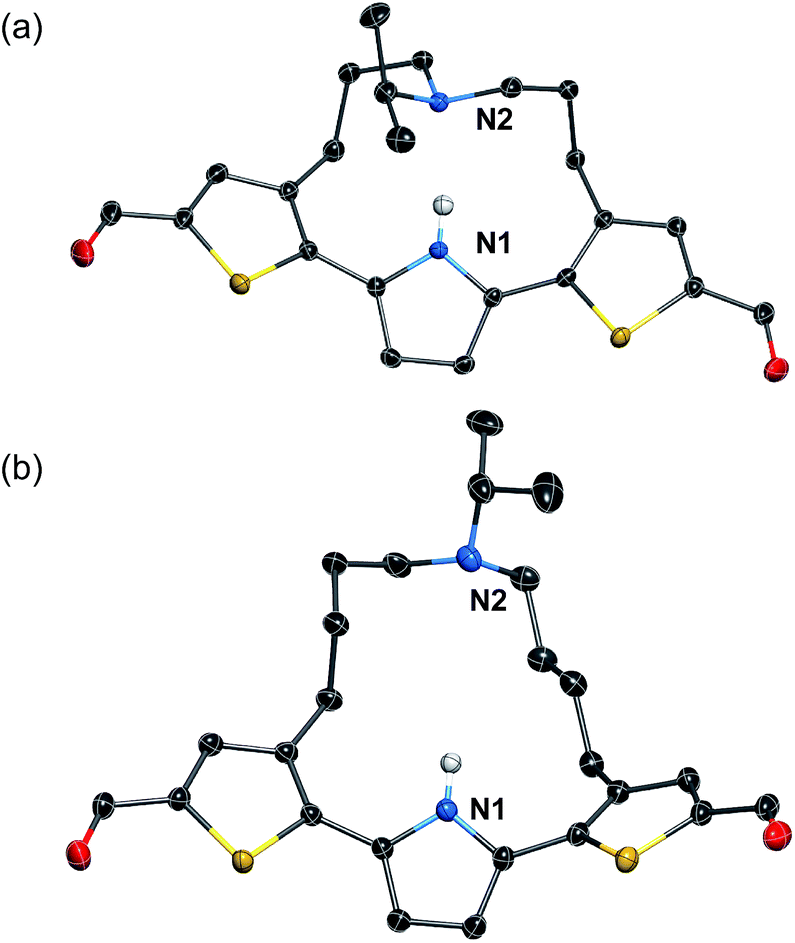 | ||
| Fig. 3 X-ray crystal structures of (a) 3 and (b) 6 (thermal ellipsoids set to 50% probability). Expect for the N–H proton of the pyrrole ring, all hydrogen atoms are omitted for clarity. | ||
The 1H NMR spectra of 3 and 6 in C6D6 revealed that this structural difference is retained in solution. While the resonance signal for the N–H proton of 6 appeared at 8.27 ppm, that of 3 was significantly downfield-shifted (13.1 ppm) (Fig. S1†). Comparisons of the ESIPT behavior and the structural features between 3 and 6 demonstrate that the preorganization of the intramolecular hydrogen bond with a strap chain of appropriate chain length enables the efficient formation of the ESIPT state.
Theoretical study of the ES
In comparison with conventional ESIPT systems that comprise 2-hydroxyphenyl-substituted azoles or related scaffolds, the most notable feature of our system is that intense emission is retained even in the ESIPT state. From this feature it can be inferred that even after the proton transfer occurs, the fluorophore skeleton retains the π-extended character of the A–π–D–π–A electronic structure. Moreover, in the ESIPT state, as the electron-donating character of the pyrrole moiety is enhanced, a quinoidal character with a more rigid planar structure is adopted, which could be the origin of the intense emission. For the theoretical elucidation of the ESIPT behavior in our system, 3 is the most suitable compound to investigate in depth, as it shows distinct emissions from the LE state in cyclohexane and from the ESIPT state in acetone. Based on this consideration, we conducted TD-DFT calculations on 3, using the CAM-B3LYP functional in order to determine the structures in the LE and ESIPT states, where solvent effects were considered using the PCM model.The geometry optimization of 3 in S1 revealed two local minima that can be associated with the LE and ESIPT states in both cyclohexane and acetone (Fig. 4). The relative energies of these two states strongly depend on the solvent polarity. In cyclohexane, the energies of the LE and ESIPT states relative to the optimized structure in S0 are +2.92 eV and +3.08 eV. In contrast, the energies of both states decrease in acetone (LE: +2.68 eV; ESIPT: +2.59 eV), and, more importantly, the ESIPT state becomes thermodynamically more stable. These results are consistent with the experimentally observed solvent effect for 3.
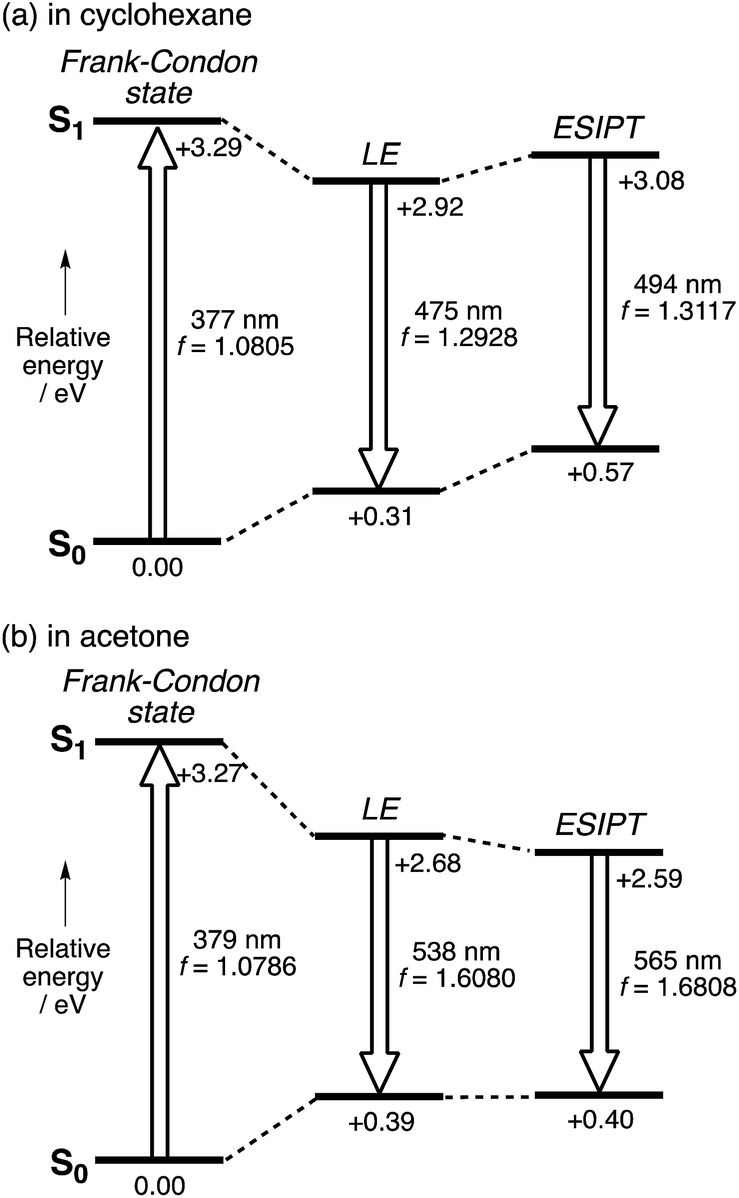 | ||
| Fig. 4 Potential energy diagrams for 3 (a) in cyclohexane and (b) in acetone. The relative energies were calculated at the CAM-B3LYP/6-31+G(d) (N and O), 6-31G(d,p) (migrating H), and 6-31G(d) (all other atoms) levels of theory, and are given in eV relative to the optimized geometry in S0. | ||
A notable characteristic of our strapped ESIPT system is the large oscillator strength (f) of the electronic transition even in the ESIPT state. The f value of 3 for the LE state in cyclohexane (1.293) increases for the ESIPT state in acetone (1.681). As the radiative decay rate constant (kr) is proportional to the ν2f value,33 where ν is discuss this ν2f value. The calculated values for the LE state in cyclohexane (2.72 × 1011 cm−2) and the ESIPT state in acetone (2.97 × 1011 cm−2) are comparable to each other. This fact demonstrates that although the red-shift in the emission from the LE state in cyclohexane to the ESIPT state in acetone significantly decreases the ν value, this is compensated by a concomitant increase in the oscillator strength in the ESIPT state by 30%. This is a rather unique feature of our system compared to conventional ESIPT systems.28
In addition, it is worth noting that the significant stabilization of the ESIPT state in acetone accompanies a substantial structural change. Fig. 5 shows the optimized geometry and plots of the variation in each bond distance from the ground state to the LE state in cyclohexane and the ESIPT state in acetone. The larger structural relaxation in acetone than in cyclohexane leads to a more pronounced quinoidal character of the ESIPT state in acetone (Fig. 5c), where the C7–C8 bond is significantly shortened, and the C6–C7 and C8–C9 bonds in the central pyrrole ring are elongated. This structural change should be, at least in part, responsible for the increase in oscillator strength, and might also be relevant to the suppression of the non-radiative decay by forming a rigid planar quinoidal structure in the ESIPT state.
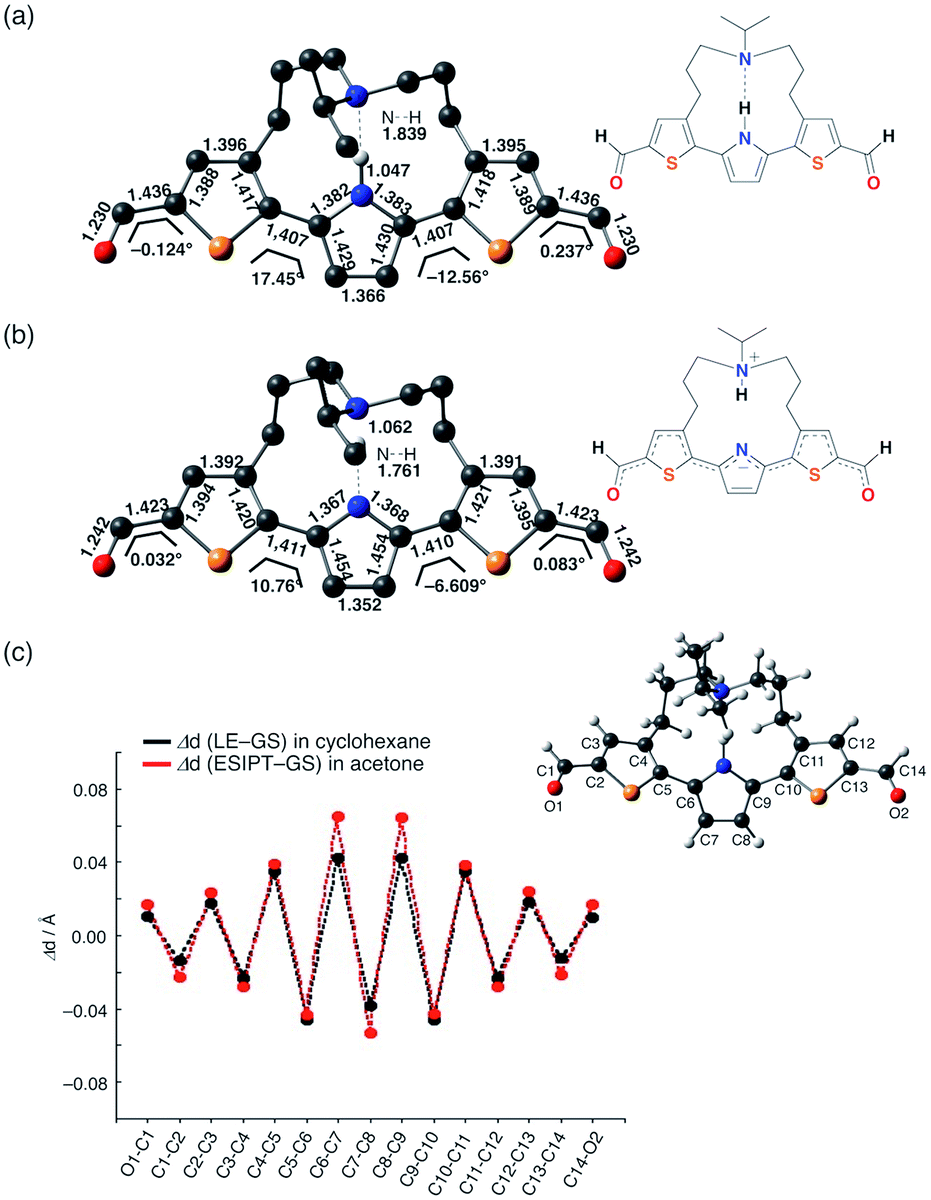 | ||
| Fig. 5 Optimized geometries of 3 for (a) the LE state in cyclohexane and (b) the ESIPT state in acetone; (c) plots of the change in each bond distance during the structural relaxation in the ES and an atom-labeling scheme. | ||
Structural modifications to attain more red-shifted and brighter NIR emission
In order to achieve efficient ESIPT emission in the NIR region, we subsequently tried to expand the π-conjugation. Among 1–3, dimesitylboryl-substituted 1 showed the longest-wavelength emission from the ESIPT state. Therefore, we used this scaffold and synthesized the corresponding π-extended derivatives 4 and 5, which contain 3,5-dimethylphenyl and ethenyl groups between the thiophene and BMes2 groups, respectively. Indeed, compounds 4 and 5 exhibited emission in the NIR region in polar solvents, whereby 4 showed a more pronounced response to the solvent polarity (Fig. 6). In cyclohexane, 4 exhibited an absorption band at 435 nm and a green fluorescence band at 513 nm with a Stokes shift of 3500 cm−1. In acetone, a significantly red-shifted ESIPT emission band appeared at 728 nm (ΦF = 0.18). The Stokes shift reached 9600 cm−1, which is substantially larger than those of other derivatives, indicative of a large structural relaxation of 4 in polar solvents.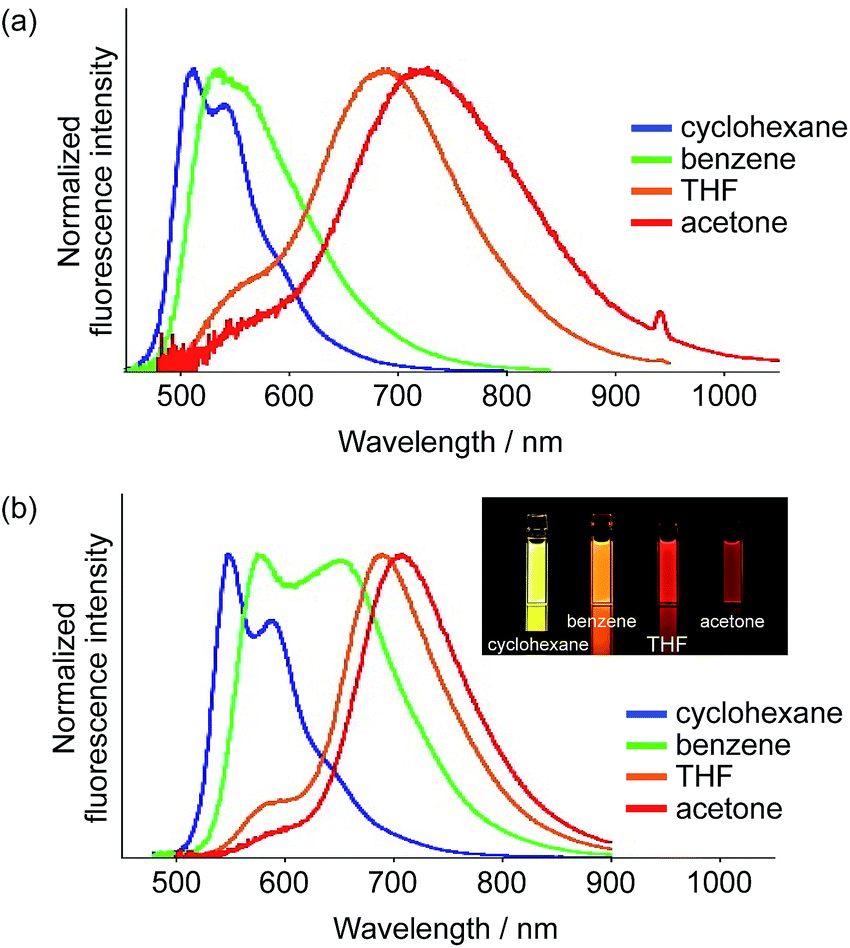 | ||
| Fig. 6 Fluorescence spectra of (a) 4 and (b) 5 in various solvents (inset in b: photographs under irradiation with λex = 365 nm). | ||
In terms of the fluorescence quantum yield, ethenyl-extended 5 exhibited more attractive properties. While 5 showed an intense emission at 547 nm from the LE state (ΦF = 0.68) in cyclohexane, it exhibited NIR fluorescence from the ESIPT state in acetone at 708 nm with a Stokes shift of 6600 cm−1. Notably, the fluorescence quantum yield remained high (ΦF = 0.55), despite the large Stokes shift. In light of the fact that examples of NIR-fluorescent ESIPT dyes with λem >700 nm and high fluorescence quantum yields are still limited,21g,34 our system can be considered as a rather unique fluorescent scaffold.
Two-photon absorption properties
It should also be noted that our system is beneficial to two-photon excitation. It has been well documented that A–π–D–π–A-type fluorophores exhibit a large two-photon absorption cross section (TPACS).31,35 To assess this feature, we conducted TPA measurements on 5 together with 1 and 7 as reference compounds (Fig. 7). The TPA spectra were recorded in THF using the open-aperture Z-scan method with a femtosecond optical parametric amplifier (SpectraPhysics TOPAS Prime pumped by a Spitfire operating at a repetition rate of 1 kHz) as the light source by varying the incident wavelength. The results revealed that 5 exhibits the largest TPACS (698 GM at 881 nm), which is almost double that of boryl-substituted 1 (322 GM at 800 nm), which demonstrates the impact of the extension of the π-conjugation in 5. This result also means that 5 can show an NIR emission at ∼700 nm by two-photon excitation in the NIR region. It is the large Stokes shift of the ESIPT that can be the mechanism to achieve such a close energy difference between the emission and the two-photon excitation.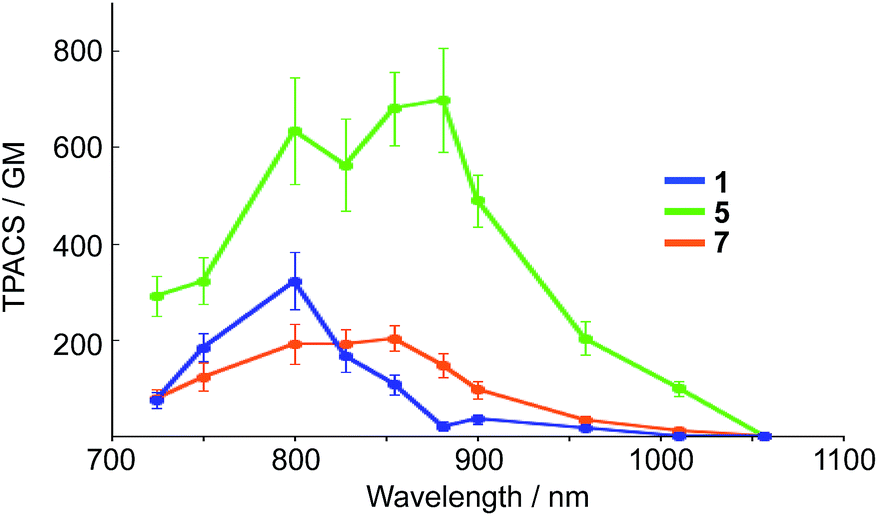 | ||
| Fig. 7 Two-photon absorption cross section (TPACS) of 1, 5, and 7 in THF (solid circles). All data were corrected based on MPPBT/DMSO as the in-house standard.35 | ||
A comparison between 5 and 7 revealed the crucial role of the amine strap in 5 for its TPA properties. Compound 7, which contains an alkyl chain strap instead of an amine, exhibited a TPACS value that was one third smaller that of 5, despite the fact that these compounds have the same π-conjugated scaffold. The temperature-dependent absorption spectra of 7 afforded insight into this difference (Fig. S21†). Compound 7 showed a broad absorption band at 300–550 nm at room temperature. Upon decreasing the temperature, the absorbance at 396 nm increased gradually, while that at 490 nm decreased with an isosbestic point. This spectral change indicates that 7 comprises two conformers in the ground state. The conformer exhibiting the absorption band at the longer wavelength, which most likely has a more planar structure, is thermodynamically less stable than the other. In sharp contrast, 5 only showed a single absorption band at 490 nm in solution. These differences suggest that the intramolecular hydrogen bond in 5 helps to constrain the conformation of the 2,5-dithienylpyrrole moiety in a planar fashion in the ground state. The population of the planar conformer is probably responsible for the increased TPACS of 5 compared to that of 7.
Conclusions
We have demonstrated that the nature of the terminal electron-accepting groups plays a crucial role for the ESIPT fluorescence properties of an alkylamine-strapped ESIPT chromophore based on 2,5-dithienylpyrrole. A diformyl derivative represents a suitable model to study the emission properties from the LE and ESIPT states. TD-DFT calculations revealed that in the ESIPT state the π-conjugated part of the molecule adopts a significant quinoidal structure. Of particular note is that a borylethenyl-substituted derivative 5 showed an intense NIR emission (λem = 708 nm, ΦF = 0.55) in acetone. Moreover, 5 can be excited by two-photon absorption, as it exhibits high two-photon absorption cross-section values. Notably, it is the ESIPT mechanism with a large Stokes shift that enables the close energy between the NIR two-photon excitation and the NIR emission. These features should offer significant merits in further applications of NIR fluorescent dyes, such as a deep-tissue fluorescence imaging and NIR organic light-emitting diodes.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was partly supported by JSPS KAKENHI grants 15H02163 (S. Y.), 24109007 and 25248007 (K. K.), and by a Grant-in-Aid for Scientific Research on Innovative Areas, Photosynergetics (JP26107004, K. K.) from MEXT, Japan, as well as by the Nagase Science and Technology Foundation and the Naito Foundation (S. Y.). ITbM is supported by the World Premier International Research Center (WPI) Initiative, Japan.Notes and references
- For recent reviews, see: (a) S. Luo, E. Zhang, Y. Su, T. Cheng and C. Shi, Biomaterials, 2011, 32, 7127–7138 CrossRef CAS PubMed; (b) V. Pansare, S. Hejazi, W. Faenza and R. K. Prud’homme, Chem. Mater., 2012, 24, 812–827 CrossRef CAS PubMed; (c) X. Yi, F. Wang, W. Qin, X. Yang and J. Yuan, Int. J. Nanomed., 2014, 9, 1347–1365 CrossRef PubMed; (d) F. Anzengruber, P. Avci, L. Freitas de Freitas and M. R. Hamblin, Photochem. Photobiol. Sci., 2015, 14, 1492–1509 RSC; (e) G. Hong, A. L. Antaris and H. Dai, Nature Biomedical Engineering, 2017, 1, 0010 CrossRef; (f) A. Haque, M. S. H. Faizi, J. A. Rather and M. S. Khan, Bioorg. Med. Chem., 2017, 25, 2017–2034 CrossRef CAS PubMed.
- For reviews, see: (a) K.-Y. Law, Chem. Rev., 1993, 93, 449–486 CrossRef CAS; (b) Y. Yang, Y. H. Zhang, W. Z. Shen and H. C. Liu, Prog. Quantum Electron., 2011, 35, 77–108 CrossRef.
- (a) N. Tessler, V. Medvedev, M. Kazes, S. Kan and U. Banin, Science, 2002, 295, 1506–1508 CrossRef PubMed; (b) E. L. Williams, J. Li and G. E. Jabbour, Appl. Phys. Lett., 2006, 89, 083506 CrossRef; (c) D. Y. Kim, D. W. Song, N. Chopra, P. D. Somer and F. So, Adv. Mater., 2010, 22, 2260–2263 CrossRef CAS PubMed; (d) J. Fischer, M. Tietke, F. Fritze, O. Muth, M. Paeschke, D. Han, J. Kwack, T. Kim, J. Lee, S. Kim and H. Chung, J. Soc. Inf. Disp., 2011, 19, 163–169 CrossRef CAS; (e) B. Stender, S. F. Völker, C. Lambert and J. Pflaum, Adv. Mater., 2013, 25, 2943–2947 CrossRef CAS PubMed; (f) P. A. Haigh, F. Bausi, Z. Ghassemlooy, I. Papakostantinou, H. L. Minh, C. Fléchon and F. Cacialli, Opt. Express, 2014, 22, 2830–2838 CrossRef PubMed; (g) H. Tachibana, N. Aizawa, Y. Hidaka and T. Yasuda, ACS Photonics, 2017, 4, 223–227 CrossRef CAS.
- R. Weissleder, Nat. Biotechnol., 2001, 19, 316–317 CrossRef CAS PubMed.
- (a) C. Bremer, C.-H. Tung and R. Weissleder, Nat. Med., 2001, 7, 743–748 CrossRef CAS PubMed; (b) V. Ntziachristos, Nat. Methods, 2010, 7, 603–614 CrossRef CAS PubMed; (c) K. Welsher, S. P. Sherlock and H. Dai, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 8943–8948 CrossRef CAS PubMed; (d) N. Zhang, K. P. Francis, A. Prakash and D. Ansaldi, Nat. Med., 2013, 19, 500–506 CrossRef CAS PubMed; (e) D. M. Shcherbakova, M. Baloban, A. V. Emelyanov, M. Brenowitz, P. Guo and V. V. Verkhusha, Nat. Commun., 2015, 7, 12405 CrossRef PubMed.
- (a) C. M. Lochner, Y. Khan, A. Pierre and A. C. Arias, Nat. Commun., 2014, 5, 5745 CrossRef CAS PubMed; (b) T. Yamanaka, H. Nakanotani, S. Hara, T. Hirohata and C. Adachi, Appl. Phys. Express, 2017, 10, 074101 CrossRef.
- (a) G. Qian, Z. Zhong, M. Luo, D. Yu, Z. Zhang, Z. Y. Wang and D. Ma, Adv. Mater., 2009, 21, 111–116 CrossRef CAS; (b) A. L. Antaris, H. Chen, K. Cheng, Y. Sun, G. Hong, C. Qu, S. Diao, Z. Deng, X. Hu, B. Zhang, X. Zhang, O. K. Yaghi, Z. R. Alamparambli, X. Hong, Z. Cheng and H. Dai, Nat. Mater., 2016, 15, 235–242 CrossRef CAS PubMed; (c) Y. Sun, C. Qu, H. Chen, M. He, C. Tang, K. Shou, S. Hong, M. Yang, Y. Jiang, B. Ding, Y. Xiao, L. Xing, X. Hong and Z. Cheng, Chem. Sci., 2016, 7, 6203–6207 RSC; (d) P. Murto, A. Minotto, A. Zampetti, X. Xu, M. R. Andersson, F. Cacialli and E. Wang, Adv. Opt. Mater., 2016, 4, 2068–2076 CrossRef CAS; (e) Q. Yang, Z. Ma, H. Wang, B. Zhou, S. Zhu, Y. Zhang, J. Wang, H. Wan, A. Antaris, R. Ma, X. Zhang, J. Yang, X. Zhang, H. Sun, W. Liu, Y. Liang and H. Dai, Adv. Mater., 2017, 29, 1605497 CrossRef PubMed.
- (a) A. Wakamiya, K. Mori and S. Yamaguchi, Angew. Chem., Int. Ed., 2007, 46, 4273–4276 CrossRef CAS PubMed; (b) Z. Zhang, R. M. Edkins, J. Nitsch, K. Fucke, A. Steffen, L. E. Longobardi, D. W. Stephan, C. Lambert and T. B. Marder, Chem. Sci., 2015, 6, 308–321 RSC; (c) X. Yin, K. Liu, Y. Ren, R. A. Lalancette, Y.-L. Loo and F. Jäkle, Chem. Sci., 2017, 8, 5497–5505 RSC; (d) K. Liu, R. A. Lalancette and F. Jäkle, J. Am. Chem. Soc., 2017, 139, 18170–18173 CrossRef CAS PubMed.
- S. Wang, X. Yan, Z. Cheng, H. Zhang, Y. Liu and Y. Wang, Angew. Chem., Int. Ed., 2015, 54, 13068–13072 CrossRef CAS PubMed.
- M. Luo, H. Shadnia, G. Qian, X. Du, D. Yu, D. Ma, J. S. Wright and Z. Y. Wang, Chem.–Eur. J., 2009, 15, 8902–8908 CrossRef CAS PubMed.
- For recent reviews, see: (a) W. Sun, S. Guo, C. Hu, J. Fan and X. Peng, Chem. Rev., 2016, 116, 7768–7817 CrossRef CAS PubMed; (b) C. Shi, J. B. Wu and D. Pan, J. Biomed. Opt., 2016, 21, 050901 CrossRef PubMed; (c) H. A. Shindy, Dyes Pigm., 2017, 145, 505–513 CrossRef CAS.
- For recent reviews, see: (a) Y.-Q. Sun, J. Liu, X. Lv, Y. Liu, Y. Zhao and W. Guo, Angew. Chem., Int. Ed., 2012, 51, 7634–7636 CrossRef CAS PubMed; (b) Y. Kushida, T. Nagano and K. Hanaoka, Analyst, 2015, 140, 685–695 RSC.
- A. Fukazawa, J. Usuba, R. A. Alder and S. Yamaguchi, Chem. Commun., 2017, 53, 8565–8568 RSC.
- (a) X. Chai, X. Cui, B. Wang, F. Yang, Y. Cai, Q. Wu and T. Wang, Chem.–Eur. J., 2015, 21, 16754–16758 CrossRef CAS PubMed; (b) Y.-J. Gong, X.-B. Zhang, G.-J. Mao, L. Su, H.-M. Meng, W. Tan, S. Feng and G. Zhang, Chem. Sci., 2016, 7, 2275–2285 RSC; (c) X. Zhou, R. Lai, J. R. Beck, H. Li and C. I. Stains, Chem. Commun., 2016, 52, 12290–12293 RSC; (d) J. Liu, Y.-Q. Sun, H. Zhang, H. Shi, Y. Shi and W. Guo, ACS Appl. Mater. Interfaces, 2016, 8, 22953–22962 CrossRef CAS PubMed.
- (a) M. V. Sednev, C. A. Wurm, V. N. Belov and S. W. Hell, Bioconjugate Chem., 2013, 24, 690–700 CrossRef CAS PubMed; (b) A. Roth, H. Li, C. Anorma and J. Chan, J. Am. Chem. Soc., 2015, 137, 10890–10893 CrossRef CAS PubMed; (c) M. Grzybowski, M. Taki and S. Yamaguchi, Chem.–Eur. J., 2017, 23, 13028–13032 CrossRef CAS PubMed.
- For recent reviews, see: (a) A. Loudet and K. Burgess, Chem. Rev., 2007, 107, 4891–4932 CrossRef CAS PubMed; (b) Y. Ni and J. Wu, Org. Biomol. Chem., 2014, 12, 3774–3791 RSC.
- (a) J. Bartelmess, M. Baldrighi, V. Nardone, E. Parisini, D. Buck, L. Echegoyen and S. Giordani, Chem.–Eur. J., 2015, 21, 9727–9732 CrossRef CAS PubMed; (b) S. Cherumukkil, S. Ghosh, V. K. Praveen and A. Ajayaghosh, Chem. Sci., 2017, 8, 5644–5649 RSC; (c) A. Zampetti, A. Minotto, B. M. Squeo, V. G. Gregoriou, S. Allard, U. Scherf, C. L. Chochos and F. Cacialli, Sci. Rep., 2017, 7, 1611 CrossRef PubMed.
- (a) C. Wei, M. Gao, F. Hu, J. Yao and Y. S. Zhao, Adv. Opt. Mater., 2016, 4, 1009–1014 CrossRef CAS; (b) A. Jana, L. Bai, X. Li, H. Ågren and Y. Zhao, ACS Appl. Mater. Interfaces, 2016, 8, 2336–2347 CrossRef CAS PubMed.
- (a) Q. Zhao, K. Li, S. Chen, A. Qin, D. Ding, S. Zhang, Y. Liu, B. Liu, J. Z. Sun and B. Z. Tang, J. Mater. Chem., 2012, 22, 15128–15135 RSC; (b) X. Du, J. Qi, Z. Zhang, D. Ma and Z. Y. Wang, Chem. Mater., 2012, 24, 2178–2185 CrossRef CAS; (c) K. Cai, J. Xie and D. Zhao, J. Am. Chem. Soc., 2014, 136, 28–31 CrossRef CAS PubMed; (d) G. M. Paternò, L. Moretti, A. J. Barker, C. D'Andrea, A. Luzio, N. Barbero, S. Galliano, C. Barolo, G. Lanzani and F. Scotognella, J. Mater. Chem. C, 2017, 5, 7732–7738 RSC; (e) F. S. Freyria, J. M. Cordero, J. R. Caram, S. Doria, A. Dodin, Y. Chen, A. P. Willard and M. G. Bawendi, Nano Lett., 2017, 17, 7665–7674 CrossRef CAS PubMed.
- For recent reviews, see: (a) J. E. Kwon and S. Y. Park, Adv. Mater., 2011, 23, 3615–3642 CrossRef CAS PubMed; (b) J. Zhao, S. Ji, Y. Chen, H. Guo and P. Yang, Phys. Chem. Chem. Phys., 2012, 14, 8803–8817 RSC; (c) A. P. Demochenko, K.-C. Tang and P.-T. Chou, Chem. Soc. Rev., 2013, 42, 1379–1408 RSC; (d) V. S. Padalkar and S. Seki, Chem. Soc. Rev., 2016, 45, 169–202 RSC.
- (a) D. L. Williams and A. Heller, J. Phys. Chem., 1970, 74, 4473–4480 CrossRef; (b) K. Sakai, H. Kawamura, N. Kobayashi, T. Ishikawa, C. Ikeda, T. Kikuchi and T. Akutagawa, CrystEngComm, 2014, 16, 3180–3185 RSC; (c) H.-W. Tseng, J.-Q. Liu, Y.-A. Chen, C.-M. Chao, K.-M. Liu, C.-L. Chen, T.-C. Lin, C.-H. Hung, Y.-L. Chou, T.-C. Lin, T.-L. Wang and P.-T. Chou, J. Phys. Chem. Lett., 2015, 6, 1477–1486 CrossRef CAS PubMed; (d) W. Zhang, Y. Yan, J. Gu, J. Yao and Y. S. Zhao, Angew. Chem., Int. Ed., 2015, 54, 7125–7129 CrossRef CAS PubMed; (e) P. Xu, T. Gao, M. Liu, H. Zhang and W. Zeng, Analyst, 2015, 140, 1814–1816 RSC; (f) T. Gao, P. Xu, M. Liu, A. Bi, P. Hu, B. Ye, W. Wang and W. Zeng, Chem.–Asian J., 2015, 10, 1142–1145 CrossRef CAS PubMed; (g) D. Dahal, L. McDonald, X. Bi, C. Abeywickrama, F. Gombedza, M. Konopka, S. Paruchuri and Y. Pang, Chem. Commun., 2017, 53, 3697–3700 RSC.
- (a) C. H. Kim, J. Park, J. Seo, S. Y. Park and T. Joo, J. Phys. Chem. A, 2010, 114, 5618–5629 CrossRef CAS PubMed; (b) Y. Xu and Y. Pang, Chem. Commun., 2010, 46, 4070–4072 RSC; (c) K. Benelhadj, W. Muzuzu, J. Massue, P. Retailleau, A. Charaf-Eddin, A. D. Laurent, D. Jacquemin, G. Ulrich and R. Ziessel, Chem.–Eur. J., 2014, 20, 12843–12857 CrossRef CAS PubMed.
- (a) C. J. Fahrni, M. M. Henary and D. G. VanDerveer, J. Phys. Chem. A, 2002, 106, 7655–7663 CrossRef CAS; (b) M. M. Henary, Y. Wu, J. Cody, S. Sumalekshmy, J. Li, S. Mandal and C. J. Fahrni, J. Org. Chem., 2007, 72, 4784–4797 CrossRef CAS PubMed; (c) S. Park, J. E. Kwon and S. Y. Park, Phys. Chem. Chem. Phys., 2012, 14, 8878–8884 RSC; (d) K. Skonieczny, J. Yoo, J. M. Larsen, E. M. Espinoza, M. Barbasiewicz, V. I. Vullev, C.-H. Lee and D. T. Gryko, Chem.–Eur. J., 2016, 22, 7485–7496 CrossRef CAS PubMed.
- For a recent review, see: A. J. Stasyuk, P. J. Cywiński and D. T. Gryko, J. Photochem. Photobiol., C, 2016, 28, 116–137 CrossRef CAS.
- (a) H. Shono, T. Ohkawa, H. Tomoda, T. Mutai and K. Araki, ACS Appl. Mater. Interfaces, 2011, 3, 654–657 CrossRef CAS PubMed; (b) A. J. Stasyuk, M. Banasiewicz, M. K. Cyrański and D. T. Gryko, J. Org. Chem., 2012, 77, 5552–5558 CrossRef CAS PubMed; (c) T. Mutai, H. Sawatani, T. Shida, H. Shono and K. Araki, J. Org. Chem., 2013, 78, 2482–2489 CrossRef CAS PubMed; (d) A. J. Stasyuk, M. Banasiewicz, B. Ventura, M. K. Cyrański and D. T. Gryko, New J. Chem., 2014, 38, 189–197 RSC; (e) T. Mutai, T. Ohkawa, H. Shono and K. Araki, J. Mater. Chem. C, 2016, 4, 3599–3606 RSC.
- (a) E. L. Roberts, P. T. Chou, T. A. Alexander, R. A. Agbaria and I. M. Warner, J. Phys. Chem., 1995, 99, 5431–5437 CrossRef CAS; (b) J. Piechowska and D. T. Gryko, J. Org. Chem., 2011, 76, 10220–10228 CrossRef CAS PubMed; (c) J. Piechowska, K. Huttunen, Z. Wróbel, H. Lemmetyinen, N. V. Tkachenko and D. T. Gryko, J. Phys. Chem. A, 2012, 116, 9614–9620 CrossRef CAS PubMed; (d) K.-Y. Chen, H.-Y. Tsai, W.-C. Lin, H.-H. Chu, Y.-C. Weng and C.-C. Chan, J. Chem. Sci., 2014, 126, 955–966 CrossRef CAS; (e) J. Piechowska, K. Virkki, B. Sadowski, H. Lemmetyinen, N. V. Tkachenko and D. T. Gryko, J. Phys. Chem. A, 2014, 118, 144–151 CrossRef CAS PubMed; (f) S. Hristova, G. Dobrikov, F. S. Kamounah, S. Kawauchi, P. E. Hansen, V. Deneva, D. Nedeltcheva and L. Antonov, RSC Adv., 2015, 5, 102495–102507 RSC; (g) H. Marciniak, S. Hristova, V. Deneva, F. S. Kamounah, P. E. Hansen, S. Lochbrunner and L. Antonov, Phys. Chem. Chem. Phys., 2017, 19, 26621–26629 RSC.
- S. Kim, J. S. Seo and S. Y. Park, J. Photochem. Photobiol., A, 2007, 191, 19–24 CrossRef CAS.
- (a) A. V. Gaenko, A. Devarajan, I. V. Tselinskii and U. Ryde, J. Phys. Chem. A, 2006, 110, 7935–7942 CrossRef CAS PubMed; (b) L. Xie, Y. Chen, W. Wu, H. Guo, J. Zhao and X. Yu, Dyes Pigm., 2012, 92, 1361–1369 CrossRef CAS; (c) H. Roohi, F. Hejazi, N. Mohtamedifar and M. Jahantab, Spectrochim. Acta, Part A, 2014, 118, 228–238 CrossRef CAS PubMed; (d) R. Daengngern and N. Kungwan, J. Lumin., 2015, 167, 132–139 CrossRef CAS; (e) I. Deperasińska, A. Makarewicz, M. Krzeszewski, D. T. Gryko and B. Kozankiewicz, J. Phys. Chem. A, 2015, 119, 9051–9058 CrossRef PubMed.
- (a) S. H. Lin, J. Chem. Phys., 1970, 53, 3766–3767 CrossRef CAS; (b) J. V. Caspar, E. M. Kober, B. P. Sullivan and T. J. Meyer, J. Am. Chem. Soc., 1982, 104, 630–632 CrossRef; (c) J. V. Caspar and T. J. Meyer, J. Phys. Chem., 1983, 87, 952–957 CrossRef CAS.
- N. Suzuki, A. Fukazawa, K. Nagura, S. Saito, H. Kitoh-Nishioka, D. Yokogawa, S. Irle and S. Yamaguchi, Angew. Chem., Int. Ed., 2014, 53, 8231–8235 CrossRef CAS PubMed.
- For a recent review, see: (a) M. Pawlicki, H. A. Collins, R. G. Denning and H. L. Anderson, Angew. Chem., Int. Ed., 2009, 48, 3244–3266 CrossRef CAS PubMed; (b) Q. Zhang, X. Tian, H. Zhou, J. Wu and Y. Tian, Materials, 2017, 10, 223 CrossRef PubMed.
- For a review, see: T. Steiner, Angew. Chem., Int. Ed., 2002, 41, 48–76 CrossRef CAS.
- N. J. Turro, in Modern Molecular Photochemistry, University Science Books, Sausalito, CA, 1991, ch. 5, pp. 76–152 Search PubMed.
- (a) X. Cheng, K. Wang, S. Huang, H. Zhang, H. Zhang and Y. Wang, Angew. Chem., Int. Ed., 2015, 54, 8369–8373 CrossRef CAS PubMed; (b) X. Cheng, Y. Zhang, S. Han, F. Li, H. Zhang and Y. Wang, Chem.–Eur. J., 2016, 22, 4899–4903 CrossRef CAS PubMed; (c) R. Li, L. Yan, Z. Wang and Z. Qi, J. Mol. Struct., 2017, 1136, 1–6 CrossRef CAS.
- Y. Iwase, K. Kamada, K. Ohta and K. Kondo, J. Mater. Chem., 2003, 13, 1575–1581 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed synthetic protocols and characterization data of all new compounds, crystal structures of 3 and 6, photophysical properties of 2–7, details of theoretical calculations for 2 and 3, including Schemes S1–S6 and Fig. S1–S83, and X-ray crystallographic data for 3 (CCDC 1813601) and 6 (CCDC 1813602). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8sc00066b |
| This journal is © The Royal Society of Chemistry 2018 |