
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Asymmetric synthesis of multiple quaternary stereocentre-containing cyclopentyls by oxazolidinone-promoted Nazarov cyclizations†
Rohan
Volpe
 a,
Romain J.
Lepage
b,
Jonathan M.
White
c,
Elizabeth H.
Krenske
b and
Bernard L.
Flynn
*a
a,
Romain J.
Lepage
b,
Jonathan M.
White
c,
Elizabeth H.
Krenske
b and
Bernard L.
Flynn
*a
aMonash Institute of Pharmaceutical Sciences, Monash University, 381 Royal Parade, Parkville, Victoria 3052, Australia. E-mail: bernard.flynn@monash.edu
bSchool of Chemistry and Molecular Biosciences, The University of Queensland, Brisbane, QLD 4072, Australia
cBio21 Institute, School of Chemistry, University of Melbourne, Parkville, VIC 3010, Australia
First published on 20th April 2018
Abstract
Carbometalation of oxazolidinone (Ox)-substituted ynamides is used to generate highly substituted Ox-divinyl (and aryl vinyl) ketones for use in Nazarov cyclizations. The Ox-group serves as a remarkably effective chiral activating group, enabling the torquoselective Nazarov cyclization of these sterically congested substrates to be performed under mild conditions. It also serves as a charge-stabilizing group in the intermediate oxyallyl cation, suppressing undesired [1,2]-sigmatropic shifts of neighboring substituents and facilitating the regio- and stereoselective incorporation of nucleophiles to yield cyclopentanoids containing up to three contiguous all-carbon quaternary (4°) stereocentres.
Introduction
The enantioselective synthesis of quaternary (4°) stereocentres is a major challenge in organic synthesis, hindering access to sp3-rich scaffolds in drug discovery and natural products synthesis.1,2 Particularly problematic is the enantioselective formation of multiple 4°-stereocentres, which requires control over both relative and absolute stereochemistry.The Nazarov cyclization offers inherent control over relative stereochemistry through conservation of orbital symmetry and constitutes an attractive route to multistereocentre-containing cyclopentanoids.3 However, the potential of the Nazarov cyclization for 4°-stereocentre formation has not yet been fully realized due to two significant challenges: (i) stereoselective access to highly substituted divinyl (and aryl vinyl) ketone substrates4 and (ii) torquoselective5 ring closure. In a landmark study, Tius and co-workers6 reported chiral Brønsted acid-catalyzed Nazarov cyclizations of divinyl ketones 1 (Scheme 1a) leading to cyclopentenols 3 containing two new vicinal 4°-stereocentres (R1–3 ≠ H) with high enantioselectivities (often er > 97![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3). Careful design of the divinyl ketone 1 with dual-activating electron donor (OCHPh2) and acceptor (CO2R) elements was key to attaining efficient cyclization.6 Electrofugal release of Ph2HC+ from the intermediate oxyallyl cation 2 further promoted the cyclization and suppressed competing Wagner–Meerwein rearrangements ([1,2]-sigmatropic shifts of R1–3 within 2). Herein, we report that highly substituted aryl vinyl and divinyl ketones 5 can be readily accessed through carbometalations of oxazolidinone (Ox)-substituted ynamides 4 (Scheme 1b).7 The Ox-group proves to be remarkably effective as a single chiral activating group for the Nazarov cyclizations of these highly substituted and sterically congested substrates 5, giving exo-methylene cyclopentanones 7 under remarkably mild conditions, with excellent and predictable enantiocontrol. Furthermore, since no electrofugal release is required for substrate activation or suppression of Wagner–Meerwein rearrangements, the oxyallyl cation 6 can be exploited in nucleophilic trapping7,8 to afford multistereocentre-centre-containing products 8 with up to three all-carbon 4°-stereocentres. The rapid assembly of such levels of complexity from a prochiral starting material highlights the powerful activating and stereocontrolling influence of the Ox group. Using theoretical calculations, we show that the exceptional activating properties of Ox originate from a combination of covalent and non-covalent transition-state stabilizing effects.
:3). Careful design of the divinyl ketone 1 with dual-activating electron donor (OCHPh2) and acceptor (CO2R) elements was key to attaining efficient cyclization.6 Electrofugal release of Ph2HC+ from the intermediate oxyallyl cation 2 further promoted the cyclization and suppressed competing Wagner–Meerwein rearrangements ([1,2]-sigmatropic shifts of R1–3 within 2). Herein, we report that highly substituted aryl vinyl and divinyl ketones 5 can be readily accessed through carbometalations of oxazolidinone (Ox)-substituted ynamides 4 (Scheme 1b).7 The Ox-group proves to be remarkably effective as a single chiral activating group for the Nazarov cyclizations of these highly substituted and sterically congested substrates 5, giving exo-methylene cyclopentanones 7 under remarkably mild conditions, with excellent and predictable enantiocontrol. Furthermore, since no electrofugal release is required for substrate activation or suppression of Wagner–Meerwein rearrangements, the oxyallyl cation 6 can be exploited in nucleophilic trapping7,8 to afford multistereocentre-centre-containing products 8 with up to three all-carbon 4°-stereocentres. The rapid assembly of such levels of complexity from a prochiral starting material highlights the powerful activating and stereocontrolling influence of the Ox group. Using theoretical calculations, we show that the exceptional activating properties of Ox originate from a combination of covalent and non-covalent transition-state stabilizing effects.
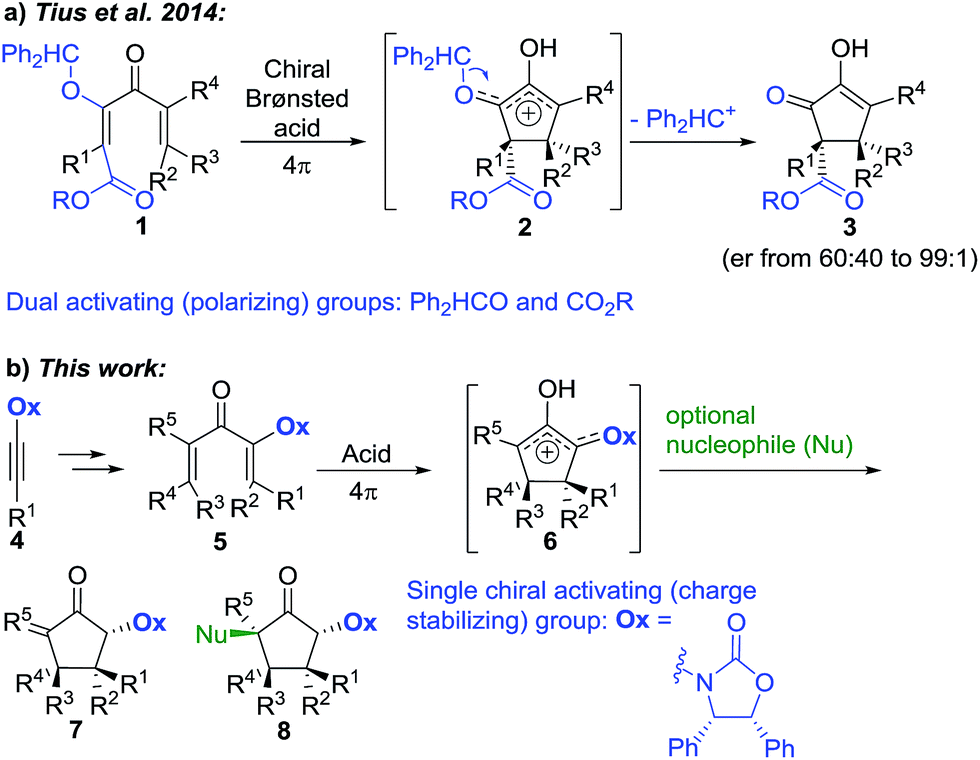 | ||
| Scheme 1 Nazarov substrate activation modes for the enantioselective synthesis of 4°-stereocentres. | ||
Results and discussion
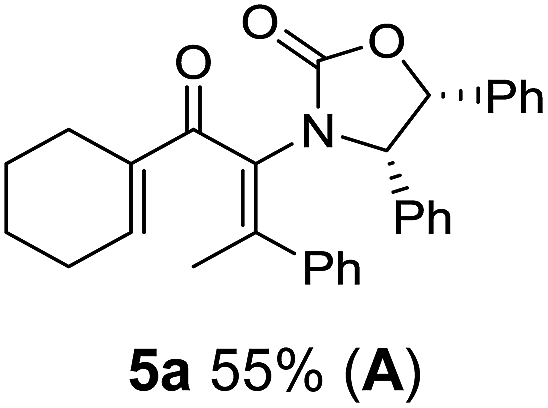
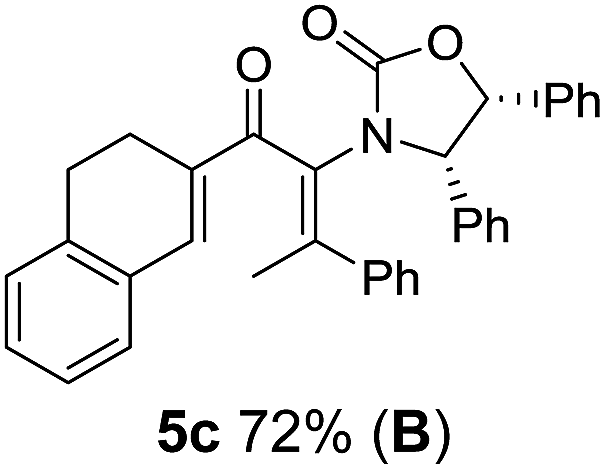
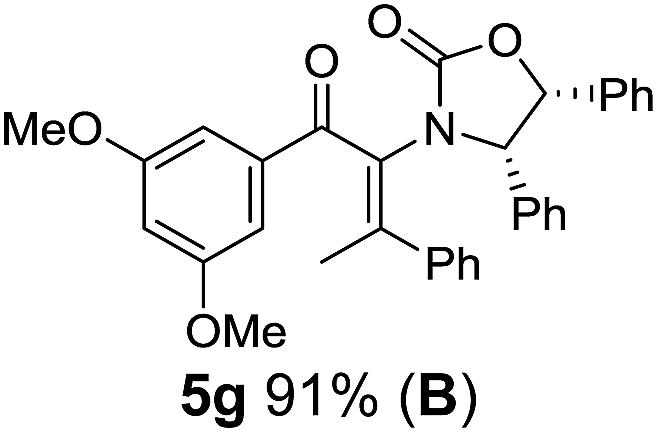
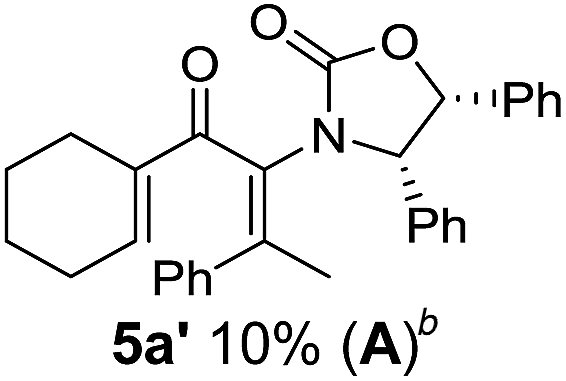
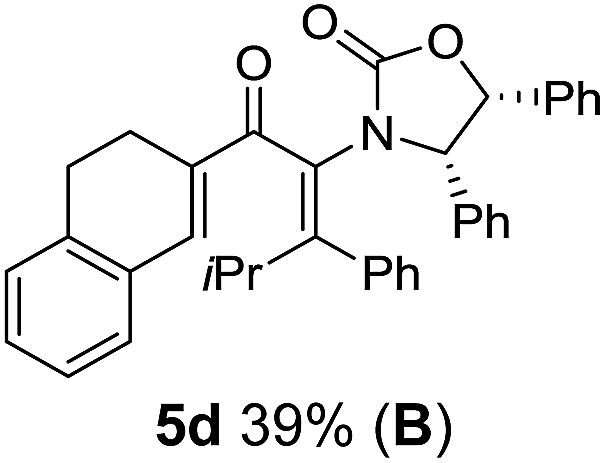
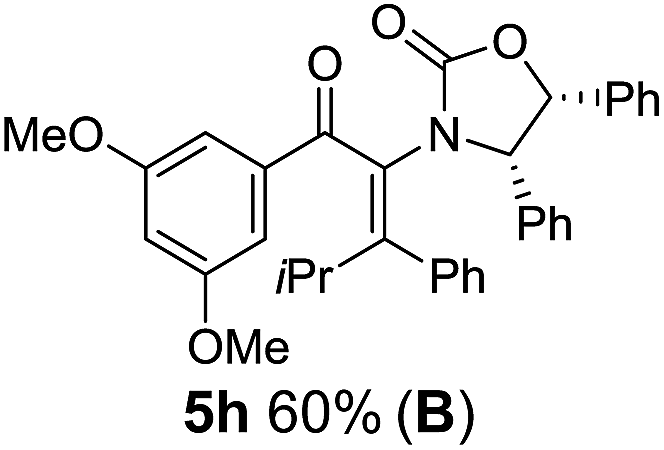
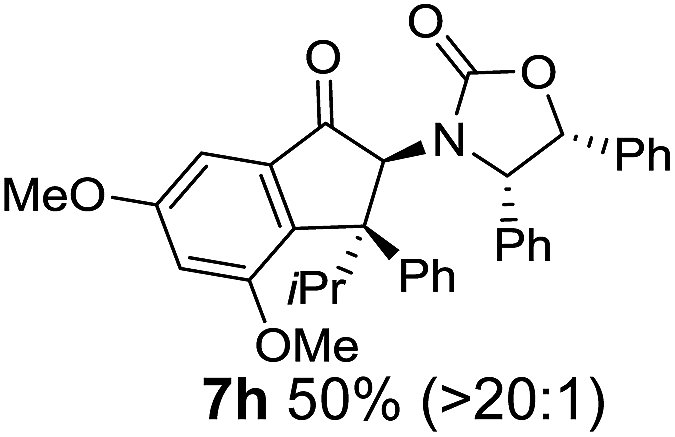
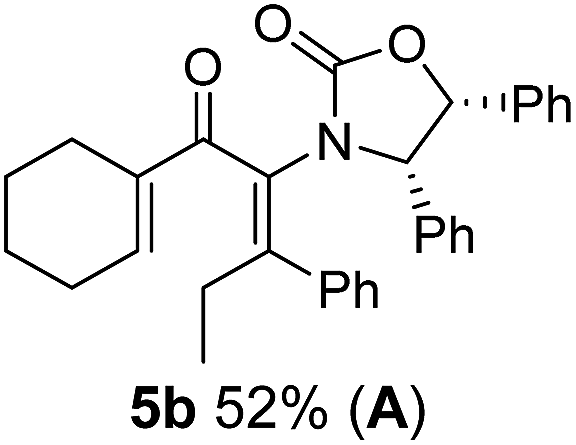
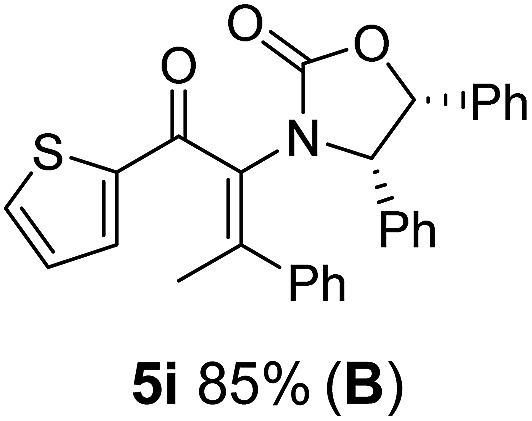
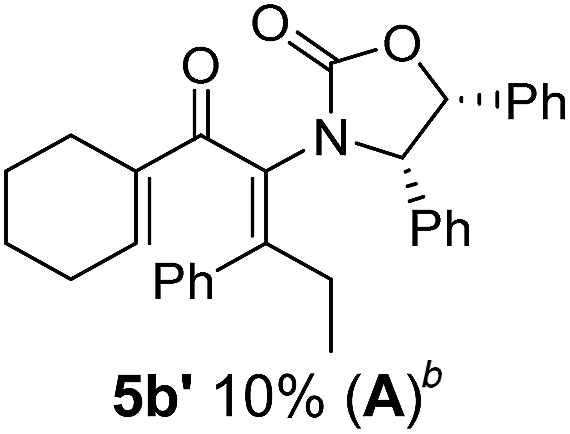
Two different carbometalation strategies were developed to give access to Ox-containing divinyl and aryl vinyl ketones 5 (R2 = alkyl/aryl, Table 1). Firstly, Cu-catalyzed carbomagnesiation of Ox-ynamides 4 with Grignard reagents gave 9 (M = MgBr);9 alternatively, Rh-catalyzed carbozincation of 4 with ZnEt2 gave 9 (M = ZnEt).10 Addition of iodine to organometallics 9 (M = MgBr or ZnEt) gave the key building block alkenyliodides 10a (68%) and 10b (79%). Carbonylative Stille coupling (Method A) of 10a and 10b with tributyl(cyclohexen-1-yl)stannane afforded divinyl ketones 5a/a′ and 5b/b′, respectively, each as a 5:1 mixture of E/Z-isomers about the Ox-substituted double bond (entries 1–4).11 Despite this partial isomerization, the major isomers, 5a and 5b, were isolated in 55% and 52% yield, respectively. All other divinyl and aryl vinyl ketones 5 shown in Table 1 were accessed by reaction of 9 (M = MgBr) with the corresponding aldehyde followed by Dess–Martin periodinane oxidation of the crude alcohols (Method B) giving 5c–j in yields of 31–91% (entries 5–12).
|
|
||||||||
|---|---|---|---|---|---|---|---|---|
| 4/10 → 5a | 5 → 7c (dr)d | 4/10 → 5a | 5 → 7c (dr)d | 4/10 → 5a | 5 → 7c (dr)d | |||
|
a Nazarov substrates 5 formed from 4 using Method A (A) or Method B (B), as indicated.
b Isolated as a minor isomer using Method A.
c Cyclized with BF3·THF or TfOH in CH2Cl2 at various temperatures (ranging from −78 °C to 40 °C) depending on acid and substrate; see text and ESI for details.
d Diastereomeric ratio (dr) refers to stereochemistry at C1 relative to Ox (determined by 1H NMR). Some products 7 were isolated as a mixture of C2-epimers, indicated by a wavy bond (see ESI for ratio), these give a single enantiomer upon Ox removal (eqn (1), ref. 7).
e Isolated yield of C1-(S) isomer, an additional 24% was isolated as a 3:1 (R):(S)-C1 mix.
|
||||||||
| 1 |
|
|
5 |
|
|
9 |
|
|
| 2 |
|
|
6 |
|
|
10 |
|
|
| 3 |
|
|
7 |
|
|
11 |
|
|
| 4 |
|
|
8 |
|
12 |
|
|
|
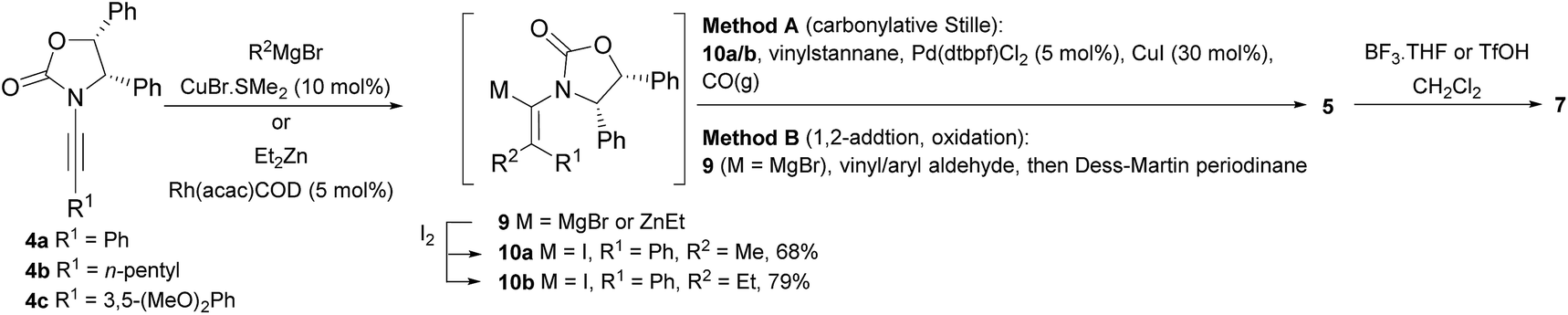
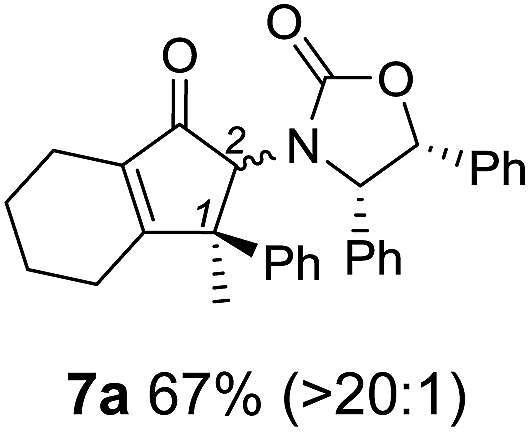
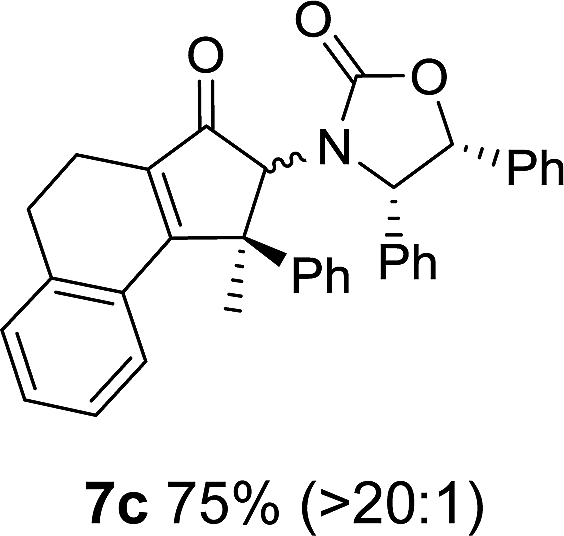
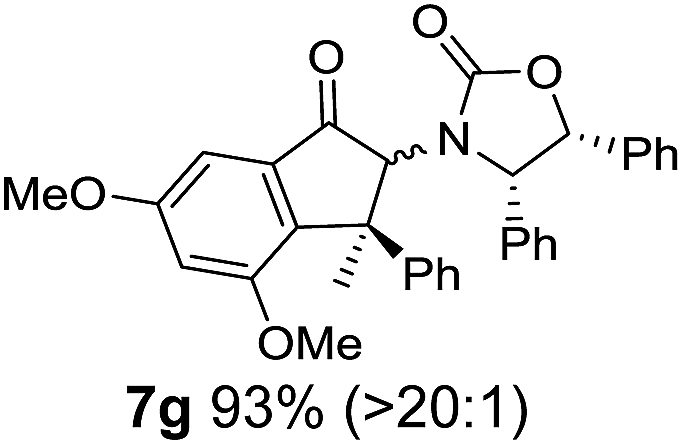
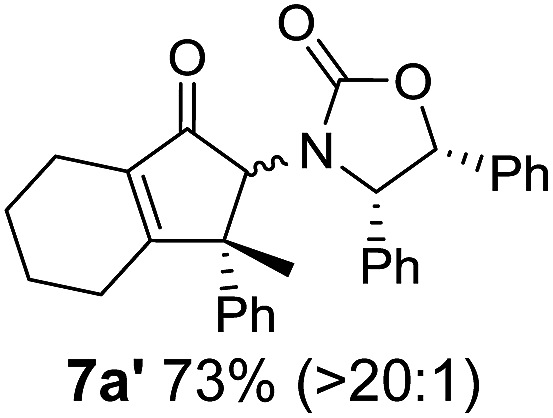
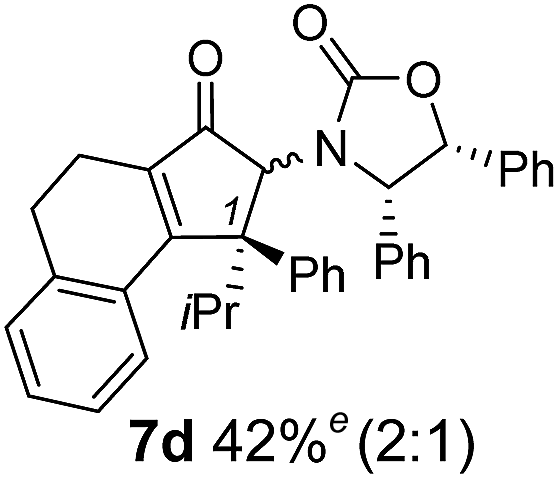
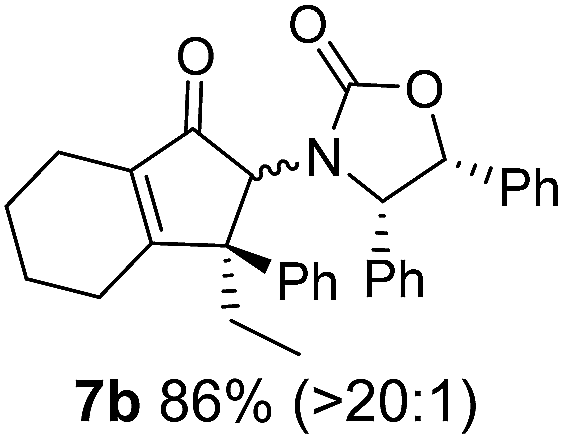
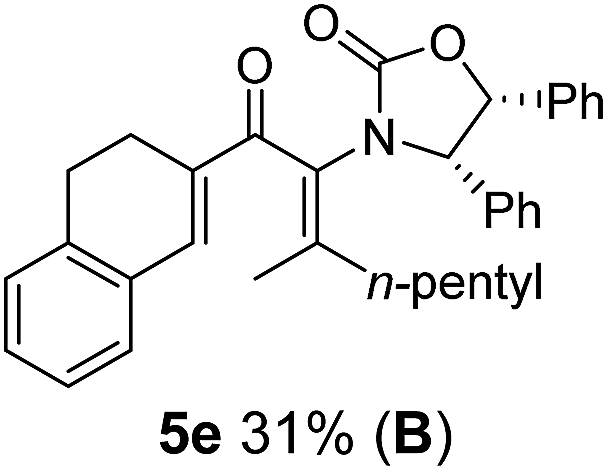
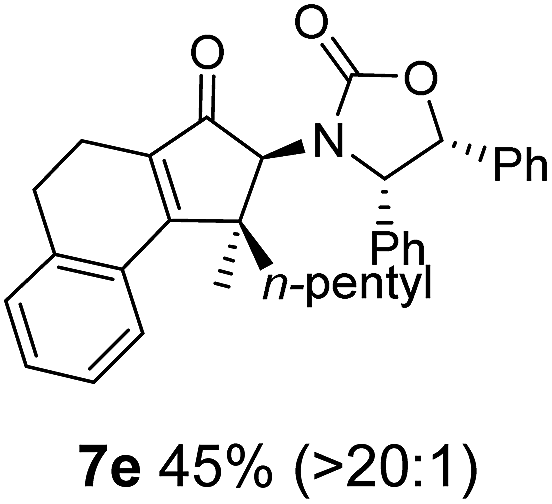
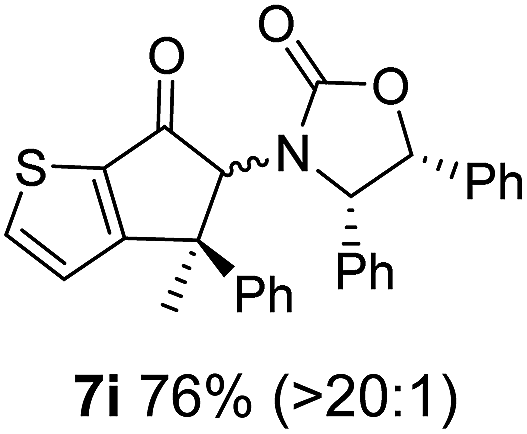
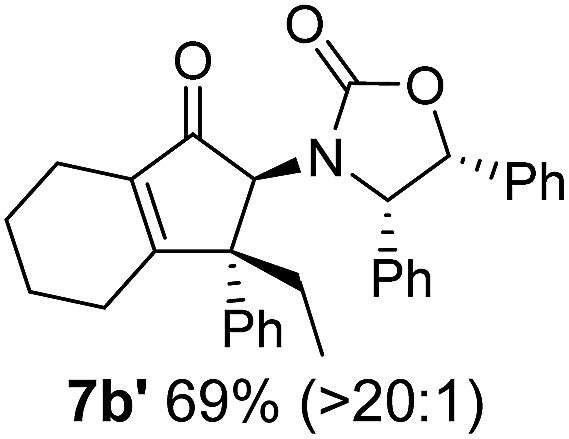
Nazarov cyclizations of divinyl and aryl vinyl ketones 5a–j were performed using either BF3·THF or TfOH as catalyst in CH2Cl2, giving cyclopentanoids 7a–i (5f and 5j did not cyclize) containing one new 4°-stereocentre (Table 1). Broadly speaking, these Nazarov cyclizations performed very well, particularly where the “inner” substituent (R2) in 5 was Me, Et or Ph (BF3·THF or TfOH). Use of TfOH as catalyst allowed the Nazarov cyclization to be conducted at temperatures as low as −78 °C, but generally the reactions were performed at 0 °C to rt or in refluxing CH2Cl2 (40 °C) using either TfOH or BF3·THF.‡ The torquoselectivities were very high (dr > 20:1 for C1 relative to Ox), with the sole exception of 7d (dr = 2:1 (S):(R)-C1, entry 6). X-ray crystal structure and density functional theory (DFT) studies have shown that Ox auxiliaries of this configuration consistently favor anticlockwise conrotation leading to R1-β stereochemistry (see below);7b we have therefore assigned this stereochemistry to each product in Table 1. Most likely, the cyclization of 5d, which required heating to 40 °C due to the sterically encumbering isopropyl group (R2 = iPr), gave lower selectivity due to partial Z/E-isomerization of the oxazolidinyl-alkene prior to cyclization, rather than because of poor stereoinduction by the auxiliary (see also below).
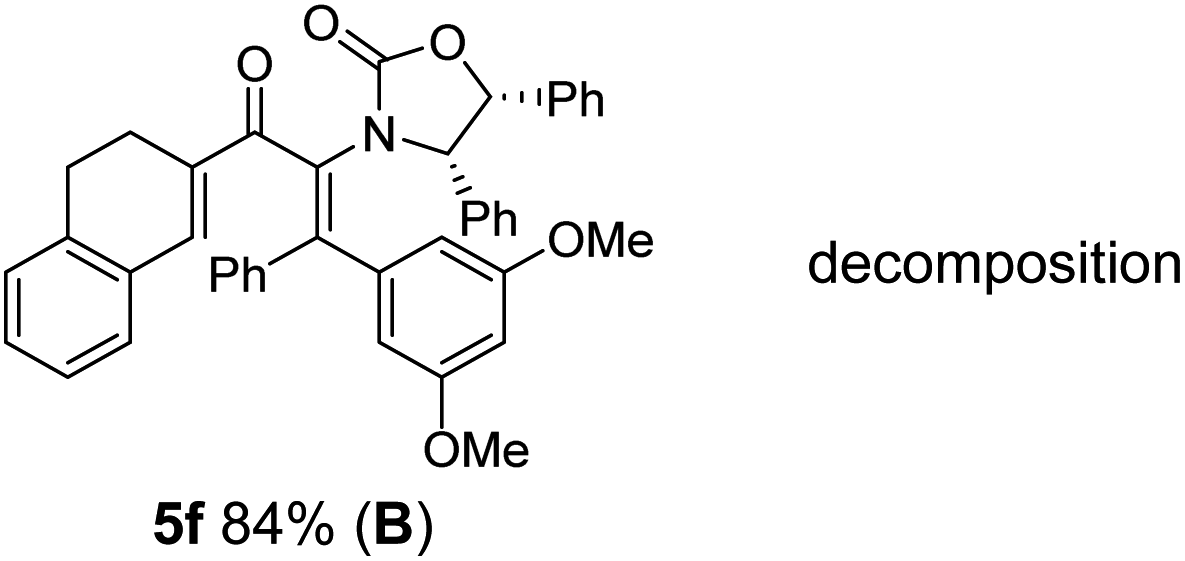
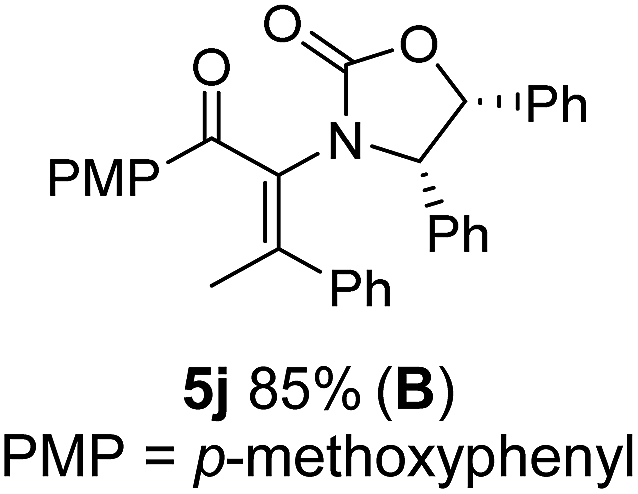
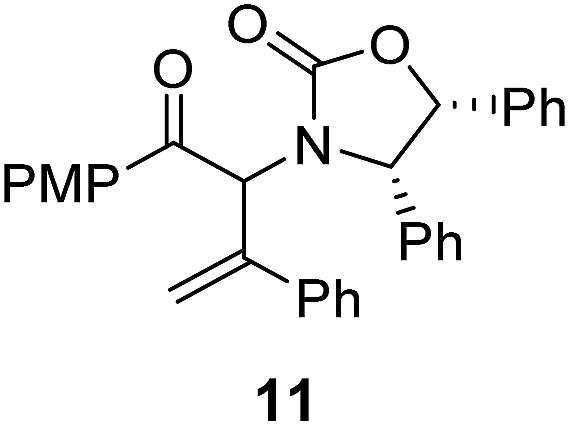
The presence of two aliphatic substituents on the tetrasubstituted alkene terminus, as in 5e, led to slower cyclization, but the stereoinduction remained high (entry 7). Diaryl-substituted alkene 5f underwent undesired side reactions to give multiple minor products along with return of starting material (entry 8). In a number of cases, the presence of epimers at C2 (the carbon bearing Ox) was apparent, but both epimers lead to the same product once the auxiliary is removed by reductive cleavage (see below). Cyclizations of electron-rich aryl vinyl ketones were successful (entries 9–11), even for the very hindered substrate 5h where R2 = iPr. For the less activated aryl vinyl ketone 5j, alkene isomerization to form β,γ-unsaturated ketone 11 became the dominant pathway and no Nazarov cyclization was observed.
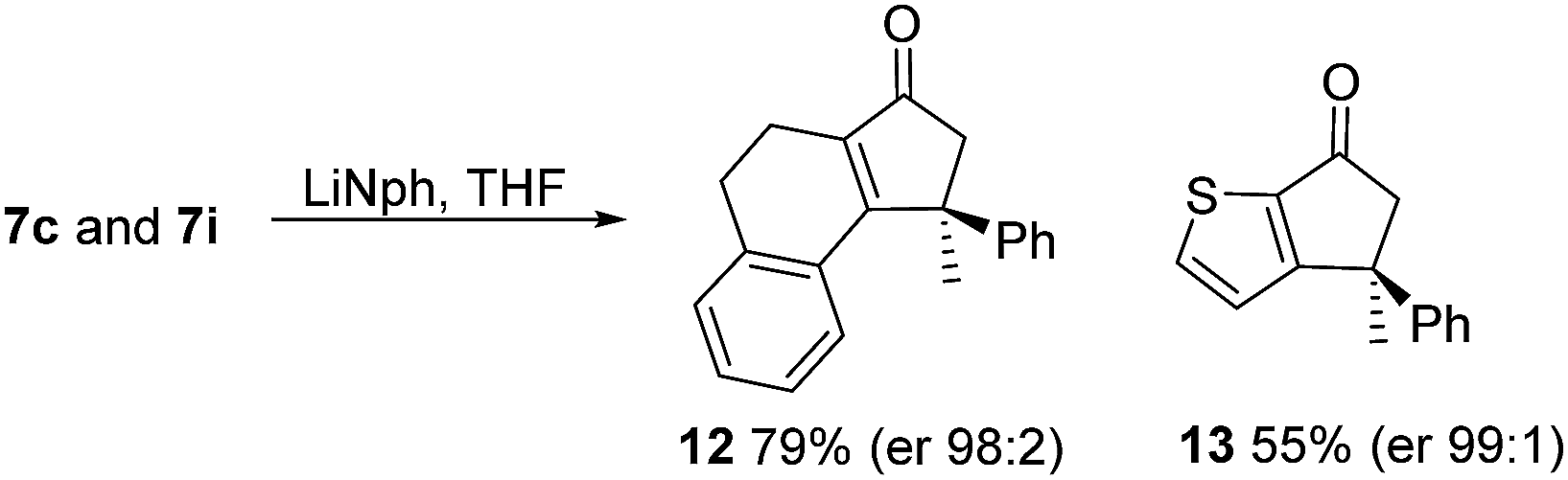
As has been demonstrated in our previous study utilizing a diverse array of less substituted Nazarov products 7 (R2 = R3 = H), the oxazolidinone can be removed by reductive-cleavage using lithium naphthalenide (LiNph).7c Two examples are given as part of this work (eqn (1)): reductive cleavage of the Ox group in 7c and 7i gave 12 (79%) and 13 (55%), respectively, both in high enantiomeric purity (er > 98:2).
 | (1) |
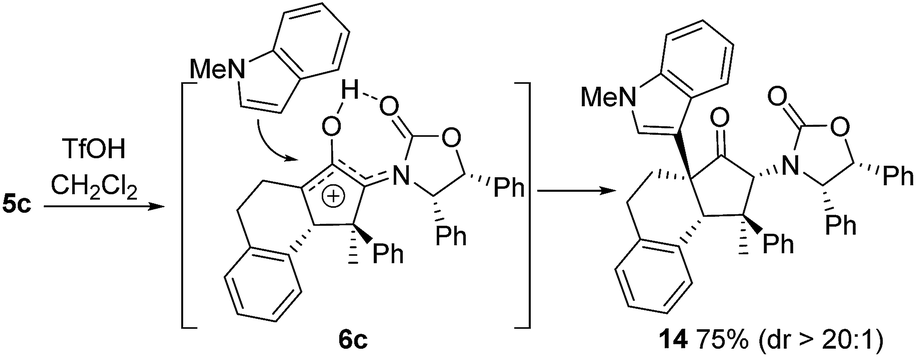
Also, as per our previous work, additional stereochemical complexity can be built up by nucleophilic trapping of the intermediate oxyallyl cations 6.7c Accordingly, the highly substituted divinyl ketone 5c was converted into the indole-trapped product 14 (75%) (eqn (2)). Notably, this tandem sequence generates four new contiguous stereocentres, including two 4°-centres, with excellent control over both relative and absolute stereochemistry: only a single isomer was observed.
 | (2) |
Having achieved stereoselective Nazarov cyclizations leading to products with adjacent 3° and 4°-stereocentres, we next addressed the formation of vicinal 4°-stereocentres. To prepare the fully substituted Nazarov substrate 17 we developed a convergent carbometalation approach starting from two alkynes: ynamide 4a and 3-hexyne (Scheme 2a). Cu-catalyzed addition of MeMgBr to 4a, followed by in situ formylation with ethylformate, afforded 15 (52%) stereoselectively. Carboalumination of 3-hexyne to give 16,11 followed by 1,2-addition of 16 to 15 and oxidation with DMP, gave divinyl ketone 17 (71%). The C2–C3 double bond retained its Z stereochemistry while the C5–C6 double bond was formed as a 3:1 E:Z mixture.12§ Separation of these isomers proved challenging; however, a pure sample of (2Z,5E)-17 was isolated in 24% yield (from 15). We also prepared the fully substituted ketone 20 (Scheme 2b) bearing a tethered nucleophile (electron-rich aryl group). Access to 20 commenced with formation of vinyl bromide 19 from bromoalcohol 18.13 Lithiation of 19, followed by addition to a solution of 15 and AlMe3 (Lewis acid) and DMP oxidation of the crude carbinol (not shown) afforded 20 (44%) as a single alkene-stereoisomer.¶
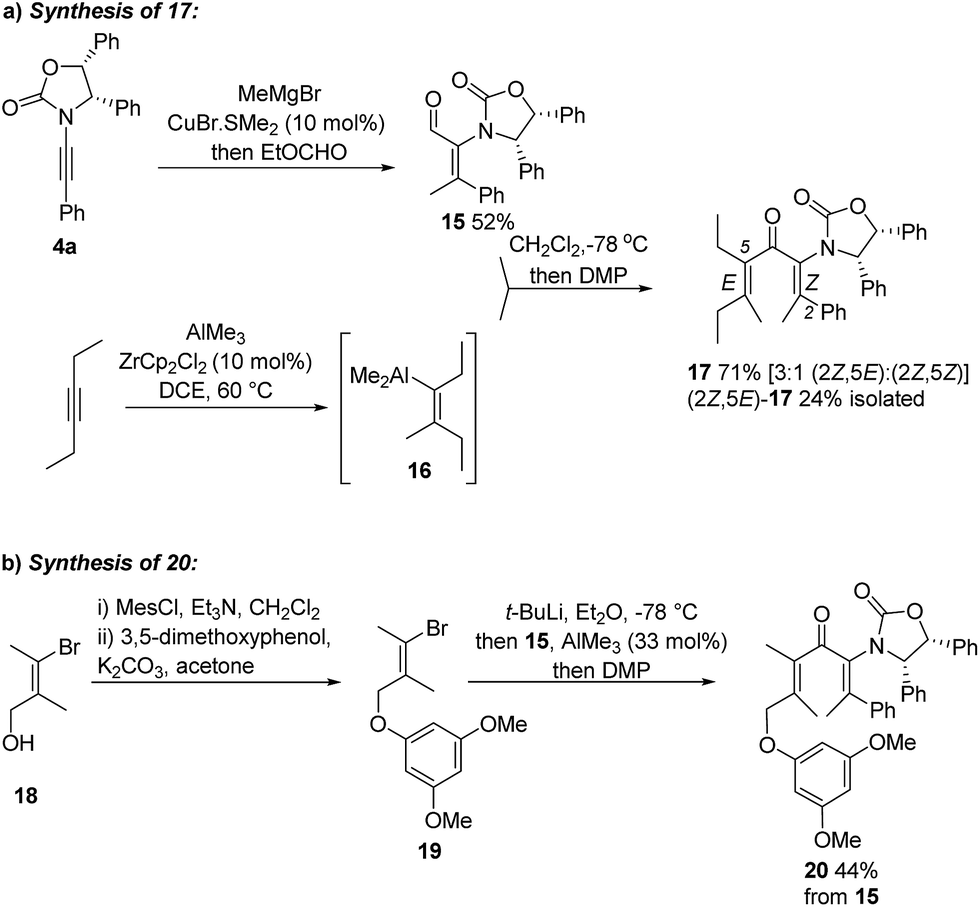 | ||
| Scheme 2 Syntheses of fully substituted Ox-divinyl ketones 17 and 20. | ||
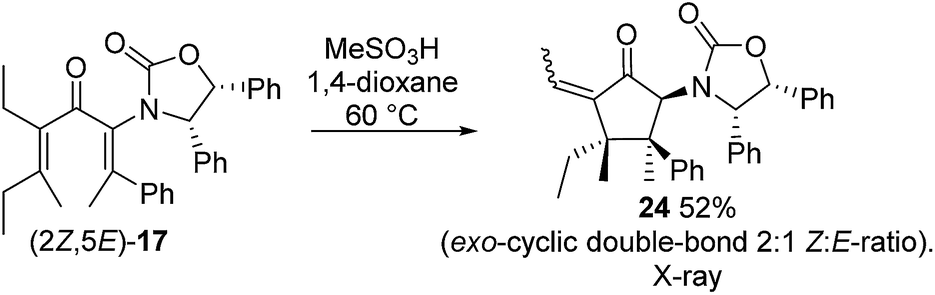
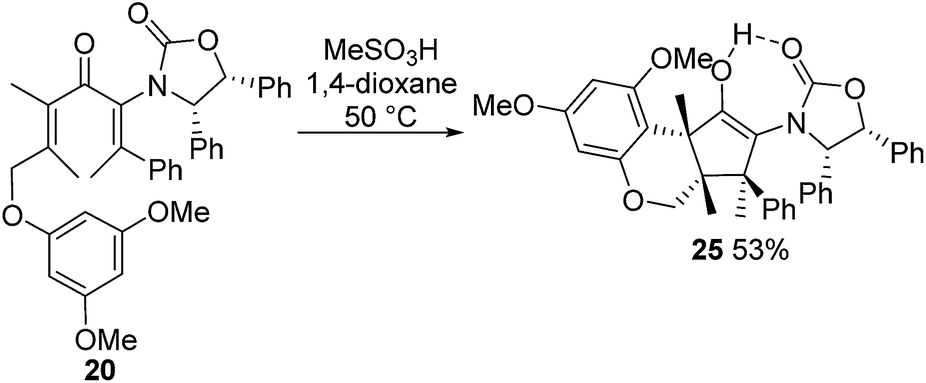
Nazarov cyclization of (2Z,5E)-17 with MeSO3H (CH2Cl2, −78 °C) gave 21 as a complex mixture of C2,3-diastereomers, keto/enol-tautomers and E/Z-isomers (Scheme 3). Warming the mixture to ambient temperature resulted in a double Wagner–Meerwein shift of the C3-ethyl and C2-phenyl substituents in the reversibly formed oxyallyl cation 22 to give (3R)-23 and (3S)-23 in a 2:5 ratio (Scheme 3).14 The stereochemistry of these products was confirmed by X-ray crystallography of (3S)-23.‡ We believe that the origin of this epimeric mixture is partial double-bond isomerization of (2Z,5E)-17 to (2E,5E)-17 under the acidic conditions prior to Nazarov cyclization. While this isomerization was undesired, the rapid (<2 h) cyclisation of both isomers of 17 at −78 °C demonstrates the remarkable ability of the Ox group to activate the Nazarov reaction. Upon further experimentation with reaction conditions (acids and solvents) to avoid double-bond isomerization of (2Z,5E)-17 to (2E,5E)-17, we found that treatment of (2Z,5E)-17 with MeSO3H in 1,4-dioxane with mild heating gave cyclopentanone 24 stereoselectively in 52% isolated yield (eqn (3)). The stereochemistry of (E)- and (Z)-24 were confirmed by X-ray crystallography and 2D NMR, respectively.‡ Replacing CH2Cl2 with 1,4-dioxane as solvent appears to exert different effects on the rates of the various competing reactions involved in the formation of 21, 23 and 24 (Scheme 3 and eqn (3)). Solvation of MeSO3H by 1,4-dioxane likely reduces the rates of all of these reactions, however, its strongest effects appear to be the suppression of C2–C3 double-bond isomerization in 17 and Wagner–Meerwein rearrangement in 22, leading to the observed stereo- and chemoselective formation of 24.15 Cyclization of 20 (eqn (4)) under these conditions was also successful, yielding the intramolecularly trapped product 25 as the only product discernable by 1H-NMR (53% isolated yield). Conversion of 20 to 25 forms two new rings and three contiguous 4°-stereocentres, underscoring the effectiveness of the Ox-controlled Nazarov reaction for synthesis of structurally complex, 4°-stereocentre-containing scaffolds. The asymmetric formation of three contiguous 4°-stereocentres entirely from prochiral carbons is a rare transformation; a Diels–Alder reaction reported by Nicolaou et al. is the only other example known to us.16
 | (3) |
 | (4) |
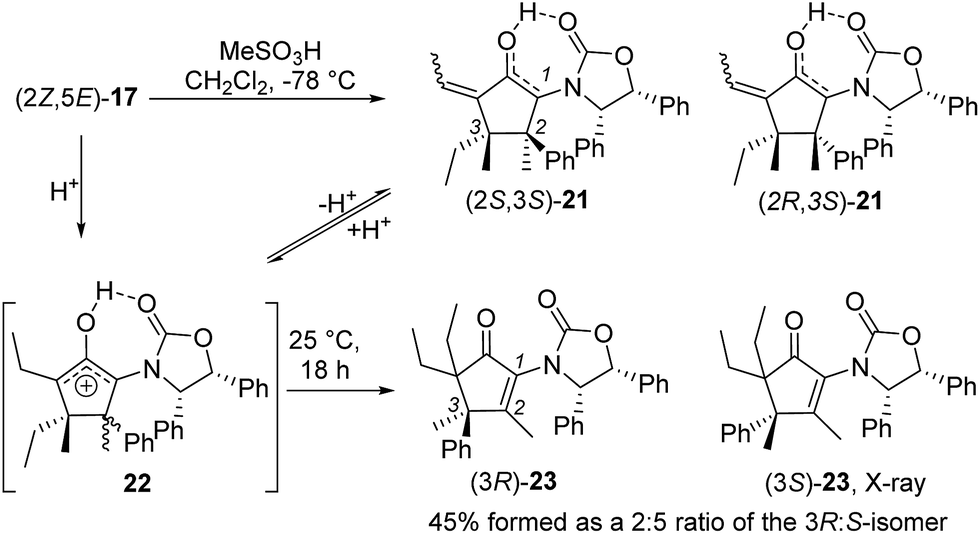 | ||
| Scheme 3 Nazarov cyclization of 17 in CH2Cl2. | ||
These Ox-promoted Nazarov cyclizations are remarkably facile, allowing efficient generation of sterically congested products at temperatures as low as −78 °C. This points to a powerful activating influence of the Ox auxiliary. In order to determine the origins of this activation, we performed DFT calculations (Fig. 1).‡ Calculations with M06-2X show that in the absence of an oxazolidinone, the activation energies (ΔG‡) for Nazarov cyclizations of 26–28 leading to zero, one, or two 4°-centres are 16.8, 22.5, and 28.9 kcal mol−1, respectively. Each new 4°-stereocentre raises the barrier by 6 kcal mol−1.‡ An achiral oxazolidinone devoid of Ph substituents (OxH2, see 29–31) lowers the cyclization barrier by 7–13 kcal mol−1 (ΔG‡ = 9.7–15.6 kcal mol−1) relative to the oxazolidinone-free substrates, while the diphenyl-oxazolidinone (Ox, see 32–34) provides further activation still, leading to cyclization barriers of only 8.5–10.9 kcal mol−1. These very low barriers are consistent with the facile ring closures observed for 5, 17, and 20.
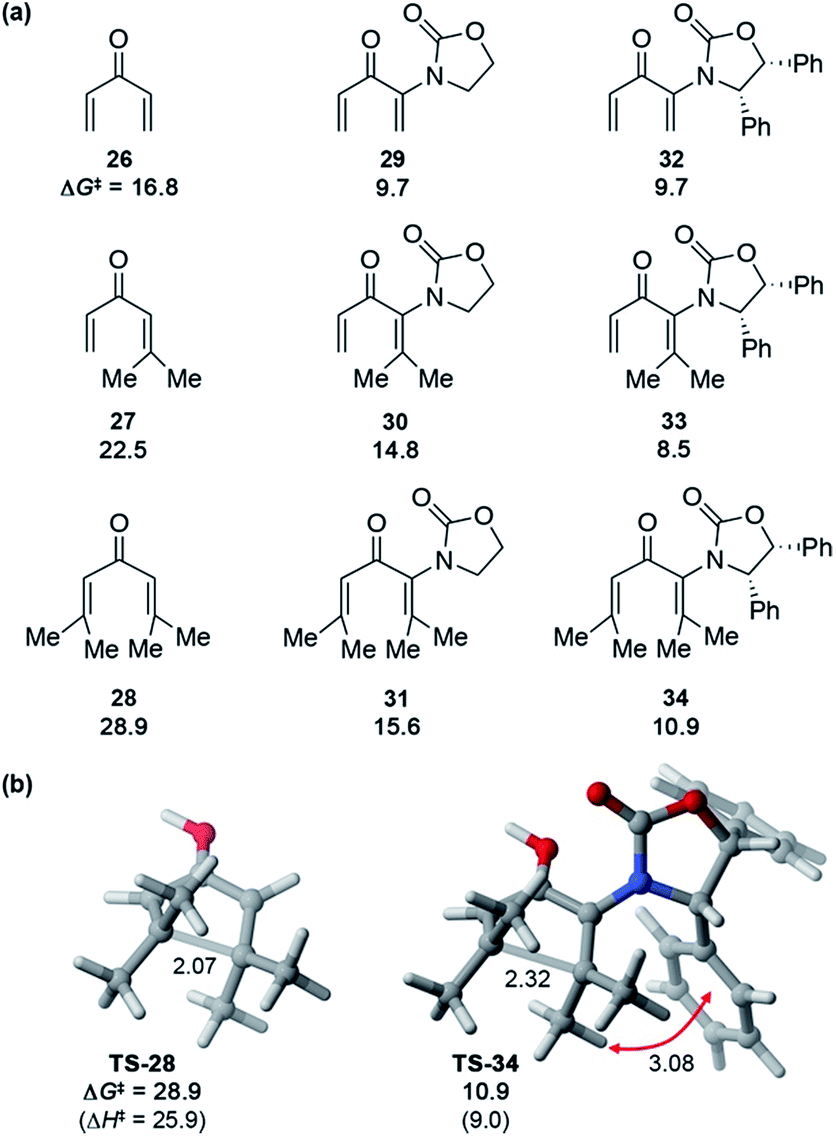 | ||
| Fig. 1 (a) Activation barriers for H+-catalyzed Nazarov cyclizations of model divinyl ketones 26–34 and (b) transition states for cyclizations of 28 and 34, calculated with M06-2X/6-311+G(d,p)//M06-2X/6-31G(d) in implicit (SMD) dichloromethane. Distances in Å, ΔH‡ and ΔG‡ in kcal mol−1. | ||
The transition states (TSs) for OxH2- and Ox-promoted cyclizations benefit from several stabilizing effects. Firstly, the nitrogen lone pair affords resonance stabilization of the incipient oxyallyl cation. Secondly, the oxazolidinone-containing TSs feature a longer forming C–C bond than the corresponding oxazolidinone-free TSs, leading to reduced steric repulsion between the Me groups about the forming C–C bond (see Fig. 1b). A third activating influence of Ox is evident from a comparison of the cyclizations of 33 and 34 (containing Ox) with those of 30 and 31 (containing OxH2). The two Ox-substituted TSs have ΔG‡ values about 6 kcal mol−1 lower than those of the corresponding OxH2 derivatives. The additional activation by Ox can be traced to a CH–π interaction in the TS between the “inner” substituent on C2 (R2, rotating downwards) and the nearby Ph substituent on Ox (see red arrow in Fig. 1). Together, these three TS-stabilizing influences of Ox make it an exceptionally powerful activating group, capable of reducing the barrier for vicinal 4°-centre formation by almost 18 kcal mol−1 (28vs.34). Indeed, computations predict that when the R1 substituent is an aryl group, like in many of our substrates (5, 17, and 20) (with R2 = alkyl) the barrier for cyclization is even lower still.‡
Conclusions
To conclude, carbometalation of Ox-ynamides affords direct access to highly substituted Ox-divinyl and -aryl vinyl ketones, which undergo exceptionally facile Nazarov cyclizations leading to 4°-stereocentre-containing cyclopentanoids. In addition to the powerful activating and stereodirecting influence of Ox in the Nazarov cyclization, the Ox auxiliary helps suppress undesired Wagner–Meerwein rearrangements in the intermediate oxyallyl cations, and facilitates nucleophilic trapping of these intermediates enabling rapid assembly of multiple stereocentres (including vicinal 4°-stereocentres) with excellent stereochemical control. Theoretical studies allowed us to discover the electronic origin of the strong activating effect of the Ox, which is traced to a combination of covalent (lone pair donation to the incipient oxyallyl cation and reduced steric crowding about the newly forming bond) and non-covalent (CH–π interaction) effects which are generally applicable across most of the divinyl (or aryl vinyl) ketones reported here.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research has been supported by the Australian Research Council (DP150103131 and FT120100632). Computer resources were provided by the Australian National Computational Infrastructure National Facility and by the University of Queensland Research Computing Centre.Notes and references
- For a review on asymmetric quaternary stereocentre synthesis, see: K. W. Quasdorf and L. E. Overman, Nature, 2014, 516, 181 CrossRef CAS PubMed.
- For a review on sp3-rich scaffolds in drug discovery, see: A. Karawajczyk, F. Giordanetto, J. Benningshof, D. Hamza, T. Kalliokoski, K. Pouwer, R. Morgentin, A. Nelson, G. Müller, A. Piechot and D. Tzalis, Drug Discovery Today, 2015, 20, 1310 CrossRef CAS PubMed.
- For reviews on the Nazarov reaction, see: D. R. Wenz and J. Read de Alaniz, Eur. J. Org. Chem., 2015, 2015, 23 CrossRef CAS; M. A. Tius, Chem. Soc. Rev., 2014, 43, 2979 RSC; F. G. West, O. Scadeng, Y.-K. Wu, R. J. Fradette and S. Joy in Comprehensive Organic Synthesis II, ed. P. Knochel, Elsevier, 2014, pp. 827–866 CrossRef; T. Vaidya, R. Eisenberg and A. J. Frontier, ChemCatChem, 2011, 3, 1531 CrossRef; N. Shimada, C. Stewart and M. A. Tius, Tetrahedron, 2011, 67, 5851 CrossRef PubMed; W. T. Spencer III, T. Vaidya and A. J. Frontier, Eur. J. Org. Chem., 2013, 2013, 3621 CrossRef PubMed.
- For a review on stereoselective synthesis of tetrasubstited alkenes, see: P. Polak, H. Vanova, D. Dvorak and T. Tobrman, Tetrahedron Lett., 2016, 57, 3684 CrossRef CAS.
- S. E. Denmark, M. A. Wallace and C. B. Walker, J. Org. Chem., 1990, 55, 5543 CrossRef CAS.
- (a) A. Jolit, P. M. Walleser, G. P. A. Yap and M. A. Tius, Angew. Chem., Int. Ed., 2014, 53, 6180 CrossRef CAS PubMed; (b) A. Jolit, C. F. Dickinson, K. Kitamura, P. M. Walleser, G. P. A. Yap and M. A. Tius, Eur. J. Org. Chem., 2017, 2017, 6067 CrossRef CAS.
- For earlier studies employing less substituted Ox-divinyl ketones, see: (a) D. J. Kerr, M. Miletic, J. H. Chaplin, J. M. White and B. L. Flynn, Org. Lett., 2012, 14, 1732 CrossRef CAS PubMed; (b) B. L. Flynn, N. Manchala and E. H. Krenske, J. Am. Chem. Soc., 2013, 135, 9156 CrossRef CAS PubMed; (c) N. Manchala, H. Y. L. Law, D. J. Kerr, R. Volpe, R. J. Lepage, J. M. White, E. H. Krenske and B. L. Flynn, J. Org. Chem., 2017, 82, 6511 CrossRef CAS PubMed.
- (a) T. N. Grant, C. J. Rieder and F. G. West, Chem. Commun., 2009, 5676 RSC; (b) Y. K. Wu and F. G. West, Org. Lett., 2014, 16, 2534 CrossRef CAS PubMed; (c) Y. Kwon, R. McDonald and F. G. West, Angew. Chem., Int. Ed., 2013, 52, 8616 CrossRef CAS PubMed; (d) Y.-K. Wu, C. R. Dunbar, R. McDonald, M. J. Ferguson and F. G. West, J. Am. Chem. Soc., 2014, 136, 14903 CrossRef CAS PubMed.
- Y. Minko, M. Pasco, H. Chechik and I. Marek, Beilstein J. Org. Chem., 2013, 9, 526 CrossRef CAS PubMed.
- B. Gourdet and H. W. Lam, J. Am. Chem. Soc., 2009, 131, 3802 CrossRef CAS PubMed.
- (a) A. M. Echavarren and J. K. Stille, J. Am. Chem. Soc., 1988, 110, 1557 CrossRef CAS; (b) S. Ceccarelli, U. Piarulli and C. Gennari, J. Org. Chem., 2000, 65, 6254 CrossRef CAS PubMed; (c) R. D. Mazzola, S. Giese, C. L. Benson and F. G. West, J. Org. Chem., 2004, 69, 220 CrossRef CAS PubMed.
- (a) E. Negishi, J. P. Maye and D. Choueiry, Tetrahedron, 1995, 51, 4447 CrossRef CAS; (b) E. Negishi, D. E. Van Horn and T. Yoshida, J. Am. Chem. Soc., 1985, 107, 6639 CrossRef CAS.
- D. P. Curran and S.-C. Kuo, Tetrahedron, 1987, 43, 5653 CrossRef CAS.
- J. Huang, D. Lebœuf and A. J. Frontier, J. Am. Chem. Soc., 2011, 133, 6307 CrossRef CAS PubMed.
- Ethereal solvents are known to suppress Wagner-Meerwein rearrangement of the oxyallyl cation intermediate of Nazarov cyclization, see: ref. 14 and P. Chiu and S. Li, Org. Lett., 2004, 6, 613 CrossRef CAS PubMed.
- K. C. Nicolaou, J. Wang, Y. Tang and L. Botta, J. Am. Chem. Soc., 2010, 132, 11350 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details and NMR spectra of all newly synthesized compounds. X-ray crystal data for (3S)-23 and E-24. Details and further discussion of computational studies. CCDC 1814626 and 1814627. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8sc00031j |
| ‡ See the ESI.† |
| § The basis of the isomerization of the C5-alkenyl unit has not yet been fully discerned. Treatment of 16 with I2 gave only the expected E-iodoalkene, whereas 1,2-addition of 16 to 15 gives the corresponding carbinol (not shown) as ∼3:1 mixture of the E- and Z-isomer. |
| ¶ In the absence of AlMe3 the reaction affords mostly an acyl migration product involving ring opening of the oxazolidinone: |
| This journal is © The Royal Society of Chemistry 2018 |