
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSuper electron donors derived from diboron†
Li
Zhang
and
Lei
Jiao
 *
*
Center of Basic Molecular Science (CBMS), Department of Chemistry, Tsinghua University, Beijing 10084, China. E-mail: Leijiao@mail.tsinghua.edu.cn
First published on 12th February 2018
Abstract
Single-electron transfer is an important process in organic chemistry, in which a single-electron reductant (electron donor) acts as a key component. Compared with metal-based electron donors, organic electron donors have some unique advantages, such as tunable reduction ability and mild reaction conditions. The development of novel organic electron donors with good reduction ability together with ease of preparation is in high demand. Based on the pyridine-catalyzed radical borylation reaction developed in our laboratory, we have discovered that, the reaction system consisting of a diboron(4) compound, methoxide and a pyridine derivative could smoothly produce super electron donors in situ. Two boryl–pyridine based species, the major one being a trans-2H,2′H-[2,2′-bipyridine]-1,1′-diide borate complex and the minor one being a pyridine radical anion–borate complex, were observed and carefully characterized. These complexes were found to be organic super electron donors unprecedented in literature, and their formation mechanisms were studied by DFT calculations. The diboron/methoxide/pyridine system enables the preparation of organic super electron donors from easily accessible starting materials under mild conditions, which has the potential to be a general and practical single-electron reducing agent in organic synthesis.
Introduction
Single-electron transfer (SET) is an important process in various redox- and radical-type organic reactions,1 where a single-electron reductant (electron donor) plays an important role. Usually, single-electron reductants involved in these reactions include alkali metals, alkali earth metals, low valent metallic reagents (e.g., SmI2 and TiCl3) and organometallic reagents (e.g., sodium naphthalide, Cp2TiIII),2 which have been used in organic chemistry for more than half a century. Compared with these traditional electron donors, organic electron donors have attracted the attention of organic chemists more recently due to their tunable reducing ability and mild reduction conditions.Tetrathiafulvalene (TTF) and tetrakis(dimethylamino)-ethylene (TDAE) have long been known as organic electron donors, but they exhibit moderate reducing ability and can only be used in the reduction of a limited number of substrates.3 Since 1993, Murphy and co-workers have developed a series of organic electron donors based on electron-rich alkenes (e.g., 1), which exhibit potent reduction abilities (Scheme 1a).4 The sufficiently low oxidation potential even allows them to promote single-electron reduction of aryl iodides, and thus they are regarded as “super electron donors” (SEDs).4,5 These structurally well-defined SEDs can efficiently promote a series of single-electron reductions and radical-type reactions;4 however, the structural complexity and the requirement for multi-step synthesis limit their utility in synthesis to some extent. Due to the importance of single-electron reducing agents in organic chemistry, the development of novel organic electron donors with good reduction ability, tunable structure, and a simple preparation procedure compared to traditional metal-based electron donors will significantly advance organic redox chemistry.
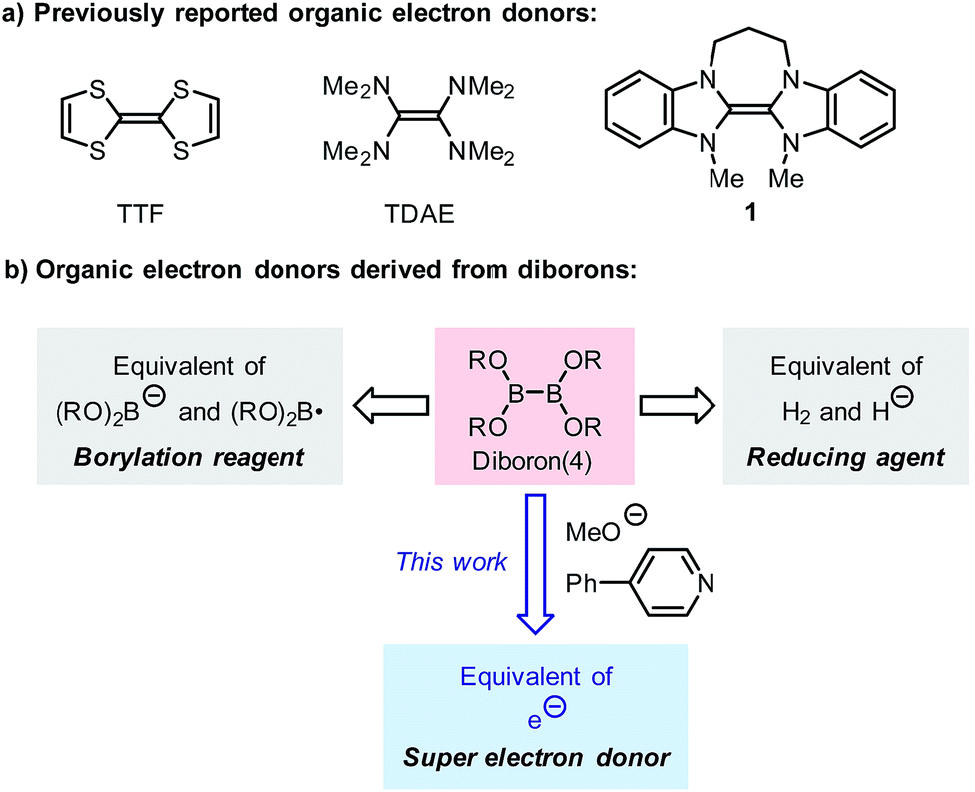 | ||
| Scheme 1 Organic electron donors. | ||
Recently, we reported a pyridine-catalyzed radical borylation reaction of aryl halides employing diboron(4) compounds as the boron source.6 The preliminary mechanistic study revealed that diboron, methoxide and the pyridine catalyst formed a reductive mixture, which promoted single-electron reduction of the aryl halide to produce an aryl radical as the key intermediate. The electron donor formed in the reaction system exhibited comparable reduction ability to previously reported SEDs and is rather attractive due to the ease of preparation. However, the structure of the electron donor was not clearly elucidated in the previous study, nor was its synthetic potential extensively exploited.
Although diborons have been employed as reducing agents in several reduction reactions as either hydrogen or hydride equivalents,7 to the best of our knowledge, there is no precedent to the use of diboron as the precursor to a structurally well-defined single-electron reductant. Thus, the investigation into the SEDs derived from diborons is significant. In this work, we demonstrate that the combination of diboron, pyridine and a base can be used as an efficient source of structurally well-defined SEDs (Scheme 1b). The structures of the electron donors in the reaction system have been clearly elucidated, leading to the discovery of two novel boron–pyridine based SEDs. The detailed mechanism of their formation and interconversion has also been disclosed, which represents a novel activation mode of the boron–boron bond in diboron(4) compounds. This reaction system is able to promote single-electron reduction of a variety of substrates and able to trigger radical chain reactions initiated by SET. Due to this feature, the SEDs derived from diboron have the potential to be used as a general and practical single-electron reducing agent in organic synthesis. Herein we report the details of our investigation on SEDs derived from diborons via a combined experimental and DFT computational study.
Results and discussion
Since the reaction between diboron, methoxide and a pyridine derivative was found to exhibit electron-donating ability in the radical borylation reaction, we sought to make clear the following points:(1) The structure of the electron donor produced in the diboron/methoxide/pyridine system;
(2) The formation mechanism of the electron donor;
(3) The potential application of the reduction system to other electron-transfer-related reactions.
Because 4-phenylpyridine (2) showed the best performance in the borylation reaction of aryl halides, we chose the reaction system consisting of bis(pinacolato)diborane, methoxide and pyridine derivative 2 as a model throughout our study.
1. Elucidation of electron donor structure
It was found in our previous study that by simply mixing B2pin2, MeOK and 2 in DMSO, a deep purple color emerged and an intense EPR signal was detected, which was attributed to the formation of the electron donor in this reductive mixture.6 However, the poor solubility of methoxide in DMSO led to a limited concentration of the electron donor, which hampered further structural characterization. To prepare a solution of the SED with a reasonable concentration, we used 18-crown-6 (18-C-6) as an additive and performed the reaction in THF. To our delight, the mixture of B2pin2, MeOK, 18-C-6 and 2 in THF formed a homogenous deep purple solution. Quite unexpectedly, the solution exhibited well-resolved signals in both NMR and EPR spectra (Scheme 2). This observation was in contrast with the previous proposal, where only two radical species were generated in this reaction mixture. Therefore, we set out to carefully characterize the observed species.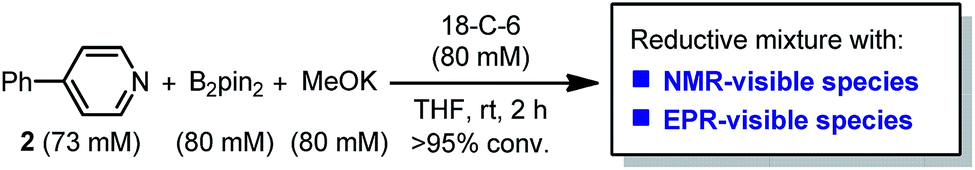 | ||
| Scheme 2 Reaction conditions of the model study. | ||
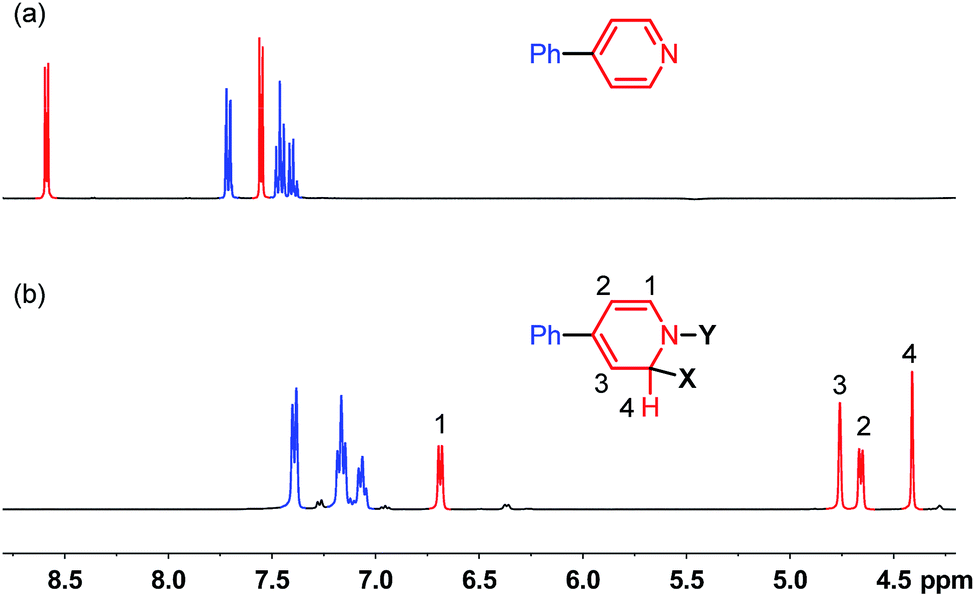 | ||
| Fig. 1 1H-NMR spectra (400 MHz) of (a) 4-phenylpyridine and (b) reaction mixture shown in Scheme 2 at room temperature in THF. | ||
We sought to characterize the structure of this species by single-crystal X-ray diffraction (XRD) analysis, but it was difficult to directly crystallize it from the reaction mixture. Fortunately, we discovered that the reaction of 4-phenylpyridine radical anion (3) with B2pin2 in the presence of excess 2 in DME could afford a pale yellow crystalline solid upon the addition of hexane. After treatment with 18-C-6 in THF, it produced identical 1H-, 13C- and 11B-NMR spectra as the previously acquired ones of the reaction mixture, confirming the same identity of the NMR-visible species and the crystal after treatment. Single-crystal XRD analysis elucidated the structure of the crystal as the ate complex 4·DME (Scheme 3),8 which suggested that the NMR-visible species was a complex with the composition of 4·18-C-6 (Scheme 3). Meanwhile, it was found that in DMSO without 18-C-6, the B2pin2/MeOK/2 reaction system also produced the same complex 4 as the major species, since the recorded 1H-NMR spectrum was identical to that of independently prepared 4·DME recorded in DMSO (see the ESI for details†). The structure of 4 well accounted for the dearomatized pyridine ring revealed by NMR analysis.
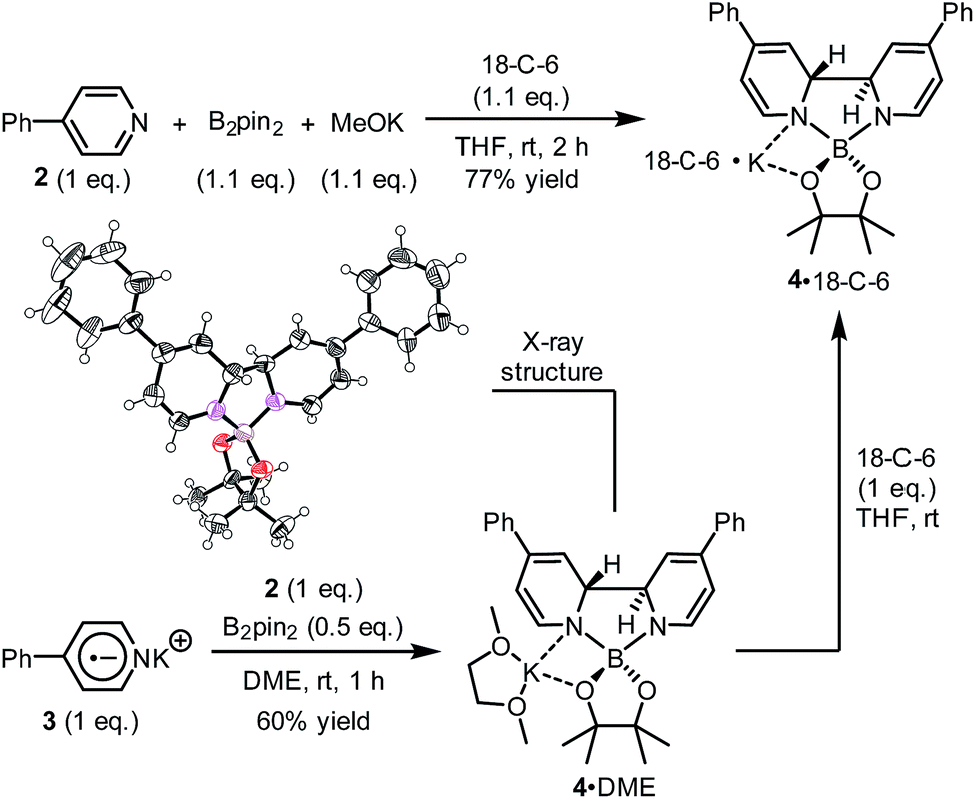 | ||
| Scheme 3 Independent preparation of the NMR-visible species. In the X-ray structure of compound 4, the DME part and the potassium cation are omitted for clarity. Thermal ellipsoids are drawn at 50% probability level. | ||
With the pure ate complex 4·DME in hand, we were able to assess its redox properties. Electrochemical study revealed that the THF solution of complex 4·DME exhibited an irreversible oxidation wave at −1.11 V versus the Fc+/0 couple, as determined by both cyclic voltammetry (CV) and differential pulse voltammetry (DPV) experiments (Fig. 2). The oxidation potential was similar to that of many reported SEDs,3c–e which allows for electron transfer to a variety of substrates.
 | ||
| Fig. 2 CV and DPV plots of 2 mM complex 4·DME in THF (0.1 M n-Bu4N+PF6− as the supporting electrolyte). The blue solid line: CV plot (scan rate = 0.1 V s−1); the red dashed line: DPV plot. | ||
The reaction between complex 4·DME and 4-iodoanisole (5) also verified its electron donor character. By addition of 4 to excess iodoarene 5 in THF under room temperature, anisole (6) was produced in a good yield rather quickly (Scheme 4). The observation was in agreement with a mechanism involving SET-initiated carbon–iodine bond activation in iodoarene 5 to form an aryl radical, which then underwent a hydrogen atom transfer (HAT) from the solvent THF to form the reduction product 6. More interestingly, both N,N′-diboryl-2H,2′H-2,2′-bipyridine 7 and free pyridine 2 was observed by 1H-NMR in the crude reaction mixture as the end-product of complex 4, indicating that 4 served as an electron donor (vide infra). Notably, no trace of arylboronate was observed as the product in this reaction, even if the solvent was switched to DMSO to suppress the HAT pathway, indicating that complex 4 served merely as an electron donor, but not a borylation reagent.
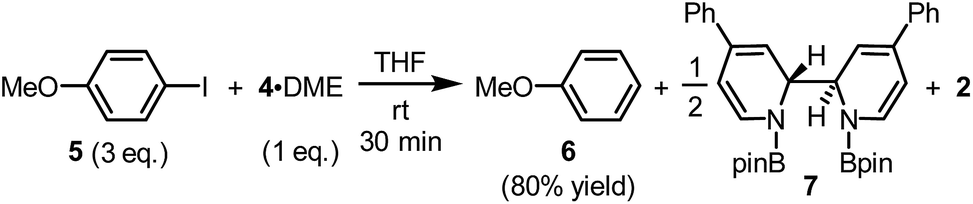 | ||
| Scheme 4 Electron donor character of complex 4. | ||
Complex 4 represents an ate complex of pinacolato boronate with a trans-2H,2′H-[2,2′-bipyridine]-1,1′-diide unit as the ligand. This ate complex has not been reported previously, though several borate compounds with a 2,2′-bipyridine skeleton were reported. In 1992, Meller reported the formation of [1,3,2]-diazaborolobipyridine in the reaction between the pyridine radical anion and dialkylaminodifluorborane;9 more recently, Tang and co-workers reported the reaction between isoquinoline and diboron to form a N,N′-diborate of 1,1′,2,2′-tetrahydro-1,1′-bisisoquinoline.10 However, these compounds were never found to exhibit any electron donor character. We attributed the electron-donating ability of complex 4 to both the dearomatized bipyridine scaffold and the negatively charged boron center. From a thermodynamic viewpoint, the cleavage of the B–B bond released the energy stored in the diboron and transferred it to the dearomatized bipyridine unit as the C–C bond formed. The negatively charged boron center may facilitate the loss of an electron from complex 4, which induced the break of the bipyridine linkage and recovered the aromaticity of the pyridine ring. The dissociation of the bipyridine scaffold and the electron transfer of this complex were similar to the behavior of dihydropyridine dimers.11
 | ||
| Fig. 3 X-band first derivative EPR spectra of (a) B2pin2/MeOK/2/18-C-6 reaction solution in THF; (b) the borylation reaction of 4-MeOPhBr employing B2pin2·MeOK/2 in DMSO; (c) the solution of complex 9 with 18-C-6 in THF; (d) the THF solution of complex 10 prepared by the reaction between 4-phenylpyridine radical anion and MeOBpin (all acquired at room temperature). (e) Is the simulated EPR spectrum with parameters listed in Table 1. | ||
A hint for resolving the structure of the EPR-visible species came from a crystal structure obtained unexpectedly. When pyridine radical anion 8 (generated by treatment of pyridine 2 with sodium metal) was reacted with B2pin2 in THF, a deep purple crystal precipitated quickly instead of the light-yellow crystal of 4·DME obtained by the same reaction using DME as the solvent (Scheme 5). XRD analysis of the crystal revealed that it was a simple Lewis acid–base complex of the pyridine radical anion and the diboron derivative, denoted as 4-PhPy˙−·B2pin2 (9). Complex 9 could be re-dissolved in THF by the addition of 18-C-6, and EPR analysis of the solution showed a similar but significantly broadened spectrum compared to that of the reductive mixture (Fig. 3c). The formation of 9 indicated that the radical anion of pyridine could coordinate with a boron Lewis acid center,12 and the similar EPR spectrum also implied the structural relevance of the unknown radical species to complex 9.
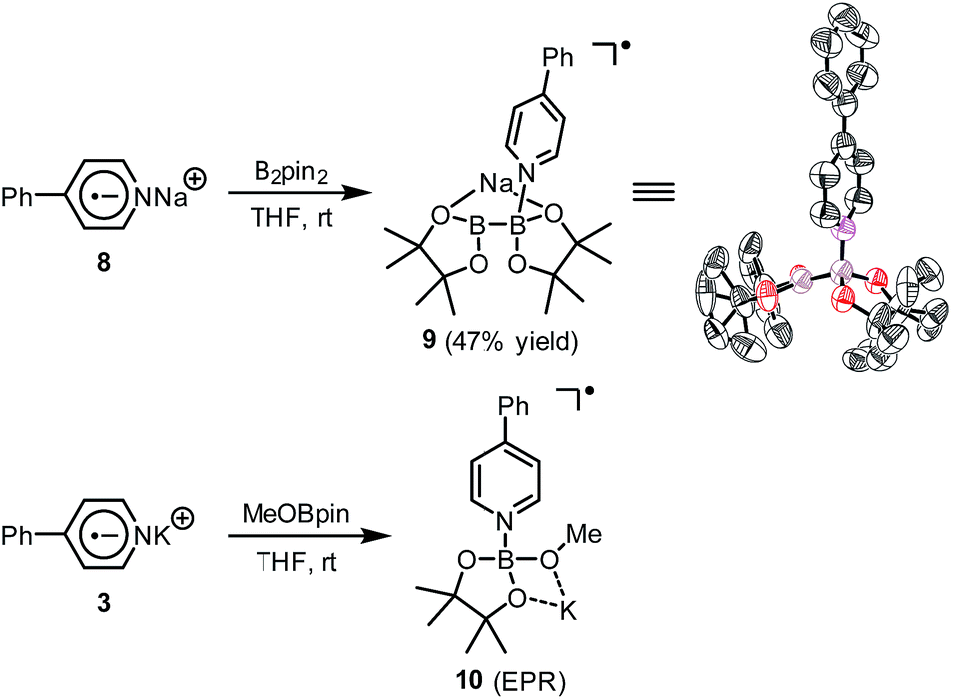 | ||
| Scheme 5 Preparation of complexes 9 and 10. In the X-ray structure of 9, the sodium cation, the coordinating THF molecule, and hydrogen atoms are omitted for clarity. Thermal ellipsoids are drawn at 50% probability level. | ||
We assumed that the EPR-visible species was also a complex of the 4-phenylpyridine radical anion and a boron Lewis acid. Methyl pinacolyl borate (MeOBpin) was the most probable one, because it existed as the byproduct in the B2pin2/MeOK/2 reaction system. To confirm this hypothesis, we conducted the reaction between pyridine radical anion 3 and MeOBpin (Scheme 5). It was found that upon addition of MeOBpin to a THF solution of 3, the color of the solution changed immediately from deep blue to deep purple, and it showed a well-resolved EPR spectrum, which exhibited similar spectral pattern and identical peak positions to the one obtained for the B2pin2/MeOK/2/18-C-6 mixture (Fig. 3d). Meanwhile, the addition of excess MeOBpin to a THF solution of complex 9 and 18-C-6 resulted in a similar EPR spectrum to that shown in Fig. 3a (Fig. S3 in the ESI†). These results suggested that the complex between the 4-phenylpyridine radical anion and MeOBpin, denoted as 4-PhPy˙−·B(OMe)pin (10), accounted for the EPR-visible radical species. Computer simulation of the recorded EPR spectrum13 afforded the experimental hyperfine coupling constants (hfcs) of the proposed radical species (Fig. 3e), and the comparison with the DFT-calculated hfcs further supported the identity of this species (Table 1). The slightly broadened peak shape observed for the B2pin2/MeOK/2 mixture (Fig. 3a) could be attributed to the existence of both complexes 9 and 10 in the reaction system, as a combination of the EPR spectra of complexes 9 and 10 well reproduced the measured spectrum (Fig. S7 in the ESI†).
|
|
|||||||
|---|---|---|---|---|---|---|---|
| hfcs/MHz | a (11B) | a (N) | a (2Ha) | a (2Hb) | a (2Hc) | a (2Hd) | a (He) |
| a The hfcs obtained from simulation of the experimental EPR spectrum (g = 2.0030) using EasySpin software. Only the magnitudes, but not the signs of the hfcs could be derived. b DFT calculated hfcs at the UB3LYP/6-31+G(d) level for the model structure of 10·THF. | |||||||
| Exptl.a | 3.9 | 10.2 | 12.5 | 1.3 | 8.1 | 3.1 | 8.3 |
| DFTb | −4.8 | 12.1 | −10.4 | 1.2 | −9.7 | 3.8 | −12.5 |

The comparison of the UV-vis spectrum of the reductive mixture and that of complex 9 also supported this structural assignment (Fig. 4a). In the UV-vis spectrum of the mixture containing complexes 4 and 10,14 two absorptions were observed at 377 and 567 nm. These absorption bands were attributed to complex 10 due to the absence of corresponding absorptions for complex 4. The two absorption bands fitted well with those of the radical anion complex 9 and were attributed to the same pyridine substructure. This observation confirmed the structural relevance of complexes 9 and 10 and the nature of complex 10 as a pyridine radical anion complex.
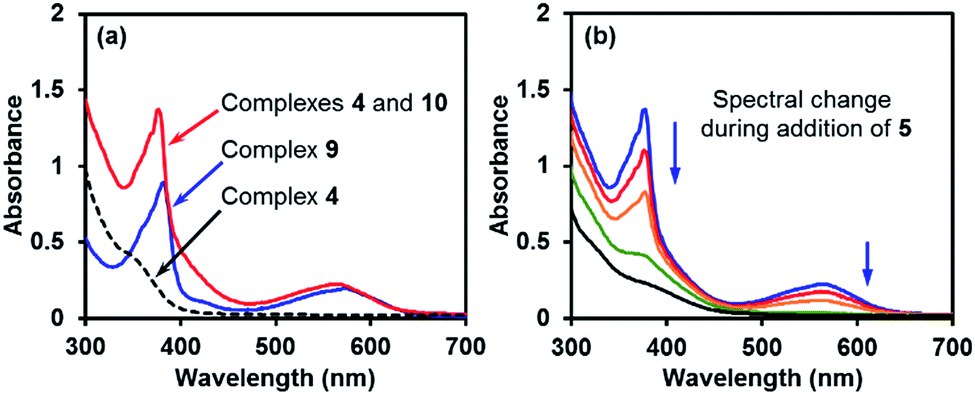 | ||
| Fig. 4 (a) UV-vis spectra of the boryl–pyridine complexes in THF (red line: reaction mixture containing complexes 4 and 10; blue line: solution of complex 9 and 18-C-6; dashed line: solution of complex 4·DME). (b) UV-vis spectral change in the reaction mixture during the addition of iodoarene 5. | ||
Similar to ate complex 4, complex 10 was found to be a good electron donor. During the addition of iodoarene 5 to the mixture of complexes 4 and 10, the bleaching of the two characteristic absorption bands (Fig. 4b) and the disappearance of the EPR signal were observed, which was indicative of the consumption of complex 10 by the iodoarene as an electron acceptor.
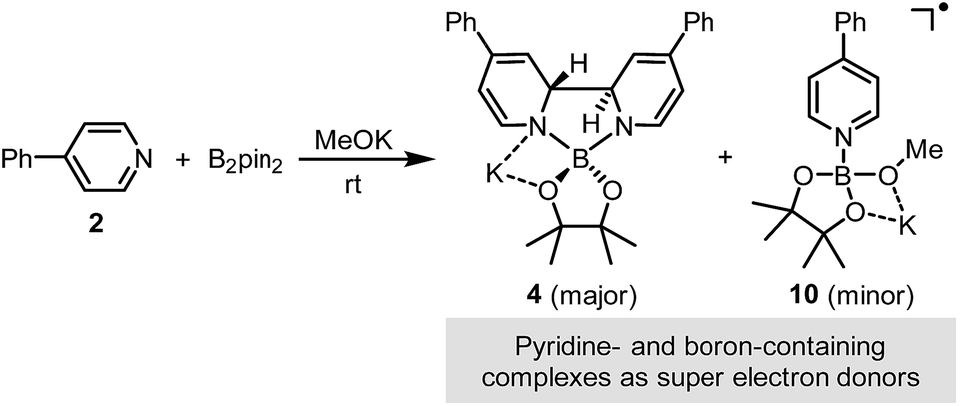 | ||
| Scheme 6 Super electron donors in the reaction system. | ||
2. Mechanism for the formation of super electron donors
The key mechanistic problem for the formation of the two SED complexes in the reaction system was the mode of boron–boron bond cleavage. To solve this problem and to approach a clear mechanistic picture of this conversion, DFT computational study was conducted using the Gaussian 09 (ref. 15) software package. The geometric optimization was conducted at the M06-2X16/6-31+G(d) level of theory, and the frequency analysis was performed to verify the located structure to be either a minimum or a saddle point. Single-point energies were calculated at the M06-2X/6-311+G(d,p) level of theory, and the CPCM17 method was used to evaluate the solvation effect in DMSO.This homolytic cleavage pathway was evaluated by DFT calculations in the present study (Fig. 5). The diboron first formed ate complex IN1 with methoxide, which was already confirmed by the experimental studies of Marder et al.19 Then pyridine 2 coordinated to the other vacant boron center in IN1 to produce complex IN2, and this step is endergonic by 15.2 kcal mol−1 However, the proposed homolytic cleavage of the B–B bond in IN2 was highly endergonic (by >30 kcal mol−1 from IN1), indicating that the formation of radical anion IN3 and pyridine-stabilized boryl radical IN4 from IN2 was not feasible. Therefore, the mode of diboron cleavage promoted by 4-phenylpyridine and methoxide should be rather different from that promoted by 4-cyanopyridine.18
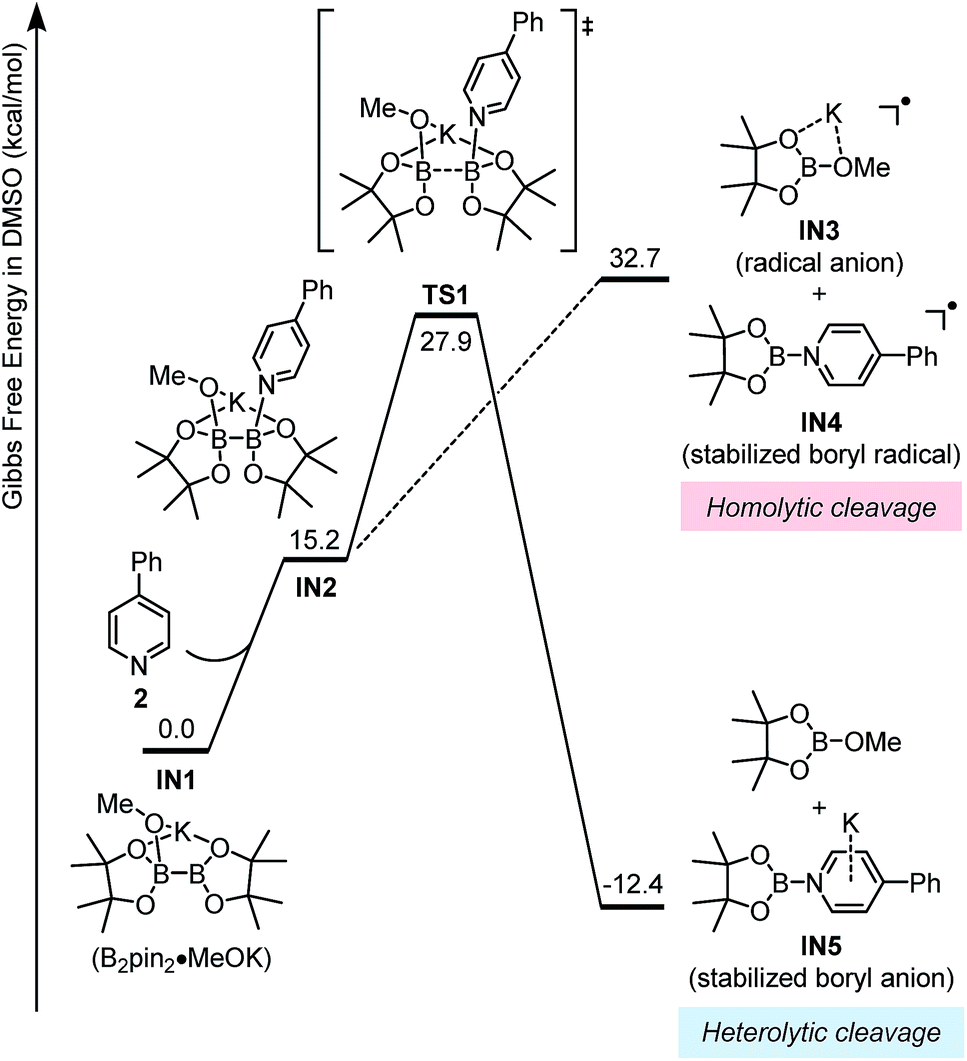 | ||
| Fig. 5 Gibbs free energy (ΔGsolv 298 K) profile for the homolytic and heterolytic cleavage pathways of diboron. | ||
Intermediate IN5 could be viewed as a complex between a boryl anion and a 4-phenylpyridine molecule or a so-called pyridine-stabilized boryl anion.21 We proposed that IN5 will be further transformed to complex 4, and the potential energy surface for this transformation was calculated (Fig. 6). First, pyridine 2 in the reaction system could coordinate with the boron center of IN5 through TS2 to form a new complex IN6. Subsequently, intramolecular C–C bond formation occurs with a reasonable overall free energy barrier (ΔG≠ = 23.1 kcal mol−1 from IN5) to form the observed complex 4. The overall conversion from IN5 to complex 4 is exergonic by 2.1 kcal mol−1.
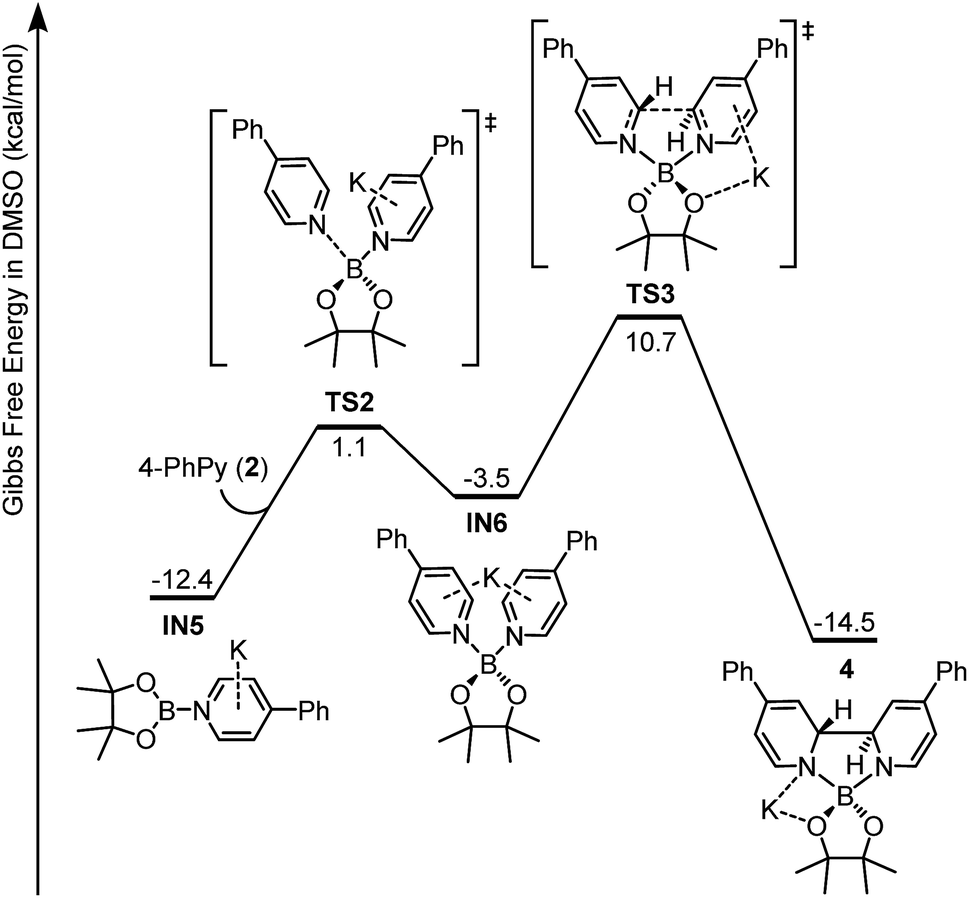 | ||
| Fig. 6 Gibbs free energy (ΔGsolv 298 K) profile for the formation of ate complex 4. | ||
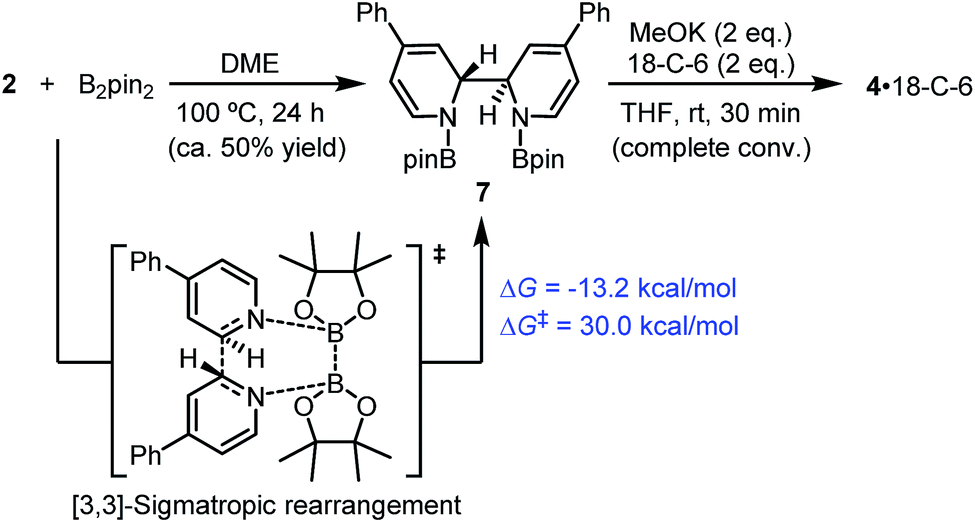 | ||
| Scheme 7 [3,3]-Sigmatropic rearrangement pathway. DFT-calculated thermodynamic and kinetic parameters are shown in blue. | ||
In fact, it was found that heating a mixture of B2pin2 and pyridine 2 in DME resulted in the formation of 7, together with some unidentified byproducts. Upon treatment with MeOK, 7 was smoothly transformed to complex 4, as confirmed by 1H-NMR analysis (Scheme 7). This indicated that compound 7 could act as a precursor to SED. However, the [3,3]-sigmatropic rearrangement pathway was not likely to operate in the present reaction system, because the reaction between B2pin2 and pyridine 2 proceeded rather sluggishly even at 100 °C, while the B2pin2/MeOK/2/18-C-6 reaction system showed >95% conversion within 2 h at room temperature. DFT calculation of the [3,3]-sigmatropic rearrangement mechanism also supported this conclusion, since a higher activation free energy barrier (ΔG≠ = 30.0 kcal mol−1) rendered this pathway unfavorable compared with the heterolytic cleavage pathway (ΔG≠ = 27.9 kcal mol−1).
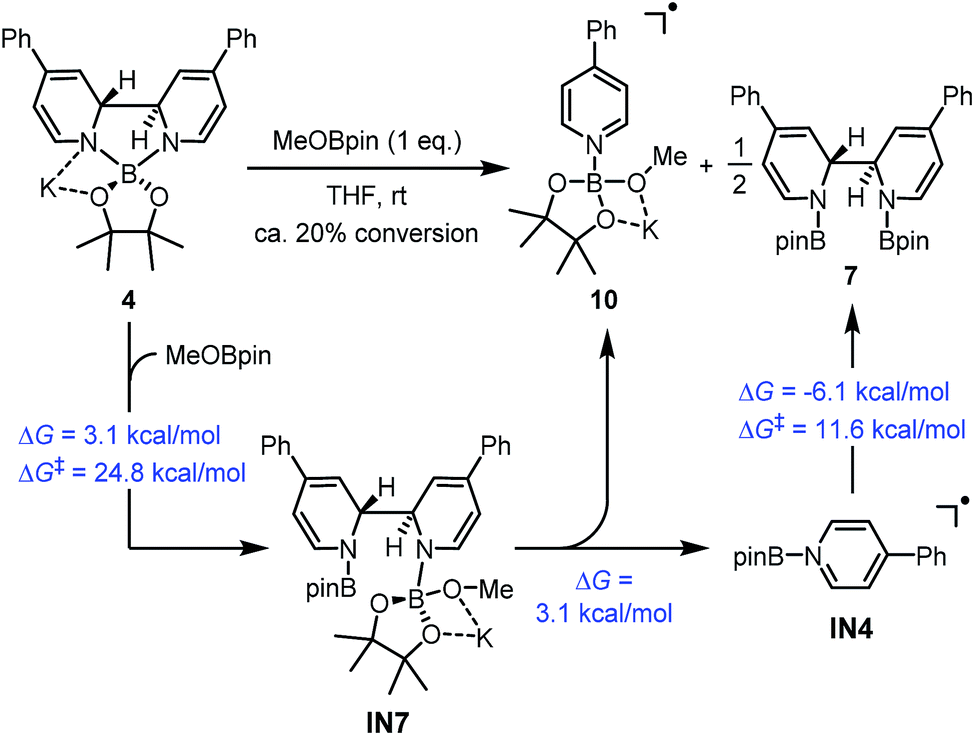 | ||
| Scheme 8 Formation of complex 10 from complex 4. DFT calculated thermodynamic and kinetic parameters are shown in blue. | ||
A reasonable mechanism was proposed for this conversion with the aid of DFT calculation (Scheme 8). First, the Lewis acidic MeOBpin reacted with complex 4 to form a new complex, IN7, and this transformation was slightly endergonic. Then the homolytic cleavage of the bipyridine C2–C2′ bond led to both radical anion complex 10 and pyridine-stabilized boryl radical IN4, and this step was also slightly endergonic. Subsequently, radical species IN4 dimerized to compound 7 rather facilely, as judged by the low activation free energy barrier and the thermodynamic driving force. The overall change in terms of Gibbs free energy was 0.1 kcal mol−1, which was in agreement with the observed incomplete conversion.
In addition to this cleavage pathway, another pathway leading to complex 10 by direct SET was considered. Given that the anionic species IN5 was highly electron rich, it may also act as an electron donor. We proposed that IN5 could undergo SET with the complex of MeOBpin and pyridine 2 to produce complex 10 directly, and meanwhile IN5 was transformed to radical IN4 and then dimerized to 7. This pathway was also feasible according to the DFT calculations (Scheme 9).
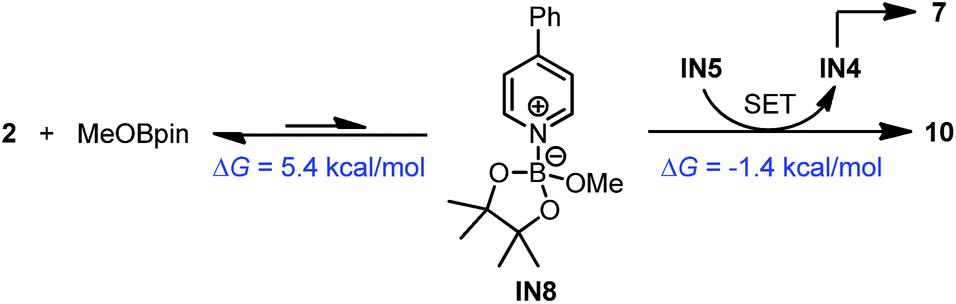 | ||
| Scheme 9 Formation of complex 10 from IN5. DFT-calculated thermodynamic and kinetic parameters are shown in blue. | ||
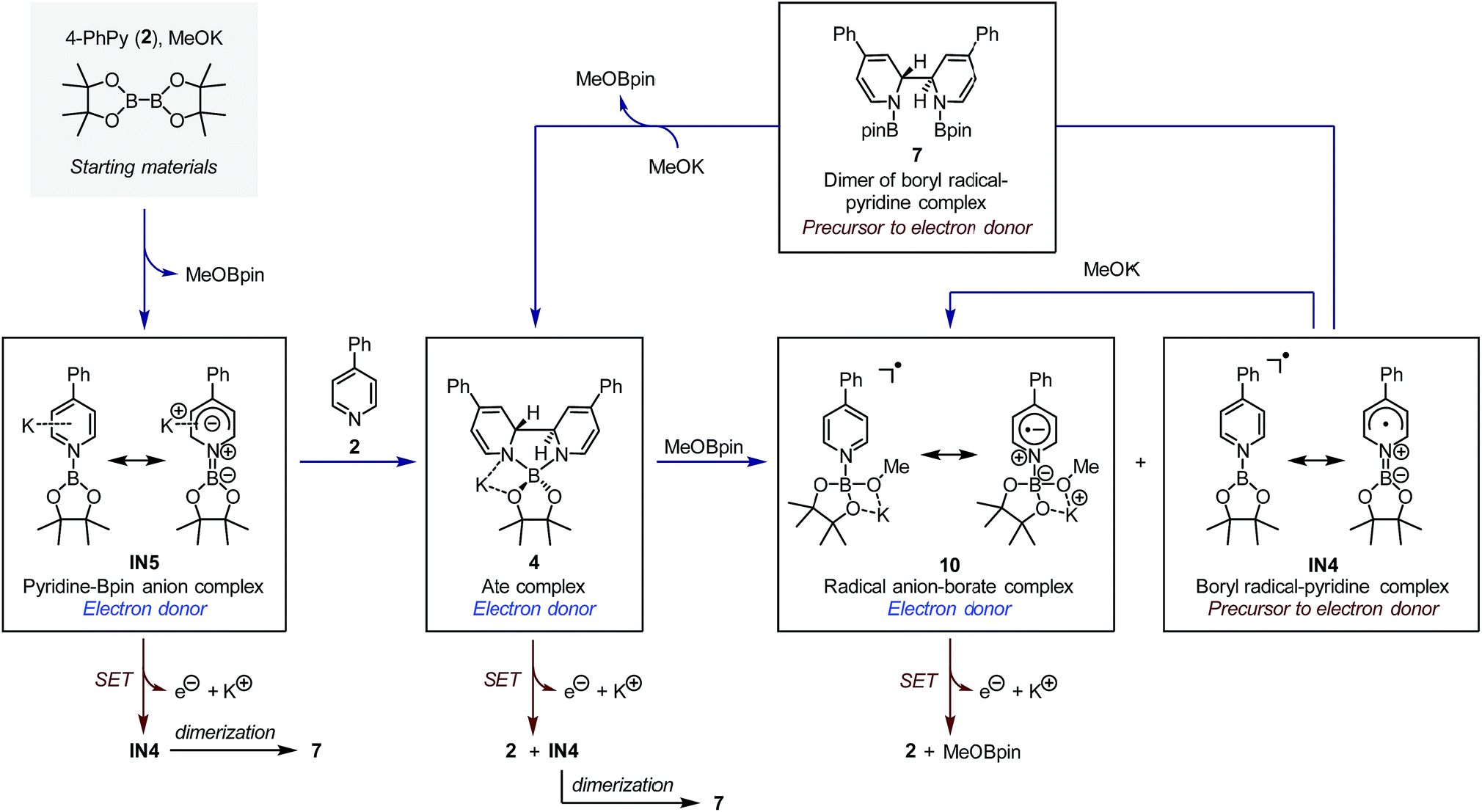 | ||
| Fig. 7 Boryl–pyridine species involved in the reaction system and their interconversion. The blue arrows indicate the formation and interconversion between each species, while the red arrows indicate the single-electron transfer reaction of the electron donors. | ||
The SET pathways of the proposed SEDs are also summarized Fig. 7. After the electron transfer process, complex IN5 produced pyridine-stabilized boryl radical IN4, which dimerized to afford compound 7. Electron transfer from ate complex 4 resulted in the formation of both pyridine 2 and IN4, and then IN4 produced dimer 7 rapidly. Radical anion complex 10 generated free pyridine 2 and MeOBpin upon losing an electron. Because compound 7 was proved a precursor to complex 4, in the reaction system, 7 could be transformed to SED again with sufficient amount of methoxide. This transformation network realized an overall pyridine-catalyzed electron transfer process of B2pin2 + 2MeO− → 2MeOBpin + 2e−, which rendered the diboron an efficient electron source.
3. Revised mechanism of the radical borylation reaction
The discovery of the real SED species in the diboron/methoxide/pyridine system prompted us to reconsider the mechanism of the radical borylation reaction based on this system. The generation of SEDs and the formation of aryl radical from the haloarene by SET have been made clear through the study presented above; however, the absence of boryl radical species IN4 in the reaction mixture questioned the previously proposed cross-coupling mechanism between aryl radical and IN4.6Interestingly, the reaction of iodoarene 5 with complex 4 in DMSO could produce the borylation product 11 only when B2pin2 was added. Crossover experiment using bis(hexylene glycolato)diboron in place of B2pin2 afforded the corresponding hexylene glycolyl boronate as the only borylation product (see the ESI for details†). These results indicated that diboron or its related species served as the borylation reagent. To figure out the very species that participates in the C–B bond formation event with the aryl radical, we conducted a series of competition experiments using the aryl radical cleanly generated from complex 4 and iodoarene 5 (Fig. 8). The experiments were conducted in a mixed solvent of THF and DMSO, and the ratio of arylboronate 11 (from borylation) to reduction product 6 (from HAT) was used as a measure of relative borylation rate. It was found that the concentration of pyridine 2 exhibited a neglectable effect (Fig. 8a), while the concentration of B2pin2 had a perfect linear relationship with the ratio of 11 to 6 (Fig. 8b). In this kinetic competition scenario, the change in the relative borylation rate was affected merely by [B2pin2], clearly indicating that the diboron itself rather than pyridine-containing boryl species (such as the complex between 2 and B2pin2 (ref. 22)) dictated the C–B bond formation step with the aryl radical.
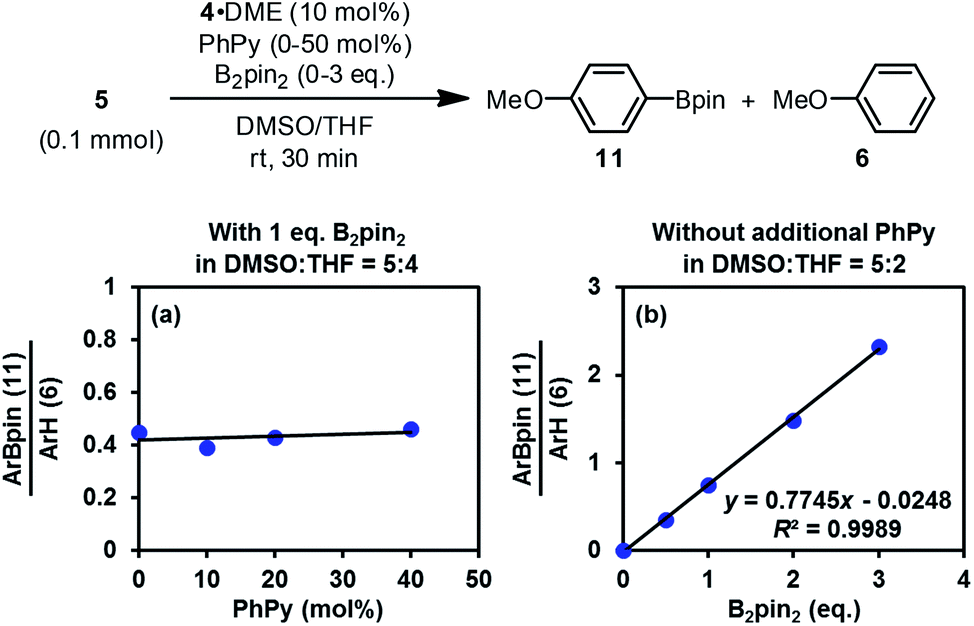 | ||
| Fig. 8 Competition experiments. | ||
That conclusion was distinct to the one drawn in the present study. By comparison of the reaction conditions used in both studies, we were able to figure out the reason for the difference. In the previous complex reaction system, MeOBpin was produced after the heterolytic cleavage of diboron by pyridine 2 and methoxide, and the effect of pyridine concentration on product distribution could be attributed to this Lewis acid: a higher concentration of pyridine 2 led to the production of more MeOBpin, which could release more B2pin2 from the MeOK·B2pin2 complex via ligand exchange equilibrium. The released diboron acted as the borylation reagent to favor the formation of the borylation product. In this way, the concentration of the free diboron was dictated by the concentration of pyridine 2, which might be the reason for the apparent dependence of product distribution on pyridine 2. In the present study, the clean and simple reaction system for aryl radical generation allowed for the investigation of the borylation process without interference of other factors, and thus we believed that the results obtained here are more reasonable.
With these new results in hand, currently, we propose a revised mechanism for the pyridine-catalyzed radical borylation (Scheme 10): the combination of diboron, MeOK and pyridine 2 produced SEDs 4 and 10, which transformed a haloarene to an aryl radical by SET; the aryl radical reacted with the free diboron to produce arylboronate, which was proposed in several previous reports.23 It was noteworthy that in the revised reaction mechanism, the borylation process was not subject to the persistent radical effect24 but proceeded via a simple radical trapping mechanism. This feature would provide an opportunity for utilizing the present reaction system as an efficient tool for radical generation to access various functionalization products other than boronate, if the concentration of the free diboron could be controlled.
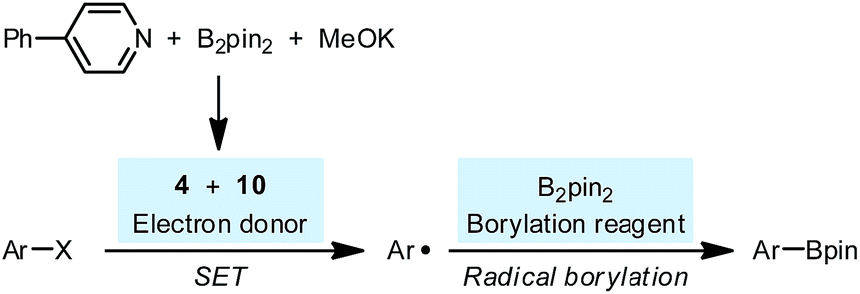 | ||
| Scheme 10 Revised radical borylation mechanism. | ||
4. Application of the diboron-derived super electron donors in synthesis
The feature that super electron donors could be produced by simply mixing easily available and stable compounds would enable the application of the diboron/methoxide/pyridine system to various synthetically useful reactions, such as electron reduction reactions and electron-transfer-initiated radical chain reactions. In this study, we briefly explore its potential use in the synthesis as a proof of concept.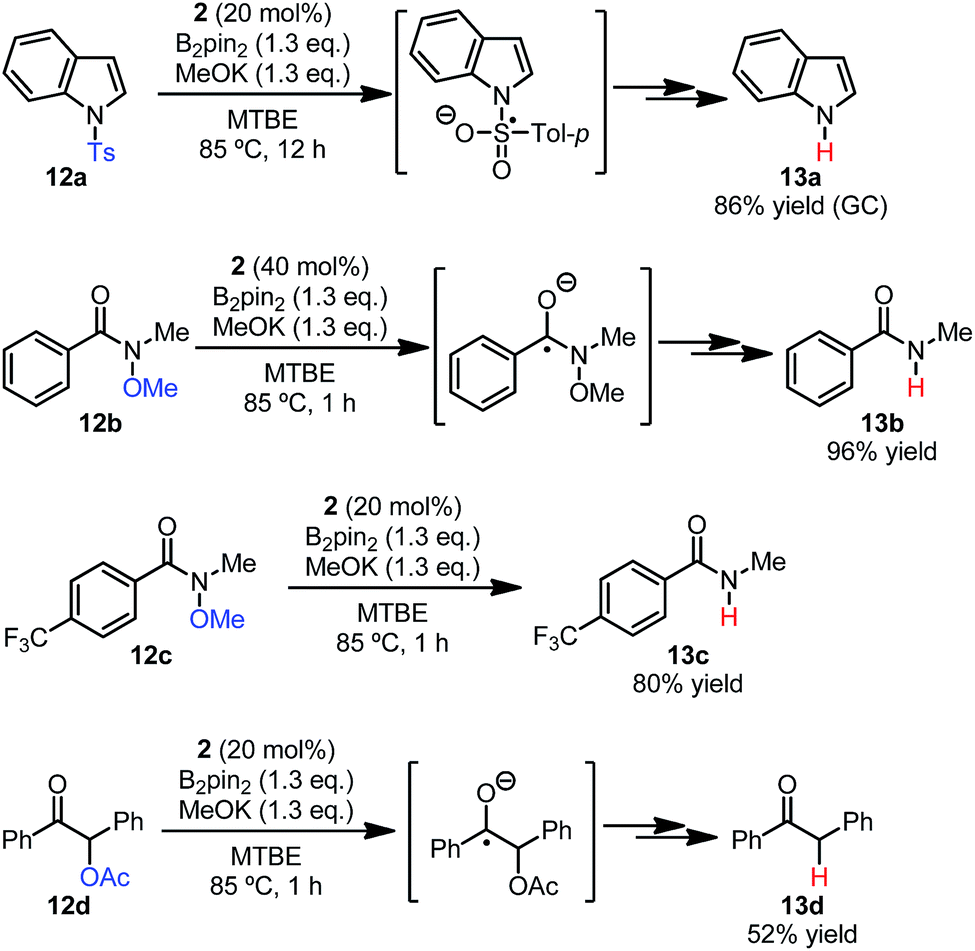 | ||
| Scheme 11 Reductive cleavage reactions employing the diboron/MeOK/2 system. | ||
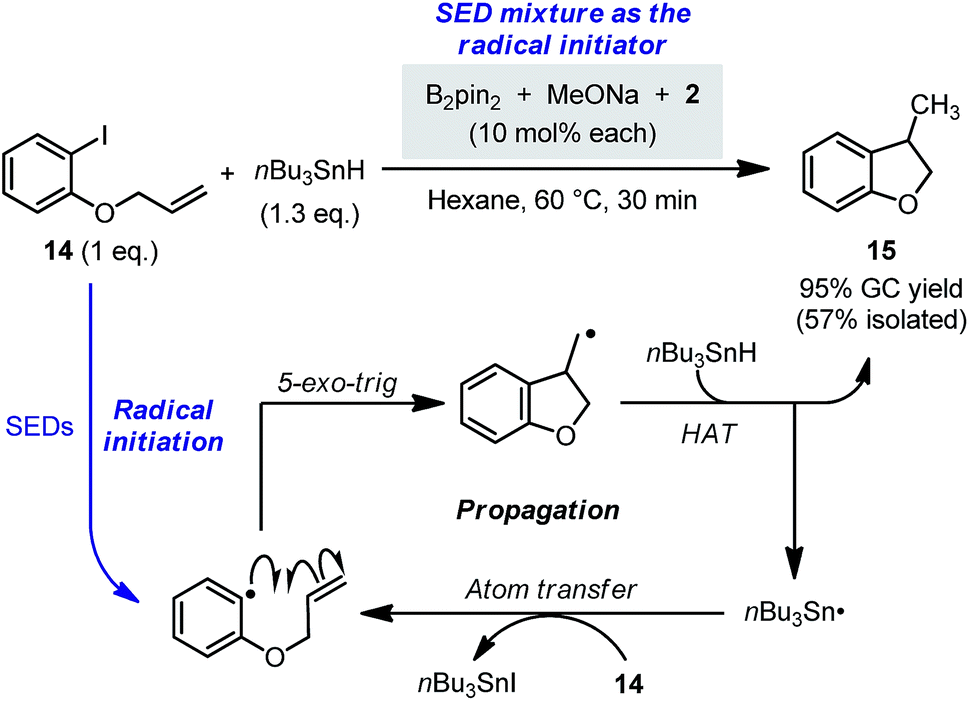 | ||
| Scheme 12 Radical chain reaction initiated by the diboron-derived SEDs. | ||
Compared with the radical initiation by thermolabile initiators, the present method allowed for fine-tuning of the initiation rate by controlling more factors (e.g., concentration of each component, structure of each component, and temperature), which provides a more flexible option for the initiation of radical chain processes. Although with this single experiment we have merely showcased a simple proof of concept instead of extensively testing its usefulness, we think that the present method could contribute to the toolbox of radical chemistry.
Conclusions
In this work, a detailed study on the reaction system containing diboron, methoxide and 4-phenylpyridine was performed. This mixture was previously found to be effective for the borylation of haloarenes via a radical mechanism. With the discovery of two novel electron-donating boryl–pyridine species, the present study not only revealed the formation of super electron donors in the reaction system, but also showcased the value of this system as a practical single electron reducing agent.Through a combined experimental and DFT computational study, we made clear the structures of two SEDs in the reaction system, as well as the mechanism for their formations. It was found that both the pyridine derivative and the methoxide anion were essential for the cleavage of the B–B bond in the diboron. In a complex of diboron with pyridine and methoxide coordinating to the two boron centers, heterolytic cleavage of the B–B bond, rather than previously assumed homolytic cleavage, occurred to form a methyl borate and a pyridine–boryl anion complex. In the presence of excess pyridine, these two intermediates were transformed to a trans-2H,2′H-[2,2′-bipyridine]-1,1′-diide borate complex and a pyridine radical anion–borate complex. These complexes were the major species observed in the reaction system, and their structures were determined by NMR, EPR and UV-vis spectroscopy and XRD analysis. Both complexes exhibited good electron-donating ability and were found to be novel SEDs not reported previously in literature. On the basis of these new findings, the mechanism of the pyridine-catalyzed radical borylation was revised.
The present reaction system represents the first example of utilizing diboron(4) compounds as the precursor to structurally well-defined single-electron reducing agents, and the SEDs derived from diboron have the potential to be used as a common single-electron reductant in organic synthesis. As our preliminary exploration revealed, in addition to the borylation reaction, this reaction system could be successfully applied to reductive cleavage reactions and radical chain cyclization reactions to replace common single-electron reductants. The features of these SEDs, such as the ease of preparation and the flexibility in structural tuning, make the present system a practical and promising complement to traditional electron donors. We seek to further investigate the structure–activity relationship of the reaction system, and expect that more applications in synthesis could be realized.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The National Natural Science Foundation of China (Grant No. 21772110 and 21390403) and the Thousand Talents Plan for Young Professionals are acknowledged for financial support. The technology platform of CBMS and the Tsinghua Xuetang Talents Program are acknowledged for providing instrumentation and computational resources.Notes and references
- (a) L. Eberson, Electron Transfer Reactions in Organic Chemistry, Springer-Verlag, Berlin, 1987 Search PubMed; (b) Encyclopedia of Radicals in Chemistry, Biology and Materials, ed. C. Chatgilialoglu and A. Studer, Wiley, New York, 2012 Search PubMed.
- For selected examples and reviews, see: (a) N. D. Scott, J. F. Walker and V. L. Hansley, J. Am. Chem. Soc., 1936, 58, 2442 CrossRef CAS; (b) P. Girard, J. L. Namy and H. B. Kagan, J. Am. Chem. Soc., 1980, 102, 2693 CrossRef CAS; (c) W. A. Nugent and T. V. RajanBabu, J. Am. Chem. Soc., 1988, 110, 8561 CrossRef CAS; (d) D. J. Procter, R. A. Flowers and T. Skrydstrup, Organic Synthesis Using Samarium Diiodide: A Practical Guide, RSC Publishing, Cambridge, 2010 Search PubMed; (e) M. Szostak, N. J. Fazakerley, D. Parmar and D. J. Procter, Chem. Rev., 2014, 114, 5959 CrossRef CAS PubMed; (f) A. Gansäuer and H. Bluhm, Chem. Rev., 2000, 100, 2771 CrossRef.
- For earliest examples and reviews of electron donors, see: (a) C. Lampard, J. A. Murphy and N. Lewis, J. Chem. Soc., Chem. Commun., 1993, 295 RSC; (b) N. Wiberg and J. W. Buchler, Chem. Ber., 1963, 96, 3223 CrossRef CAS; (c) J. Broggi, T. Terme and P. Vanelle, Angew. Chem., Int. Ed., 2014, 53, 384 CrossRef CAS PubMed; (d) J. A. Murphy, J. Org. Chem., 2014, 79, 3731 CrossRef CAS PubMed; (e) E. Doni and J. A. Murphy, Chem. Commun., 2014, 50, 6073 RSC.
- For selected examples of “super electron donors” as reagents, see: (a) J. A. Murphy, T. A. Khan, S. Z. Zhou, D. W. Thomson and M. Mahesh, Angew. Chem., Int. Ed., 2005, 44, 1356 CrossRef CAS PubMed; (b) J. A. Murphy, S.-Z. Zhou, D. W. Thomson, F. Schoenebeck, M. Mohan, S. R. Park, T. Tuttle and L. E. A. Berlouis, Angew. Chem., Int. Ed., 2007, 46, 5178 CrossRef CAS PubMed; (c) F. Schoenebeck, J. A. Murphy, S. Z. Zhou, Y. Uenoyama, Y. Miclo and T. Tuttle, J. Am. Chem. Soc., 2007, 129, 13368 CrossRef CAS PubMed; (d) P. I. Jolly, S. Zhou, D. W. Thomson, J. Garnier, J. A. Parkinson, T. Tuttle and J. A. Murphy, Chem. Sci., 2012, 3, 1675 RSC; (e) J. A. Murphy, F. Schoenebeck, N. J. Findlay, D. W. Thomson, S. Z. Zhou and J. Garnier, J. Am. Chem. Soc., 2009, 131, 6475 CrossRef CAS PubMed; (f) E. Cahard, F. Schoenebeck, J. Garnier, S. P. Y. Cutulic, S. Zhou and J. A. Murphy, Angew. Chem., Int. Ed., 2012, 51, 3673 CrossRef CAS PubMed; (g) E. Doni, S. O'Sullivan and J. A. Murphy, Angew. Chem., Int. Ed., 2013, 52, 2239 CrossRef CAS PubMed; (h) S. O'Sullivan, E. Doni, T. Tuttle and J. A. Murphy, Angew. Chem., Int. Ed., 2014, 53, 474 CrossRef PubMed; (i) E. Doni, B. Mondal, S. O'Sullivan, T. Tuttle and J. A. Murphy, J. Am. Chem. Soc., 2013, 135, 10934 CrossRef CAS PubMed; (j) S. S. Hanson, E. Doni, K. T. Traboulsee, G. Coulthard, J. A. Murphy and C. A. Dyker, Angew. Chem., Int. Ed., 2015, 54, 11236 CrossRef CAS PubMed.
- For selected mechanistic study of “super electron donors” as reactive intermediates, see: (a) A. Studer and D. P. Curran, Angew. Chem., Int. Ed., 2011, 50, 5018 CrossRef CAS PubMed; (b) A. Studer and D. P. Curran, Nat. Chem., 2014, 6, 765 CrossRef CAS PubMed; (c) S. Zhou, G. M. Anderson, B. Mondal, E. Doni, V. Ironmonger, M. Kranz, T. Tuttle and J. A. Murphy, Chem. Sci., 2014, 5, 476 RSC; (d) S. Zhou, E. Doni, G. M. Anderson, R. G. Kane, S. W. MacDougall, V. M. Ironmonger, T. Tuttle and J. A. Murphy, J. Am. Chem. Soc., 2014, 136, 17818 CrossRef CAS PubMed; (e) H. Yi, A. Jutand and A. Lei, Chem. Commun., 2015, 51, 545 RSC; (f) M. Patil, J. Org. Chem., 2016, 81, 632 CrossRef CAS PubMed; (g) J. P. Barham, G. Coulthard, R. G. Kane, N. Delgado, M. P. John and J. A. Murphy, Angew. Chem., Int. Ed., 2016, 55, 4492 CrossRef CAS PubMed; (h) L. Zhang, H. Yang and L. Jiao, J. Am. Chem. Soc., 2016, 138, 7151 CrossRef CAS PubMed; (i) J. P. Barham, G. Coulthard, K. J. Emery, E. Doni, F. Cumine, G. Nocera, M. John, L. E. A. Berlouis, T. McGuire, T. Tuttle and J. Murphy, J. Am. Chem. Soc., 2016, 138, 7402 CrossRef CAS PubMed; (j) M. Li, O. Gutierrez, S. Berritt, A. Pascual-Escudero, A. Yeşilçimen, X. Yang, J. Adrio, G. Huang, E. Nakamura-Ogiso, M. C. Kozlowski and P. J. Walsh, Nat. Chem., 2017, 9, 997 CrossRef CAS PubMed; (k) M. Li, S. Berritt, L. Matuszewski, G. Deng, A. Pascual-Escudero, G. B. Panetti, M. Poznik, X. Yang, J. J. Chruma and P. J. Walsh, J. Am. Chem. Soc., 2017, 139, 16327 CrossRef CAS PubMed; (l) H. Yang, L. Zhang and L. Jiao, Chem.–Eur. J., 2017, 23, 65 CrossRef CAS PubMed.
- L. Zhang and L. Jiao, J. Am. Chem. Soc., 2017, 139, 607 CrossRef CAS PubMed.
- For a review, see: (a) E. C. Neeve, S. J. Geier, I. A. Mkhalid, S. A. Westcott and T. B. Marder, Chem. Rev., 2016, 116, 9091 CrossRef CAS PubMed; For examples employing diborons as reducing agents, see: (b) S. Bae and M. K. Lakshman, J. Org. Chem., 2008, 73, 1311 CrossRef CAS PubMed; (c) H. P. Kokatla, P. F. Thomson, S. Bae, V. R. Doddi and M. K. Lakshman, J. Org. Chem., 2011, 76, 7842 CrossRef CAS PubMed; (d) H. Lu, Z. Geng, J. Li, D. Zou, Y. Wu and Y. Wu, Org. Lett., 2016, 18, 2774 CrossRef CAS PubMed; (e) K. Yang, F. Zhou, Z. Kuang, G. Gao, T. G. Driver and Q. Song, Org. Lett., 2016, 18, 4088 CrossRef CAS PubMed; (f) M. Flinker, H. Yin, R. W. Juhl, E. Z. Eikeland, J. Overgaard, D. U. Nielsen and T. Skrydstrup, Angew. Chem., Int. Ed., 2017, 56, 15910 CrossRef CAS PubMed.
- Single crystals of 4·DME were obtained by diffusion of solutions of B2pin2 (in hexane) and radical anion 3 (contained excess 2, in DME) at 25 °C. Due to the labile nature of this complex, the obtained crystals are of insufficient qualities despite several attempts. Thus the quality of the acquired diffraction data was slightly below the average level (see the attached CIF file for details). Despite that, the present X-ray diffraction data confirmed the chemical structure of 4·DME unambiguously.
- W. Maringgele, A. Meller, S. Dielkus and R. Herbst-Irmer, Z. Naturforsch., B: J. Chem. Sci., 1993, 48, 561 CAS.
- D. Chen, G. Xu, Q. Zhou, L. W. Chung and W. Tang, J. Am. Chem. Soc., 2017, 139, 9767 CrossRef CAS PubMed.
- For a review, see: (a) E. M. Kosower, Top. Curr. Chem., 1983, 112, 117 CrossRef CAS; For examples, see: (b) E. M. Kosower and J. L. Cotter, J. Am. Chem. Soc., 1964, 86, 5524 CrossRef CAS; (c) R. J. Gale and R. A. Osteryoung, J. Electrochem. Soc., 1980, 127, 2167 CrossRef CAS; (d) F. Pragst and R. Stößer, Z. Chem., 1985, 25, 222 CrossRef CAS; (e) F. Pragst and M. Šantrůček, Adv. Synth. Catal., 1987, 329, 67 CAS.
- M. Grandl, B. Rudolf, Y. Sun, D. F. Bechtel, A. J. Pierik and F. Pammer, Organometallics, 2017, 36, 2527 CrossRef CAS.
- The simulation of the experimental EPR spectra was performed using the EasySpin software package. See: S. Stoll and A. Schweiger, J. Magn. Reson., 2006, 178, 42 CrossRef CAS PubMed.
- A mixed solution of complexes 4 and 10, prepared by the reaction between complex 4 and MeOBpin (see Scheme 8 and the ESI for details†), was used as the UV-vis sample in this study, because we were not able to isolate the pure form of complex 10. Despite that, the existence of other species did not interfere with the assignment of the characteristic absorption bands of complex 10..
- For full citation of Gaussian 09, see the ESI.†.
- (a) Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215 CrossRef CAS; (b) Y. Zhao and D. G. Truhlar, Acc. Chem. Res., 2008, 41, 157 CrossRef CAS PubMed.
- (a) V. Barone and M. Cossi, J. Phys. Chem. A, 1998, 102, 1995 CrossRef CAS; (b) M. Cossi, N. Rega, G. Scalmani and V. Barone, J. Comput. Chem., 2003, 24, 669 CrossRef CAS PubMed.
- (a) G. Wang, H. Zhang, J. Zhao, W. Li, J. Cao, C. Zhu and S. Li, Angew. Chem., Int. Ed., 2016, 55, 5985 CrossRef CAS PubMed; (b) G. Wang, J. Cao, L. Gao, W. Chen, W. Huang, X. Cheng and S. Li, J. Am. Chem. Soc., 2017, 139, 3904 CrossRef CAS PubMed.
- (a) C. Kleeberg, L. Dang, Z. Li and T. B. Marder, Angew. Chem., Int. Ed., 2009, 48, 5350 CrossRef CAS PubMed; (b) S. Pietsch, E. C. Neeve, D. C. Apperley, R. Bertermann, F. Mo, D. Qiu, M. S. Cheung, L. Dang, J. Wang, U. Radius, Z. Lin, C. Kleeberg and T. B. Marder, Chem.–Eur. J., 2015, 21, 7082 CrossRef CAS PubMed.
- For selected examples of the transition-metal-free heterolytic cleavage of boron–boron bond, see: (a) K. Lee, A. R. Zhugralin and A. H. Hoveyda, J. Am. Chem. Soc., 2009, 131, 7253 CrossRef CAS PubMed; (b) A. Bonet, H. Gulyás and E. Fernández, Angew. Chem., Int. Ed., 2010, 49, 5130 CrossRef CAS PubMed; (c) J. M. O'Brien and A. H. Hoveyda, J. Am. Chem. Soc., 2011, 133, 7712 CrossRef PubMed; (d) A. Bonet, C. Pubill-Ulldemolins, C. Bo, H. Gulyás and E. Fernández, Angew. Chem., Int. Ed., 2011, 50, 7158 CrossRef CAS PubMed; (e) C. Solé, H. Gulyás and E. Fernández, Chem. Commun., 2012, 48, 3769 RSC; (f) K. Oshima, T. Ohmura and M. Suginome, Chem. Commun., 2012, 48, 8571 RSC; (g) C. Pubill-Ulldemolins, A. Bonet, C. Bo, H. Gulyás and E. Fernández, Chem.–Eur. J., 2012, 18, 1121 CrossRef CAS PubMed; (h) H. Wu, S. Radomkit, J. M. O'Brien and A. H. Hoveyda, J. Am. Chem. Soc., 2012, 134, 8277 CrossRef CAS PubMed; (i) T. P. Blaisdell, T. C. Caya, L. Zhang, A. Sanz-Marco and J. P. Morken, J. Am. Chem. Soc., 2014, 136, 9264 CrossRef CAS PubMed; (j) L. Fang, L. Yan, F. Haeffner and J. P. Morken, J. Am. Chem. Soc., 2016, 138, 2508 CrossRef CAS PubMed; (k) C. Kojima, K. H. Lee, Z. Lin and M. Yamashita, J. Am. Chem. Soc., 2016, 138, 6662 CrossRef CAS PubMed; (l) Y. Katsuma, H. Asakawa, K.-H. Lee, Z. Lin and M. Yamashita, Organometallics, 2016, 35, 2563 CrossRef CAS; (m) Y. Katsuma, H. Asakawa and M. Yamashita, Chem. Sci., 2018, 9, 1301 RSC.
- For selected examples and reviews of six-electron boryl anions, see: (a) M. Yamashita and K. Nozaki, in Synthesis and Application of Organoboron Compounds, ed. E. Fernández and A. Whiting, Springer, Cham, 2015, vol. 49, pp. 1–37 Search PubMed; (b) M. Yamashita and K. Nozaki, Bull. Chem. Soc. Jpn., 2008, 81, 1377 CrossRef CAS; (c) Y. Segawa, M. Yamashita and K. Nozaki, Science, 2006, 314, 113 CrossRef CAS PubMed; For representative work about eight-electron boryl anions, see: (d) T. Imamoto and T. Hikosaka, J. Org. Chem., 1994, 59, 6753 CrossRef CAS; (e) H. Braunschweig, C. W. Chiu, K. Radacki and T. Kupfer, Angew. Chem., Int. Ed., 2010, 49, 2041 CrossRef CAS PubMed; (f) K. Nozaki, Nature, 2010, 464, 1136 CrossRef CAS PubMed; (g) R. Bertermann, H. Braunschweig, R. D. Dewhurst, C. Hörl, T. Kramer and I. Krummenacher, Angew. Chem., Int. Ed., 2014, 53, 5453 CrossRef CAS PubMed.
- A. Fawcett, J. Pradeilles, Y. Wang, T. Mutsuga, E. L. Myers and V. K. Aggarwal, Science, 2017, 357, 283 CrossRef CAS PubMed.
- (a) A. M. Mfuh, J. D. Doyle, B. Chhetri, H. D. Arman and O. V. Larionov, J. Am. Chem. Soc., 2016, 138, 2985 CrossRef CAS PubMed; (b) A. M. Mfuh, V. T. Nguyen, B. Chhetri, J. E. Burch, J. D. Doyle, V. N. Nesterov, H. D. Arman and O. V. Larionov, J. Am. Chem. Soc., 2016, 138, 8408 CrossRef CAS PubMed; (c) W. Liu, X. Yang, Y. Gao and C. J. Li, J. Am. Chem. Soc., 2017, 139, 8621 CrossRef CAS PubMed.
- (a) H. Fischer, Chem. Rev., 2001, 101, 3581 CrossRef CAS PubMed; (b) A. Studer, Angew. Chem., Int. Ed., 2000, 39, 1108 CrossRef CAS; (c) A. Studer, Chem. Soc. Rev., 2004, 33, 267 RSC.
- C. Najera and M. Jus, Tetrahedron, 1999, 55, 10547 CrossRef CAS.
- G. E. Keck, T. T. Wager and S. F. McHardy, Tetrahedron, 1999, 55, 11755 CrossRef CAS.
- A. Fuerstner, A. Hupperts, A. Ptock and E. Janssen, J. Org. Chem., 1994, 59, 5215 CrossRef CAS.
- A. L. J. Beckwith and W. B. Gara, J. Chem. Soc., Perkin Trans. 2, 1975, 7, 795 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, characterization data, simulation of the EPR spectrum and computational details. CCDC 1578447 and 1585926. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8sc00008e |
| This journal is © The Royal Society of Chemistry 2018 |