
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Mechanochemistry as an emerging tool for molecular synthesis: what can it offer?
Joseph L.
Howard
,
Qun
Cao
and
Duncan L.
Browne
 *
*
School of Chemistry, Cardiff University, Main Building, Park Place, Cardiff, CF10 3AT, UK. E-mail: dlbrowne@cardiff.ac.uk
First published on 7th March 2018
Abstract
Mechanochemistry is becoming more widespread as a technique for molecular synthesis with new mechanochemical reactions being discovered at increasing frequency. Whilst mechanochemical methods are solvent free and can therefore lead to improved sustainability metrics, it is more likely that the significant differences between reaction outcomes, reaction selectivities and reduced reaction times will make it a technique of interest to synthetic chemists. Herein, we provide an overview of mechanochemistry reaction examples, with ‘direct’ comparators to solvent based reactions, which collectively seemingly show that solid state grinding can lead to reduced reaction times, different reaction outcomes in product selectivity and in some instances different reaction products, including products not accessible in solution.
 Joseph L. Howard | Joseph L. Howard graduated from Sidney Sussex College, University of Cambridge with a BA in Natural Sciences. He then received an MPhil in Chemistry from Cardiff University, working with Dr Duncan Browne, and is currently a PhD student in the Browne group. The focus of his research is on industrially relevant flow processing and mechanochemical synthesis. |
 Qun Cao | Qun Cao obtained a B. Eng in Chemical Engineering and Technology from China University of Mining and Technology in 2011. He obtained his PhD in 2016 from Queen's University Belfast under direction of Dr Mark Muldoon, working on developing selective catalytic methods for oxidation reactions. He then worked as a postdoctoral researcher in the University of Nottingham with Dr Victor Sans Sangorrin in June 2016. Since March 2017, he joined Dr Duncan Browne's group as a postdoctoral researcher and focused on mechanochemical synthesis. |
 Duncan L. Browne | Duncan obtained a MChem degree in Chemistry with study in Industry from the University of Sheffield (one year intern at GSK, Stevenage, UK). In 2009 he was awarded a Ph.D. in organic synthesis under the supervision of Prof. Joseph P. A. Harrity (Syngenta, CASE). He was then awarded a one year Doctoral Prize Fellowship from the EPSRC before moving in 2010 to the University of Cambridge for his Postdoctoral studies with Professor Steven V. Ley FRS CBE. In September 2014, Duncan established his independent research group and became Lecturer in Organic Synthesis & Medicinal Chemistry at Cardiff University. In 2017 he was recognised by both Green Chemistry and Reaction Chemistry and Engineering as an ‘Emerging Investigator’ and he was a recipient of a Thieme Chemistry Journal Award. |
1. Introduction
Mechanochemistry, well known in the area of crystal engineering and polymorphism, is re-emerging as a technique for organic synthesis.1,2 Whilst the technique is not new, it is now evolving beyond simply being a solvent free alternative. Despite many synthetic transformations now reported, perhaps the most interesting transformations are those that exhibit different reactivity to conventional solution-based reactions.3,4 In this perspective, we highlight a selection of such examples as well as provide a brief overview and introduction to the topic.1.1 What is mechanochemistry?
A mechanochemical reaction is defined as “a chemical reaction that is induced by the direct absorption of mechanical energy”.5 Mechanochemistry therefore complements the conventional methods of activation: heat, irradiation and electrochemistry.6 It is also related to tribochemistry, in which chemical reactions occur on the surface/boundary between different environments.7,8 Despite a recent resurgence in mechanochemistry, it remains far less well studied and understood in comparison to conventional methods of energy input.It is well understood that chemical reactions, initiated through different forms of energy input, can lead to different products and provide access to reaction manifolds that are inaccessible by other means; e.g. photochemistry and electrochemistry. For example, pericyclic reactions proceed differently depending on whether they are induced by light or heat. The reaction outcome is predictable through use of the Woodward–Hoffmann rules.9,10 Interestingly, when ultrasound is used as the input energy for a pericyclic reaction, products have been observed that are not predicted; indeed it has also been suggested that Woodward–Hoffmann rules should not necessarily be applied to mechanochemical pericyclic reactions.11,12
Mechanochemistry therefore presents an opportunity to explore a novel chemical space of reactions. Mechanochemistry can describe impact or pulling forces.13 This perspective focuses on impact caused by milling.14–19
1.2 What are the types of milling device?
The earliest mechanochemical reactions were carried out using a pestle and mortar.20 However, how these reactions behave is highly operator dependent, as each individual may impart different levels of energy. Running reactions for longer than a few minutes also becomes challenging, and depends on the operator's stamina! Therefore, electronic milling devices are commonly used.The mixer mill is one type of ball milling machine, which uses the movement of ball bearings to apply mechanical force to the reagents. In this case, reagents are loaded into jars and one or more ball bearings are added. The jars are then mounted horizontally and shaken at the desired frequency (Scheme 1). The main mechanical energy applied to the reagents is impact force. The other most common type of ball mill is a planetary mill. In this case, the reagents and ball(s) are loaded as before, but the motion is different. In this instance, the jars spin counter-directionally to the spinning disc that they are mounted on. The central spinning disc is called the ‘sun wheel’ and the jars orbit around the central spinning axis. In both cases, the material and size of the jars and balls can be altered. The main type of force applied in planetary milling is shear force. Stirred media ball mills are also available, in which the reagents and balls are stirred together. This type of mill is commonly used to create very fine powders, e.g. of coal.21,22 Stirred media reactions or slurries can also be set up in round bottom flasks or more traditional lab glassware.23
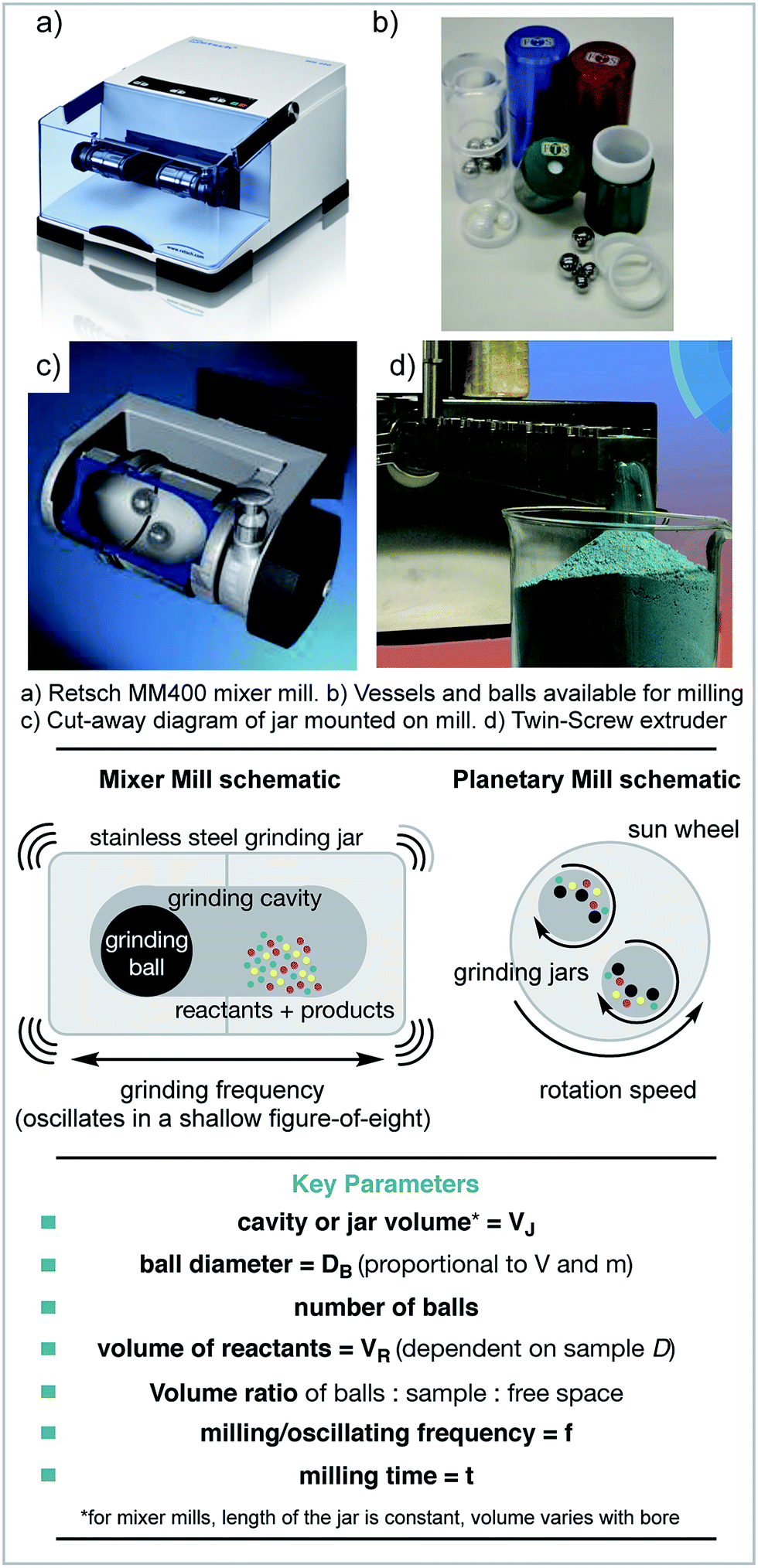 | ||
| Scheme 1 Examples of equipment used for mechanochemical reactions and key parameters. (a & c) Reproduced with permission from Retsch, (b) reproduced with permission from FormTech Scientific. (d) Reproduced with permission from Prof Stuart James and RSC. | ||
1.3 What scales?
If any synthetic procedure is to be useful to society e.g. in molecule discovery or manufacture it must be achievable across a variety of scales. For mechanochemistry, each type of milling device can achieve different scales. The mixer mill can achieve gram scales, which are suitable for lab investigations. However, for anything larger, other types of mill must be used, and different sizes of planetary mill are available. However, for pilot and manufacture scales, stirred media ball mills are used. For example, the Outotec HIGMill has a volume of 30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 litres and can be used at >1000 kg scales.24
000 litres and can be used at >1000 kg scales.24
Stolle et al. demonstrated the scalability of the mechanochemical Knoevenagel condensation between vanillin and barbituric acid in a planetary mill, scaling from 20 to 300 mmol.25 This resulted in the ability to obtain quantitative yields on approximately 80 g scales within short reaction times.
A different approach to scalability can be attempted if the process is modified from batch to continuous. This can be achieved by using extruders instead of mills. Extruders continuously force material through confined spaces and apply shear and compression forces. James, Crawford and co-workers have synthesised MOFs at rates of kilograms per hour by making use of twin-screw extruders.26
1.4 What are the variables?
As when performing any reaction, understanding and controlling the variables is important. However, when using a ball mill it can be challenging to control or even measure the variables individually. There are three main variables that affect how mechanochemical reactions perform: the kinetic energy of the ball(s) prior to collision, how that energy is transferred to the reagents and the frequency of collisions.The amount of kinetic energy that the ball(s) possess prior to collision is the maximum amount of energy that can be transferred to the reagents per collision.
In a collision, how that energy is transferred can have an effect on whether or how a reaction occurs. This can be by direct impact, under which the material is locally compressed, or by shear force in which a reactive face is exposed. It has been shown that these different types of energy absorption can lead to different outcomes.27 Different types of ball mill achieve different ratios of impact and shear forces.
Differences in mixing or mass transfer, can affect the outcome of any reaction. In solution, this is easily controlled by stirring, mass transfer is rarely a problem on small scale. However, in milled reactions, this can be a difficult variable to control and may have a dramatic effect on the outcome of a reaction. To help homogeneity and mixing, grinding agents can be used (see Section 1.6).
Finally, those variables that are usually changed in any standard reaction optimisation also apply, such as stoichiometry, reaction time and temperature. However, temperature is not a parameter that is easily controlled, the reaction vessel heats up due to the collisions and has a dependence on the filling degree volumes of grinding balls, sample and vessel size as well as oscillating frequency. Cryo-mills are available, which consist of pausing the grinding process to automatically cool the jars with liquid nitrogen but are not widely explored for chemistry purposes.28
The previous section discussed the variables that affect mechanochemical reactions. However, these variables cannot be directly controlled. The variables that can be controlled often have multiple influences on a reaction. One of the first variables to be decided is what type of ball mill is to be used. As discussed previously, there are three main types: the mixer mill, the planetary mill and the stirred media mill. It has also been demonstrated that different mills can lead to differences in reaction performance.29–32
The next consideration is usually given to the filling degree. This is a measure of how full the jars are, considering the volumes occupied by the reagents and balls compared to the total cavity volume of the jars. This has a significant effect on the trajectories of the balls, and therefore the energy transfer and mixing. The effect of the milling ball filling degree has been investigated for the Knoevenagel condensation of vanillin with barbituric acid in a planetary mill.23 For this specific example it was observed that having the balls filling approximately 25% of the total volume was optimal. However, this may not be the case for other reactions and/or other types of mills.
The milling frequency is perhaps the easiest variable to control and can be changed simply by adjusting the settings on the mill. When increased, this increases the velocities of the balls, and so increases their kinetic energy. This is a facile way to change the energy input into a reaction.
1.5 Which reactions are best suited to mechanochemistry?
As so many of the mechanisms operating mechanochemically remain elusive, it can be difficult to decide when to use mechanochemistry. Certainly, many of the examples presented here, which exhibit different reactivity in solution, are unlikely to have been predicted. However, there are a few cases where definite advantages over solution-based reactions can be expected a priori.Perhaps the most obvious advantage is that reactions can be performed under solvent-free conditions. This leads to several cases where it is worth attempting mechanochemical reactions.
Firstly, reactions between solids that are not soluble (or not all components are soluble in the same solvent) are well suited to mechanochemistry. This class of reactions can be very challenging, or even impossible in solution. Reactions in which the solvent can interfere are also interesting candidates for mechanochemical investigations. For example, many catalysts and reagents can be very sensitive towards water, or solvents with Lewis basic sites. Indeed, great lengths are often taken, with expense, to dry solvents. However, in the mill the solvent is not required. Finally, reactions that require hazardous solvents could be made safer by using solvent-free conditions, such as mechanochemistry.
So far, only reactions between solid reagents have been discussed, although reactions between solids and liquids, or even liquids can be performed mechanochemically. These usually require the addition of a solid material referred to as a grinding agent or grinding auxiliary.
1.6 What is a grinding auxiliary/agent?
Reactions in the mill that feature one or more liquid component (starting material, reagent or product) often require the addition of a solid additive for efficient reactivity. It has been shown that different textures of reaction mixtures can lead to different reaction kinetics.33 When liquids are used, the reaction mixture can become sticky and resemble a paste or gum, this prevents efficient mass and energy transfer. To account for this, it is common to use an auxiliary material such as silica, alumina, talc or inorganic salts as a milling agent/auxiliary or adsorbent.28,34 In pharmaceutical formulation science these materials are termed ‘glidants’ or ‘lubricants’ and assist in the uniform passage of powdered materials through screw extruders.35It is important that such additives are chemically inert towards the desired reaction, otherwise they might interfere with the reactivity; proving inertness is difficult!
1.7 What is liquid-assisted-grinding (LAG)?
The mechanochemical reaction environment can be modified further by the addition of a small amount of liquid. This is termed “Liquid Assisted Grinding” (LAG). The amount of added liquid has been characterised by the parameter η, which is a ratio of the volume of liquid added to the total mass of reagents.36 This ratio can vary from grinding neat reagents together with no liquid, through to LAG, slurry reactions and eventually to solution reactions with increasing amounts of liquid.LAG was originally used in mechanochemical cocrystallisation and it was found to speed up cocrystal formation.37 It has also been demonstrated that LAG can lead to different results to slurry reactions and that the outcomes do not necessarily depend on the solubility of the starting materials in the liquid used.34 More recently, it has been shown that using different amounts and types of liquid can lead to the formation of different polymorphs, dispelling the commonly held belief that polymorphism depends solely on the solvent.38
These observations show that LAG can lead to different and currently unpredictable reaction outcomes compared to both neat grinding and solution based processes. It is therefore an added variable when exploring solid state grinding.
1.8 Expect the unexpected
Given the significant difference in reaction environment between conventional methods and mechanochemical methods, it is reasonable to suggest that one would expect to observe significant differences in reaction outcomes. Given the many mechanisms and processes involved, what these differences might be is not easily predicted. When one takes into account the interdependent nature of the variables in a mechanochemical reaction, and further modifications, such as LAG, it becomes even more complex and challenging to know what to expect. As such, it is likely that in many of the examples presented in this perspective, the outcomes were unexpected. However, we have attempted to combine many of the recent examples and classify them into three categories: time saving (in the form of reduced reaction time or greatly enhanced yield over a similar reaction time), selectivity enhancement and alternative reactivity. To highlight the differences these have been compared to solvent-based reactions; where these are known. This overview is to raise awareness or the capabilities of the technique and enable others to explore this exciting method rather than to necessarily advocate the use of mechanochemistry instead of solution based approaches.2. Reduced reaction times
One way in which mechanochemistry offers apparent advantages over solution based reactions is a reduction in reaction times. Clearly this in part derives from a large increase in concentration. It may also be true that in some cases there is a temperature difference between the solution based reaction and the mechanochemical process. In general the control of instantaneous temperatures under milling conditions is difficult to achieve, and control over the bulk temperature relies on bespoke equipment. Nonetheless there are many reactions where the comparison to running a solvent based reaction to an ‘ambient’ mechanochemical reaction demonstrate significant reductions in reaction time and we highlight some of those examples in this section.An example of an inorganic reaction with increased reactivity is the synthesis of Cu–NHC complexes (NHC = N-heterocyclic carbene). These are used widely as organometallic catalysts for a variety of reactions and various methods have been developed for the synthesis of Cu–NHC complexes.39 Under solvent-based conditions, Cu–NHC complexes can be synthesized by the reaction of metallic Cu(0) with imidazolium salts, although these reactions require a large excess of insoluble Cu(0) and long reaction times.40 Recently, Lamaty and co-workers reported that Cu–NHC complexes (2) could be synthesized from imidazolium salts (1) and metallic copper using a planetary ball mill (Scheme 2).41 The rate of reactions was enhanced due to the high concentration of reagents and the highly efficient mixing under mechanochemical conditions. Using this novel method, five Cu–NHC complexes with different counter ions (Cl−, BF4−, and PF6−) were successfully synthesized in improved yields compared to the analogous reactions in solution.
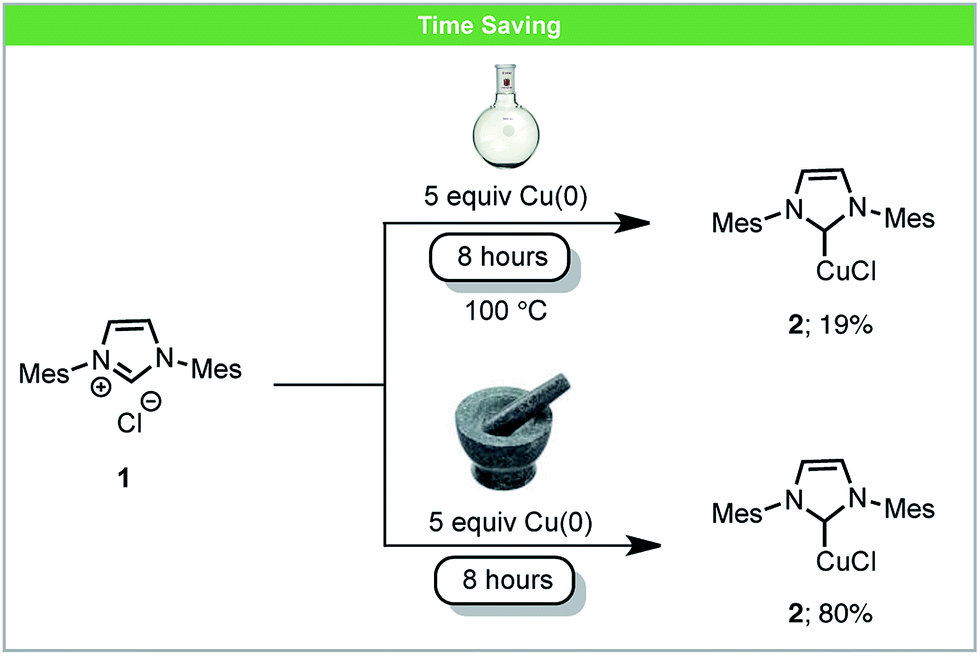 | ||
| Scheme 2 Formation of Cu–NHC complexes; Lamaty and co-workers.41 | ||
One of the most powerful tools for chemical synthesis is the selective functionalization of C–H bonds. Such methods allow the formation of C–C bonds without prefunctionalisation of the starting materials.42 There are already several known methods for C–H activation and functionalisation using mechanochemical conditions, some of which provide a time saving against the analogous reactions in solution.43
In 2014, Ćurić and co-workers achieved the first mechanochemical transition-metal-mediated C–H bond activation and monitored the transformation with in situ solid-state Raman spectroscopy.44 Using liquid-assisted grinding (LAG) with acetic acid, palladacycle 4 was synthesized from asymmetrically substituted azobenzene 3 and Pd(OAc)2 in 78% yield after 4.5 hours (Scheme 3). When performed in solution, this reaction required 3 days and a significantly poorer yield was achieved. Further milling of 4 with Pd(OAc)2 yielded dicyclopalladated complex 5, which was not observed after multiple attempts at the same transformation in solution. In addition to saving time, this example demonstrates that using mechanochemistry can offer novel reaction pathways for the synthesis of organometallic compounds which could not be obtained using other methods.
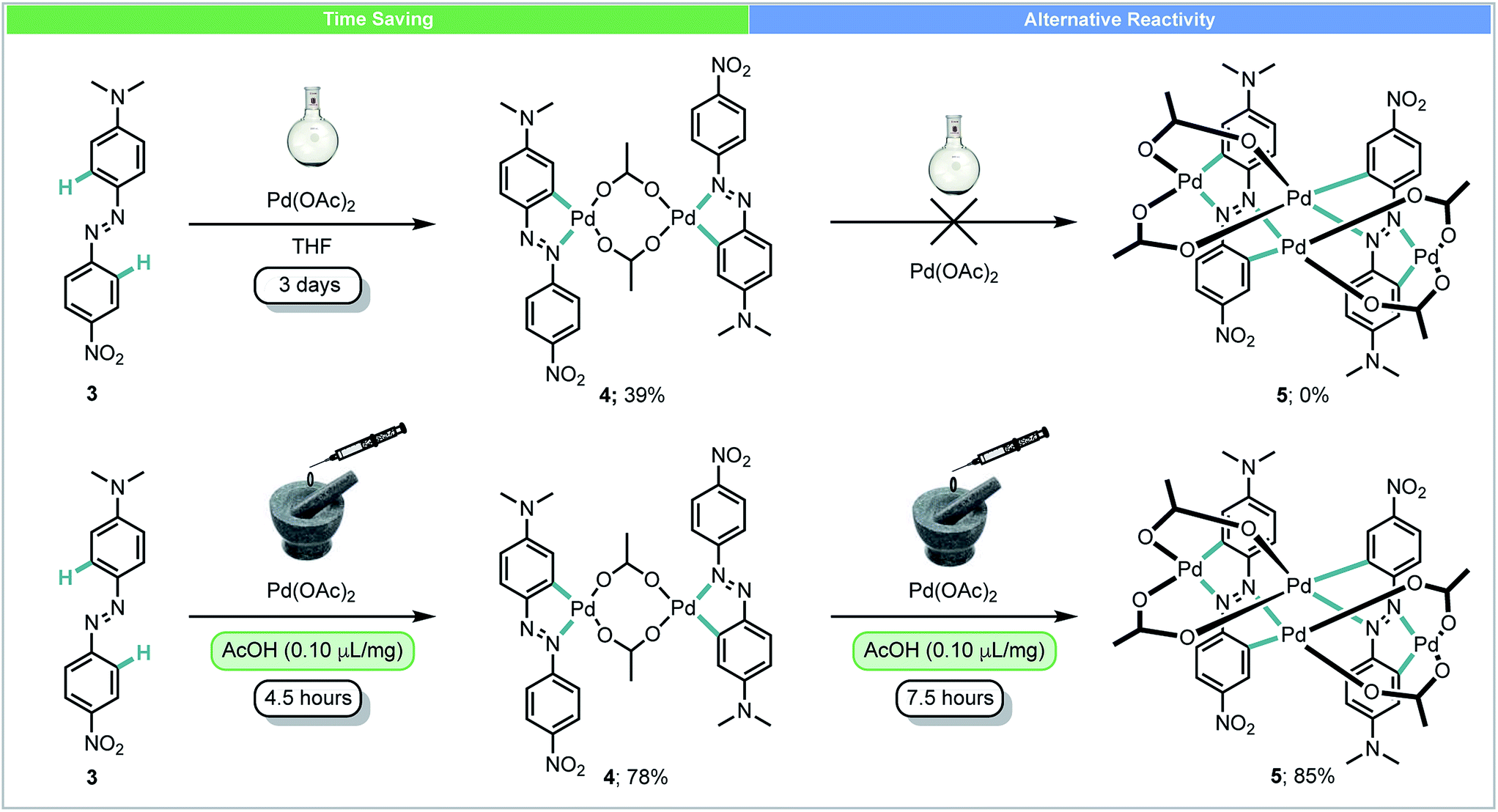 | ||
| Scheme 3 Palladacycle synthesis; Ćurić and co-workers.44 | ||
In 2016, Bolm and co-workers developed a mechanochemical iridium(III)-catalyzed C–H bond amidation of benzamides 6 with sulfonyl azides 7 (Scheme 4).45 In this study, it was demonstrated that the active cationic Ir(III) catalyst could be formed in situ in the mixer mill by reaction of [{Cp*IrCl2}2] with AgNTf2. The corresponding amidated products could be obtained in high yields with shorter reaction times (99 min) than those under a solvent-based protocol (12 hours) as reported by Chang and co-workers.46
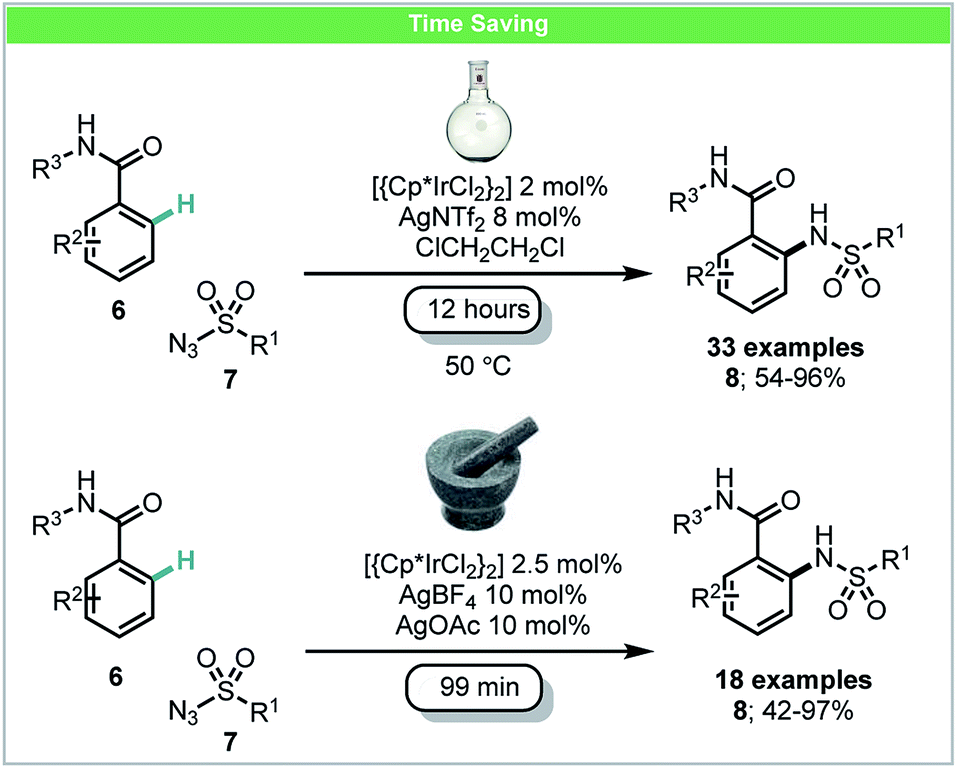 | ||
| Scheme 4 C–H amidation of benzamides; Bolm and co-workers.45 | ||
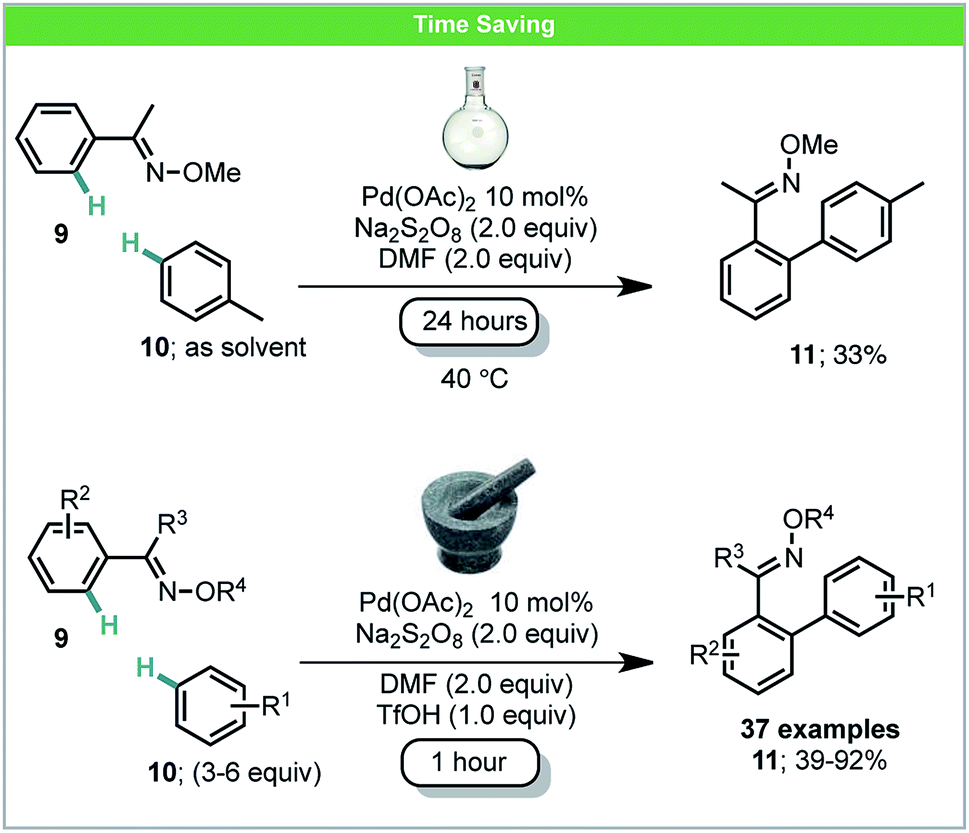
Oxidative C–H/C–H coupling has the potential to become a very powerful tool for sustainable chemical synthesis as no starting material prefunctionalization is required for either coupling partner.47 Ball-milling has also been used for dehydro-C–C coupling reactions with an illustrative example having been developed by Xu and co-workers.48 Under mechanochemical conditions, biaryl products 11 could be obtained in both high selectivity and yield within a one hour reaction time. Specifically, electron-deficient oximes 9 were treated with a variety of arenes 10 in the presence of a palladium catalyst and oxidant (Scheme 5). Anilides were also found to be competent directing groups for this transformation. The comparable reaction in solution, after stirring for 24 hours using toluene as both a solvent and reagent, achieved a poorer yield than any mechanochemical results. Similar solution based methods for coupling anilides with arenes developed by Yu,49a Dong,49b and You49c also usually require more than 16 hours for the reaction to be complete. By employing ball milling, only 3–6 equiv. of simple arenes were required. It is also worth noting that electron-deficient arenes such as acetophenone and fluorobenzene were found reactive using this method, which were less studied in other C–H/C–H reports.47
 | ||
| Scheme 5 Oxidative C–H/C–H coupling; Xu and co-workers.48 | ||
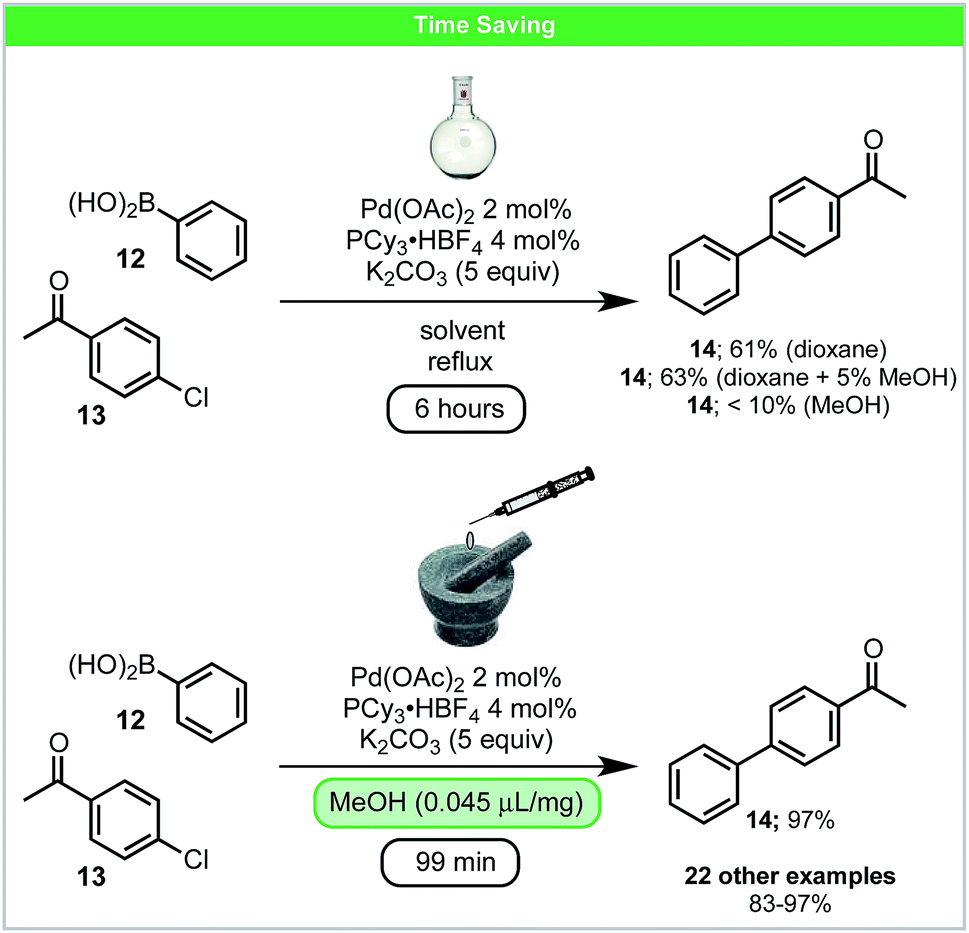
In 2016, Su and co-workers developed a LAG accelerated, palladium catalysed Suzuki–Miyaura coupling of aryl chlorides 13 with boronic acids 12 under ball-milling conditions (Scheme 6).50 Compared to the solvent-based reaction (Scheme 6), higher yields in shorter reaction times could be achieved. Adding solvents, which are commonly used in Suzuki–Miyaura reactions (THF, dioxane, DMF or MeCN), as LAG agents, did not lead to improved results.51 However, protic solvents such as alcohols/H2O led to improved reactivity. It was proposed that under these conditions alcohols form alkoxides in situ, which could participate in both ligand exchange and boronic acid activation. This may explain the improved reactivity observed using LAG. In addition, it was also shown that much lower catalyst loading, 0.5 mol% Pd with 2.5 equiv. K2CO3, could be used when the reaction was scaled up to gram scale.
 | ||
| Scheme 6 Suzuki–Miyaura cross coupling; Su and co-workers.50 | ||
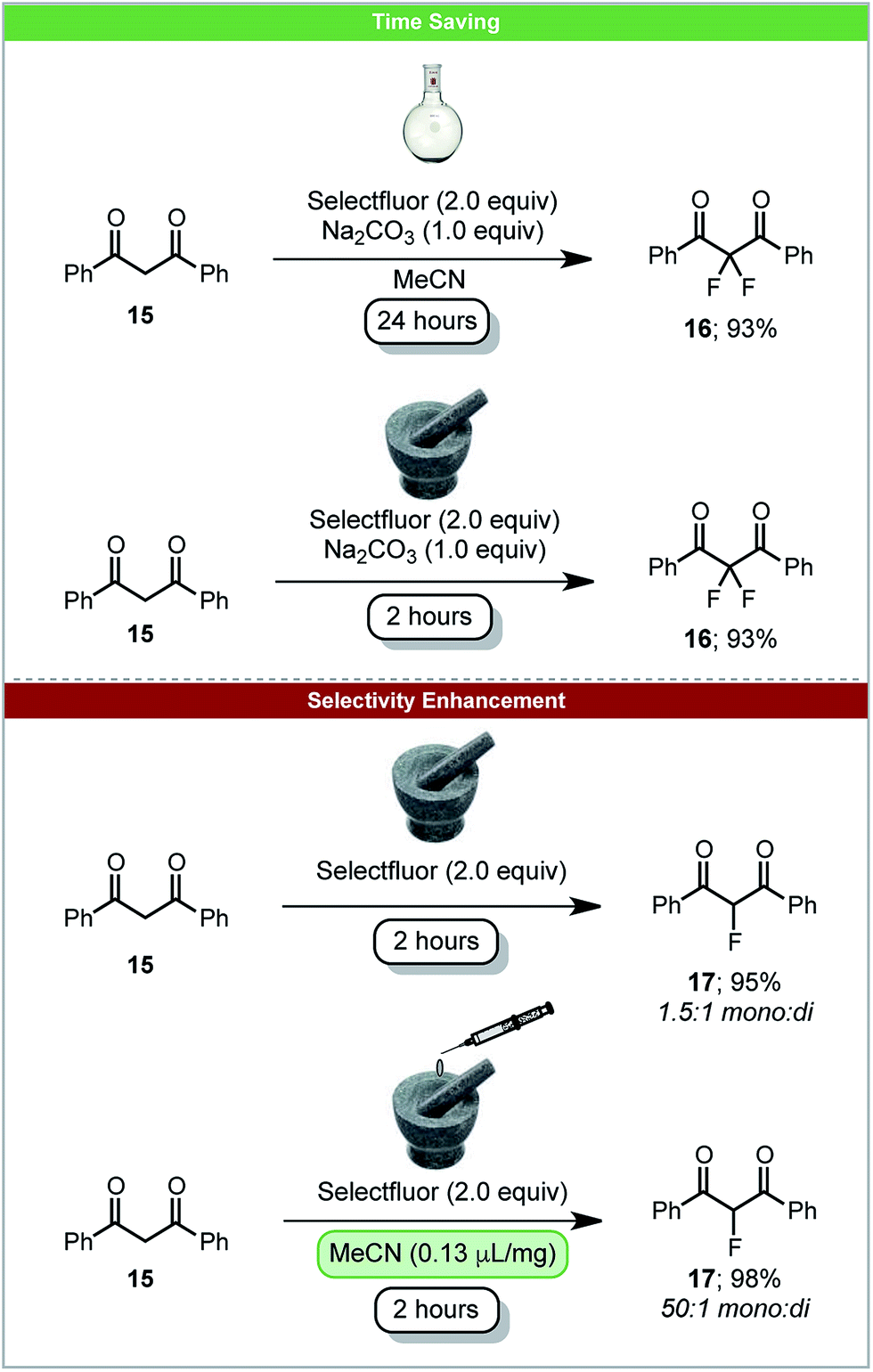
The carbon–fluorine bond is present in a wide range of valuable chemical products, such as pharmaceuticals and agrochemicals. In 2017 the first carbon–fluorine bond formation under solid-state mechanochemical milling conditions was reported. Simple C–F bond formation was achieved by fluorinating 1,3-diketones 15 using Selectfluor as an electrophilic source of fluorine (Scheme 7).52 It was observed that by using LAG with acetonitrile, the selectivity of monofluorinated products 17 was enhanced in comparison to neat grinding. This demonstrates the significant effects LAG can have on a reaction's outcome. Difluorination could be achieved by the addition of sodium carbonate under solvent free conditions within two hours. The analogous solvent based reaction requires 24 hours (Scheme 7). When using the less reactive β-keto ester, solvent based reactions take five days to complete, whereas under ball-milling conditions reaction is complete within two hours.53
 | ||
| Scheme 7 Fluorination of 1,3-diketones; Browne and co-workers.52 | ||
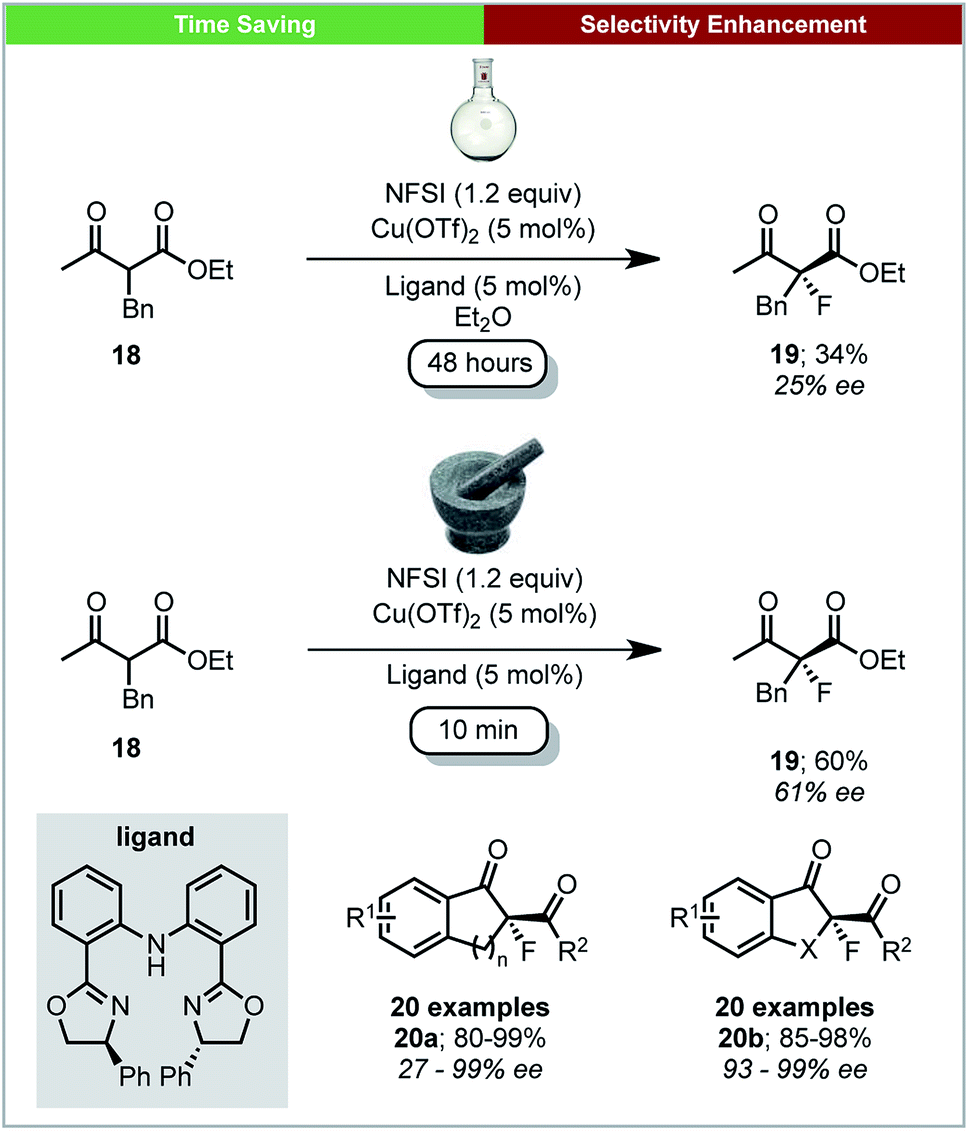
Further work on mechanochemical fluorination was performed by Xu and co-workers, who developed a method for the copper catalysed enantioselective fluorination of β-keto esters using N-fluorobenzenesulfonimide (NFSI) under ball milling conditions (Scheme 8).54 A selection of chiral bis(oxazolines) and bis(azoline) Cu(II) complexes were generated in situ by milling for five minutes before the addition of substrates and NFSI. Using ball-milling, the reaction was complete within four minutes for most substrates, with high enantioselectivities and yields. Using the acyclic β-keto ester 18, the solution reaction required 48 hours to achieve a yield of 34% and an ee of 24%. This is in stark contrast to the mechanochemical conditions, which give a significantly improved yield (60%) and ee (61%) after only 10 minutes.
 | ||
| Scheme 8 Enantioselective fluorination of β-ketoesters; Xu and co-workers.54 | ||
The use of organocatalysts negates the need for a metal catalyst, which can lead to greener synthetic processes.55 However, there are still limitations such as high catalyst loadings (often 10–20 mol%), limited solvent choice (chlorinated solvents are commonly used), catalyst recovery and long reaction times. In the last decade, ball-milling has been used to improve the performance of organocatalysts under solvent free/LAG conditions, particularly in the area of secondary amine organocatalysis.
Pioneering investigations of mechanochemical (S)-proline-catalyzed asymmetric aldol reactions were carried out by Bolm and co-workers, affording anti-aldol products in high yield and with up to 99% ee (Scheme 9).56 When comparing the reaction of 4-nitrobenzaldehyde 21 with tetrahydrothiopyran-4-one 22 to analogous solution reactions, a higher yield was achieved with shorter reaction time and similar ee.57 Following this pioneering work, several other reports on secondary amine mechanochemical organocatalysis have been published, reporting similar improvements in comparison to solution based reactions.58
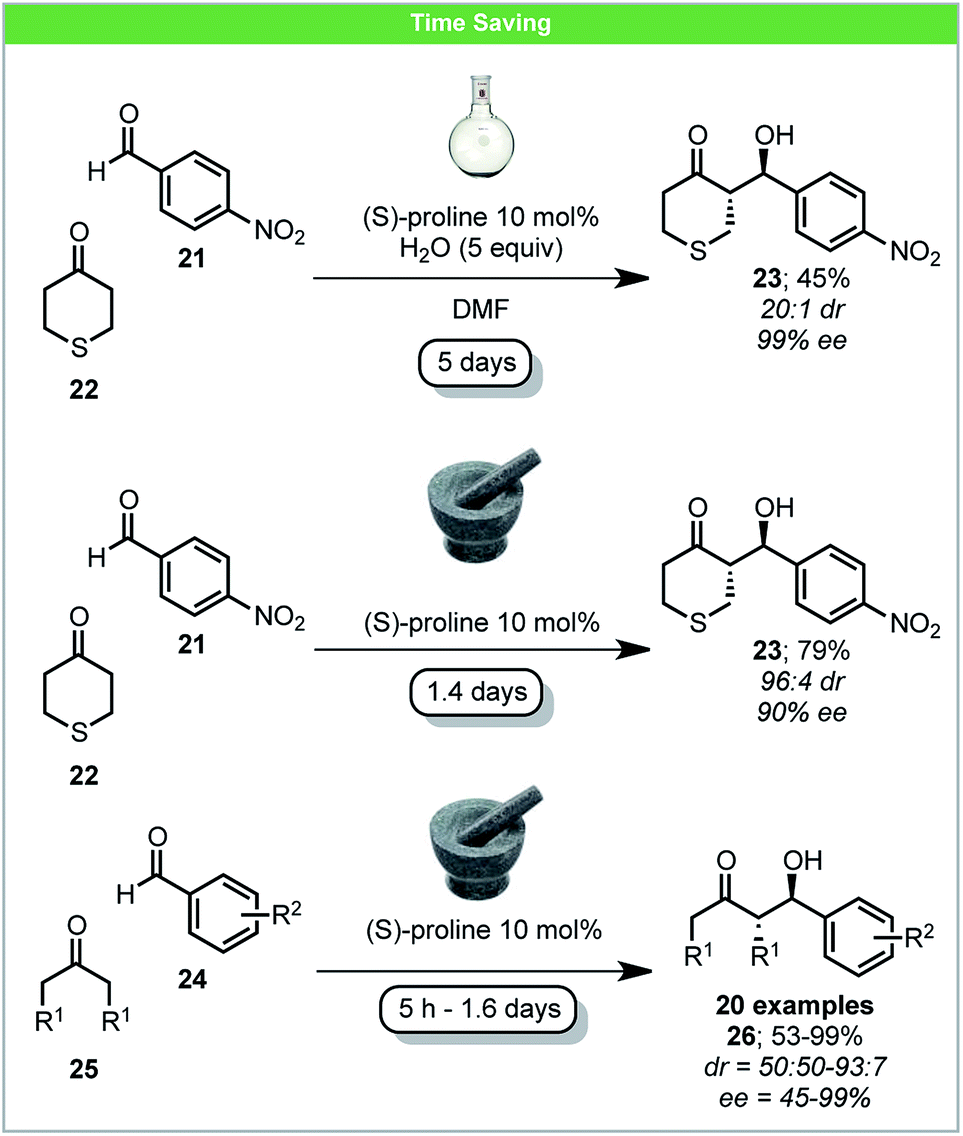 | ||
| Scheme 9 Proline catalysed enantioselective aldol reaction; Bolm and co-workers.56 | ||
Metal complexes containing macrocyclic polyamine ligands have a range of applications such as medical imaging agents, protein binding agents, antimalarial drugs and catalysis.59 In 2016, Archibald and co-workers developed a novel method for N-alkylation of glyoxal-bridged bisaminal derivatives of cyclam (1,4,8,11-tetraazacyclotetradecane) 27 and cyclen (1,4,7,10-tetraazacyclododecane) 28 under mechanochemical LAG conditions (Scheme 10).60 Most of the monofunctionalized quaternary ammonium salts could be formed in good to excellent yields by using stoichiometric amounts of alkyl bromides within 30 minutes. Yields of bis-N-alkylated products could be improved by increasing the relative quantity of bromide reagent or the reaction time. Compared to conventional solution methods, using mechanochemistry resulted in a five-fold reduction in the reaction time. It was further suggested that CB-TE2A, widely used to form stable 64Cu complexes for PET imaging in vivo, could be synthesized within five days using this method. This approach offers a much faster synthetic route compare to the conventional six-step process which takes 35 days developed by Weisman and co-workers.61
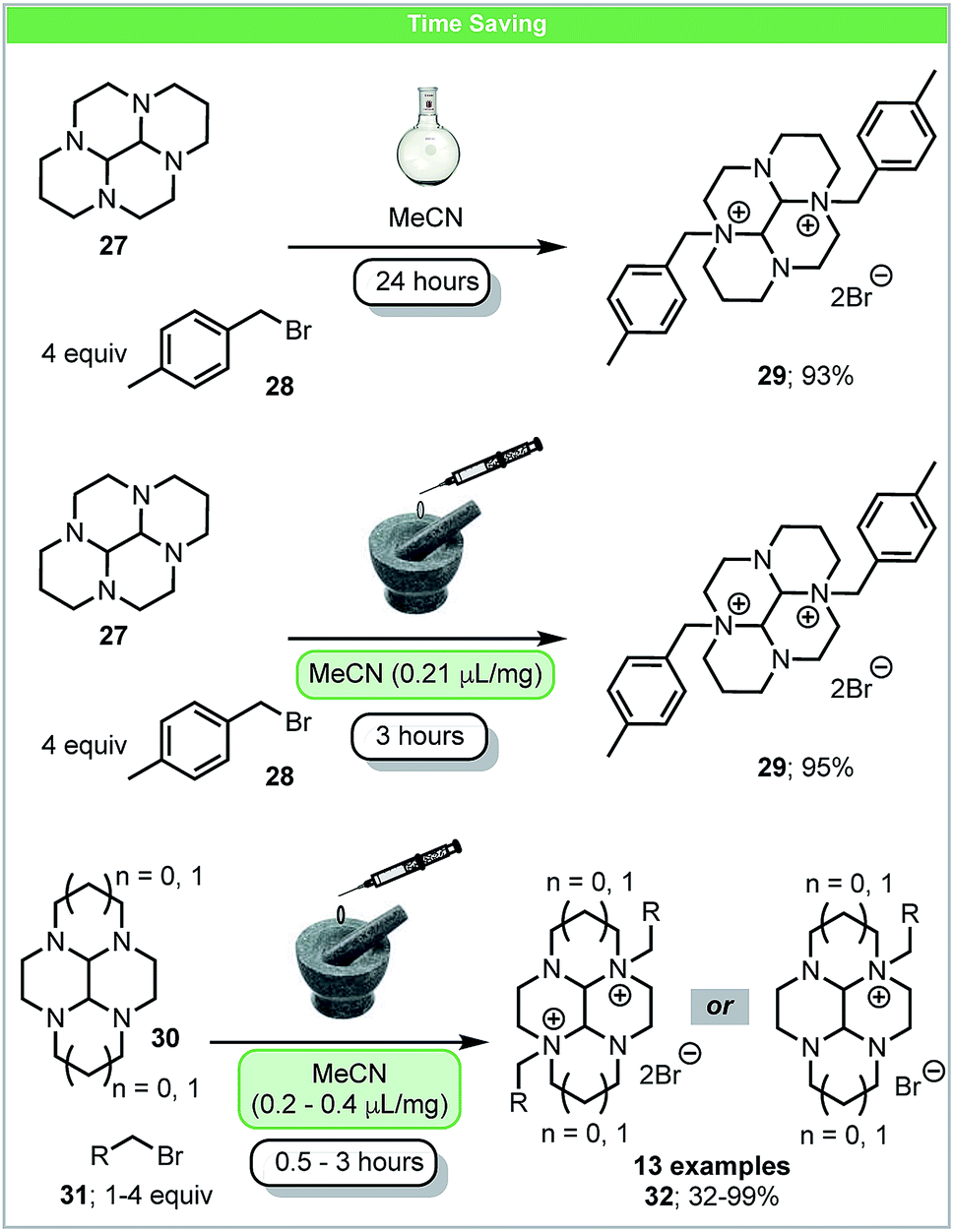 | ||
| Scheme 10 N-Alkylation of bridged cyclam and cyclen derivatives; Archibald and coworkers.60 | ||
The examples described above demonstrate that mechanochemical conditions can, in several instances, lead to greatly reduced reaction times compared to the comparable reaction in solution. In some instances this is coupled with an increase in yield. This is an interesting general observation to note, and could be due to running reactions between neat reagents, increased energy input or a contribution from both. Instantaneous localised temperatures in the mill have not been accounted for and could also play a role. Regardless of the explanation, the experimental observation holds true that the use of mechanochemistry can reduce reaction times.
3. Selectivity enhancement
As well as reducing reaction times, mechanochemistry has been used to alter or control the selectivity of reaction outcomes. This section describes a few examples of reactions exhibiting a change in selectivity compared to reactions performed in solution.Plant biomass is a potential feedstock for the production of fuels and chemicals.62 The degradation of lignin is one such method to convert the feedstock into commodity chemicals. Mechanochemistry could have the potential to be used in industry for the degradation of lignin, cellulose and chitin.63 In 2013, Anastas, Crabtree, Hazari and co-workers reported the mechanochemical oxidation of lignin-like methoxylated aromatic substrates 33 (Scheme 11) using Oxone (potassium peroxymonosulfate) as the oxidant.64 When this reaction was carried out in aqueous solution, the major product was 2,3,4-trimethoxyphenol 34, with several other side-products observed. In contrast, under mechanochemical conditions, quinone 35 was the only product formed. Making use of a rock tumbler/polisher, seven days were required for this transformation.
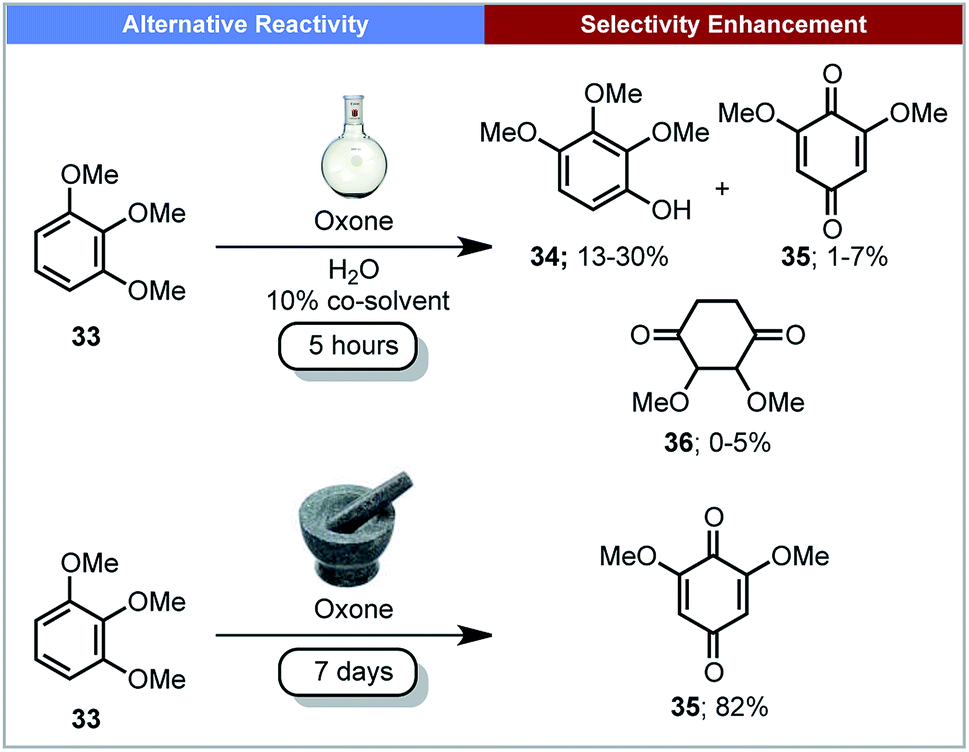 | ||
| Scheme 11 Oxidation of lignin-like methoxylated aromatics; Anastas, Crabtree, Hazari and co-workers.64 | ||
In 2014, Friščić demonstrated that ball milling techniques could offer stereoselective control for the synthesis of organometallic compounds.65 The oxidative halogenation of cyclopentadienyl tricarbonyl Re(I) complexes 38 was studied under ball-milling conditions (Scheme 12). The rhenium complexes [CpReX2(CO)2] 37 and [Cp*ReX2(CO)2] could be selectively formed in either diag (trans) or lat (cis) forms in one step by milling their corresponding cyclopentadienyl tricarbonyl Re(I) complexes with a metal halide (MXn). It was reported that diag-37 was more thermally stable in the solid state or in solution than its lat-37 form, and that this transformation was irreversible in solution.66 It is notable that isomerization of both diag-37 and lat-37 can occur by mechanical treatment instead of thermal treatment. Indeed after 1 h of milling of diag-37 at low temperature it isomerised to a 64:36 mixture of diag-37:lat-37.
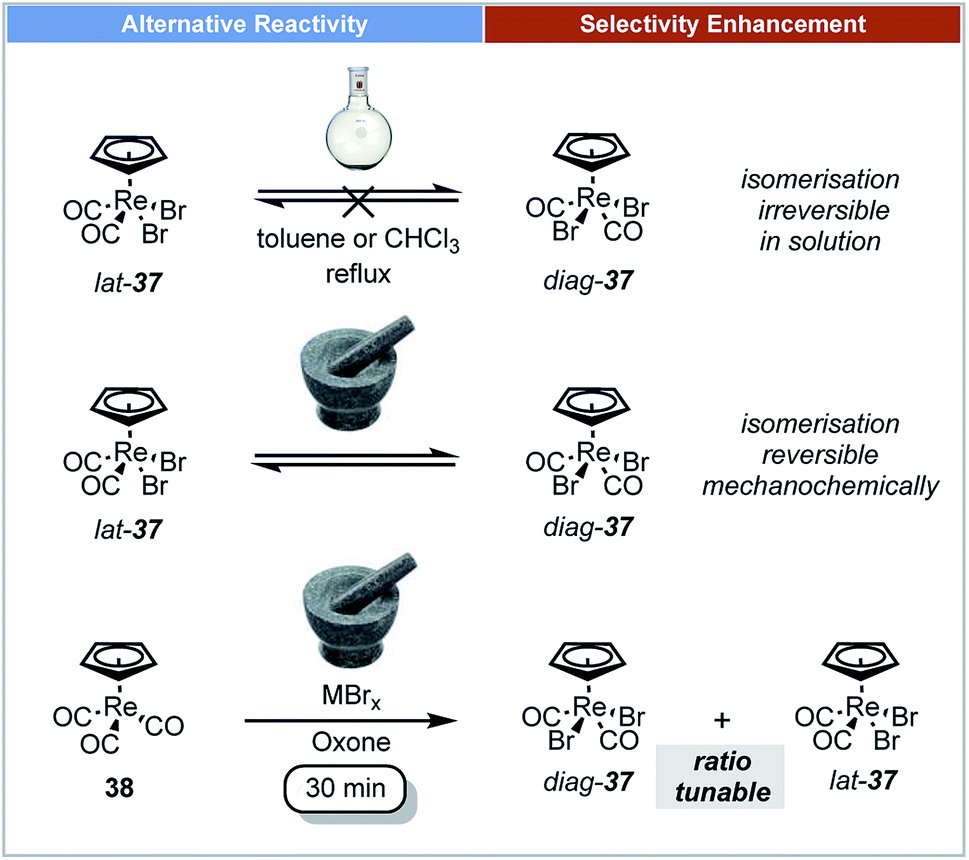 | ||
| Scheme 12 Oxidative halogenation of Re(I) complexes; Friščić and co-workers.65 | ||
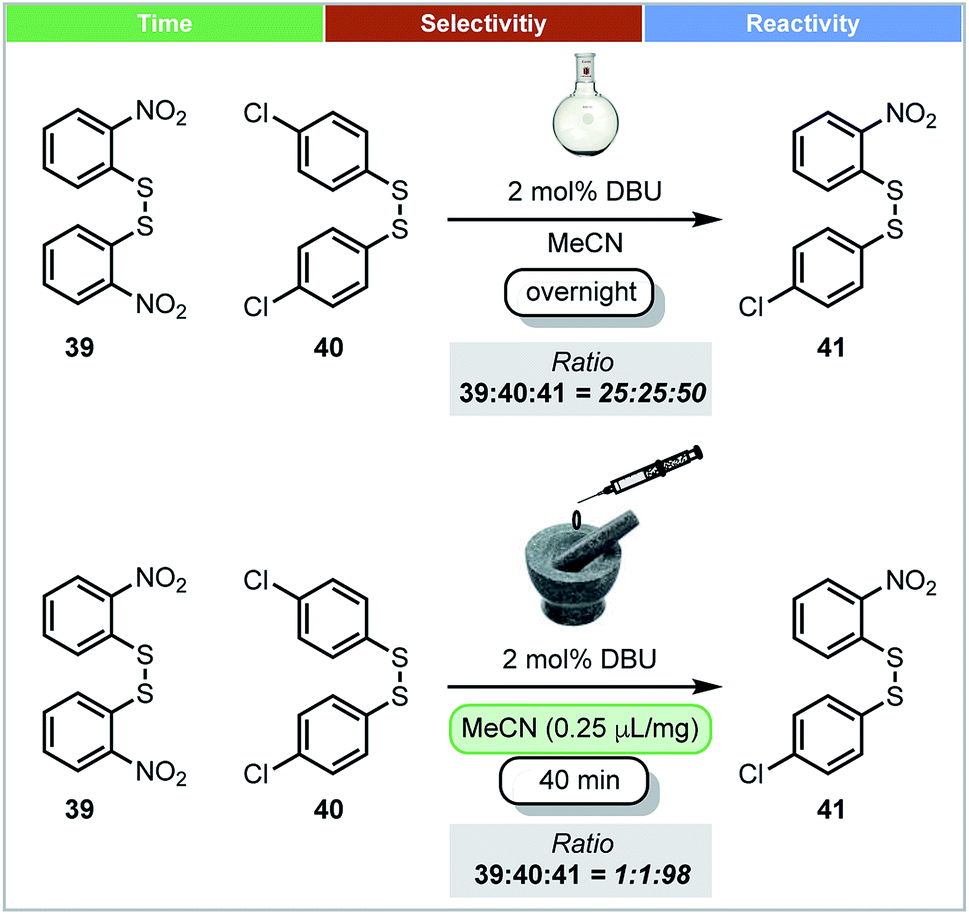
In 2010, Friščić and co-workers demonstrated that thermodynamic equilibrium could be obtained under mechanochemical conditions. Using the base catalysed metathesis of aromatic disulfides as a model reaction (Scheme 13), it was shown that there was a significant difference in the position of equilibrium under mechanochemical and solution-based conditions.67 In dilute acetonitrile solution, reactions afforded a ratio of 1:1:2 between homodimers (39 and 40) and heterodimer 41. However, both LAG (with MeCN) or neat grinding led to almost complete conversion of homodimers, and afforded almost 98% heterodimer 41 (Scheme 13). These different equilibrium compositions could be explained by crystal packing effects, which do not exist in solution, but are a factor for consideration under mechanochemical conditions.
 | ||
| Scheme 13 Disulfide metathesis; Friščić and co-workers.67 | ||
4. Different reaction products or different reactivity
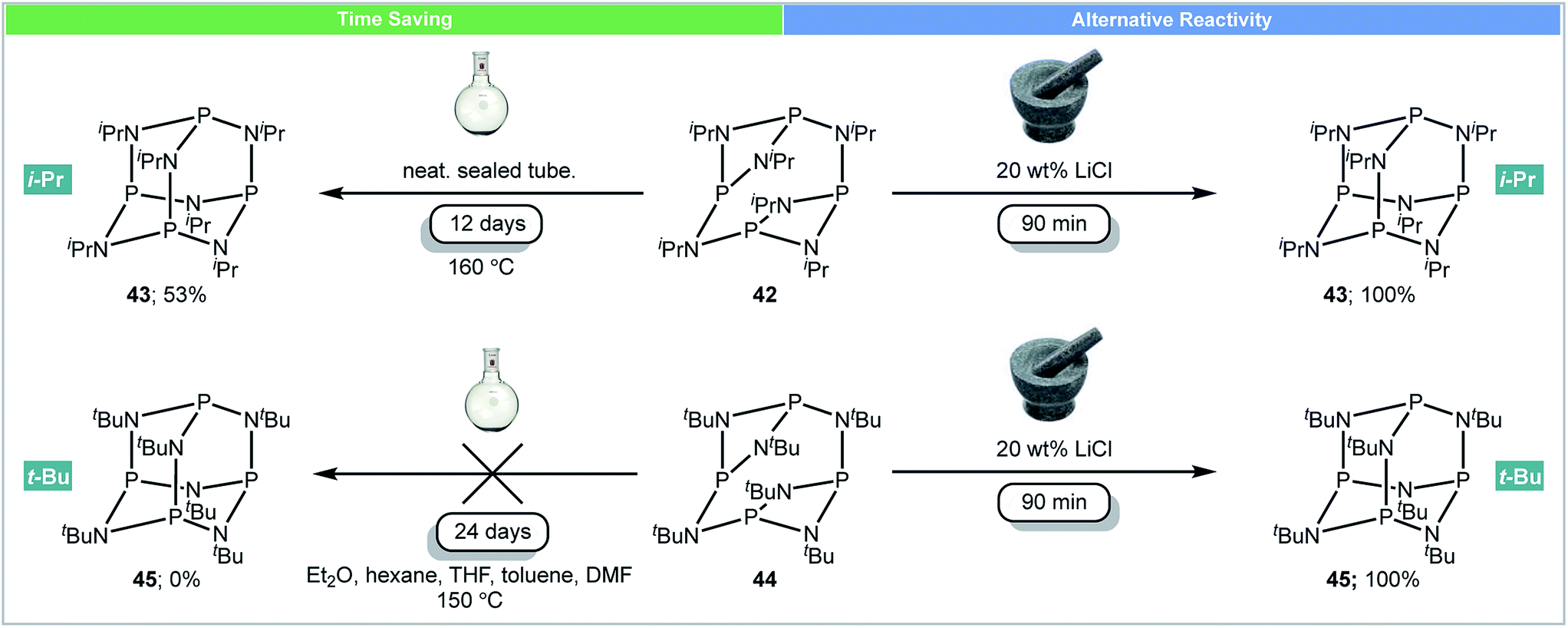
Perhaps the most interesting observation arising from running reactions under mechanochemical conditions is that different products can be achieved. This suggests that the kinetics and thermodynamics of some reactions can be significantly altered by using a mill, and leads to the tempting supposition that some molecules are only accessible by this method.In 2016, García and co-workers developed the first example of using mechanochemistry for the synthesis of adamantoid substituted cyclophosphazenes (Scheme 14).68 As strong non-carbon covalent backbones, phosphazanes (P–N) are interesting compounds, as they are used as multi-dentate ligands,69 catalysts70 and antitumor drugs.71 It was reported that subjecting the isopropyl-substituted ring [{P(μ-NiPr)2}2(μ-NiPr)]242 to mechanochemical milling caused rearrangement to its adamantoid isomer P4(NiPr)643 in 90 minutes.66 However, the same rearrangement under high temperature (160 °C) conditions required 12 days (Scheme 14).72 The analogous tert-butyl-substituted ring [{P(μ-NtBu)2}2(μ-NtBu)]244 could not be converted to its adamantoid isomer P4(NtBu)645 using solution methods (reflux in DMF, THF, toluene etc.) or prolonged heating.73 However, this compound was synthesized for the first time under mechanochemical conditions. Both adamantoid P4(NiPr)643 and P4(NtBu)645 could be synthesized in the mixer mill within 90 minutes in the presence of LiCl (20 wt%).
 | ||
| Scheme 14 Synthesis of adamantoid cyclophosphazenes; García and co-workers.68 | ||
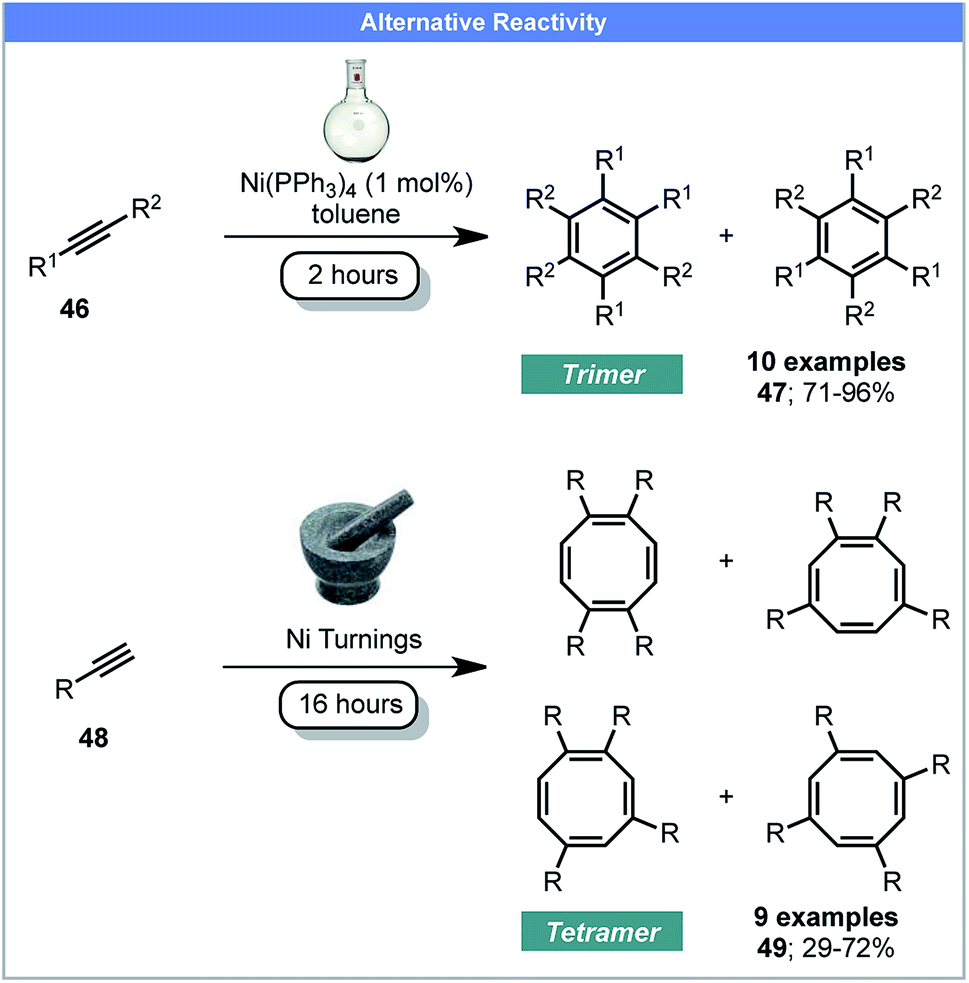
It has been shown by Mack and co-workers that using a copper vial or balls instead of conventional milling materials, or adding silver foil enables elemental Cu(0) and Ag(0) to be used as active recyclable catalysts under ball milling conditions.74 In 2016, Guan, Mack and co-workers developed a method for the cyclotetramerisation of alkynes to afford cyclooctatetraenes (COT) 49 using recyclable Ni(0) pellets as the catalyst under ball milling conditions (Scheme 15).75 Conversely, reactions performed in solution, catalyzed by Ni(0) complexes, yielded the major products as aromatic trimers 47.76 This proof-of-principle study demonstrates the potential of using Ni(0) pellets as active catalyst under mechanochemical conditions, which avoids the use of air-sensitive nickel complexes. It also demonstrates that mechanochemistry may achieve different reaction products compared to conventional solution methods, although grinding with a Ni(PPh3)4 catalyst would help confirm this.
 | ||
| Scheme 15 Nickel catalysed cyclotri/tetra-merisation of alkynes; Guan, Mack and co-workers.75 | ||
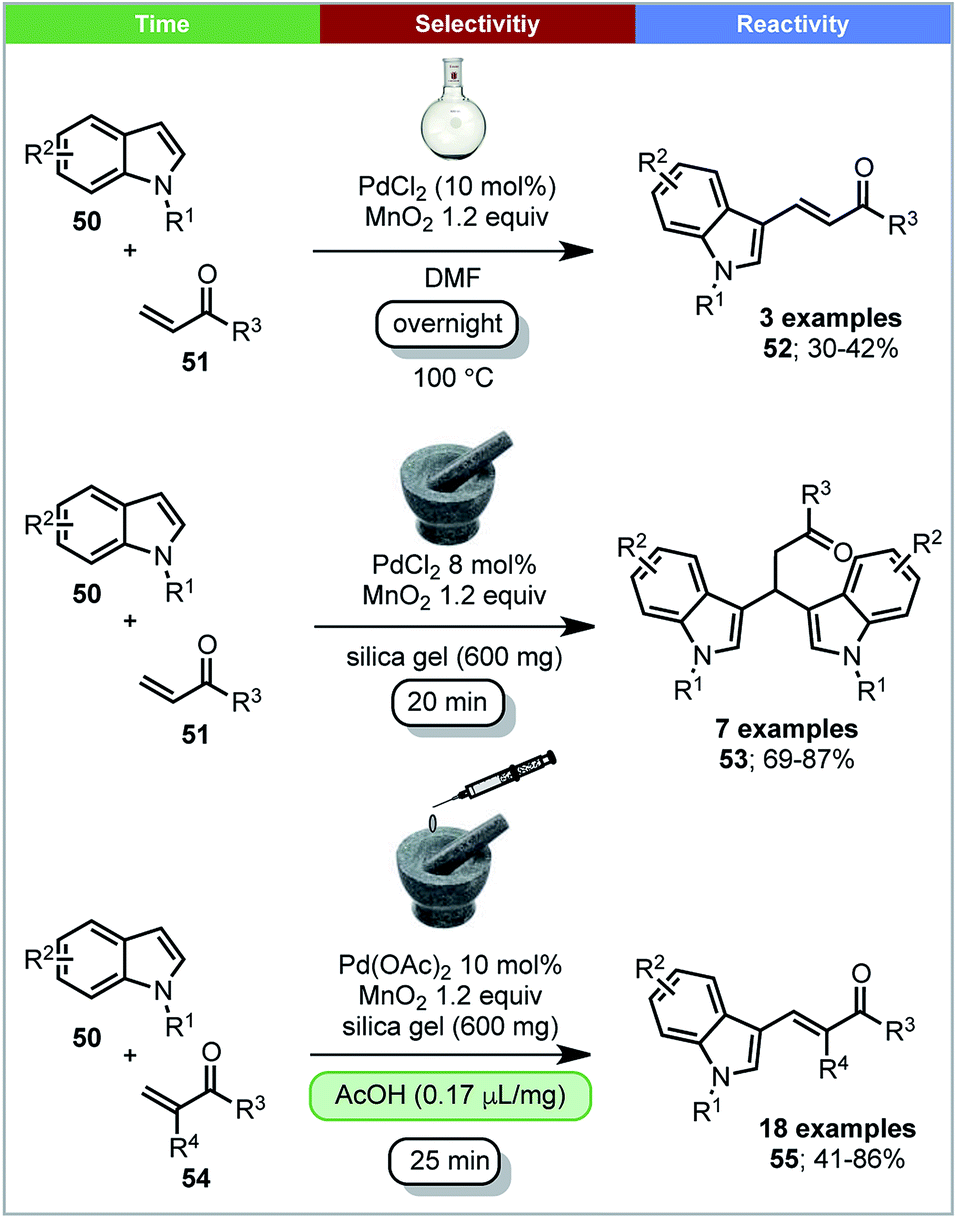
Su and coworkers reported that 3-vinylindoles 55 and β,β-diindolyl propionates 53 could be synthesized by Pd(II) catalyzed oxidative coupling reactions between indoles and acrylates with MnO2 as oxidant under ball milling conditions (Scheme 16).77 It was found that the selectivity of the reaction is influenced by the Pd(II) source. When Pd(OAc)2 was used with acetic acid as additive, high yields of 3-vinylindoles (55) were obtained. However, when using PdCl2 as a catalyst without a liquid additive, β,β-diindolyl propionates were formed selectively. In contrast, solution conditions; DMF as solvent, 100 °C, overnight, only the 3-substituted-vinylindoles 52 were formed, with no trace of β,β-diindolyl propionates 53 being detected (Scheme 16). A control experiment in solution with added silica and/or acetic acid is not reported. Mechanistic studies using ESI-MS indicated that the formation of a dimeric palladium intermediate under ball milling conditions may be causing the differences in selectivity under solvent based and ball milling conditions.
 | ||
| Scheme 16 Palladium catalysed oxidative coupling of indoles and acrylates; Su and co-workers.77 | ||
In solution, anilines react with bis(benzotriazolyl)methanethiones 57 and form their corresponding isothiocyanates 59 and benzotriazoles through intermediate N-(thiocarbamoyl)benzotriazoles 58 (Scheme 17).78 Due to the high reactivity of N-(thiocarbamoyl)benzotriazoles 58 in solution, they were not isolable using conventional solution chemistry. In 2015, Friščić and co-workers reported the first isolation of N-(thiocarbamoyl)benzotriazoles 58 using mechanochemistry.79 Excellent yields (>97%) of N-(thiocarbamoyl)benzotriazoles could be obtained by milling anilines 56 with bis(benzotriazolyl)methanethiones 57 under LAG conditions within 10 minutes. N-(Thiocarbamoyl)benzotriazoles were found to be bench stable in the solid state and could be used for further synthesis of both symmetrical and nonsymmetrical thioureas. These compounds were characterized by solid state magic angle spinning 13C NMR spectroscopy. This novel example shows that mechanochemistry could be used to isolate reactive intermediates, which are not isolable in solution. It further demonstrates the ability to form different reaction products, isothiocyanates in solution and thioureas mechanochemically.
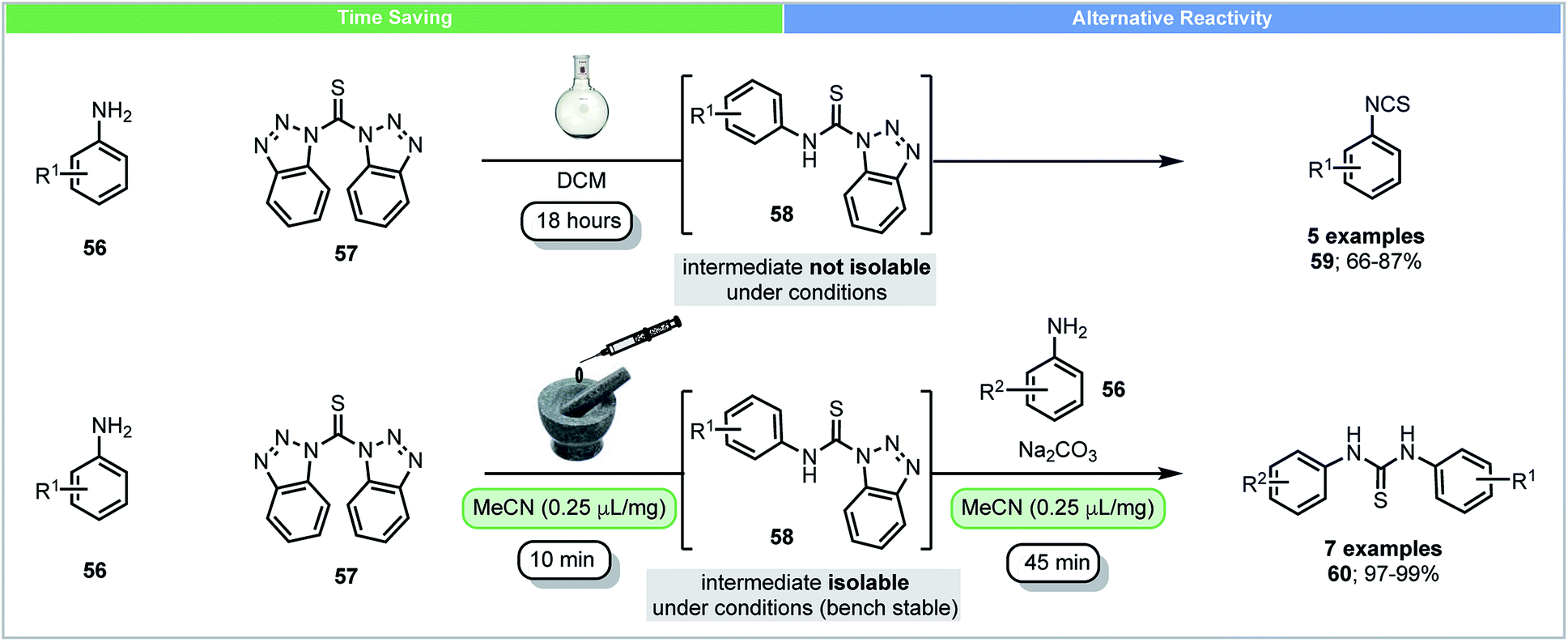 | ||
| Scheme 17 Reaction of anilines with bis(benzotriazolyl)methanethiones and isolation of intermediate; Friščić and co-workers.79 | ||
4.1 Reactivity not possible in solution
Due to the unique electronic and structural properties of fullerene and its functionalized derivatives, a number of methods for the functionalization of fullerene have been developed.80 However, fullerene and fullerene-related materials, such as carbon nanotubes and graphite, often have low solubility in organic solvents and water, which can make their functionalization and application challenging. Mechanochemical techniques offer advantages to reactions of these carbon rich nanostructure materials and have been used to tackle these problems.81 In 1997, Komatsu and co-workers developed the first method for the synthesis of a fullerene dimer, C12063 by the use of high speed vibration milling (HSVM) (Scheme 18).82 Fullerene dimer C120 could be obtained in 30 minutes with 29% yield by milling fullerene C60 with KCN at 2800 cycles per minute followed by a trifluoroacetic acid wash. A similar yield could also be achieved by replacing KCN with other reagents such as K2CO3, KOAc, alkali metals (Li, Na, K) and 4-aminopyridine. A later study revealed that 4% fullerene trimer C180 could also be obtained when 4-aminopyridine was used as catalyst.83 These compounds could not be obtained using liquid-phase protocols. Under solution based reaction conditions, Wudl and co-workers only obtained cyano functionalised fullerene 62 by stirring fullerene with NaCN in a solvent mixture of 1,2-dichlorobenzene and DMF (Scheme 18).84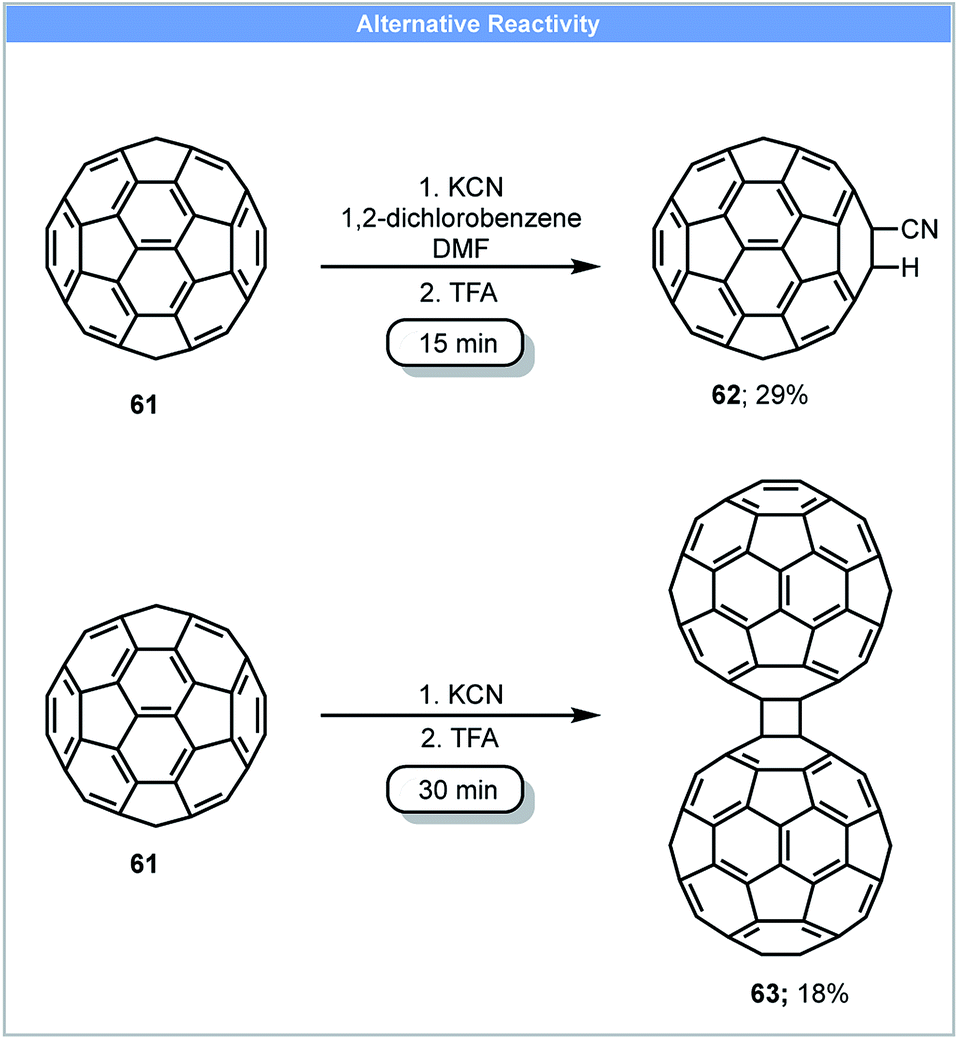 | ||
| Scheme 18 Dimerisation of fullerene; Komatsu and co-workers.82 | ||
Another example of fullerene functionalisation was reported in 2013 by Wang and co-workers. C60-Fused indanes 65 could be synthesised by the reaction of fullerene 61 with N-benzhydryl sulphonamides 64 and FeCl3 in a ball mill (Scheme 19).85 C60-Fused indanes (yield: 15–41%) could be obtained within 1 hour under ball milling, whereas no product was formed using solution-based methods (Scheme 19).
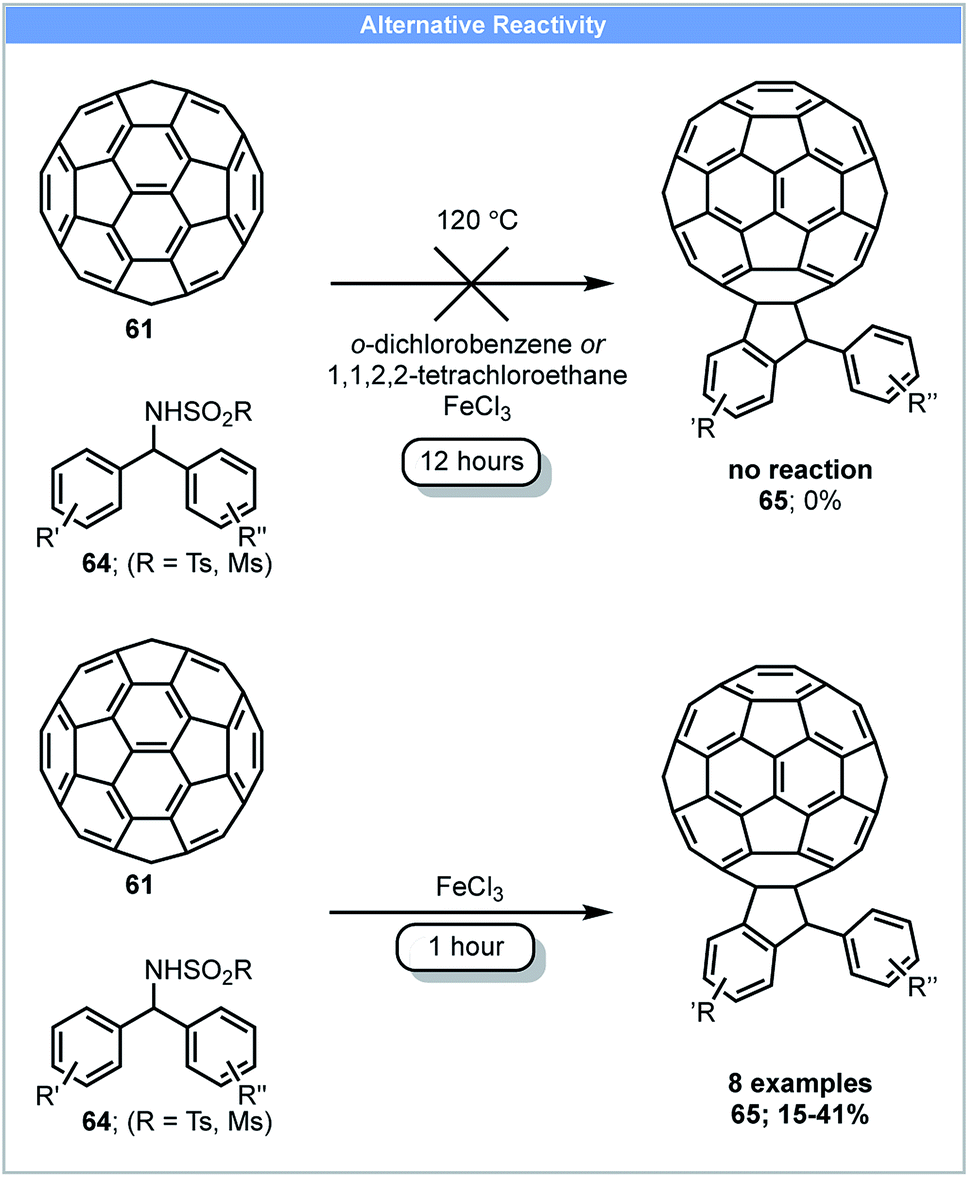 | ||
| Scheme 19 Fullerene functionalisation; Wang and co-workers.85 | ||
In 2014, Friščić and co-workers demonstrated that sulfonyl-(thio)ureas could be synthesized from sulfonamides and isocyanates using catalytic CuCl under ball milling conditions.86 They subsequently reported that N-sulfonylguanidines could be synthesized via the copper-catalyzed coupling of arylsulfonamides 66 and carbodiimides 67 under nitromethane LAG conditions (Scheme 20).87 Further optimization showed that acetone was the best LAG agent and good yields were obtained after milling for two hours. In contrast, refluxing arylsulfonamides and carbodiimides in solvents (DCM or acetone) overnight with or without CuCl led to no product formation.
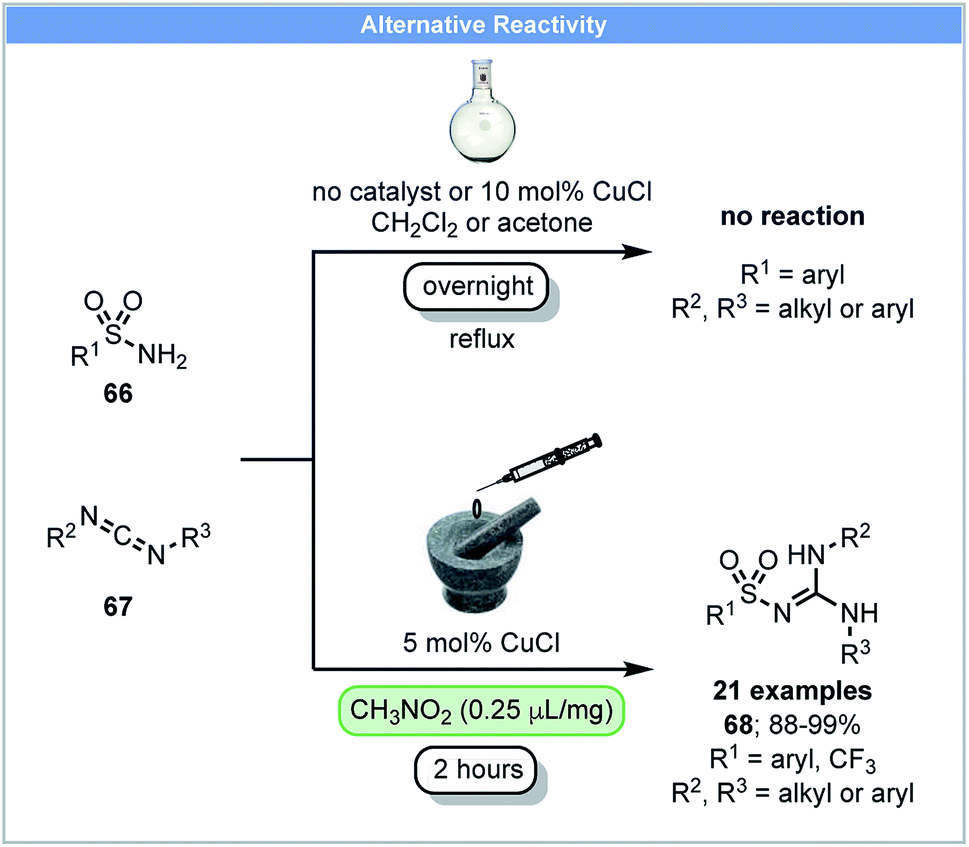 | ||
| Scheme 20 Synthesis of N-sulfonylguanidines; Friščić and co-workers.87 | ||
Su and co-workers developed an Fe(III)-catalyzed cross-dehydrogenative coupling (CDC) of 3-benzylic indoles 72 with compounds bearing acidic methylene groups (73 & 74) under ball-milling conditions (Scheme 21).88 This catalytic system was also successfully used for the synthesis of bisindoles 71 (yield: 24–77%). This was in contrast to the comparable solution-based reaction carried out at 100 °C under N2 gas atmosphere using DCE as solvent (Scheme 21), under which conditions, only trace amounts of product were observed.
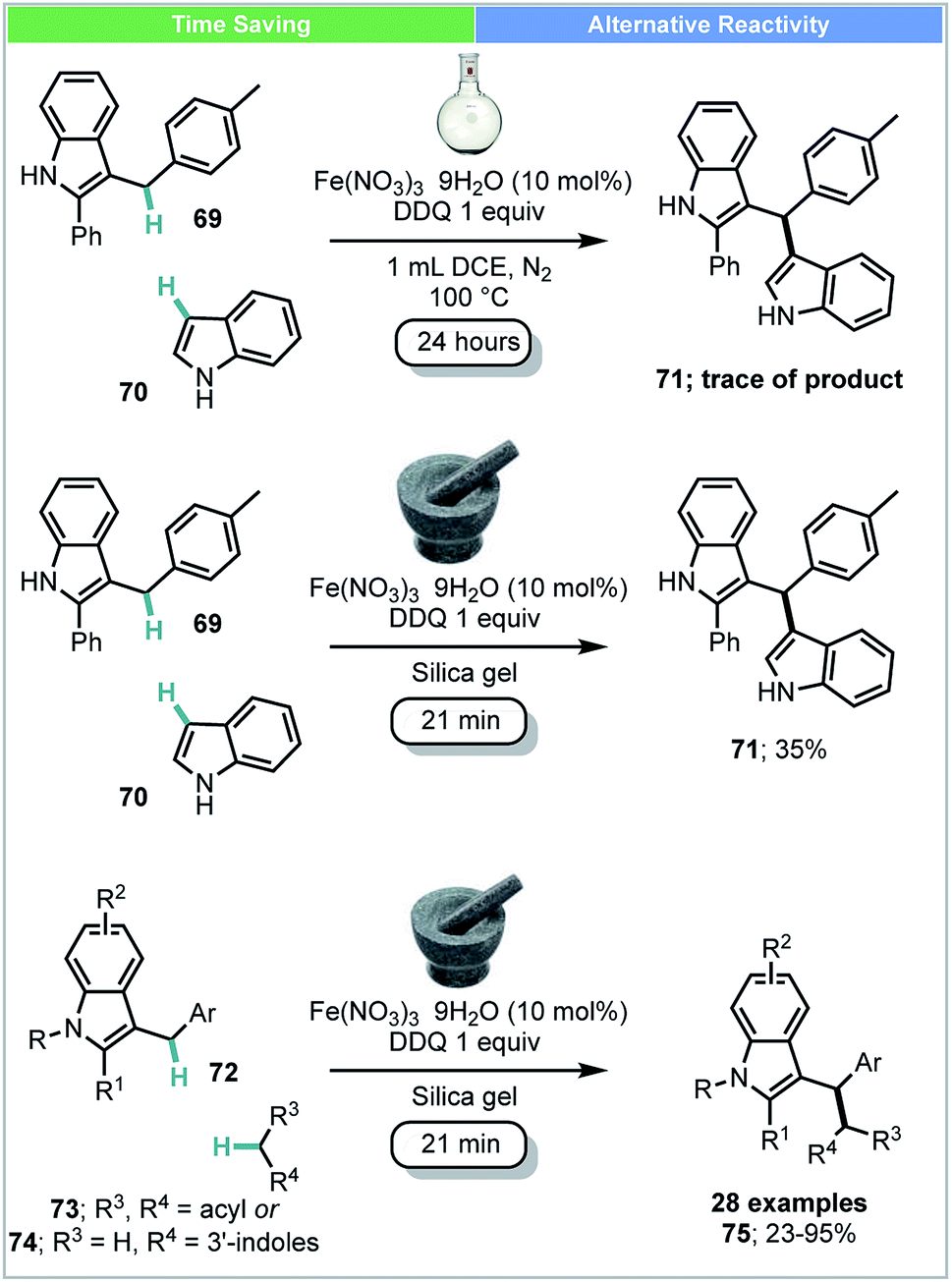 | ||
| Scheme 21 Iron catalysed cross dehydrogenative coupling; Su and co-workers.88 | ||
Synthesising organometallics under mechanochemical conditions can extend the scope of suitable reactants without concern for solubility or potential interference from solvent, such as through coordination or quenching.
In 2014, an unsolvated tris(allyl)aluminium complex 77 was first isolated and reported by Hanusa and co-workers using mechanochemical conditions (Scheme 22).89 Attempts to synthesise this complex by stirring K[1,3-(SiMe3)2C3H3] 76 with AlX3 (X = Cl, I) in different solvents (Et2O, THF, hexane) led to a mixture of unidentified products. The unsolvated tris(allyl)aluminium complex 77 (yield up to 88%) could be obtained within five minutes using a planetary ball mill. The reactivity of this newly synthesized complex 77 was tested by reaction with benzophenone in hexane at −78 °C. Compared to the THF adduct (C3H5)3Al(THF), this unsolvated tris(allyl)aluminium complex 77 showed a faster reaction rate, despite its bulkier ligand.
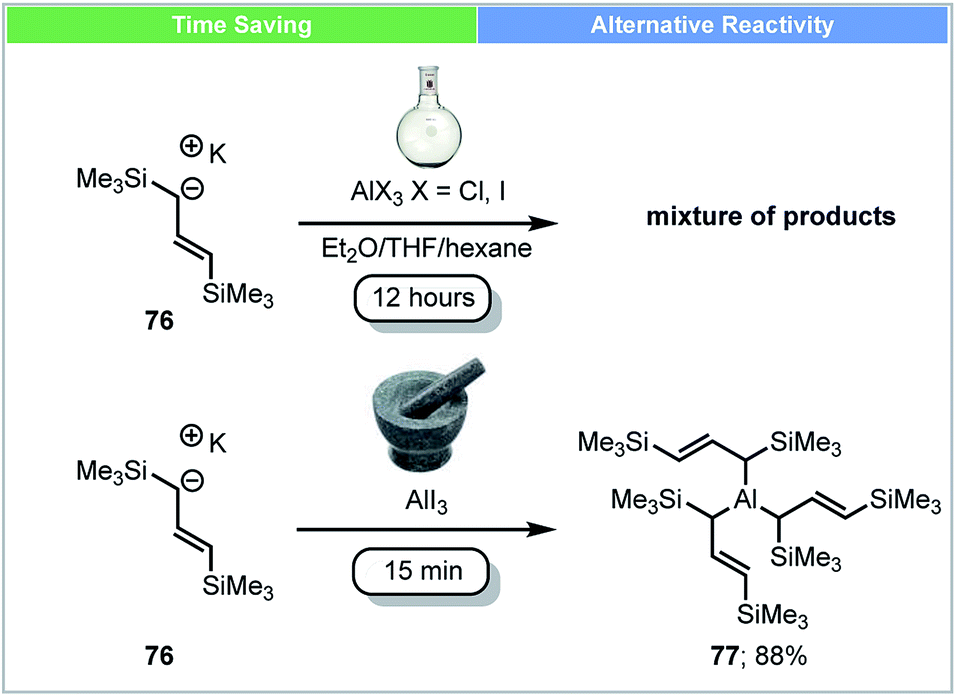 | ||
| Scheme 22 Synthesis of unsolvated trisallylaluminium complex; Hanusa and co-workers.89 | ||
Iptycenes have three dimensional rigid molecular architectures, and potentially have multiple applications, such as molecular machines, novel liquid crystals and porous polymers.90 In 2016, Swager and co-workers showed that highly functionalized iptycenes (molecular weight > 2000 g mol−1) could be synthesized by iterative Diels–Alder/aromatisation reactions under solvent-free conditions (Scheme 23).91 A good yield (87%) of adduct 80 could be achieved in a ball mill using ZnCl2 as a Lewis acid. In contrast, only 5% of this material was obtained when the reaction was carried out in solution in the presence of ZnCl2 at 80 °C after 24 h. To achieve extended iptycenes 82, perfluorononanoic acid (C8F17COOH) was used as an additive to increase the acidity and catalytic performance of the milled reaction. This multistep solvent free synthesis demonstrates the strengths of mechanochemical methods for the synthesis of large functionalized extended iptycenes over traditional methods.
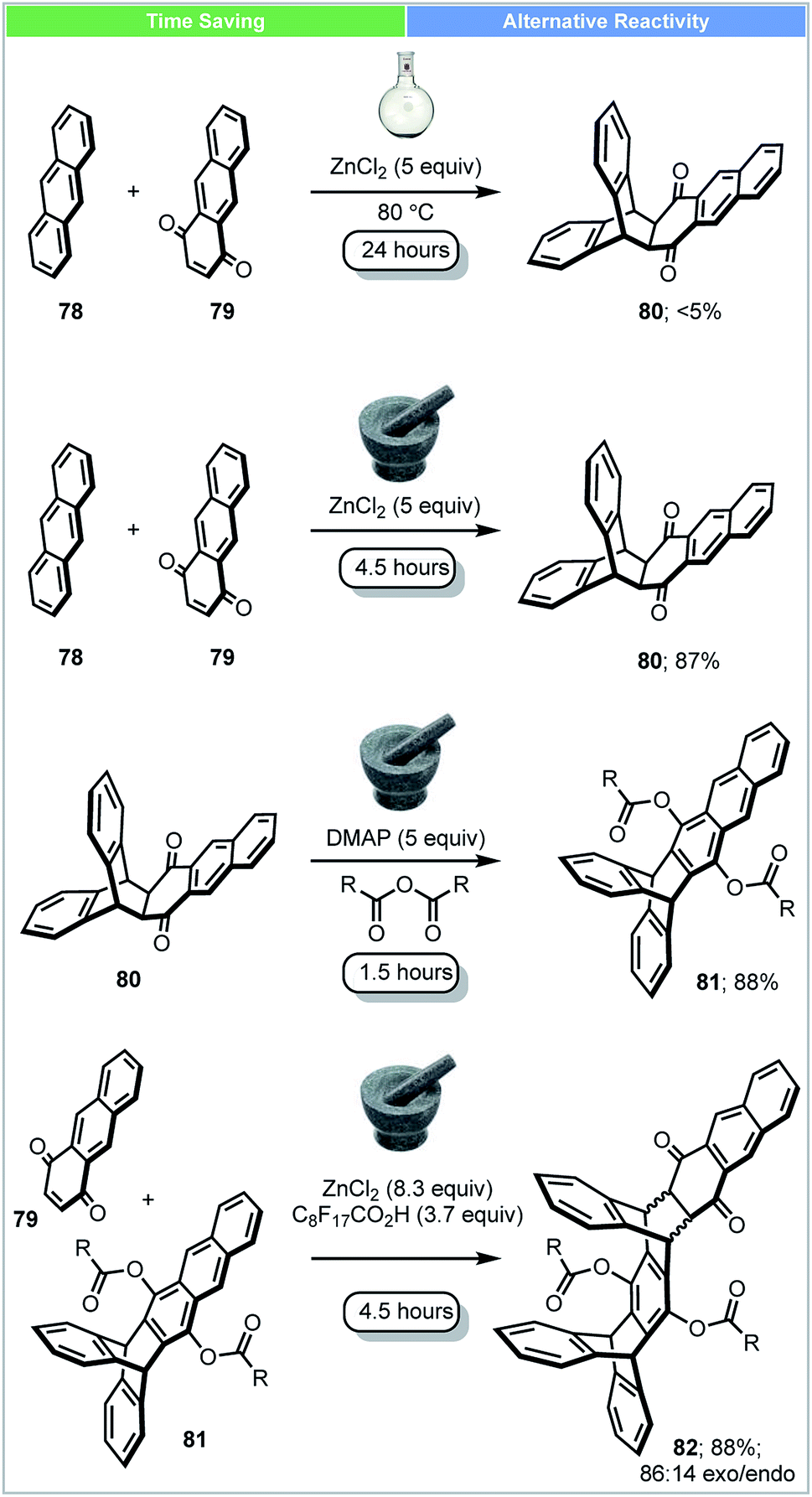 | ||
| Scheme 23 Synthesis of extended iptycenes; Swager and co-workers.91 | ||
5. Conclusions and outlook
We hope that this overview will provide inspiration for others to explore the relatively uncharted territory of mechanochemistry for organic synthesis. Mechanochemistry is still in the early phase of its development, especially as a technique for organic synthesis. However, it has already been shown that a wide variety of important transformations are possible in the absence of solvent. Furthermore, it has been demonstrated that it can be scaled to manufacture levels. Not only driven by the inherent appeal from a sustainability perspective, we have highlighted some examples that are particularly interesting when compared to reactions in solution. Notably, we have shown that there exist a number of examples where using mechanochemistry enables faster, more selective and novel reactivity, much of which is not yet predictable or expected, but is certainly exciting, intriguing and sometimes perplexing! It is currently standard practice during reaction optimisation to screen solvents. However, as shown here, a lack of solvent could well lead to improved or completely unexpected results.Therefore, we are optimistic that there remains a large part of unexplored ‘chemical space’ and fundamental ‘chemical understanding’ hidden within this technique. With much experimentation, more will be discovered about what is possible mechanochemically and the technique will build into one which is predictable. For now, why don't you have a go?92
Conflicts of interest
There are no conflicts to declare.Acknowledgements
D. L. B. thanks Cambridge Reactor Design for a Ph.D. award to J. L. H., the EPSRC First Grant Scheme for funding Q. C. (D. L. B. EP/P002951/1) and the School of Chemistry at Cardiff University for generous support.Notes and references
- J.-L. Do and T. Friščić, Synlett, 2017, 28, 2066–2092 CrossRef CAS.
- D. Tan and T. Friščić, Eur. J. Org. Chem., 2018, 18–33 CrossRef CAS.
- J.-L. Do and T. Friščić, ACS Cent. Sci., 2017, 3, 13–19 CrossRef CAS PubMed.
- J. G. Hernandez and C. Bolm, J. Org. Chem., 2017, 82, 4007–4019 CrossRef CAS PubMed.
- IUPAC Compendium of Chemical Terminology, ed. M. Nič, J. Jirát, B. Košata, A. Jenkins and A. McNaught, IUPAC, 2009 Search PubMed.
- P. Baláž, Mechanochemistry in Nanoscience and Minerals Engineering, Springer-Verlag, Berlin, Heidelberg, 2008 Search PubMed.
- S. M. Hsu, J. Zhang and Z. Yin, Tribol. Lett., 2002, 13, 131–139 CrossRef CAS.
- F. Galembeck, T. A. L. Burgo, L. B. S. Balestrin, R. F. Gouveia, C. A. Silva and A. Galembeck, RSC Adv., 2014, 4, 64280–64298 RSC.
- R. Hoffmann and R. B. Woodward, Acc. Chem. Res., 1968, 1, 17–22 CrossRef CAS.
- R. B. Woodward and R. Hoffmann, J. Am. Chem. Soc., 1965, 87, 395–397 CrossRef CAS.
- M. Wollenhaupt, M. Krupička and D. Marx, ChemPhysChem, 2015, 1593–1597 CrossRef CAS PubMed.
- C. R. Hickenboth, J. S. Moore, S. R. White, N. R. Sottos, J. Baudry and S. R. Wilson, Nature, 2007, 446, 423–427 CrossRef CAS PubMed.
- R. Stevenson and G. De Bo, J. Am. Chem. Soc., 2017, 139, 16768–16771 CrossRef CAS PubMed.
- G.-W. Wang, Chem. Soc. Rev., 2013, 42, 7668–7700 RSC.
- S. L. James, C. J. Adams, C. Bolm, D. Braga, P. Collier, T. Friščić, F. Grepioni, K. D. M. Harris, G. Hyett, W. Jones, A. Krebs, J. Mack, L. Maini, a G. Orpen, I. P. Parkin, W. C. Shearouse, J. W. Steed and D. C. Waddell, Chem. Soc. Rev., 2012, 41, 413–447 RSC.
- D. Margetić and V. Štrukil, Mechanochemical Organic Synthesis, Elsevier, Oxford, 2016 Search PubMed.
- E. Boldyreva, Chem. Soc. Rev., 2013, 42, 7719–7738 RSC.
- A. Stolle, T. Szuppa, S. E. S. Leonhardt and B. Ondruschka, Chem. Soc. Rev., 2011, 40, 2317–2329 RSC.
- A. Stolle and B. Ranu, Ball Milling Towards Green Synthesis: Applications, Projects, Challenges, Royal Society of Chemistry, Cambridge, 2014 Search PubMed.
- L. Takacs, J. Therm. Anal. Calorim., 2007, 90, 81–84 CrossRef CAS.
- H. Choi, W. Lee, J. Lee, H. Chung and W. S. Choi, Met. Mater. Int., 2007, 13, 353–358 CrossRef.
- M. J. Mankosa, G. T. Adel and R. H. Yoon, Powder Technol., 1989, 59, 255–260 CrossRef CAS.
- W. L. Noorduin, T. Izumi, A. Millemaggi, M. Leeman, H. Meekes, W. J. P. Van Enckevort, R. M. Kellogg, B. Kaptein, E. Vlieg and D. G. Blackmond, J. Am. Chem. Soc., 2008, 130, 1158–1159 CrossRef CAS PubMed.
- http://new.outotec.com/products/grinding/higmill-high-intensity-grinding-mill/, accessed March 2017.
- A. Stolle, R. Schmidt and K. Jacob, Faraday Discuss., 2014, 170, 267–286 RSC.
- (a) D. Crawford, J. Casaban, R. Haydon, N. Giri, T. McNally and S. L. James, Chem. Sci., 2015, 6, 1645–1649 RSC; (b) D. E. Crawford, C. K. G. Miskimmin, A. B. Albadarin, G. Walker and S. L. James, Green Chem., 2017, 19, 1507–1518 RSC.
- A. A. L. Michalchuk, I. a. Tumanov, V. A. Drebushchak and E. V. Boldyreva, Faraday Discuss., 2014, 170, 311–335 RSC.
- N. Kumar and K. Biswas, Rev. Sci. Instrum., 2015, 86, 83903 Search PubMed.
- F. Schneider, T. Szuppa, A. Stolle, B. Ondruschka and H. Hopf, Green Chem., 2009, 11, 1894 RSC.
- R. Schmidt, R. Thorwirth, T. Szuppa, A. Stolle, B. Ondruschka and H. Hopf, Chem.–Eur. J., 2011, 17, 8129–8138 CrossRef CAS PubMed.
- R. Thorwirth, F. Bernhardt, A. Stolle, B. Ondruschka and J. Asghari, Chem.–Eur. J., 2010, 16, 13236–13242 CrossRef CAS PubMed.
- R. Trotzki, M. M. Hoffmann and B. Ondruschka, Green Chem., 2008, 10, 767 RSC.
- B. P. Hutchings, D. E. Crawford, L. Gao, P. Hu and S. L. James, Angew. Chem., Int. Ed., 2017, 56, 15252–15256 CrossRef CAS PubMed.
- J. L. Do, C. Mottillo, D. Tan, V. Štrukil and T. Friščić, J. Am. Chem. Soc., 2015, 137, 2476–2479 CrossRef CAS PubMed.
- J. Li and Y. Wu, Lubricants, 2014, 2, 21–43 CrossRef.
- T. Friščić, S. L. Childs, S. A. A. Rizvi and W. Jones, CrystEngComm, 2009, 11, 418–426 RSC.
- N. Shan, F. Toda and W. Jones, Chem. Commun., 2002, 2372–2373 RSC.
- D. Hasa, E. Miniussi and W. Jones, Cryst. Growth Des., 2016, 16, 4582–4588 CAS.
- (a) J. D. Egbert, C. S. J. Cazin and S. P. Nolan, Catal. Sci. Technol., 2013, 3, 912 RSC; (b) J. C. Y. Lin, R. T. W. Huang, C. S. Lee, A. Bhattacharyya, W. S. Hwang and I. J. B. Lin, Chem. Rev., 2009, 109, 3561 CrossRef CAS PubMed; (c) F. Lazreg, F. Nahra and C. S. J. Cazin, Coord. Chem. Rev., 2015, 293–294, 48 CrossRef CAS; (d) S. Díez-González and S. Nolan, Synlett, 2007, 2158 CrossRef.
- (a) B. Liu, X. Ma, F. Wu and W. Chen, Dalton Trans., 2015, 44, 1836 RSC; (b) D. N. Barsoum, N. Okashah, X. Zhang and L. Zhu, J. Org. Chem., 2015, 80, 9542 CrossRef CAS PubMed; (c) B. R. M. Lake, E. K. Bullough, T. J. Williams, A. C. Whitwood, M. A. Little and C. E. Willans, Chem. Commun., 2012, 48, 4887 RSC.
- A. Beillard, T.-X. Métro, X. Bantreil, J. Martinez and F. Lamaty, Chem. Sci., 2017, 8, 1086 RSC.
- (a) T. W. Lyons and M. S. Sanford, Chem. Rev., 2010, 110, 1147–1169 CrossRef CAS PubMed; (b) H. M. L. Davies, J. Du Bois and J.-Q. Yu, Chem. Soc. Rev., 2011, 40, 1855–1856 RSC.
- J. G. Hernández, Chem.–Eur. J., 2017, 1–10 Search PubMed.
- M. Juribašić, K. Užarević, D. Gracin and M. Ćurić, Chem. Commun., 2014, 50, 10287 RSC.
- G. N. Hermann, P. Becker and C. Bolm, Angew. Chem., Int. Ed., 2016, 55, 3781 CrossRef CAS PubMed.
- D. Lee, Y. Kim and S. Chang, J. Org. Chem., 2013, 78, 11102 CrossRef CAS PubMed.
- Y. Yang, J. Lan and J. You, Chem. Rev., 2017, 117, 8787–8863 CrossRef CAS PubMed.
- S.-J. Lou, Y.-J. Mao, D.-Q. Xu, J.-Q. He, Q. Chen and Z.-Y. Xu, ACS Catal., 2016, 6, 3890 CrossRef CAS.
- (a) H. Xu, M. Shang, H.-X. Dai and J.-Q. Yu, Org. Lett., 2015, 17, 3830 CrossRef CAS PubMed; (b) C. S. Yeung, X. Zhao, N. Borduas and V. M. Dong, Chem. Sci., 2010, 1, 331 RSC; (c) F. Yang, F. Song, W. Li, J. Lan and J. You, RSC Adv., 2013, 3, 9649 RSC.
- Z.-J. Jiang, Z.-H. Li, J.-B. Yu and W.-K. Su, J. Org. Chem., 2016, 81, 10049–10055 CrossRef CAS PubMed.
- (a) F. Proutiere and F. Schoenebeck, Angew. Chem., Int. Ed., 2011, 50, 8192 CrossRef CAS PubMed; (b) A. F. Littke and G. C. Fu, Angew. Chem., Int. Ed., 1998, 37, 3387 CrossRef CAS.
- J. L. Howard, Y. Sagatov, L. Repusseau, C. Schotten and D. L. Browne, Green Chem., 2017, 41, 413 Search PubMed.
- J. L. Howard, Y. Sagatov and D. L. Browne, Tetrahedron, 2017 DOI:10.1016/j.tet.2017.11.066.
- Y. Wang, H. Wang, Y. Jiang, C. Zhang, J. Shao and D. Xu, Green Chem., 2017, 19, 1674–1677 RSC.
- I. McCort-Tranchepain, M. Petit and P. I. Dalko, in Handbook of Green Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2010, pp. 255–318 Search PubMed.
- B. Rodríguez, A. Bruckmann and C. Bolm, Chem.–Eur. J., 2007, 13, 4710 CrossRef PubMed.
- A. I. Nyberg, A. Usano and P. M. Pihko, Synlett, 2004, 1891–1896 CAS.
- (a) J. G. Hernández and E. Juaristi, J. Org. Chem., 2011, 76, 1464 CrossRef PubMed; (b) J. G. Hernández and E. Juaristi, Tetrahedron, 2011, 67, 6953 CrossRef; (c) J. G. Hernández, V. García-López and E. Juaristi, Tetrahedron, 2012, 68, 92 CrossRef.
- (a) P. Caravan, J. J. Ellison, T. J. McMurry and R. B. Lauffer, Chem. Rev., 1999, 99, 2293 CrossRef CAS PubMed; (b) P. Caravan, Acc. Chem. Res., 2009, 42, 851 CrossRef CAS PubMed; (c) C. M. Fisher, E. Fuller, B. P. Burke, V. Mogilireddy, S. J. A. Pope, A. E. Sparke, I. Déchamps-Olivier, C. Cadiou, F. Chuburu, S. Faulkner and S. J. Archibald, Dalton Trans., 2014, 43, 9567 RSC; (d) M. Tropiano, C. J. Record, E. Morris, H. S. Rai, C. Allain and S. Faulkner, Organometallics, 2012, 31, 5673 CrossRef CAS; (e) R. Smith, D. Huskens, D. Daelemans, R. E. Mewis, C. D. Garcia, A. N. Cain, T. N. C. Freeman, C. Pannecouque, E. D. Clercq, D. Schols, T. J. Hubin and S. J. Archibald, Dalton Trans., 2012, 41, 11369 RSC; (f) A. Khan, J. D. Silversides, L. Madden, J. Greenman and S. J. Archibald, Chem. Commun., 2007, 42, 416 RSC; (g) T. J. Hubin, P. N. A. Amoyaw, K. D. Roewe, N. C. Simpson, R. D. Maples, T. N. Carder Freeman, A. N. Cain, J. G. Le, S. J. Archibald, S. I. Khan, B. L. Tekwani and M. O. F. Khan, Bioorg. Med. Chem., 2014, 22, 3239 CrossRef CAS PubMed; (h) T. J. Hubin, J. M. McCormick, S. R. Collinson, M. Buchalova, C. M. Perkins, N. W. Alcock, P. K. Kahol, A. Raghunathan and D. H. Busch, J. Am. Chem. Soc., 2000, 122, 2512 CrossRef CAS.
- B. H. Abdulwahaab, B. P. Burke, J. Domarkas, J. D. Silversides, T. J. Prior and S. J. Archibald, J. Org. Chem., 2016, 81, 890 CrossRef CAS PubMed.
- E. H. Wong, G. R. Weisman, D. C. Hill, D. P. Reed, M. E. Rogers, J. S. Condon, M. A. Fagan, J. C. Calabrese, K.-C. Lam, I. A. Guzei and A. L. Rheingold, J. Am. Chem. Soc., 2000, 122, 10561 CrossRef CAS.
- G. W. Huber, S. Iborra and A. Corma, Chem. Rev., 2006, 106, 4044 CrossRef CAS PubMed.
- (a) X. Chen, H. Yang, Z. Zhong and N. Yan, Green Chem., 2017, 19, 2783–2792 RSC; (b) T. Kleine, J. Buendia and C. Bolm, Green Chem., 2013, 15, 160 RSC; (c) S. M. Hick, C. Griebel, D. T. Restrepo, J. H. Truitt, E. J. Buker, C. Bylda and R. G. Blair, Green Chem., 2010, 12, 468 RSC; (d) N. Meine, R. Rinaldi and F. Schüth, ChemSusChem, 2012, 5, 1449 CrossRef CAS PubMed; (e) F. Schüth, R. Rinaldi, N. Meine, M. Käldström, J. Hilgert and M. D. K. Rechulski, Catal. Today, 2014, 234, 24 CrossRef; (f) F. Boissou, N. Sayoud, K. De Oliveira Vigier, A. Barakat, S. Marinkovic, B. Estrine and F. Jérôme, ChemSusChem, 2015, 8, 3263 CrossRef CAS PubMed; (g) M. D. Kaufman Rechulski, M. Käldström, U. Richter, F. Schüth and R. Rinaldi, Ind. Eng. Chem. Res., 2015, 54, 4581 CrossRef CAS; (h) F. Jérôme, G. Chatel and K. De Oliveira Vigier, Green Chem., 2016, 18, 3903 RSC.
- S. L. Collom, P. T. Anastas, E. S. Beach, R. H. Crabtree, N. Hazari and T. J. Sommer, Tetrahedron Lett., 2013, 54, 2344 CrossRef CAS.
- J. G. Hernández, N. A. J. Macdonald, C. Mottillo, I. S. Butler and T. Friščić, Green Chem., 2014, 16, 1087 RSC.
- (a) L. Cheng and N. J. Coville, Organometallics, 1996, 15, 867 CrossRef CAS; (b) R. B. King, R. H. Reimann and D. J. Darensbourg, J. Organomet. Chem., 1975, 93, C23 CrossRef CAS; (c) R. B. King and R. H. Reimann, Inorg. Chem., 1976, 15, 179 CrossRef CAS.
- A. M. Belenguer, T. Friščić, G. M. Day and J. K. M. Sanders, Chem. Sci., 2011, 2, 696 RSC.
- Y. X. Shi, K. Xu, J. K. Clegg, R. Ganguly, H. Hirao, T. Friščić and F. García, Angew. Chem., Int. Ed., 2016, 55, 12736 CrossRef CAS PubMed.
- (a) M. S. Balakrishna, D. Suresh and J. T. Mague, Inorg. Chim. Acta, 2011, 372, 259 CrossRef CAS; (b) T. Roth, H. Wadepohl, D. S. Wright and L. H. Gade, Chem.–Eur. J., 2013, 19, 13823–13837 CrossRef CAS PubMed.
- H. Klare, J. M. Neudörfl and B. Goldfuss, Beilstein J. Org. Chem., 2014, 10, 224 CrossRef PubMed.
- D. Suresh, M. S. Balakrishna, K. Rathinasamy, D. Panda and S. M. Mobin, Dalton Trans., 2008, 241, 2812 RSC.
- O. J. Scherer, K. Andres, C. Krüger, Y. H. Tsay and G. Wolmerhäser, Angew. Chem., Int. Ed., 1980, 19, 571 CrossRef.
- J. K. Brask, T. Chivers, M. L. Krahn and M. Parvez, Inorg. Chem., 1999, 38, 290 CrossRef CAS.
- (a) D. A. Fulmer, W. C. Shearouse, S. T. Medonza and J. Mack, Green Chem., 2009, 11, 1821 RSC; (b) T. L. Cook, J. A. Walker and J. Mack, Green Chem., 2013, 15, 617 RSC; (c) L. Chen, M. O. Bovee, B. E. Lemma, K. S. M. Keithley, S. L. Pilson, M. G. Coleman and J. Mack, Angew. Chem., Int. Ed., 2015, 54, 11084 CrossRef CAS PubMed.
- R. A. Haley, A. R. Zellner, J. A. Krause, H. Guan and J. Mack, ACS Sustainable Chem. Eng., 2016, 4, 2464–2469 CrossRef CAS.
- S. K. Rodrigo, I. V. Powell, M. G. Coleman, J. A. Krause and H. Guan, Org. Biomol. Chem., 2013, 11, 7653 CAS.
- K.-Y. Jia, J.-B. Yu, Z.-J. Jiang and W.-K. Su, J. Org. Chem., 2016, 81, 6049 CrossRef CAS PubMed.
- (a) A. R. Katritzky, S. Ledoux, R. M. Witek and S. K. Nair, J. Org. Chem., 2004, 69, 2976 CrossRef CAS PubMed; (b) A. R. Katritzky, R. M. Witek, V. Rodriguez-Garcia, P. P. Mohapatra, J. W. Rogers, J. Cusido, A. A. A. Abdel-Fattah and P. J. Steel, J. Org. Chem., 2005, 70, 7866 CrossRef CAS PubMed.
- V. Štrukil, D. Gracin, O. V. Magdysyuk, R. E. Dinnebier and T. Friščić, Angew. Chem., Int. Ed., 2015, 54, 8440 CrossRef PubMed.
- W. Yan, S. M. Seifermann, P. Pierrat and S. Bräse, Org. Biomol. Chem., 2015, 13, 25 CAS.
- S.-E. Zhu, F. Li and G.-W. Wang, Chem. Soc. Rev., 2013, 42, 7535 RSC.
- (a) G.-W. Wang, K. Komatsu, Y. Murata and M. Shiro, Nature, 1997, 387, 583 CrossRef CAS; (b) K. Komatsu, G.-W. Wang, Y. Murata, T. Tanaka, K. Fujiwara, K. Yamamoto and M. Saunders, J. Org. Chem., 1998, 63, 9358 CrossRef CAS.
- K. Komatsu, K. Fujiwara and Y. Murata, Chem. Lett., 2000, 1016 CrossRef CAS.
- M. Keshavarz-K, B. Knight, G. Srdanov and F. Wudl, J. Am. Chem. Soc., 1995, 117, 11371 CrossRef CAS.
- Y.-T. Su and G.-W. Wang, Org. Lett., 2013, 15, 3408 CrossRef CAS PubMed.
- D. Tan, V. Štrukil, C. Mottillo and T. Friščić, Chem. Commun., 2014, 50, 5248 RSC.
- D. Tan, C. Mottillo, A. D. Katsenis, V. Štrukil and T. Friščić, Angew. Chem., Int. Ed., 2014, 53, 9321 CrossRef CAS PubMed.
- J.-B. Yu, Y. Zhang, Z.-J. Jiang and W.-K. Su, J. Org. Chem., 2016, 81, 11514 CrossRef CAS PubMed.
- N. R. Rightmire, T. P. Hanusa and A. L. Rheingold, Organometallics, 2014, 33, 5952–5955 CrossRef CAS.
- J. H. Chong and M. J. MacLachlan, Chem. Soc. Rev., 2009, 38, 3301 RSC.
- Y. Zhao, S. V. Rocha and T. M. Swager, J. Am. Chem. Soc., 2016, 138, 13834 CrossRef CAS PubMed.
- Ball mills are used throughout the sciences for powdering materials into close to uniform particle sizes, for example you may find them in laboratories concerned with formulation science, forensic science, geology, earth science, food science – there is likely to be one already in your institute, company or university.
| This journal is © The Royal Society of Chemistry 2018 |