
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Catalytic vinylogous cross-coupling reactions of rhenium vinylcarbenoids†
Ji
Chen
and
Jimmy
Wu
 *
*
Department of Chemistry, Dartmouth College, Hanover, New Hampshire 03755, USA. E-mail: jimmy.wu@dartmouth.edu
First published on 30th January 2018
Abstract
We report the first example of the rhenium-catalyzed allylation reaction of indolyl compounds by means of cross-coupling with propargyl ethers as non-obvious allylating reagents. Data from isotope-labeling and kinetic isotopic studies are consistent with a mechanism that proceeds by vinylidene formation as the rate determining step, followed by 1,5-hydride shift to generate a key rhenium vinylcarbenoid complex. Bond formation occurs at the vinylogous site and the reaction is conveniently carried out in air.
Introduction
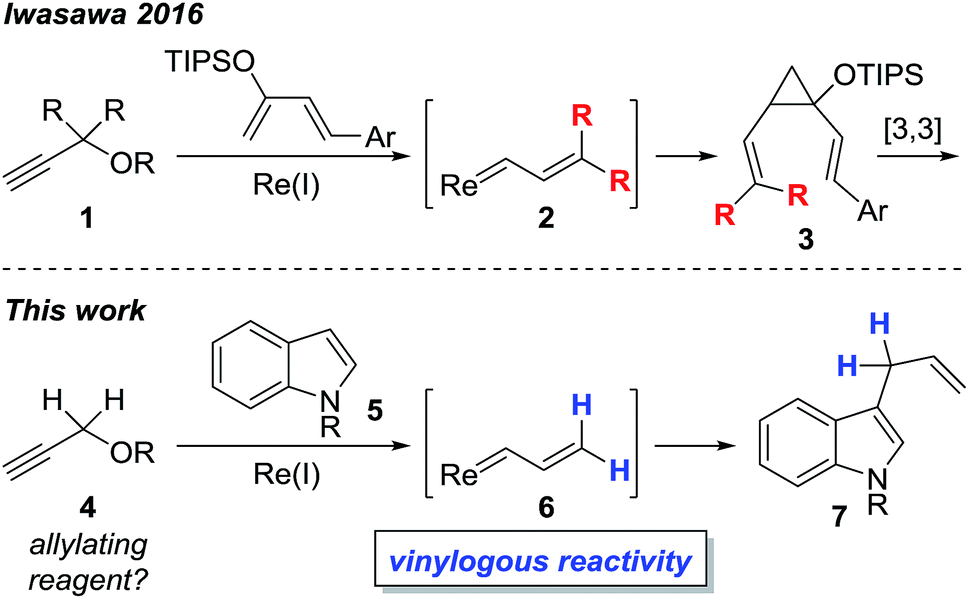
Transition metal vinylcarbenoids are multifaceted species that can engage in either C–H activation1,2 or cyclopropanation3,4 processes (Fig. 1). In the presence of nucleophiles, addition usually occurs at the carbenoid center; however, in recent years, several groups have reported on the vinylogous electrophilicity of these complexes.5–11 Although the most common metal vinylcarbenoids are those derived from rhodium, tungsten, and copper, complexes involving silver, gold, and zinc are also known. These species are often stabilized by a flanking acceptor (i.e. carbonyl) or donor group (i.e. alkoxy) and are typically prepared by diazoacetate decomposition,1,3,12 metal insertion into cyclopropenes,13,14 rearrangement of propargyl carbonates,15 or metathesis reactions.16,17 | ||
| Fig. 1 Transition metal vinylcarbenoids. | ||
In comparison, the synthesis, physical properties, and reactivity, of rhenium vinylcarbenoids are considerably less well-studied.18–23 Iwasawa recently reported a novel method for generating rhenium vinylcarbenoids 2 starting from propargyl ethers.24 This process is thought to proceed through vinylidene formation and 1,5-hydride transfer. In the presence of siloxybutadienes, cyclopropanation occurs at the carbenoid center to give 3 followed by [3,3]-sigmatropic rearrangement to produce cycloheptadiene derivatives (Scheme 1). Inspired by their work, we wondered whether it would be possible to selectively access the electrophilic nature of rhenium vinylcarbenoids, thereby allowing propargyl ethers to be used as non-obvious synthons for allylating reagents (Scheme 1). The paucity of precedents for this type of strategy likely reflects the challenge that propargyl ethers are in the wrong oxidation state for allylation and would require some form of reductive step.
 | ||
| Scheme 1 Reactivity of rhenium vinylcarbenoids. | ||
Herein, we report the first example of a rhenium-catalyzed, vinylogous, allylation of indoles with propargyl ethers. The transformation is complementary to Pd-, Ir-, Rh-based protocols that make use of other types of allyl precursors.25–28 Moreover, instances of completely unsubstituted vinylcarbene complexes, as exemplified by 6, are quite unusual as only a handful of examples, even of other metals, have been previously reported.16,17,24,29–34
We began by examining the reaction between N-methylindole 5a and propargyl tetrahydropyranyl ether 4a in the presence of various rhenium catalysts. A few representative reactions from the optimization study are summarized in Table 1. The use of Re2(CO)10 gave no reaction (entry 1), but we were pleased that a range of Re(I) species, including the commercially-available compound ReBr(CO)5 efficiently catalyzed the formation of the desired product 7a in good yield (entry 2). After some experimentation, we identified the use of ReI(CO)5 in 1,4-dioxane at 100 °C to be optimal. The reaction requires only a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 stoichiometry of indole versus propargyl ether, and is carried out in air with no precautions necessary for the removal of oxygen or water.
:1 stoichiometry of indole versus propargyl ether, and is carried out in air with no precautions necessary for the removal of oxygen or water.
|
|
||||
|---|---|---|---|---|
| Entry | Catalyst | Solvent | T [°C] | Yieldb (%) |
| a Reaction conditions: 1 equiv. of 4a, 1 equiv. of 5a, and 5 mol% of catalyst (0.2 M) for 10 h in air. b Isolated yield. c With activated 4 Å MS. | ||||
| 1 | Re2(CO)10 | 1,4-Dioxane | 100 | 0 |
| 2 | ReBr(CO)5 | 1,4-Dioxane | 100 | 81 |
| 3 | ReI(CO)5 | 1,4-Dioxane | 100 | 91 |
| 4 | [ReBrCO3thf]2 | 1,4-Dioxane | 100 | 65 |
| 5 | ReI(CO)5 | THF | 60 | 0 |
| 6 | ReI(CO)5 | Toluene | 80 | 0 |
| 7 | ReI(CO)5 | DMF | 100 | 0 |
| 8 | ReI(CO)5 | 1,4-Dioxane | 60 | 0 |
| 9 | ReI(CO)5 | 1,4-Dioxane | 80 | Trace |
| 10 | ReI(CO)5 | 1,4-Dioxane | 100 | 90c |

We then explored the reaction scope with respect to the indole substrates which exhibited good compatibility with a range of indoles bearing substituents such as halogen, amide, ester, nitro, boronate ester, and alkoxy (Table 2). The efficiency of the reaction is diminished somewhat when electron-deficient indoles are used (products 7e–g). Both N–Me and N–Bn derivatives were amenable; however, indoles bearing substituents at the C3 position were found to be inert, indicating a likely steric bias. The compatibility of this reaction with a range of functional groups makes it competitive as a new type of catalytic cross-coupling protocol.
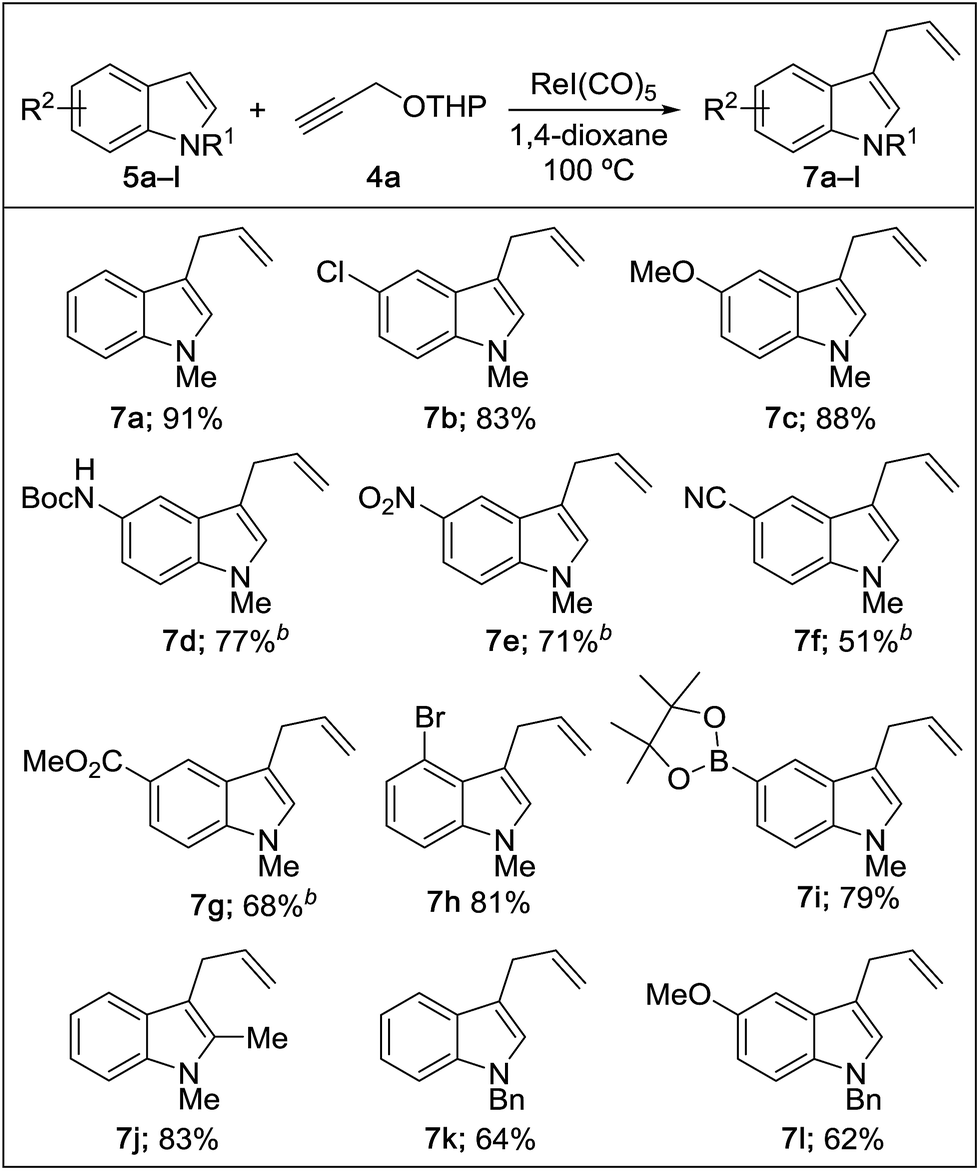
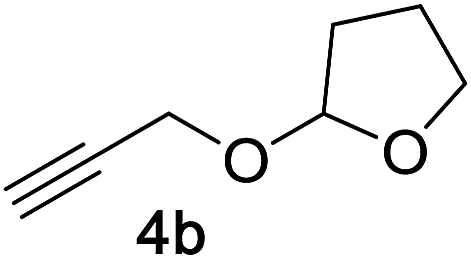
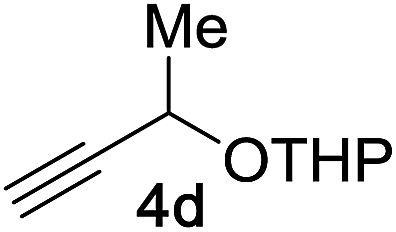
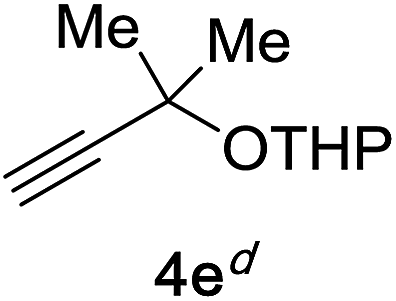
Next, we examined the reaction scope with respect to the propargyl ether (Table 3). The acetal type precursors 4a and 4b did not exhibit much difference in either reactivity or reaction rate. However, the benzyl ether precursor 4c proved to be less efficient and required 1.5 equiv. of 4c. We believe that the reason for this difference is that the use of 4c forms benzaldehyde as a side-product, which can react with indole in unproductive pathways. This may be aided by the rhenium catalyst which is able to function as a hard Lewis acid.35,36 Intriguingly, we found that substituents at the propargylic position have profound effects on the title reaction. The use of monosubstituted acetal 4d furnished no desired product, whereas the geminally disubstituted acetal 4e gave an unexpected product assigned as the bis-prenylated compound 8. That excess amounts of 4a–c did not lead to the corresponding bis-allylated products suggests a fundamental difference in the reactivity profile of 4e. Our group is presently studying this reaction in greater detail.





The proposed mechanism of the cross-coupling, along with salient features, is outlined in Fig. 2. The reaction begins with coordination of the propargyl ether 4c with the rhenium center followed by vinylidene formation as the rate-determining-step to give B. 1,5-Hydride shift generates the zwitterionic species C. Elimination of benzaldehyde generates rhenium carbenoid D, in the s-trans conformation, which can react at the vinylogous position with indole 5a to give E. It should be noted that when using 4a, δ-valerolactone is produced as the reaction by-product instead of benzaldehyde. Stereospecific protodemetallation followed by rearomatization of E would provide the observed product 7a and regenerate the rhenium catalyst.
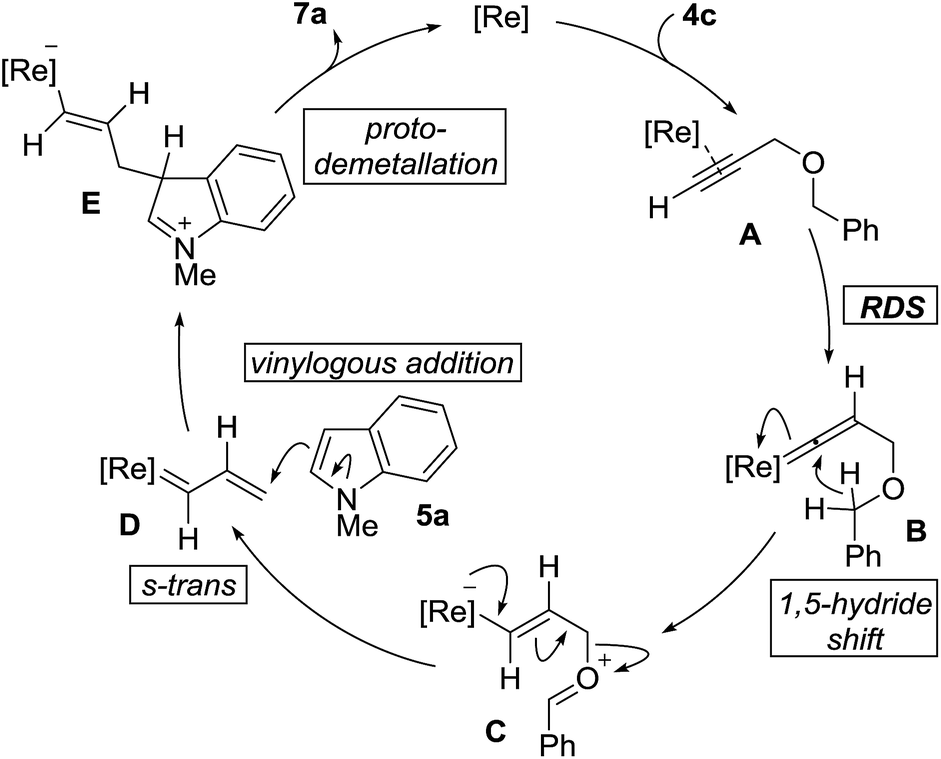 | ||
| Fig. 2 Proposed mechanism. | ||
We carried out a series of isotope-labeling and kinetic isotope studies designed to support our hypotheses and establish a clearer understanding of the reaction mechanism. We began by preparing doubly deuterated propargyl ether 9 which, under the reaction conditions, should generate vinylcarbenoid 10 that is isotopically-labeled at the carbene center (Scheme 2). This intermediate will preferentially adopt the lower energy s-trans conformation, to which vinylogous addition by indole leads to formation of terminally-labeled (Z)-12 in 77% yield with 89% deuterium incorporation. The reduced level of deuterium incorporation of the product vis-à-vis 9 (D, 98%) may be attributable in part to a primary kinetic isotope effect (KIE) incurred due to the presence of monodeuterated compound 17 (see Scheme 4). We did not observe the formation of configurational isomer (E)-12 which may form by reaction with (s-cis)-10, or evidence of regioisomer 13 which may arise by 1,2-addition to 10.
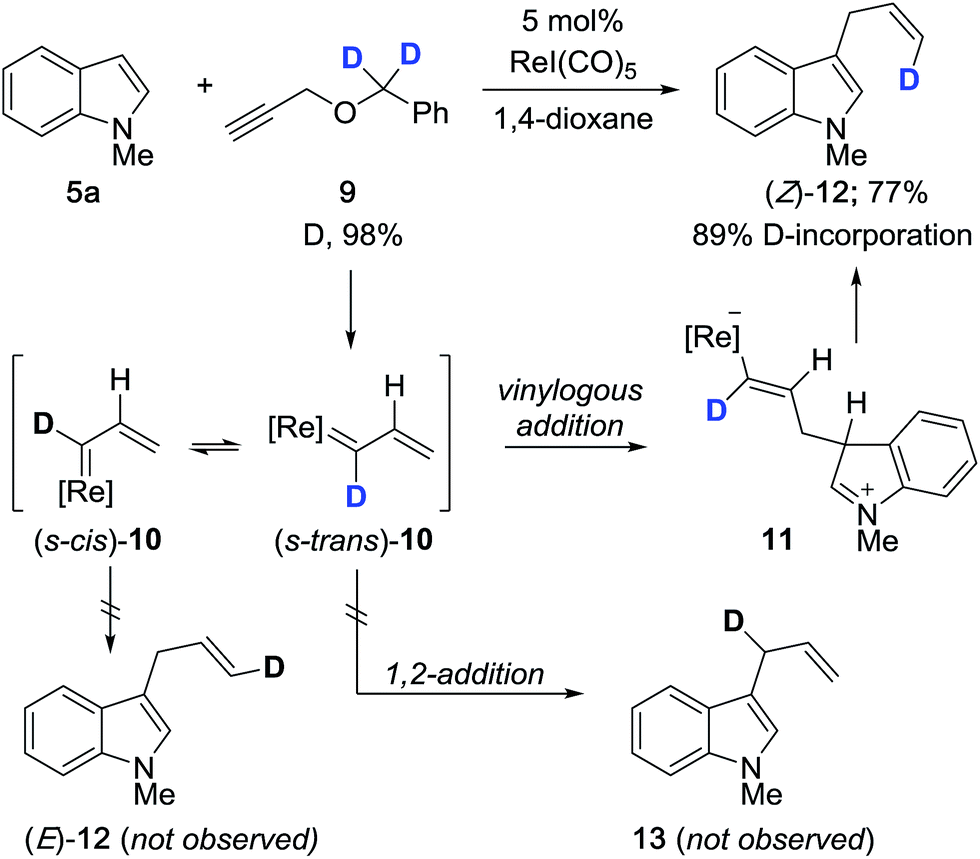 | ||
| Scheme 2 Regioselective and stereoselective cross-coupling with isotopically-labeled propargyl ether 9. | ||
We then performed the cross-coupling reaction between 1 equiv. of 5a and a 1:1 mixture of 4c and isotopically-labeled propargyl ether 14 (total 4 equiv.) (Scheme 3). Here, we expect formation of both protiated and deuterated vinylcarbenoids, 15 and D-15, which will react with indole 5a to give 7a and 16. The reaction was purposefully quenched at partial completion (∼53%), at which point we measured a product ratio of 7avs.16 of 2.0. We interpreted this value as the magnitude of the primary KIE, kH/kD (corrected for % D of 14), thereby implicating vinylidene formation as the rate determining step (RDS). The extent of this KIE is consistent with an expected non-linear trajectory for the 1,2-hydride shift during vinylidene formation.37 The magnitude of the measured KIE is also comparable to values reported for the formation of Ru- and Mo-based vinylidene complexes starting from terminal alkynes.38,39
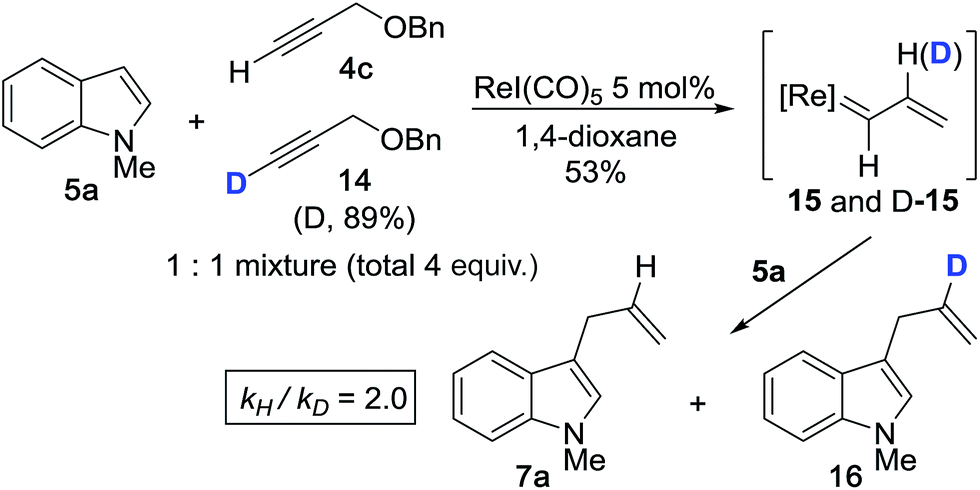 | ||
| Scheme 3 Kinetic isotope effect with isotopically-labeled propargyl ether 14. | ||
Next, based on the product ratio of 7avs. (Z)-12 for the reaction between 5a and monodeuterated propargyl ether 17, we measured an additional primary KIE of kH/kD = 1.9 (corrected for % D of 17) (Scheme 4). We expect that treating 17 with ReI(CO)5 should induce the formation of 18 which will result in 1,5-transfer of either hydrogen or deuterium. The presence of a discernible primary KIE for this step, which ostensibly occurs after the rate limiting step, suggests that 1,5-hydride transfer is irreversible and product-determining.
 | ||
| Scheme 4 Kinetic isotope effect with monodeuterated propargyl ether 17. | ||
Conclusions
In conclusion, we have reported the first instance of the rhenium catalyzed cross-coupling of indoles with propargyl ethers to give allylated products. The reaction is compatible with a range of functional groups on the indole component. Isotopic labeling studies and KIE measurements establish a mechanistic framework by which vinylidene formation occurs as the rate limiting step followed by product-determining 1,5-hydride shift. Vinylogous attack on the resultant rhenium vinylcarbenoid by indole then furnishes the desired cross-coupled product.Conflicts of interest
There are no conflicts to declare.Acknowledgements
J. W. acknowledges the generous support of the NIH NIGMS (R01GM111638).Notes and references
- H. M. L. Davies and R. E. J. Beckwith, Chem. Rev., 2003, 103, 2861–2904 CrossRef PubMed.
- M. P. Doyle, R. Duffy, M. Ratnikov and L. Zhou, Chem. Rev., 2010, 110, 704–724 CrossRef PubMed.
- H. M. L. Davies and E. G. Antoulinakis, ed. L. E. Overman, Intermolecular Metal-Catalyzed Carbenoid Cyclopropanations, in Organic Reactions, John Wiley & Sons, Inc., New York, NY, 2001, vol. 57, pp. 1–326 Search PubMed.
- H. Lebel, J.-F. Marcoux, C. Molinaro and A. B. Charette, Chem. Rev., 2003, 103, 977–1050 CrossRef CAS PubMed.
- H. M. L. Davies, D. G. Stafford and T. Hansen, Org. Lett., 1999, 1, 233–236 CrossRef CAS PubMed.
- J. Barluenga, G. Lonzi, L. Riesgo, L. A. López and M. Tomás, J. Am. Chem. Soc., 2010, 132, 13200–13202 CrossRef CAS PubMed.
- J. H. Hansen and H. M. L. Davies, Chem. Sci., 2011, 2, 457–461 RSC.
- V. V. Pagar, A. M. Jadhav and R.-S. Liu, J. Am. Chem. Soc., 2011, 133, 20728–20731 CrossRef CAS PubMed.
- X. Wang, X. Xu, P. Y. Zavalij and M. P. Doyle, J. Am. Chem. Soc., 2011, 133, 16402–16405 CrossRef CAS PubMed.
- Y. Lian and H. M. L. Davies, Org. Lett., 2012, 14, 1934–1937 CrossRef CAS PubMed.
- C. Qin and H. M. L. Davies, Org. Lett., 2013, 15, 6152–6154 CrossRef CAS PubMed.
- A. Padwa, D. J. Austin, Y. Gareau, J. M. Kassir and S. L. Xu, J. Am. Chem. Soc., 1993, 115, 2637–2647 CrossRef CAS.
- B. T. Flatt, R. H. Grubbs, R. L. Blanski, J. C. Calabrese and J. Feldman, Organometallics, 1994, 13, 2728–2732 CrossRef CAS.
- A. Archambeau, F. Miege, C. Meyer and J. Cossy, Acc. Chem. Res., 2015, 48, 1021–1031 CrossRef CAS PubMed.
- K. Miki, K. Ohe and S. Uemura, J. Org. Chem., 2003, 68, 8505–8513 CrossRef CAS PubMed.
- P. Schwab, M. B. France, J. W. Ziller and R. H. Grubbs, Angew. Chem., Int. Ed., 1995, 34, 2039–2041 CrossRef CAS.
- P. Schwab, R. H. Grubbs and J. W. Ziller, J. Am. Chem. Soc., 1996, 118, 100–110 CrossRef CAS.
- M. Green, A. G. Orpen, C. J. Schaverien and I. D. Williams, J. Chem. Soc., Chem. Commun., 1983, 1399–1401 RSC.
- N. Mantovani, L. Marvelli, R. Rossi, V. Bertolasi, C. Bianchini, I. de los Rios and M. Peruzzini, Organometallics, 2002, 21, 2382–2394 CrossRef CAS.
- J. Barluenga, J. Santamaría and M. Tomás, Chem. Rev., 2004, 104, 2259–2284 CrossRef CAS PubMed.
- F. You, L. A. Friedman, K. C. Bassett, Y. Lin, M. Sabat and W. D. Harman, Organometallics, 2005, 24, 2903–2912 CrossRef CAS.
- Y. Jiang, O. Blacque, T. Fox, C. M. Frech and H. Berke, Organometallics, 2009, 28, 4670–4680 CrossRef CAS.
- M. Valla, D. Stadler, V. Mougel and C. Copéret, Angew. Chem., Int. Ed., 2016, 55, 1124–1127 CrossRef CAS PubMed.
- H. Sogo and N. Iwasawa, Angew. Chem., Int. Ed., 2016, 55, 10057–10060 CrossRef CAS PubMed.
- M. Kimura, M. Futamata, R. Mukai and Y. Tamaru, J. Am. Chem. Soc., 2005, 127, 4592–4593 CrossRef CAS PubMed.
- A. B. Zaitsev, S. Gruber and P. S. Pregosin, Chem. Commun., 2007, 4692 RSC.
- I. Usui, S. Schmidt, M. Keller and B. Breit, Org. Lett., 2008, 10, 1207–1210 CrossRef CAS PubMed.
- F. A. Cruz, Y. Zhu, Q. D. Tercenio, Z. Shen and V. M. Dong, J. Am. Chem. Soc., 2017, 139, 10641–10644 CrossRef PubMed.
- M. A. Esteruelas, F. J. Lahoz, E. Onate, L. A. Oro and B. Zeier, Organometallics, 1994, 13, 1662–1668 CrossRef CAS.
- Y.-M. Chen and P. B. Armentrout, J. Am. Chem. Soc., 1995, 117, 9291–9304 CrossRef CAS.
- X. Niu, L. Gopal, M. P. Masingale, D. A. Braden, B. S. Hudson and M. B. Sponsler, Organometallics, 2000, 19, 649–660 CrossRef CAS.
- Y. Sheng, Y.-D. Wu and J. Leszczynski, Organometallics, 2004, 23, 3189–3196 CrossRef CAS.
- M. Gandelman, K. M. Naing, B. Rybtchinski, E. Poverenov, Y. Ben-David, N. Ashkenazi, R. M. Gauvin and D. Milstein, J. Am. Chem. Soc., 2005, 127, 15265–15272 CrossRef CAS PubMed.
- S. L. Bolton, D. E. Schuehler, X. Niu, L. Gopal and M. B. Sponsler, J. Organomet. Chem., 2006, 691, 5298–5306 CrossRef CAS.
- C. P. Casey, Science, 1993, 259, 1552 CAS.
- Y. Kuninobu and K. Takai, Chem. Rev., 2011, 111, 1938–1953 CrossRef CAS PubMed.
- R. M. O'Ferrall, J. Chem. Soc. B, 1970, 785–790 RSC.
- J. Ipaktschi, J. Mohsseni-Ala and S. Uhlig, Eur. J. Inorg. Chem., 2003, 2003, 4313–4320 CrossRef.
- M. Bassetti, V. Cadierno, J. Gimeno and C. Pasquini, Organometallics, 2008, 27, 5009–5016 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and characterization of all new compounds, selected 2D-NOESY, 2D-COSY, HMBC, HMQC data. See DOI: 10.1039/c7sc05477g |
| This journal is © The Royal Society of Chemistry 2018 |