
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceC4–H indole functionalisation: precedent and prospects
Jagadeesh
Kalepu
a,
Parthasarathy
Gandeepan
 b,
Lutz
Ackermann
*b and
Lukasz T.
Pilarski
*a
b,
Lutz
Ackermann
*b and
Lukasz T.
Pilarski
*a
aDepartment of Chemistry – BMC, Uppsala University, Box 576, 75-123 Uppsala, Sweden. E-mail: Lukasz.Pilarski@kemi.uu.se; Web: https://www.pilarskigroup.org/
bInstitut für Organische und Biomolekulare Chemie, Georg-August-Universität Göttingen, Tammannstraße 2, 37077 Goettingen, Germany. E-mail: Lutz.Ackermann@chemie.uni-goettingen.de
First published on 20th April 2018
Abstract
C4-decorated indoles feature in a plethora of bioactive and functional compounds of importance to natural product synthesis, material sciences, as well as crop protection and pharmaceutical industries. Traditionally, their syntheses largely involved harsh stoichiometric metalations and radical reactions. However, transition metal catalysed C–H activation has recently evolved into a powerful strategy for the late-stage diversification of indoles at the C4–H position. Modern photoredox, enzymatic and precious transition metal catalysis represent the key stimuli for developing challenging C–C and C–Het bond forming transformations under mild reaction conditions. Herein, we discuss the evolution and application of these methods for the step-economical transformations of otherwise inert C4–H bonds up to December 2017.
1. Introduction
The ubiquity of carbon–hydrogen (C–H) bonds in organic compounds makes their selective substitution, in principle, one of the most efficient approaches to building molecular complexity. Recent years have witnessed an explosion of interest in the development of new transformations for this purpose. Consequently, C–H functionalisation has become firmly established at a frontier of synthetic methodology. It offers expedited, more efficient and previously impossible routes to a very wide variety of high-value compounds.1 Advances in this area continue apace.Indoles – which feature in many bioactive and functional molecules2 – have come to serve as staple substrates in this context; the search for methods to functionalise the six distinct C–H bonds of indole has been the subject of many studies.3 However, highly regioselective approaches to transform C4–H, indole's least intrinsically reactive site, have been notably few in number,3a–d despite the presence of C4-substituted indole motifs in many compounds of importance to chemical research (Fig. 1).
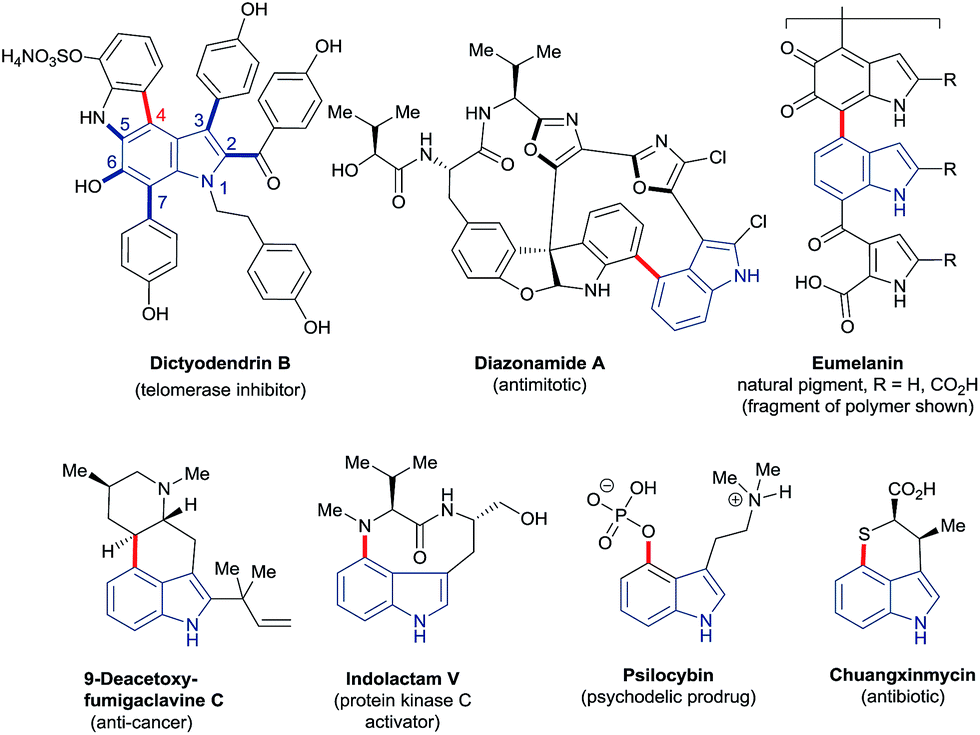 | ||
| Fig. 1 Examples of biologically relevant molecules based on the indole motif (blue) with a C4–heteroatom, C4–C(sp3) or C4–C(sp2) bond (red). | ||
The vexed relationship between the C4–H of indole and C–H functionalisation technology is exemplified by – though by no means limited to – the synthesis of the natural product dictyodendrin B (Fig. 1). Elegant recent work has shown that all but one of the bonds around its indole nucleus (C2, C3, and C5–7 – shown in blue) may be constructed using catalytic C–H functionalisation strategies.4 The remaining C4–aryl bond, by contrast (shown in red), forces disconnections to pre-functionalised substrates or requires stoichiometric palladium to assemble. More generally, there are few methods that deliver heteroatoms directly to the C4 position of indoles, despite the prominence of C4–heteroatom units in many high-value compounds. Of these, the majority are based on the use of toxic metals in stoichiometric amounts or otherwise on radical reactions with rather modest scope. The invention of new reaction systems able to overcome the comparative recalcitrance of the C4–H bond is, therefore, a highly attractive prospect.
Very recently, intriguing advances in this area have taken place. Transition metal-, enzyme-, radical- and photoredox-based approaches have suddenly made progress against indole's previously intransigent C4–H site at an unprecedented rate, expanding the scope of available transformations. This points to a bright future, not least because the progress has occurred without any strong convergence in terms of methods, metals or mechanisms. Herein, we trace the evolution of approaches to substituting indole's C4–H bond, from their early days to the current state-of-the-art. Where possible, we highlight connections in scope and the underlying principles between the new methods and the more established reactions from which they seemingly often take a cue. Various examples appear to point the way to promising new ground for prospecting.
2. Stoichiometric metalation
Early strategies for generating organometallic species reactive at the indole C4 position might seem outdated but are instructive to survey. Indeed, they often presage more advanced modern catalytic systems. Generally, one of three approaches has been pursued:(a) Selecting appropriate coordinating and/or electron-withdrawing groups, especially on the pyrrolic ring of the indole, to encourage electrophilic metalation, e.g. by salts of Tl3+ or Hg2+;
(b) Altering the reactivity of the benzenoid ring via π-complexation to an electron-withdrawing transition metal fragment, such as –Cr(CO)3;
(c) Exploiting substituents at C3 or C5 to effect ortho-directed lithiation.
The high toxicities of organo-thallium, -mercury and -chromium compounds undoubtedly preclude these methods from gaining broad acceptance. Nevertheless, such strategies offer an as-of-yet unsurpassed scope of available transformations and several seem to share their preferences, e.g. in terms of electronic or directing group effects, with more modern transition metal-catalysed reactions.
2.1 The early days: electrophilic thallation and mercuriation
Hollins and Colnago showed in 1979 that exposure of 3-carbonylindoles to Tl(TFA)3 in trifluoroacetic acid (TFA) yields isolable, exclusively C4-thallated compounds 1 (Scheme 1A). Regioselectivity was proposed to result from the carbonyl group's tempering of C2 nucleophilicity and its role as a directing group for the electrophilic Tl3+ centre. Compound 1 could be derivatised in various ways, including via iodination and reductive deuteration (2).5 3-Formyl-4-iodoindoles (2a) accessed this way underwent efficient nucleophilic aromatic substitution by alkoxy nucleophiles6 or photochemical arylation with benzene or furan7 and have been used even recently as intermediates in the construction of ergoline analogues.8 Several studies revealed that key intermediate 1 can couple via palladium catalysis with organosilanes,9 (hetero)aryl boronic acids7 or organostannanes10 (2c–h), albeit in moderate yields at best. Analogous C4 mercuriation using Hg(OAc)2 under acidic conditions has also been described (Scheme 1B), and proceeds with or without carbonyl substituents at the C3 position, but requires an electron-withdrawing 4-toluenesulfonyl (Ts) group on the indole nitrogen to proceed cleanly, again presumably to deactivate C2. Intermediate 4 was iodinated en route to Stille coupling product 5, a key intermediate for the natural product hapalindole J.11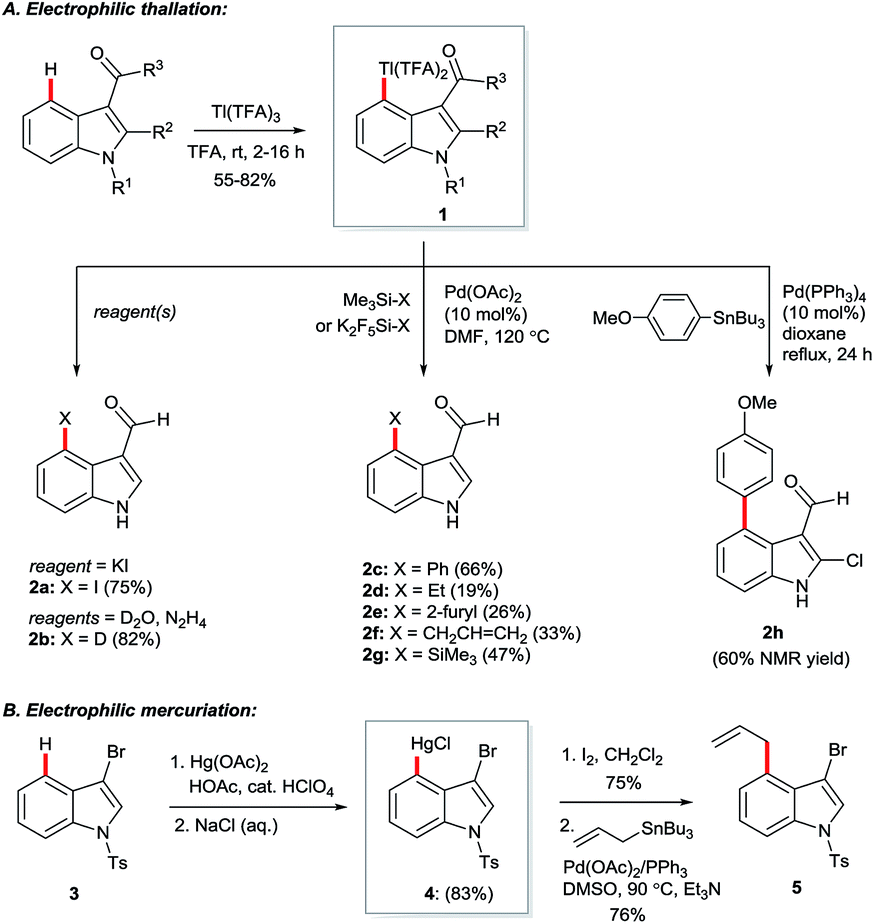 | ||
| Scheme 1 Early work on C(4)–H functionalisation via complexes of Tl and Hg. | ||
Several details in these early reports remain relevant to more recently developed systems. For example, the judicious choice of substituents at positions N1 and/or C3 to deactivate the pyrrolic ring is often of crucial importance for most transition metal-catalysed indole C4–H functionalisations. Yet more striking is the reliance of these methods specifically on carboxylate salts of thallium and mercury. The C–H mercuriations using Hg(TFA)2 were instrumental in early studies of C–H activation. Detailed mechanistic work led Roberts and co-workers to propose as early as 1980 that Ar–H mercuriation might proceed via a concerted 6-membered transition state (Fig. 2, 6a).12 Yet, it was only at the turn of the 21st century that the importance of analogous mechanisms in catalytic C–H functionalisation emerged (Fig. 2, 6b),13 eventually to include indole substrates.14 Innumerable modern transformations are now understood to proceed via concerted, carboxylate-assisted C–H metalation.15 The significance of such pathways has not yet been explicitly investigated for the C4–H activation of indoles.
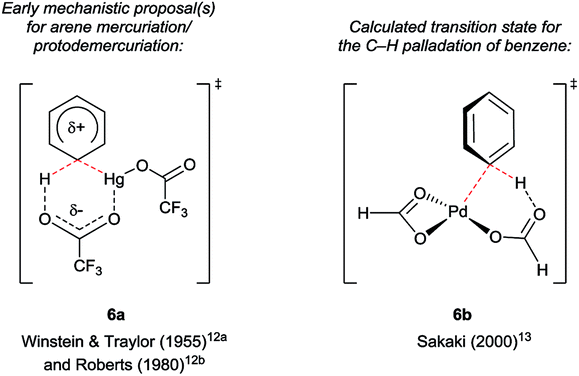 | ||
| Fig. 2 Transition states through time: proposals on the keys step in selected C–H bond activations involving carboxylate ligands. | ||
2.2 π-Complexation of indole's benzenoid ring
The π-complexation of (hetero)arenes to electron-withdrawing transition metal units can render their C–H bonds more kinetically acidic and their carbon centres more electrophilic.16 This strategy has been used only sparingly for indole derivatisation. Widdowson and co-workers showed that N-silyl indoles may be coordinated to the strongly electron-withdrawing –Cr(CO)3 fragment to give complex 7, which can undergo C4–H lithiation (8, Scheme 2A).17 Electrophilic trapping, decomplexation and desilylation gave various corresponding C4-substituted products 9. Isoelectronic complexes of manganese proved to be versatile electrophiles at the indole C4 position, but only dearomatised products were reported.18 Nearly two decades later, a related approach was used by Kamikawa and Uemura to install formyl groups (10 to 11) and effect the diastereoselective allylation of the benzenoid ring of indole to give dearomatised and axially chiral product 12a.19 The C4-substituted indole 12b was observed to form in near to equimolar amounts on failure of the allylation.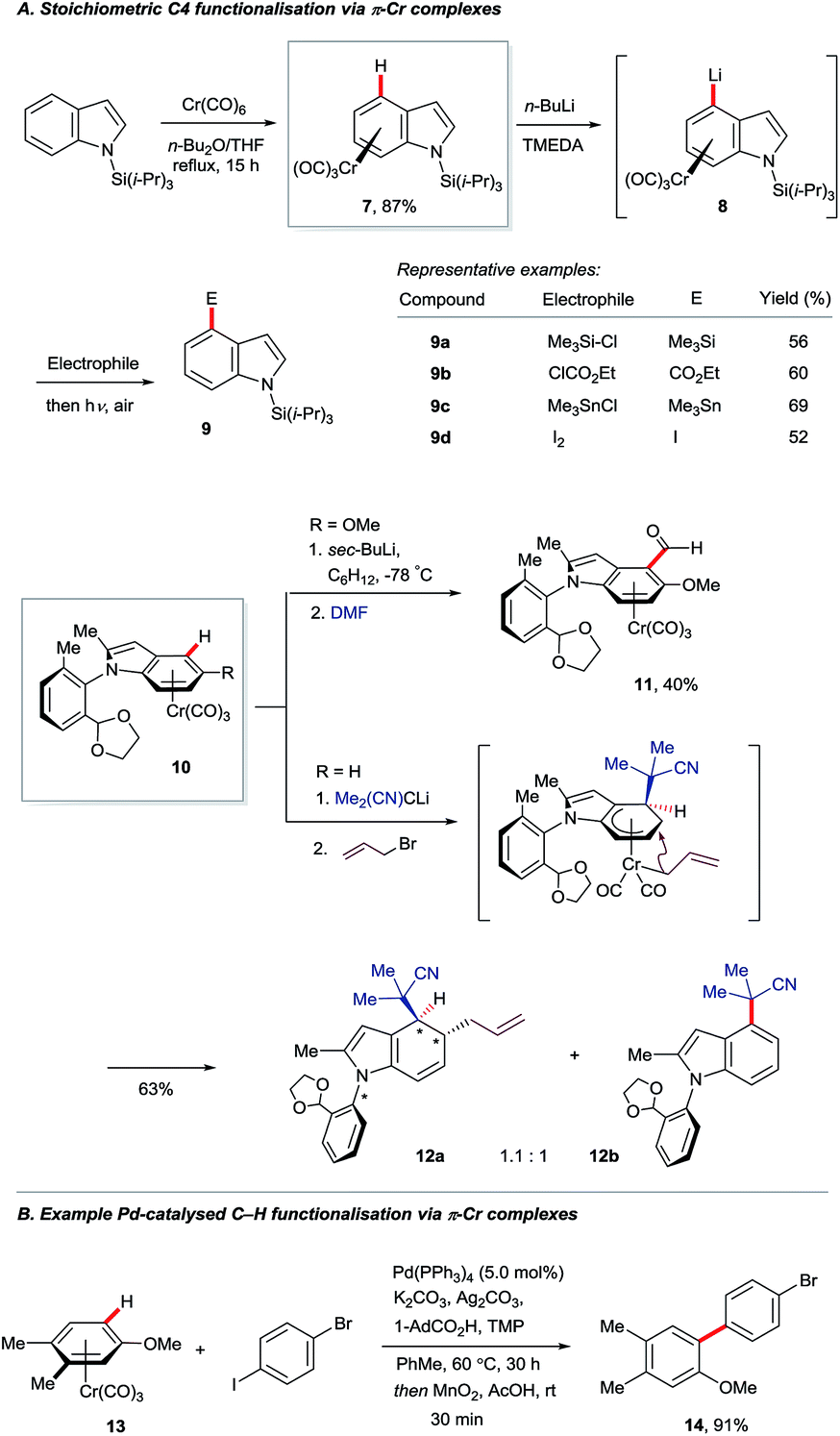 | ||
| Scheme 2 C–H functionalisations exploiting the reactivity of π–Cr complexes. | ||
Recent work by the Larrosa group has shown that η6-coordination of arenes to the –Cr(CO)3 motif can enhance by several orders of magnitude the rate of carboxylate-assisted Ar–H activation using a palladium(0) catalyst (Scheme 2B).20 Coordination to –Cr(CO)3 suppresses SEAr pathways for ordinarily electron-rich arenes and facilitates distortion of the C–H bond en route to 6-membered transition states analogous to those shown in Fig. 2. This has broadened the scope of catalytic C–H functionalisation, although indoles, to the best of our knowledge, are yet to benefit.
2.3 Directed lithiations and indolyne reactivity
Gramine is a naturally-occurring indole-based alkaloid whose dimethylaminomethylene substituent can serve as a removable directing group21 for C4 lithiation. On quarternisation, the exocyclic amine can undergo elimination–additions or, otherwise, may be replaced via a retro-Mannich pathway (Scheme 3A).22 Iwao and Ishibashi synthesised clavicipitic acid via the directed C4 lithiation of N-silyl gramine 15 to furnish intermediate 16. Subsequent electrophilic trapping gave the key alcohol intermediate 17 (Scheme 3B).23 Snieckus and co-workers demonstrated several derivatisations of 16, including its trapping with ZnBr2 followed by Negishi coupling. Mild and rapid C3 bromination of the resulting biaryl 18 gave product 19. Intermediates 16 could also undergo halogenation, hydroxylation, formylation and silylation.22b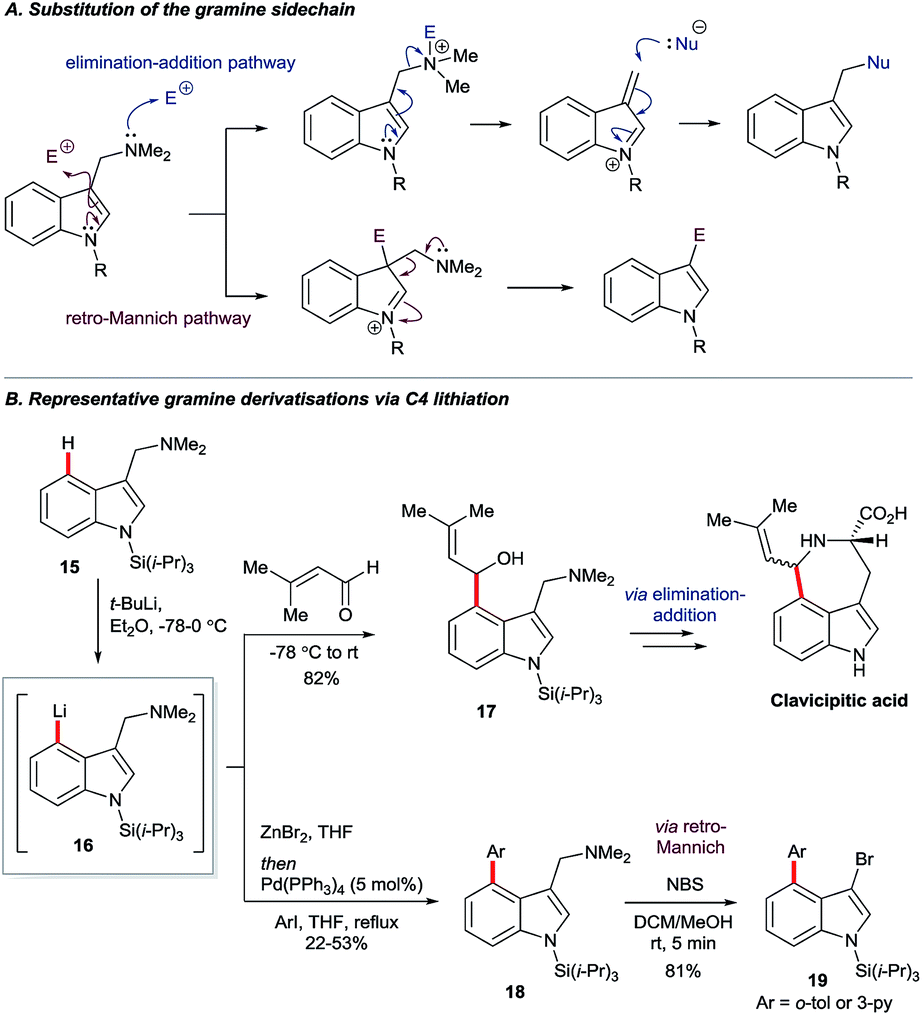 | ||
| Scheme 3 Stoichiometric C3 and C4 gramine derivatisations. | ||
Garg and co-workers reported the cine substitution of indolyl carbamates 20via their C4 lithiation (Scheme 4A).24 The resulting intermediates could be trapped, e.g. by electrophilic borylation (21), and the carbamate directing group could be removed with the aid of reductive nickel catalysis. Boronate 21 could further be coupled in Suzuki and Chan–Lam protocols to give 22a and 22b, respectively. A similar strategy based on directed lithiation at C4 was used by the Garg group in the synthesis of 4,5-indolyne precursors 24, this time with the use of Me3Si–Cl as the electrophile (Scheme 4A, transformation of 20 to 24).25
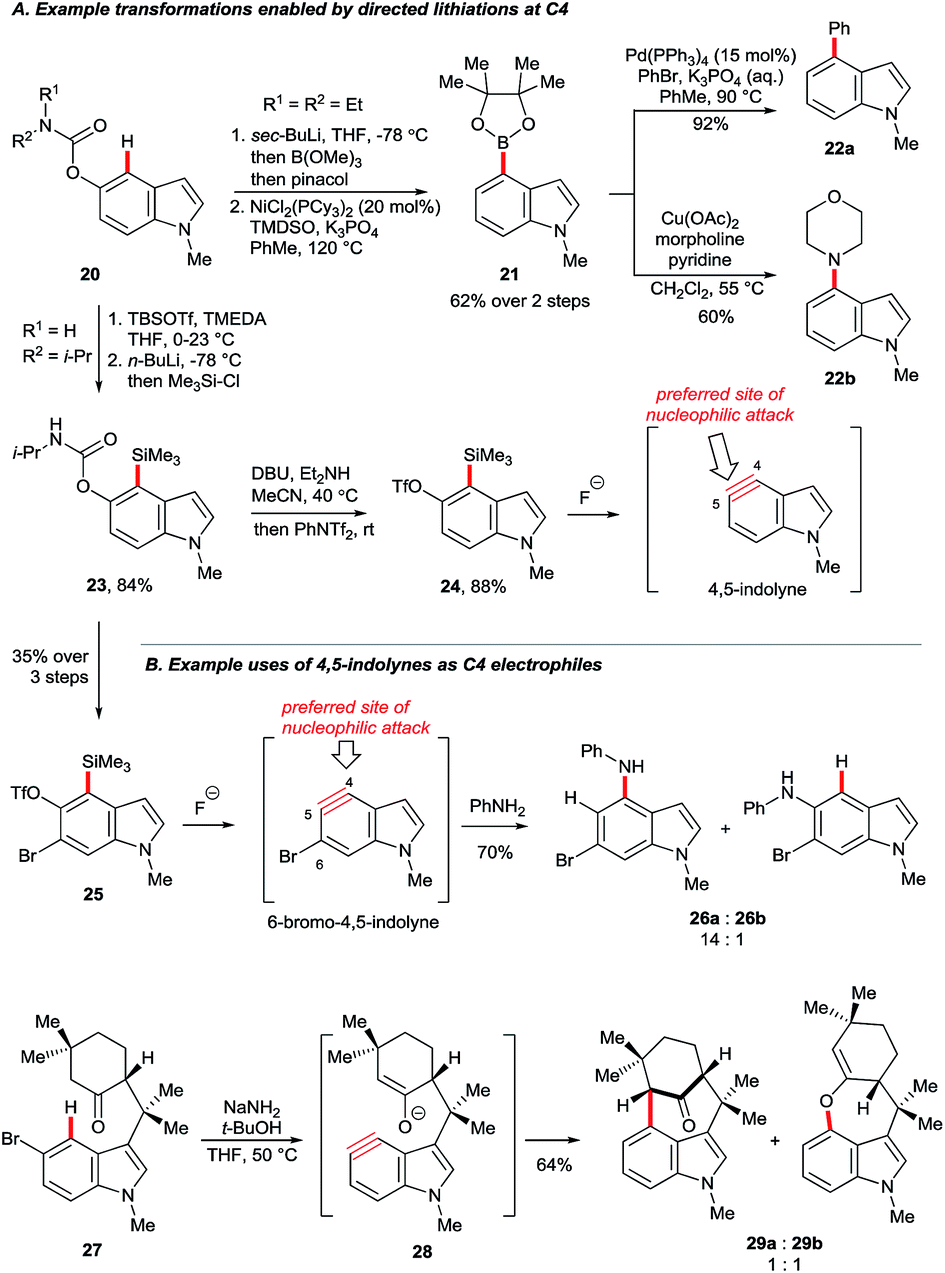 | ||
| Scheme 4 Transformations enabled by deprotonation of the C4–H. | ||
This advance worthy of note for the extraordinary synthetic versatility it unleashes. Arynes generated from fluoride-activated precursors admit a wide variety of carbon- and/or heteroatom-based groups, generally under mild conditions and with controlled regioselectivity.26 4,5-Indolyne undergoes nucleophilic attack preferentially at its more linear carbon, C5.27 However, this can be overturned by using substituent effects. Thus, precursor 25 (Scheme 4B), obtained from carbamate 23via an additional directed bromination, gave 6-bromo-4,5-indolyne upon exposure to fluoride. The electronic influence of the bromide in 6-bromo-4,5-indolyne renders C4 more linear than C5, thereby switching the aryne's preferred regioselectivity (products 26a and 26b).28 Nucleophiles may also be tethered such that they reach the C4 position, but not C5. For example, deprotonation of aryl bromide 27 at C4 leads to elimination to furnish the indolyne 28. Its attack by the enolate moiety was used to construct the N-methylwelwitindolinone C isothiocyanate scaffold 29a.29
2.4 Stoichiometric oxidative C4–H functionalisation
3-Acyl or 3-formyl indoles undergo SEAr reactions in poor yields and with low regioselectivity.30 The oxidative, manganese-mediated cyclisation of substrate 30 to arylated ketone 31 is a rare example exploiting C4 as an electron-rich site in the absence of strong electronically-biasing groups on the indole (Scheme 5).31 Regioselectivity in this instance appears to be assisted strongly by the blocking substituent at C2. However, the significance of acetate in these and closely related reactions32 has not been specifically investigated. It is noteworthy that recent work on manganese-catalysed indole C–H functionalisation33 has found that carboxylates can play a crucial role during the C–H metalation step.34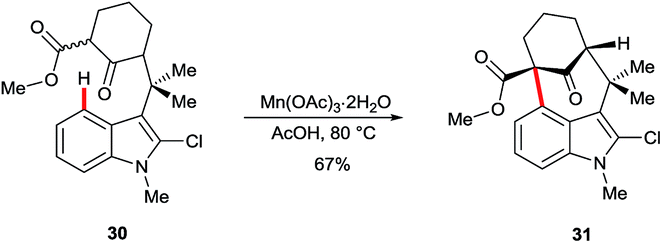 | ||
| Scheme 5 Oxidative cyclisation at C4 using stoichiometric Mn. | ||
3. Stoichiometric radical reactions
Arguably, the most spectacular C4–H indole functionalisation via a radical pathway is the Witkop cyclisation.35 This photochemically-induced intramolecular reaction of α-haloacetamides is able to close 7, 8 or 9-membered cyclic lactams with good C4 regioselectivity, even in the absence of N1 or C2 substituents. Such remarkable features have fuelled interest in the Witkop cyclisation as a strategy in complex natural product synthesis (e.g., that of diazonamide A36 – see Fig. 1). The transformation occurs most efficiently with electron-rich indole substrates, although typical yields are moderate. The most widely accepted mechanism is depicted in Fig. 3. The indole chromophore undergoes photon-induced electron transfer (PET), subsequent loss of a chloride anion, radical cyclisation and deprotonation to deliver the aromatic product. The Witkop cyclisation has been reviewed recently by Gaich and co-workers.3d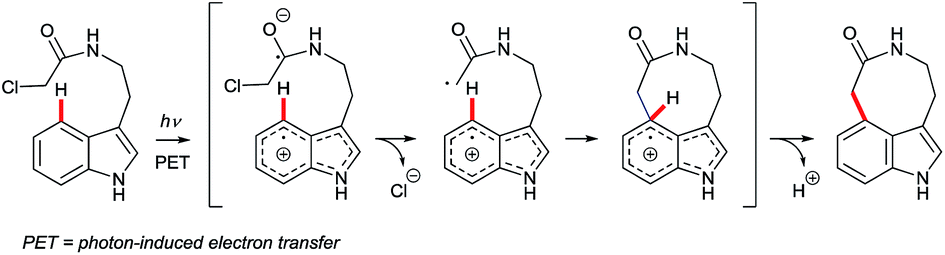 | ||
| Fig. 3 Mechanism of the Witkop cyclisation. | ||
Yi and Xiu disclosed a radical-based cross dehydrogenative coupling (CDC) of indoles and simple (cyclo)alkanes using di-tert-butyl peroxide (DTBP) as the oxidant (Scheme 6).37 C5-Substituted indoles showed a strong preference for C4 selectivity to give products 32, while C6-substituted substrates (not shown here) gave both C4- and C7-alkylation, with a slight preference for the latter.
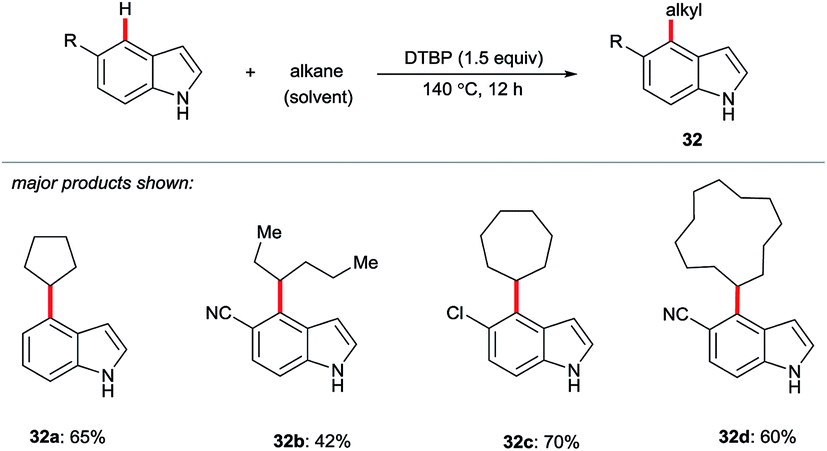 | ||
| Scheme 6 DTBP-mediated C4-selective alkylation of indoles. | ||
4. Transition metal-catalysed C–H functionalisation
The greatest number of recent advances in the functionalisation of the C4–H bond of indoles have come from transition metal catalysis.3a,3b The majority of these use a directing group (DG) at the C3 position. Interestingly, weakly-coordinating groups, such as aldehydes, ketones and amides, have proven most successful. These may be expected also to facilitate C2–H functionalisations. Thus, the most interesting cases for the purposes of this perspective are those in which the C4–H position is substituted preferentially in potential competition with C2–H. Many such transformations presumably proceed via 6-membered metallacycle intermediates of type 33a (Fig. 4), rather than the corresponding 5-membered metallacycles 33b, despite what must be inherently greater ring strain in the former. Hitherto, mechanistic insights into the principles behind such regioselectivity have been sparse.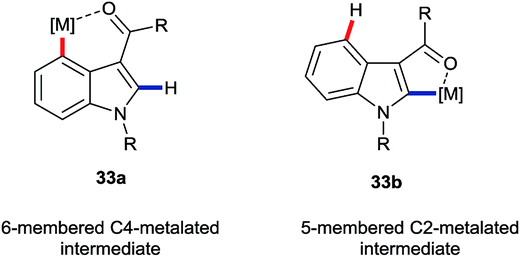 | ||
| Fig. 4 Putative metallacyclic intermediates arising from carbonyl-group directed C4–H and C2–H activation. | ||
Explicit comparisons between stoichiometric and catalytic C4–H functionalisations of indoles are rare in the literature. It seems probable, however, that some of the same factors are key to success for both. For instance, carbonyl-based DGs and high-valent, electrophilic and/or carboxylate metal salts seem at least to hint at the importance of deactivating indole's pyrrolic ring and priming C4–H for electrophilic attack or carboxylate-assisted concerted C–H activation. In the course of developing new reactions, several groups have reported unusual mechanistic data which may yet serve to expand considerably the range of possible indole C4–H functionalisations.
4.1 Ruthenium
In 2013, Prabhu and co-workers reported a regioselective C4–H alkenylation of 3-formyl indoles under ruthenium(II) catalysis (Scheme 7A).38 The reaction was accomplished with 0.5 equivalents of Cu(OAc)2·H2O under aerobic reaction conditions. A variety of activated alkenes, such as acrylates, vinyl ketone, acrylonitrile, and styrenes were effective, giving the desired products 35 in moderate to good yields. C4-Alkenyl indoles were the major products from competition experiments between 3-formyl and N-benzoyl directing groups, on the same or separate substrates (Scheme 7B). The Prabhu group also observed that, under very closely-related conditions, the electronic properties of carbonyl-based directing groups had a major influence on the site-selectivity of the C–H activation.39 For example, deuterium incorporation studies revealed that H/D exchange at C4 was much more extensive than at C2 for a 3-trifluoroacetyl indole compared to its 3-acetyl counterpart (see Section 4.2.1). This was true for both [RuCl2(p-cymene)]2 and [Cp*RhCl2]2 catalyst precursors (Scheme 8), although, in contrast to 3-formyl indoles, only [Cp*RhCl2]2 gave C4-alkenylated products for this specific substrate class.39 The authors reasoned for these observations that the trifluoroacetyl group may be unable to coordinate the metal centre prior to C–H activation, and that instead it might stabilise the σ-complex only after metalation. The formyl directing group can be removed following the C–H functionalisation.21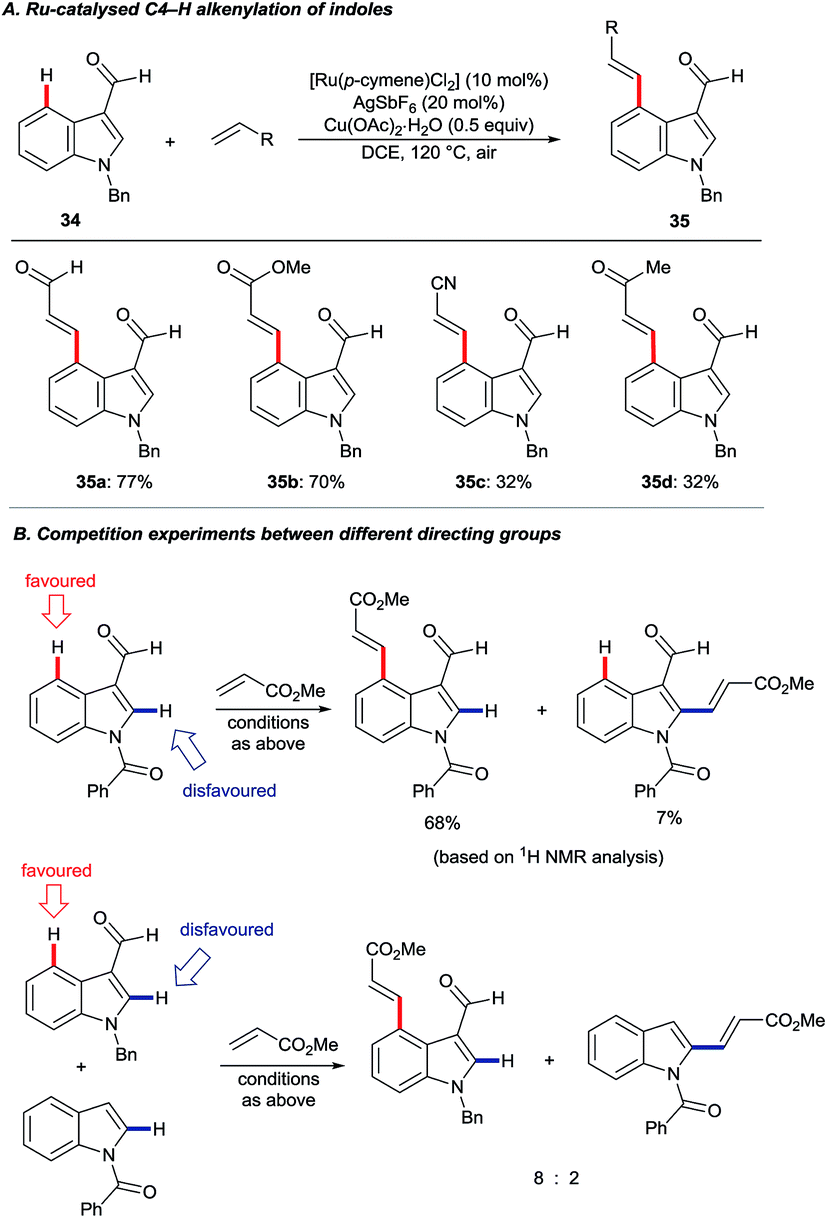 | ||
| Scheme 7 Ru(II)-catalysed C4 alkenylation of 3-formyl indoles. | ||
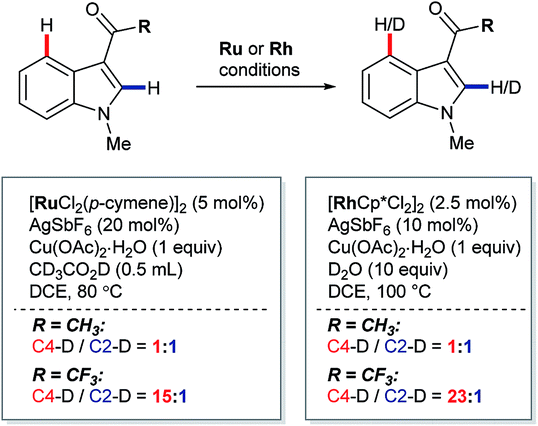 | ||
| Scheme 8 Ru(II)- and Rh(III)-catalysed C2– and C4–H/D exchange observed for electronically distinct directing groups. | ||
The use of ortho-directing groups has become the preeminent strategy for steering site selectivity in the transition metal-catalysed C–H functionalisation of (hetero)arenes.21,40 The search for complementary methods to functionalise aromatic C–H bonds meta or para with respect to a directing group has, accordingly, attracted growing levels of interest in recent years.41 Advances in this area are represented by the key findings of Frost and Ackermann on meta-selective ruthenium-catalysed C–H functionalisation.42 A range of related meta-selective C–H transformations has been disclosed since (represented by the transformation shown in Scheme 9), along with a growing body of evidence that the involved ruthenacyclic intermediates undergo functionalisation para to the C–Ru bond (meta to the directing group) via single electron transfer processes.43
 | ||
| Scheme 9 General schematic for net meta-selective C–H functionalisation of arenes via ruthenium σ-complex activation. | ||
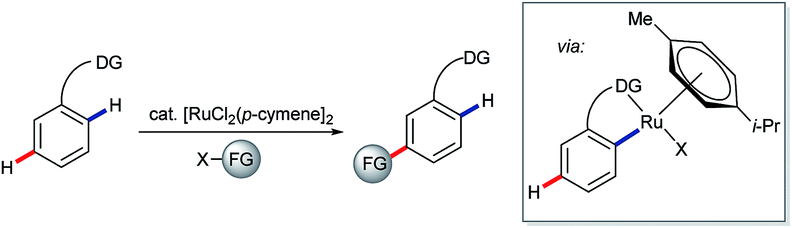
On extending their studies to indole substrates 36, the Frost group developed an elegant, catalytic C6–H selective alkylation (Scheme 10A).44 Calculated Fukui indices suggested that C6 was the most nucleophilic available position on the proposed C2-ruthenated intermediate 37b; the parent pyrimidyl indole 37a was most nucleophilic at C3 (C6 was the 5th most nucleophilic site in 37a). A further intriguing finding, however, was that C7 ruthenation would be expected to render C4 the most nucleophilic position overall in complex 37c, raising the exciting prospect of functionalising indole at C4 via the activation of remote C–H bonds.45 Frost and co-workers subsequently revealed that this strategy is effective for the analogous C4–H derivatisation of carbazoles.46
 | ||
| Scheme 10 Catalytic C6–H functionalisation of indole is enabled by C2–H ruthenation; calculations suggest analogous C4–H activation may be possible. | ||
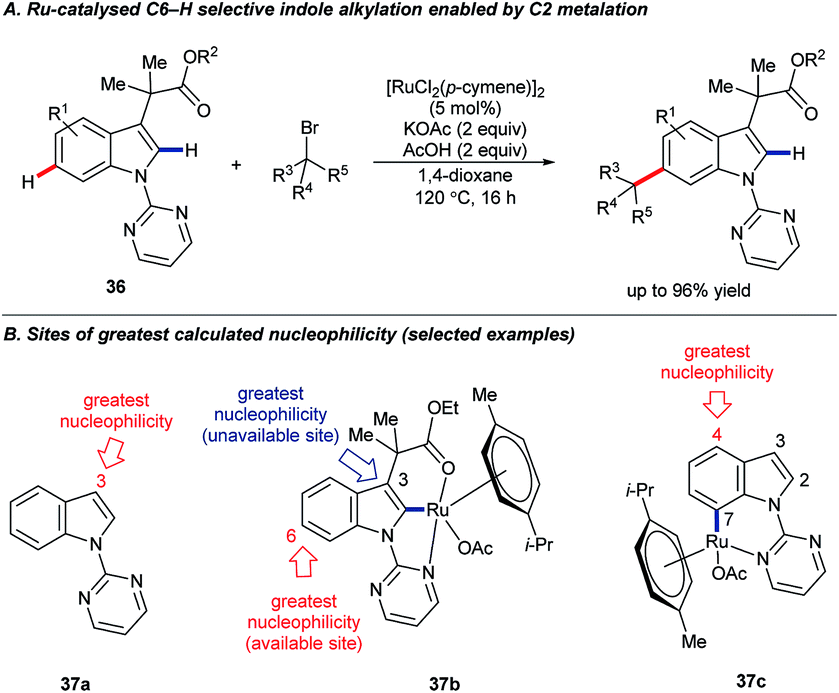
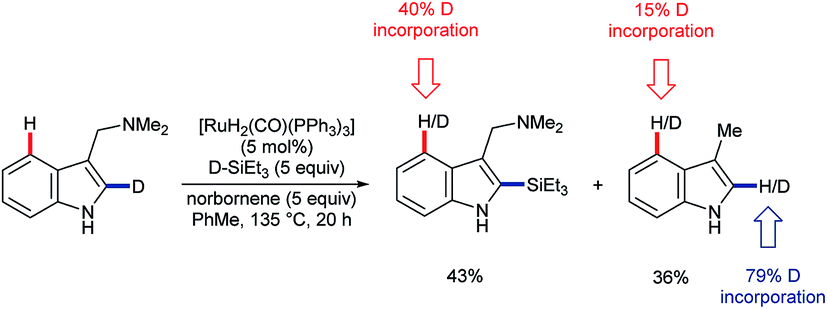
In 2016, Pilarski and co-workers reported the ruthenium-catalysed C2–H silylation of gramines.47 The reaction used electron-rich ruthenium catalysts to avoid cleavage of the directing group; electrophilic metals are known to cleave the exocyclic amine48 (cf.Scheme 3A). Significant C4–H/D exchange occurred when the reaction was conducted with deuterated versions of either or both coupling partners (Scheme 11). Although C4–H silylation was not observed, and C2–H activation was always preferred, the activation of the C4–H bond by electron-rich ruthenium species, presumably via oxidative addition, is unique. Greater levels of C4–H/D exchange found in C2-silylated products are consistent with the silane acting as a blocking group and perhaps helping to position the directing group and Ru centre over the C4 position. The C2–H selective silylation of tryptamines was also efficient, but they did not undergo the analogous C4–H/D exchange.47
 | ||
| Scheme 11 Selected aspects of C–H/D exchange observed in the silylation of gramines catalysed by electron-rich Ru species. | ||
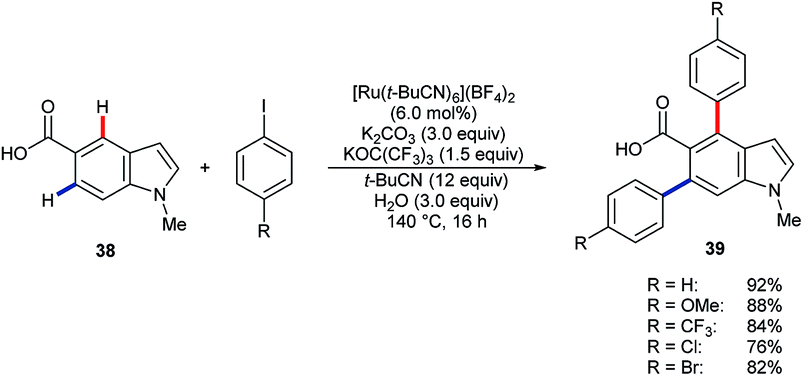
Larrosa and co-workers have shown that [Ru(t-BuCN)6](BF4)2 catalyses the efficient diarylation of carboxylic acid 38 to give products 39 (Scheme 12).49 Silver(I) or copper(II) additives, as included in many other systems, were not required. However, C2–H or C4–H arylation did not take place for the corresponding 3-indole carboxylic acid substrate.
 | ||
| Scheme 12 Ru-catalysed C4–H and C6–H arylation using a C5-based DG. | ||
4.2 Rhodium
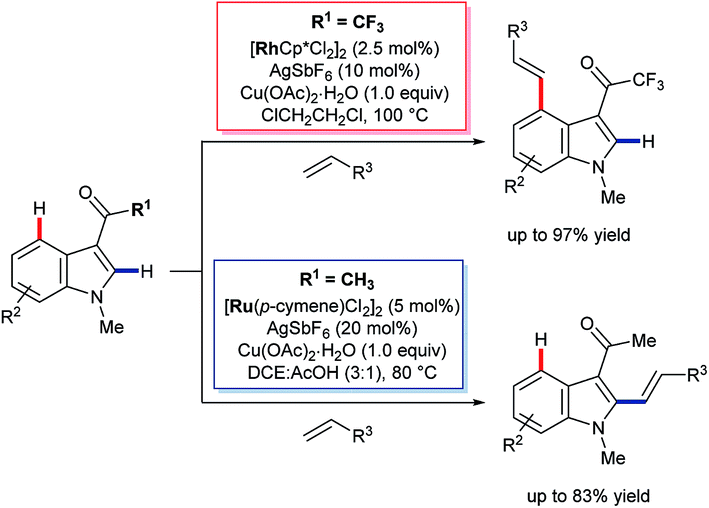 | ||
| Scheme 13 Rh(III)-catalysed C4 selective C–H alkenylation of 3-trifluoroacetyl indoles. | ||
In another contrast with ruthenium catalysis,49 rhodium-catalysed alkenylation was found by Zhang and co-workers to work on 3-indolecarboxylic acids. Here, efficient C–H activation at both the C2 and C4 sites took place, followed by in situ decarboxylation (Scheme 14).52 This builds on wider developments on the use of carboxylic acids as ‘traceless’ directing groups, which can be used to reveal decorated (hetero)arenes with otherwise difficult-to-access substitution patterns.53
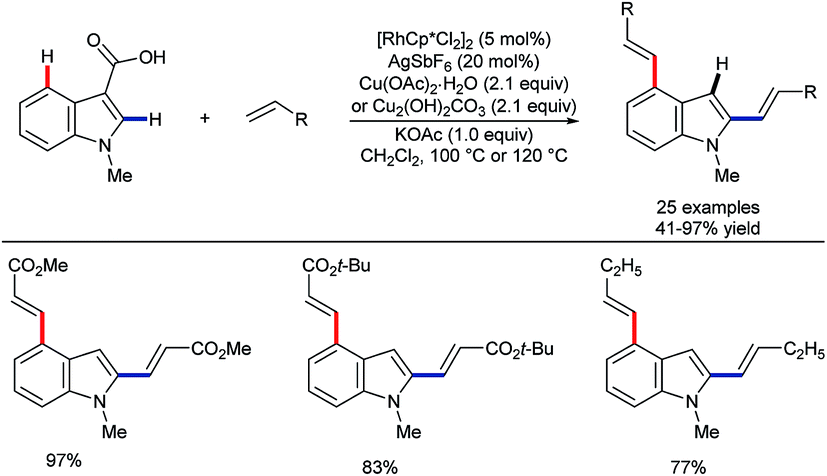 | ||
| Scheme 14 Rh(III)-catalysed C4 alkenylation using traceless carboxylic acid DG. | ||
In their recent studies on rhodium(III)-catalyzed C–H activation and hydroarylation protocol, Ellman and co-workers showed examples of the C4–H alkylation of indoles using nitroalkenes (Scheme 15).54 In these cases, regioselectivity was controlled by the amide DG at the C3 position.
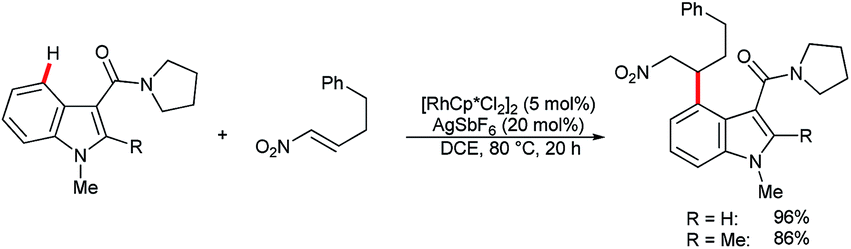 | ||
| Scheme 15 Rh(III)-catalysed C4–H alkylation with nitroalkenes. | ||
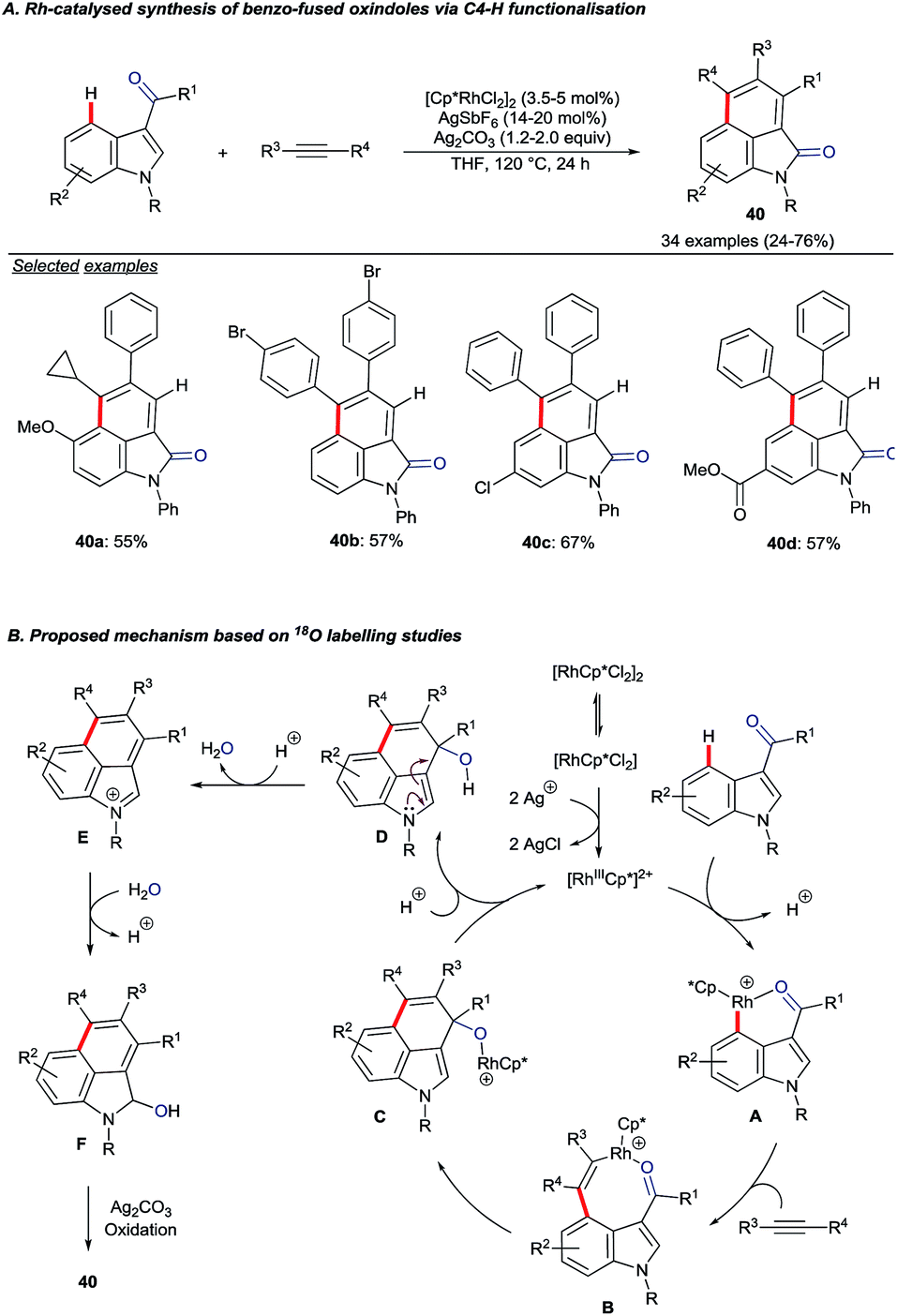 | ||
| Scheme 16 Rh(III)-catalysed C4–H activation and annulation of indolyl aldehydes or ketones with alkynes. | ||
The authors performed 18O labelling experiments to elucidate aspects of the mechanism. A key finding was that the carbonyl oxygen in the product originated from the aldehyde or ketone starting material. Addition of 18OH2 also led to greater levels of 18O incorporation, hinting at the role of water in an oxygen transposition. Based on the mechanistic studies, the catalytic cycle depicted in Scheme 16B was proposed.55 Following directed electrophilic rhodation of the starting material to give intermediate A, alkyne insertion is suggested to give 8-membered rhodacycle B. Attack onto the carbonyl group (B to C) and proto-derhodation closes the catalytic cycle. Intermediate D undergoes water transposition viaE and the resulting alcohol F is oxidised to give the product.
Whilst this manuscript was under review, Prabhu and co-workers reported an intriguing Rh(III)-catalysed formal oxidative [2 + 2 + 2] benzannulation of involving C4–H and C5–H functionalisation.56
In late 2017, Li and co-workers reported the rhodium-catalysed coupling of indoles with diazo esters (Scheme 17).57 They showed that ketone groups present at C3 could direct either a C2-selective annulation to give lactones 41 or otherwise an alkylation at C4 to provide products 42. These developments fit in as a wider set of recent work on using the migratory insertion of carbenes as a strategy in coupling reactions,58 including specifically rhodium-catalysed C–H functionalisation.58a
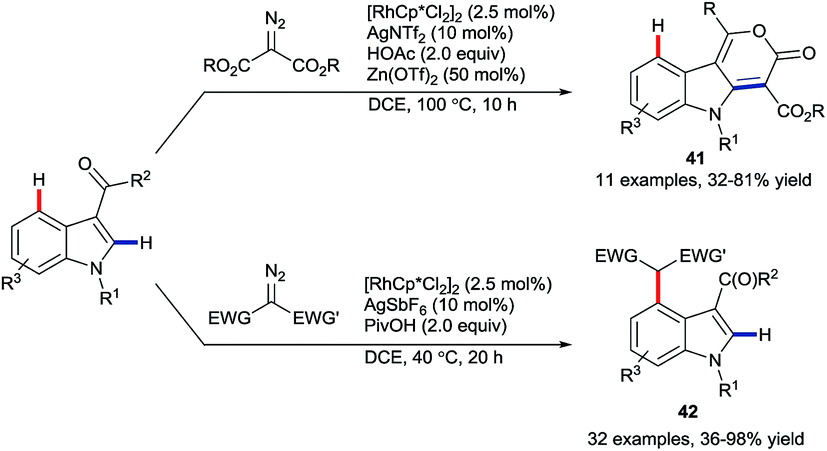 | ||
| Scheme 17 Complementary Rh(III)-catalysed C2–H and C4–H functionalisation of indoles based on diazo ester reactivity. | ||
4.3 Palladium
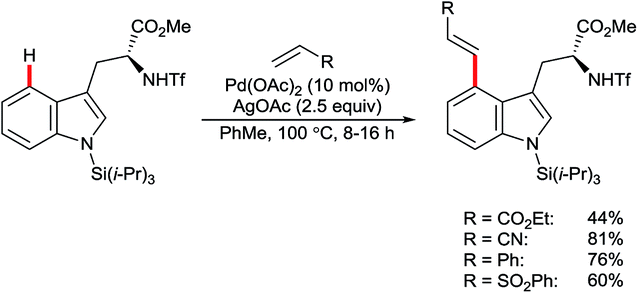 | ||
| Scheme 18 Pd-catalysed oxidative C4–H alkenylation of protected tryptophans. | ||
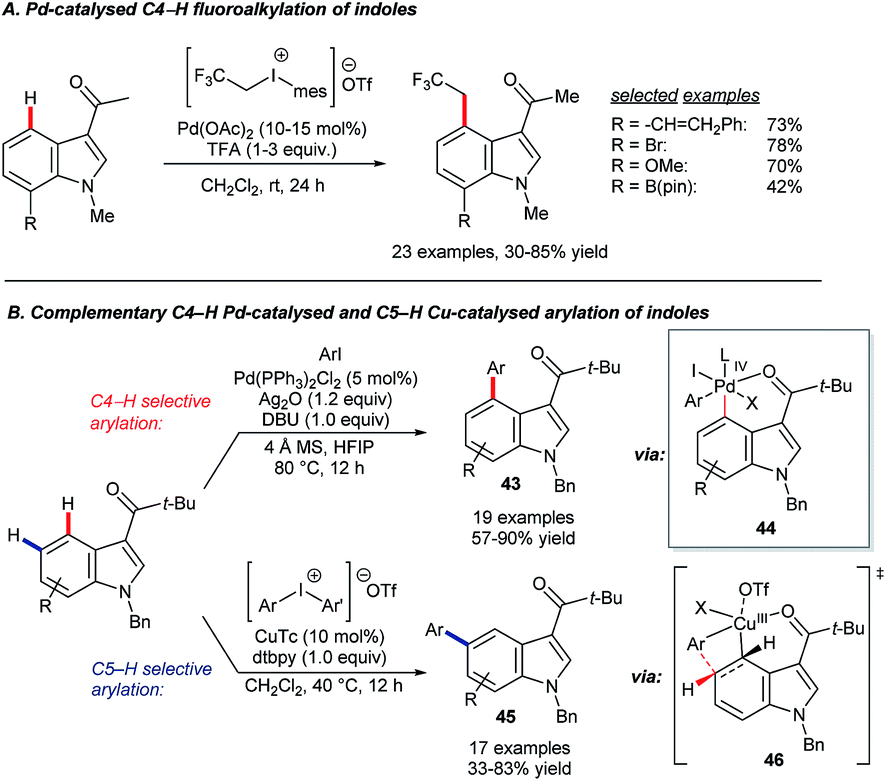 | ||
| Scheme 19 Pd-catalysed C4 alkylation and arylation. | ||
The Shi group reported two complementary systems for the arylation of 3-pivaloyl-indoles at C4 and C5 (Scheme 19B).65 The C4-selective reaction uses [PdCl2(PPh3)2] as the catalyst precursor, aryl iodides as coupling partners and Ag2O as an halogen scavenger to deliver products 43 through a key reductive elimination step at palladium(IV) centre. A combination of copper(I) thiophene-2-carboxylate (CuTc) and diaryliodonium triflate reagents gave C5-selective arylation (45). The sterically demanding pivaloyl directing group, which can be removed via a retro-Friedel–Crafts reaction, proved important to the success of both reactions. The palladium-catalysed C4–H arylation is proposed to involve electrophilic palladation at C4 and a Pd(IV) metallacycle, which forms after oxidative addition to ArI (44).66 The innovative C5–H arylation is analogous to ground-breaking work by the Gaunt group on meta-selective C–H functionalisation of arenes.67 Shi and co-workers proposed that the reaction proceeds via transition state 46.65,68 It is worth noting that, more generally, various combinations of palladium, copper and silver reagents have been used to great effect in delivering complementary regioselectivities in indole C–H functionalisation catalysis.69
Yu and co-workers reported the palladium-catalysed C4–H arylation of N-tosyl-3-formylindole using amino acid 47 as an additive to form a transient imine directing group (Scheme 20).70 Only a single example was described, but it indicates the potential for using this strategy as the basis for a broader range of transformations in the future.
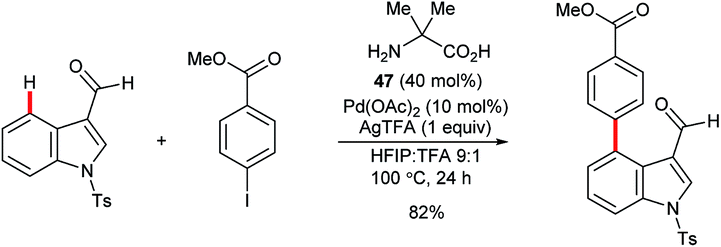 | ||
| Scheme 20 Palladium-catalysed C4–H arylation using a transient directing group. | ||
4.4 Iridium
Iridium-catalysed C–H borylation offers excellent regioselectivity at mild reaction temperatures71 without the need for preinstalled directing groups, although directed versions have also been developed.72 This, and the versatility of pinacolato boronates as chemical handles, renders iridium-catalysed C–H borylation an enormously enabling technology with wide-ranging applications. Tse and co-workers reported73 in 2007 that, whilst the C–H borylation of C2-substituted indoles occurs preferentially at C7,74 the 4,7-diborylated products 48 can be obtained if sufficient B2(pin)2 is used (Scheme 21).75 In principle, this approach could prove complementary or even superior to C4 lithiation but its potential is presently under-developed.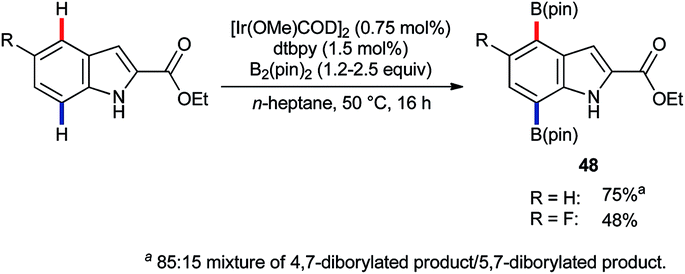 | ||
| Scheme 21 Ir-catalysed C7–H and C4–H borylation. | ||
Following the report by Tse, iridium-catalysed C4–H functionalisation of indoles experienced a decade-long hiatus. Then, a sudden flurry of reports appeared that marked a significant leap forward in indole C4–H activation methodology. The first of these was by Prabhu and co-workers, who showed that carbonyl-directed C4–H amidation was viable using [IrCp*Cl2]2 and sulfonyl azides.76 Shortly thereafter, You and co-workers described a related set of conditions for the equivalent reaction using [Cp*Ir(OAc)2]2 (Scheme 22).77 Both research groups found that the reaction was compatible with indoles that were unprotected at N1 and with a variety of directing groups at C3, including aldehydes, ketones, esters, amides and carboxylic acids. Moreover, a comparatively broad functional group tolerance was described. These features mark a significant advantage over the substrate specificity found for many of the above-mentioned ruthenium-, rhodium-, and palladium-catalysed C4–H transformations (vide supra) and mark a notable advance over preceding methods to introduce nitrogen-based groups to indole's C4 position. Both the Prabhu and You teams found that in deuterium exchange experiments H/D exchange occurs exclusively at C4. You and co-workers also found that the amidation gave useful yields even at room temperature, given sufficiently extended reaction times.
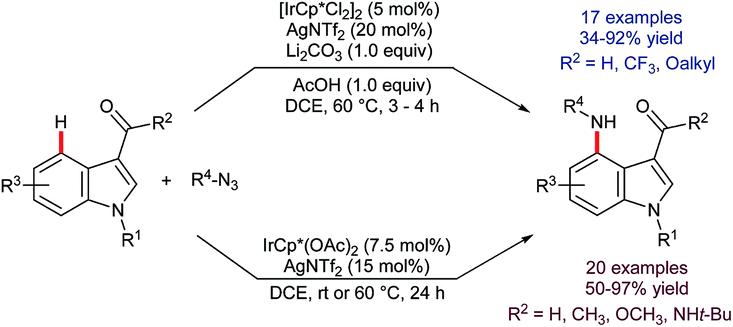 | ||
| Scheme 22 Two sets of conditions for the Ir-catalysed C4–H amidation of indoles. | ||
In recent years, photoredox catalysis has grown to offer powerful new options in molecular synthesis.78 In 2017, Xia and co-workers published79 a protocol for the C4–H trifluoromethylation of benzimidazoles under photoredox catalysis using [fac-Ir(ppy)3], and Togni's reagent64c as the –CF3 source under blue light irradiation. The substrate scope included N-methylindole, which was found to convert to a mixture of the corresponding 3- and 4-trifluoromethylated products (49) in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 ratio and a combined 52% yield (Scheme 23A).
:2 ratio and a combined 52% yield (Scheme 23A).
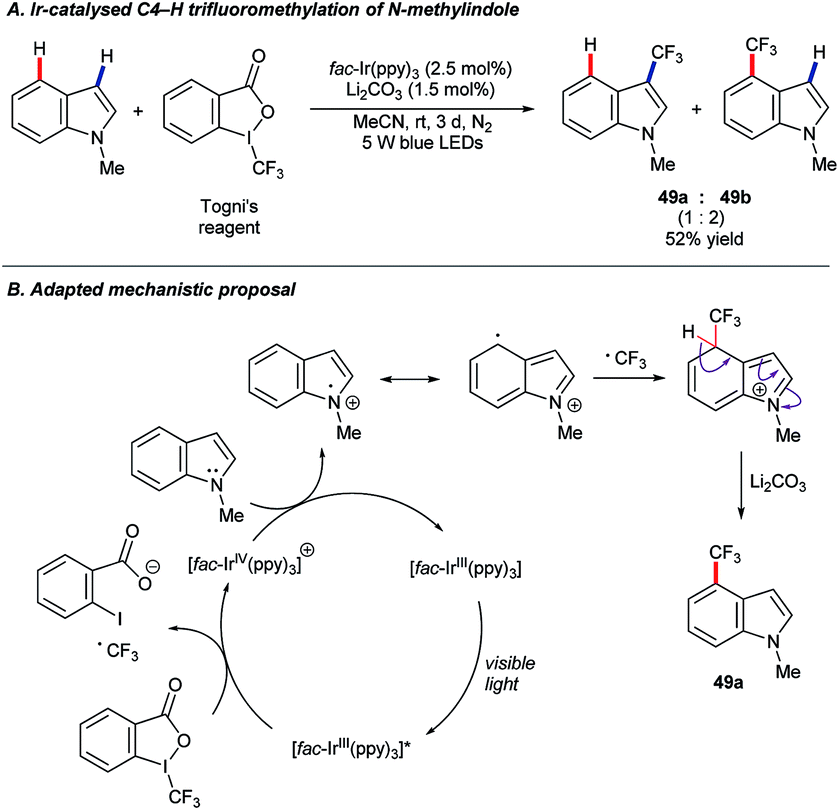 | ||
| Scheme 23 Trifluoromethylation of indoles under photoredox conditions. | ||
The importance of fluorinated compounds to pharmaceutical, agrochemical and materials chemistry, and the particularly mild conditions under which this C–H trifluoromethylation occurs, renders these findings an exciting development.64a,64b Unfortunately, only the single example shown above is known so far. A version of the proposed mechanism adapted for the N-methylindole substrate is shown in Scheme 23B.
4.5 Gold
5-Hydroxylindole derivatives are abundant motifs in nature and display enhanced C4 nucleophilicity.80 This was exploited by Sames and co-workers, who used a gold-based Lewis acid to promote the annulative hydroarylation of protected tryptamines (Scheme 24). This transformation did not proceed via C4–H auration, and decomposition products formed under equivalent conditions when indole substrates lacking C3 substituents were used, presumably via a competitive auration at C3.81 Auration of indole's C4–H bond, stoichiometrically or as part of a catalytic manifold, has not yet been reported.82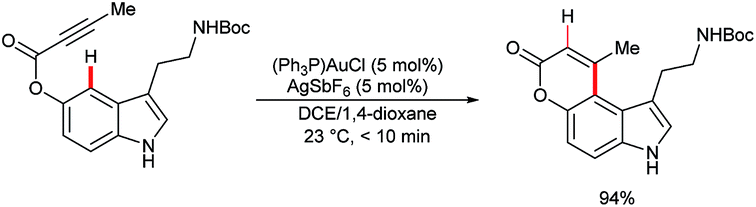 | ||
| Scheme 24 Gold-catalysed annulative hydroarylation of indole at the C4 position. | ||
4.6 Iron
Recent years have witnessed increasing attention shift towards the use of non-precious and less ‘endangered’ metals for C–H functionalisation catalysis.33,83 The natural abundance and low toxicity of iron renders it a particularly attractive metal around which to develop new catalytic technology.Xia and co-workers’ FeCl3-catalysed construction of indolofurans 50 (Scheme 25) stands, at the time of writing, as the sole example of the use of any of these more sustainable metals in the functionalisation of indole's C4–H bond.84 The reaction proceeds only in moderate yields and, unfortunately, involves toxic DDQ as an additive. However, Xia and co-workers could demonstrate its value in the concise synthesis of (±)-serotobenine.
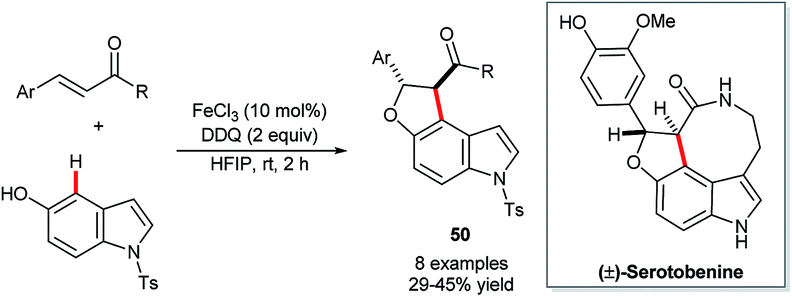 | ||
| Scheme 25 Fe-catalysed annulative synthesis of indolofurans. | ||
5. Enzymatic C4–H functionalisation of indoles
The functionalisation of otherwise inert C–H bonds using biocatalysts, such as enzymes, has been recognised as an attractive tool for chemical synthesis owing to their excellent positional selectivity and mild reaction conditions.85 For instance, 4-dimethylallyltryptophan synthases FgaPT2 efficiently catalysed the transformation of L-tryptophan with dimethylallyl diphosphate (DMAPP) to C4-dimethylallyltryptophan 51 (Scheme 26),86 a key step in the biosynthesis of ercot and clavine alkaloids.3c,87 | ||
| Scheme 26 FgaPT2-catalyzed C4 prenylation of tryptophan and attendant mechanistic proposals. | ||
Mechanistic hypotheses for this transformation initially included variations of SEAr at C4. However, recent studies have supported a route involving a C3 prenylation followed by a Cope rearrangement to give the C4 substituted product.88 Gaich and co-workers reported evidence for the Cope rearrangement pathway based on studies using the bio-inspired model substrate 52, which is designed to mimic conformational restrictions the enzyme might exert at its active site.89 That its conversion to product 53 took place spontaneously at room temperature supports the viability of the enzymatic route. The ring system of indole 53 is also present in many naturally-occurring alkaloids, access to which may be granted by the prenylation–Cope rearrangement strategy.
Recently, the cyclopiazonic acid (CPA) biosynthetic gene, CpaD was shown to act as a C4 dimethylallyltransferases (DMAT) towards tryptophan-containing hydantoins, diketopiperazines and linear peptides 54 to give the corresponding prenylated derivatives 55 (Scheme 27).90 Furthermore, findings by Loria and Challis highlighted that TxtE is a new member of the cytochrome P450 (CYP) family that catalyses the C4 selective nitration of L-tryptophan using NO and O2, which is a crucial step in the biosynthesis of thaxtomine A (Scheme 28).91
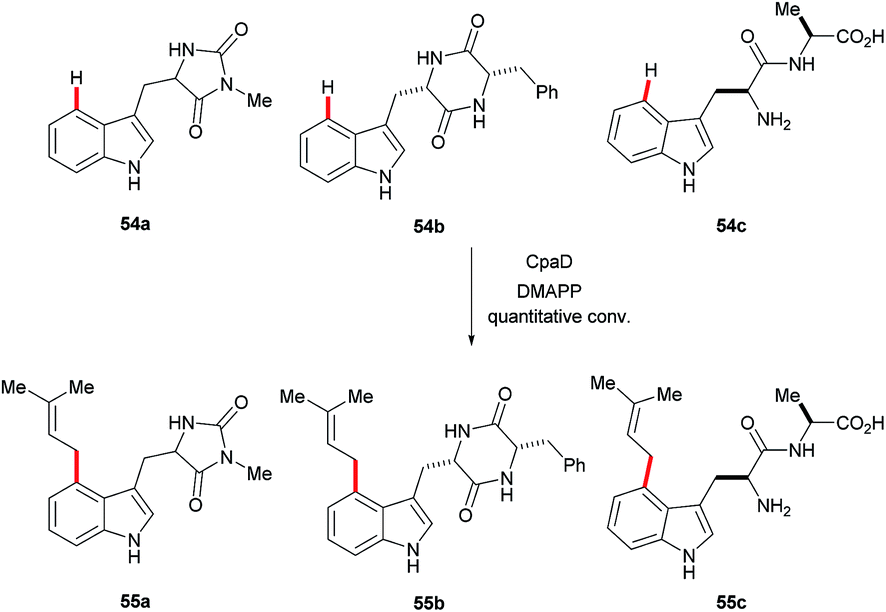 | ||
| Scheme 27 C-4 selective prenylation of tryptophan derivatives using CpaD enzyme. | ||
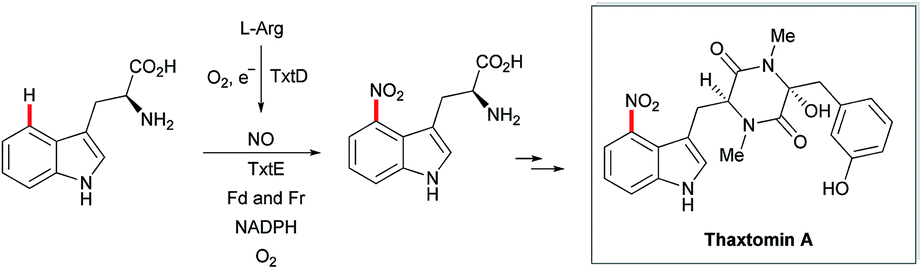 | ||
| Scheme 28 Cytochrome P450-catalyzed C-4 nitration of L-tryptophan. | ||
6. Conclusions and outlook
Indoles are key structural motifs in a plethora compounds relevant to medicinal chemistry, material sciences and crop protection. Whilst significant advances have been made in the diversification of the indole nucleus at various sites, strategies for the regioselective manipulation of the C4–H bond have lagged behind significantly and are still underdeveloped.However, some very recent and major progress has been made. The development of new, transition metal-catalysed reactions, which draw intellectual inspiration from more traditional stoichiometric reactivity, together with new enzymatic and photoredox-catalysed processes, mark the emergence of promising and powerful tools for the site-selective functionalisation of otherwise inert indole C4–H bonds. Their most pronounced advantage is that they can obviate the requirement for particularly toxic or harsh metalations, including those involving mercury and thallium compounds.
Some key challenges remain, however. Although new catalytic reactivity has enabled several previously impossible C–C and C–N bond forming processes, the chemo-selective introduction of many other heteroatoms remains a largely unsolved problem. In this specific respect, Ir-catalysed methods have shown the most versatility to date and seem to hold particular promise for further developments using photoredox catalysis (Scheme 23B). The latter usually requires notably mild conditions. Similarly, we note that very few protocols to date take advantage of the unique reactivity of iodonium reagents (Schemes 19A and 23A).92 We anticipate that more developments in these areas will emerge, as systems shift towards ever milder conditions.
More widely, systems based on several metals have shown that the C4–H bond can be addressed selectively without pre-installed blocking groups at C2. However, compromise is still required: individual systems that achieve this can seem sensitive to the precise electronic nature of the directing group and various additives. A fuller mechanistic understanding is required of how specific combinations of catalysts, additives and directing groups conspire to achieve high levels of C4–H selectivity.
Finally, the sustainability of these C–H activation strategies remains to be improved by shifting towards earth-abundant and generally less toxic 3d transition metal catalysts. Given the rapid ongoing growth of C–H functionalisation technology as an atom- and step-economical strategy in synthesis, further exciting developments are expected in this rapidly evolving research arena.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Generous support by the Alexander von Humboldt foundation (fellowship to P. G.), and the DFG (SPP1807 and Gottfried-Wilhelm-Leibniz prize) is gratefully acknowledged. We also thank the Swedish Research Council (Vetenskapsrådet) and the Wenner-Gren Foundation (postdoctoral grant to J. K.) for financial support and acknowledge the contribution of COST Action CA15106 (C–H Activation in Organic Synthesis – CHAOS).Notes and references
- (a) L. Ping, D. S. Chung, J. Bouffard and S. G. Lee, Chem. Soc. Rev., 2017, 46, 4299 RSC; (b) T. Cernak, K. D. Dykstra, S. Tyagarajan, P. Vachal and S. W. Krska, Chem. Soc. Rev., 2016, 45, 546 RSC; (c) H. M. L. Davies and D. Morton, J. Org. Chem., 2016, 81, 343 CrossRef CAS PubMed; (d) T. Gensch, M. N. Hopkinson, F. Glorius and J. Wencel-Delord, Chem. Soc. Rev., 2016, 45, 2900 RSC; (e) J. Wencel-Delord and F. Glorius, Nat. Chem., 2013, 5, 369 CrossRef CAS PubMed; (f) L. Ackermann, R. Vicente and A. R. Kapdi, Angew. Chem., Int. Ed., 2009, 48, 9792 CrossRef CAS PubMed.
- (a) N. Kaushik, N. Kaushik, P. Attri, N. Kumar, C. Kim, A. Verma and E. Choi, Molecules, 2013, 18, 6620 CrossRef CAS PubMed; (b) J. B. Chen and Y. X. Jia, Org. Biomol. Chem., 2017, 15, 3550 RSC; (c) M. A. Corsello, J. Kim and N. K. Garg, Chem. Sci., 2017, 8, 5836 RSC; (d) L. S. Hegedus, Angew. Chem., Int. Ed., 1988, 27, 1113 CrossRef; (e) E. D. Głowacki, G. Voss, L. Leonat, M. Irimia-Vladu, S. Bauer and N. S. Sariciftci, Isr. J. Chem., 2012, 52, 540 CrossRef.
- (a) J. A. Leitch, Y. Bhonoah and C. G. Frost, ACS Catal., 2017, 7, 5618 CrossRef CAS; (b) A. H. Sandtorv, Adv. Synth. Catal., 2015, 357, 2403 CrossRef CAS; (c) D. D. Schwarzer, P. J. Gritsch and T. Gaich, Synlett, 2013, 24, 1025 CrossRef CAS; (d) P. J. Gritsch, C. Leitner, M. Pfaffenbach and T. Gaich, Angew. Chem., Int. Ed., 2014, 53, 1208 CrossRef CAS PubMed; (e) G. Bartoli, G. Bencivenni and R. Dalpozzo, Chem. Soc. Rev., 2010, 39, 4449 RSC; (f) R. Dalpozzo, Chem. Soc. Rev., 2015, 44, 742 RSC; (g) S. Cacchi and G. Fabrizi, Chem. Rev., 2005, 105, 2873 CrossRef CAS PubMed; (h) H. M. Davies and J. R. Manning, J. Am. Chem. Soc., 2006, 128, 1060 CrossRef CAS PubMed.
- (a) A. K. Pitts, F. O'Hara, R. H. Snell and M. J. Gaunt, Angew. Chem., Int. Ed., 2015, 54, 5451 CrossRef CAS PubMed; (b) A. D. Yamaguchi, K. M. Chepiga, J. Yamaguchi, K. Itami and H. M. Davies, J. Am. Chem. Soc., 2015, 137, 644 CrossRef CAS PubMed; (c) An approach using stoichiometric Pd is reported: J. Liang, W. Hu, P. Tao and Y. Jia, J. Org. Chem., 2013, 78, 5810 CrossRef CAS PubMed; (d) P. Tao, J. Liang and Y. Jia, Eur. J. Org. Chem., 2014, 2014, 5735 CrossRef CAS.
- R. A. Hollins, L. A. Colnago, V. M. Salim and M. C. Seidl, J. Heterocycl. Chem., 1979, 16, 993 CrossRef CAS.
- M. Somei, F. Yamada, M. Kunimoto and C. Kaneko, Heterocycles, 1984, 22, 797 CrossRef CAS.
- M. Somei, H. Amari and Y. Makita, Chem. Pharm. Bull., 1986, 34, 3971 CrossRef CAS.
- S. Picard, F. Lecornué and G. Bashiardes, Synlett, 2014, 25, 1106 CrossRef.
- M. Somei, T. Ohta, J. Shinoda and Y. Somada, Heterocycles, 1989, 29, 653 CrossRef CAS.
- J. P. Konopelski, J. M. Hottenroth, H. M. Oltra, E. A. Véliz and Z.-C. Yang, Synlett, 1996, 1996, 609 CrossRef.
- M. A. Brown and M. A. Kerr, Tetrahedron Lett., 2001, 42, 983 CrossRef CAS.
- (a) Incredibly, the equivalent pathway for the reverse reaction was suggested in 1955! S. Winstein and T. G. Traylor, J. Am. Chem. Soc., 1955, 77, 3747 CrossRef CAS; (b) C. W. Fung, M. Khorramdel-Vahed, R. J. Ranson and R. M. G. Roberts, J. Chem. Soc., Perkin Trans. 2, 1980, 267 RSC.
- For a seminal theoretical study, see: B. Biswas, M. Sugimoto and S. Sakaki, Organometallics, 2000, 19, 3895 CrossRef CAS.
- S. Potavathri, K. C. Pereira, S. I. Gorelsky, A. Pike, A. P. LeBris and B. DeBoef, J. Am. Chem. Soc., 2010, 132, 14676 CrossRef CAS PubMed.
- (a) D. L. Davies, S. A. Macgregor and C. L. McMullin, Chem. Rev., 2017, 117, 8649 CrossRef CAS PubMed; (b) L. Ackermann, Chem. Rev., 2011, 111, 1315 CrossRef CAS PubMed; (c) S. I. Gorelsky, D. Lapointe and K. Fagnou, J. Am. Chem. Soc., 2008, 130, 10848 CrossRef CAS PubMed.
- A. R. Pape, K. P. Kaliappan and E. P. Kündig, Chem. Rev., 2000, 100, 2917 CrossRef CAS PubMed.
- P. J. Beswick, C. S. Greenwood, T. J. Mowlem, G. Nechvatal and D. A. Widdowson, Tetrahedron, 1988, 44, 7325 CrossRef CAS.
- W. J. Ryan, P. E. Peterson, Y. Cao, P. G. Williard, D. A. Sweigart, C. D. Baer, C. F. Thompson, Y. K. Chung and T. M. Chung, Inorg. Chim. Acta, 1993, 211, 1 CrossRef CAS.
- (a) K. Kamikawa, S. Kinoshita, M. Furusyo, S. Takemoto, H. Matsuzaka and M. Uemura, J. Org. Chem., 2007, 72, 3394 CrossRef CAS PubMed; (b) K. Kamikawa, S. Kinoshita, H. Matsuzaka and M. Uemura, Org. Lett., 2006, 8, 1097 CrossRef CAS PubMed.
- (a) P. Ricci, K. Krämer and I. Larrosa, J. Am. Chem. Soc., 2014, 136, 18082 CrossRef CAS PubMed; (b) P. Ricci, K. Krämer, X. C. Cambeiro and I. Larrosa, J. Am. Chem. Soc., 2013, 135, 13258 CrossRef CAS PubMed.
- F. Zhang and D. R. Spring, Chem. Soc. Rev., 2014, 43, 6906 RSC.
- (a) L. Pérez-Serrano, G. Domínguez and J. Pérez-Castells, ARKIVOC, 2010, 3, 23 Search PubMed; (b) B. Chauder, A. Larkin and V. Snieckus, Org. Lett., 2002, 4, 815 CrossRef CAS PubMed.
- M. Iwao and F. Ishibashi, Tetrahedron, 1997, 53, 51 CrossRef CAS.
- T. Mesganaw, N. F. Fine Nathel and N. K. Garg, Org. Lett., 2012, 14, 2918 CrossRef CAS PubMed.
- S. M. Bronner, K. B. Bahnck and N. K. Garg, Org. Lett., 2009, 11, 1007 CrossRef CAS PubMed.
- (a) M. Asamdi and K. H. Chikhalia, Asian J. Org. Chem., 2017, 6, 1331 CrossRef CAS; (b) J. A. Garcia-Lopez and M. F. Greaney, Chem. Soc. Rev., 2016, 45, 6766 RSC; (c) J. Shi, Y. Li and Y. Li, Chem. Soc. Rev., 2017, 46, 1707 RSC; (d) F. I. M. Idiris and C. R. Jones, Org. Biomol. Chem., 2017, 15, 9044 RSC.
- (a) A. E. Goetz, S. M. Bronner, J. D. Cisneros, J. M. Melamed, R. S. Paton, K. N. Houk and N. K. Garg, Angew. Chem., Int. Ed., 2012, 51, 2758 CrossRef CAS PubMed; (b) G. Y. Im, S. M. Bronner, A. E. Goetz, R. S. Paton, P. H. Cheong, K. N. Houk and N. K. Garg, J. Am. Chem. Soc., 2010, 132, 17933 CrossRef CAS PubMed.
- S. M. Bronner, A. E. Goetz and N. K. Garg, J. Am. Chem. Soc., 2011, 133, 3832 CrossRef CAS PubMed.
- X. Tian, A. D. Huters, C. J. Douglas and N. K. Garg, Org. Lett., 2009, 11, 2349 CrossRef CAS PubMed.
- M. Murase, T. Koike, Y. Moriya and S. Tobinaga, Chem. Pharm. Bull., 1987, 35, 2656 CrossRef CAS PubMed.
- V. Bhat, J. A. Mackay and V. H. Rawal, Org. Lett., 2011, 13, 3214 CrossRef CAS PubMed.
- J. Magolan and M. A. Kerr, Org. Lett., 2006, 8, 4561 CrossRef CAS PubMed.
- W. Liu and L. Ackermann, ACS Catal., 2016, 6, 3743 CrossRef CAS.
- (a) H. Wang, M. M. Lorion and L. Ackermann, Angew. Chem., Int. Ed., 2017, 56, 6339 CrossRef CAS PubMed; (b) W. Liu, S. C. Richter, Y. Zhang and L. Ackermann, Angew. Chem., Int. Ed., 2016, 55, 7747 CrossRef CAS PubMed; (c) L. Shi, X. Zhong, H. She, Z. Lei and F. Li, Chem. Commun., 2015, 51, 7136 RSC.
- O. Yonemitsu, P. Cerutti and B. Witkop, J. Am. Chem. Soc., 1966, 88, 3941 CrossRef CAS PubMed.
- (a) J. Li, S. Jeong, L. Esser and P. G. Harran, Angew. Chem., Int. Ed., 2001, 40, 4765 CrossRef CAS; (b) K. C. Nicolaou, D. Y. K. Chen, X. Huang, T. Ling, M. Bella and S. A. Snyder, J. Am. Chem. Soc., 2004, 126, 12888 CrossRef CAS PubMed.
- J. Xiu and W. Yi, Catal. Sci. Technol., 2016, 6, 998 CAS.
- (a) V. Lanke and K. Ramaiah Prabhu, Org. Lett., 2013, 15, 6262 CrossRef CAS PubMed; (b) For a Perspective on Ru-catalysed C–H alkenylations, see: S. I. Kozhushkov and L. Ackermann, Chem. Sci., 2013, 4, 886 RSC.
- V. Lanke, K. R. Bettadapur and K. R. Prabhu, Org. Lett., 2016, 18, 5496 CrossRef CAS PubMed.
- O. Daugulis, J. Roane and L. D. Tran, Acc. Chem. Res., 2015, 48, 1053 CrossRef CAS PubMed.
- J. Li, S. De Sarkar and L. Ackermann, Top. Organomet. Chem., 2016, 55, 217 CrossRef CAS.
- (a) O. Saidi, J. Marafie, A. E. W. Ledger, P. M. Liu, M. F. Mahon, G. Kociok-Köhn, M. K. Whittlesey and C. G. Frost, J. Am. Chem. Soc., 2011, 133, 19298 CrossRef CAS PubMed; (b) L. Ackermann, N. Hofmann and R. Vicente, Org. Lett., 2011, 13, 1875 CrossRef CAS PubMed; (c) L. Ackermann, P. Novák, R. Vicente and N. Hofmann, Angew. Chem., Int. Ed., 2009, 48, 6045 CrossRef CAS PubMed.
- (a) J. A. Leitch and C. G. Frost, Chem. Soc. Rev., 2017, 45, 7145 RSC; (b) N. Hofmann and L. Ackermann, J. Am. Chem. Soc., 2013, 135, 5877 CrossRef CAS PubMed; (c) A. J. Paterson, S. St John-Campbell, M. F. Mahon, N. J. Press and C. G. Frost, Chem. Commun., 2015, 51, 12807 RSC.
- J. A. Leitch, C. L. McMullin, M. F. Mahon, Y. Bhonoah and C. G. Frost, ACS Catal., 2017, 7, 2616 CrossRef CAS.
- Pyrimidyl indoles undergo facile C2 ruthenation. Presumably, a different directing group may be required to effect C7-directed enhancement of C4–H functionalisation, C. Sollert, K. Devaraj, A. Orthaber, P. J. Gates and L. T. Pilarski, Chem.–Eur. J., 2015, 21, 5380 CrossRef CAS PubMed.
- J. A. Leitch, C. J. Heron, J. McKnight, G. Kociok-Kohn, Y. Bhonoah and C. G. Frost, Chem. Commun., 2017, 53, 13039 RSC.
- K. Devaraj, C. Sollert, C. Juds, P. J. Gates and L. T. Pilarski, Chem. Commun., 2016, 52, 5868 RSC.
- G. de la Herrán, A. Segura and A. G. Csákÿ, Org. Lett., 2007, 9, 961 CrossRef PubMed.
- M. Simonetti, D. M. Cannas, A. Panigrahi, S. Kujawa, M. Kryjewski, P. Xie and I. Larrosa, Chem.–Eur. J., 2017, 23, 549 CrossRef CAS PubMed.
- J. Lv, B. Wang, K. Yuan, Y. Wang and Y. Jia, Org. Lett., 2017, 19, 3664 CrossRef CAS PubMed.
- X. Qi, Y. Li, R. Bai and Y. Lan, Acc. Chem. Res., 2017, 50, 2799 CrossRef CAS PubMed.
- H. Chen, C. Lin, C. Xiong, Z. Liu and Y. Zhang, Org. Chem. Front., 2017, 4, 455 RSC.
- M. Font, J. M. Quibell, G. J. P. Perry and I. Larrosa, Chem. Commun., 2017, 53, 5584 RSC.
- T. J. Potter, D. N. Kamber, B. Q. Mercado and J. A. Ellman, ACS Catal., 2017, 7, 150 CrossRef CAS PubMed.
- X. Liu, G. Li, F. Song and J. You, Nat. Commun., 2014, 5, 5030 CrossRef CAS PubMed.
- K. R. Bettadapur, R. Kapanaiah, V. Lanke and K. R. Prabhu, J. Org. Chem., 2018, 83, 1810 CrossRef CAS PubMed.
- X. Chen, G. Zheng, Y. Li, G. Song and X. Li, Org. Lett., 2017, 19, 6184 CrossRef CAS PubMed.
- (a) H. M. L. Davies and D. Morton, Chem. Soc. Rev., 2011, 40, 1857 RSC; (b) Y. Xia, D. Qiu and J. Wang, Chem. Rev., 2017, 117, 13810 CrossRef CAS PubMed.
- A. F. M. Noisier and M. A. Brimble, Chem. Rev., 2014, 114, 8775 CrossRef CAS PubMed.
- A. Schischko, H. Ren, N. Kaplaneris and L. Ackermann, Angew. Chem., Int. Ed., 2017, 56, 1576 CrossRef CAS PubMed.
- J. J. Li, T. S. Mei and J. Q. Yu, Angew. Chem., Int. Ed., 2008, 47, 6452 CrossRef CAS PubMed.
- Q. Liu, Q. Li, Y. Ma and Y. Jia, Org. Lett., 2013, 15, 4528 CrossRef CAS PubMed.
- A. J. Borah and Z. Shi, Chem. Commun., 2017, 53, 3945 RSC.
- (a) T. Furuya, A. S. Kamlet and T. Ritter, Nature, 2011, 473, 470 CrossRef CAS PubMed; (b) S. Barata-Vallejo, B. Lantaño and A. Postigo, Chem.–Eur. J., 2014, 20, 16806 CrossRef CAS PubMed; (c) J. Charpentier, N. Früh and A. Togni, Chem. Rev., 2015, 115, 650 CrossRef CAS PubMed; (d) K. Müller, C. Faeh and F. Diederich, Science, 2007, 317, 1881 CrossRef PubMed.
- Y. Yang, P. Gao, Y. Zhao and Z. Shi, Angew. Chem., Int. Ed., 2017, 56, 3966 CrossRef CAS PubMed.
- (a) G. Chen, Z. Zhuang, G.-C. Li, T. G. Saint-Denis, Y. Hsiao, C. L. Joe and J.-Q. Yu, Angew. Chem., Int. Ed., 2017, 56, 1506 CrossRef CAS PubMed; (b) P. Gandeepan, K. Parthasarathy and C.-H. Cheng, J. Am. Chem. Soc., 2010, 132, 8569 CrossRef CAS PubMed.
- (a) H. A. Duong, R. E. Gilligan, M. L. Cooke, R. J. Phipps and M. J. Gaunt, Angew. Chem., Int. Ed., 2011, 50, 463 CrossRef CAS PubMed; (b) R. J. Phipps and M. J. Gaunt, Science, 2009, 323, 1593 CrossRef CAS PubMed.
- For a related mechanistic study, see: B. Chen, X.-L. Hou, Y.-X. Li and Y.-D. Wu, J. Am. Chem. Soc., 2011, 133, 7668 CrossRef CAS PubMed.
- (a) D. R. Stuart, E. Villemure and K. Fagnou, J. Am. Chem. Soc., 2007, 129, 12072 CrossRef CAS PubMed; (b) S. Potavathri, A. S. Dumas, T. A. Dwight, G. R. Naumiec, J. M. Hammann and B. DeBoef, Tetrahedron Lett., 2008, 49, 4050 CrossRef CAS PubMed; (c) N. R. Deprez, D. Kalyani, A. Krause and M. S. Sanford, J. Am. Chem. Soc., 2006, 128, 4972 CrossRef CAS PubMed; (d) R. J. Phipps, N. P. Grimster and M. J. Gaunt, J. Am. Chem. Soc., 2008, 130, 8172 CrossRef CAS PubMed.
- (a) X.-H. Liu, H. Park, J.-H. Hu, Y. Hu, Q.-L. Zhang, B.-L. Wang, B. Sun, K.-S. Yeung, F.-L. Zhang and J.-Q. Yu, J. Am. Chem. Soc., 2017, 139, 888 CrossRef CAS PubMed; (b) For a recent review, see: P. Gandeepan and L. Ackermann, Chem, 2018, 4, 199 CrossRef CAS.
- I. A. Mkhalid, J. H. Barnard, T. B. Marder, J. M. Murphy and J. F. Hartwig, Chem. Rev., 2010, 110, 890 CrossRef CAS PubMed.
- (a) K. M. Crawford, T. R. Ramseyer, C. J. A. Daley and T. B. Clark, Angew. Chem., Int. Ed., 2014, 53, 7589 CrossRef CAS PubMed; (b) H. J. Davis, G. R. Genov and R. J. Phipps, Angew. Chem., 2017, 129, 13536 CrossRef; (c) S. D. Sarkar, N. Y. P. Kumar and L. Ackermann, Chem.–Eur. J., 2017, 23, 84 CrossRef CAS PubMed.
- W. F. Lo, H. M. Kaiser, A. Spannenberg, M. Beller and M. K. Tse, Tetrahedron Lett., 2007, 48, 371 CrossRef CAS.
- S. Paul, G. A. Chotana, D. Holmes, R. C. Reichle, R. E. Maleczka and M. R. Smith, J. Am. Chem. Soc., 2006, 128, 15552 CrossRef CAS PubMed.
- (a) For a selection of reports discussing the regioselectivity of heteroarene C–H borylation, see: M. A. Larsen and J. F. Hartwig, J. Am. Chem. Soc., 2014, 136, 4287 CrossRef CAS PubMed; (b) H. Tajuddin, P. Harrisson, B. Bitterlich, J. C. Collings, N. Sim, A. S. Batsanov, M. S. Cheung, S. Kawamorita, A. C. Maxwell, L. Shukla, J. Morris, Z. Lin, T. B. Marder and P. G. Steel, Chem. Sci., 2012, 3, 3505 RSC; (c) I. I. B. A. Vanchura, S. M. Preshlock, P. C. Roosen, V. A. Kallepalli, R. J. Staples, J. R. E. Maleczka, D. A. Singleton and I. I. I. M. R. Smith, Chem. Commun., 2010, 46, 7724 RSC.
- V. Lanke and K. R. Prabhu, Chem. Commun., 2017, 53, 5117 RSC.
- S. Chen, B. Feng, X. Zheng, J. Yin, S. Yang and J. You, Org. Lett., 2017, 19, 2502 CrossRef CAS PubMed.
- (a) M. H. Shaw, J. Twilton and D. W. C. MacMillan, J. Org. Chem., 2016, 81, 6898 CrossRef CAS PubMed; (b) N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075 CrossRef CAS PubMed.
- G. L. Gao, C. Yang and W. Xia, Chem. Commun., 2017, 53, 1041 RSC.
- (a) Y. Zhao and V. Snieckus, J. Am. Chem. Soc., 2014, 136, 11224 CrossRef CAS PubMed; (b) S. A. Monti and W. O. Johnson, Tetrahedron, 1970, 26, 3685 CrossRef CAS.
- P. A. Vadola and D. Sames, J. Org. Chem., 2012, 77, 7804 CrossRef CAS PubMed.
- S. Kramer, Chem.–Eur. J., 2016, 22, 15584 CrossRef CAS PubMed.
- (a) G. Pototschnig, N. Maulide and M. Schnurch, Chemistry, 2017, 23, 9206 CrossRef CAS PubMed; (b) R. Shang, L. Ilies and E. Nakamura, Chem. Rev., 2017, 117, 9086 CrossRef CAS PubMed; (c) G. Cera and L. Ackermann, Top. Curr. Chem., 2016, 374, 57 CrossRef PubMed; (d) M. Moselage, J. Li and L. Ackermann, ACS Catal., 2016, 6, 498 CrossRef CAS; (e) J. Yamaguchi, K. Muto and K. Itami, Top. Curr. Chem., 2016, 374, 55 CrossRef PubMed; (f) S. Z. Tasker, E. A. Standley and T. F. Jamison, Nature, 2014, 509, 299 CrossRef CAS PubMed; (g) C. Xiao-hua and B. Xie, ARKIVOC, 2015, 2015, 184 CrossRef; (h) A. E. Wendlandt, A. M. Suess and S. S. Stahl, Angew. Chem., Int. Ed., 2011, 50, 11062 CrossRef CAS PubMed; (i) X.-X. Guo, D.-W. Gu, Z. Wu and W. Zhang, Chem. Rev., 2015, 115, 1622 CrossRef CAS PubMed.
- K. Liang, T. Wu and C. Xia, Org. Biomol. Chem., 2016, 14, 4690 CAS.
- (a) J. C. Lewis, P. S. Coelho and F. H. Arnold, Chem. Soc. Rev., 2011, 40, 2003 RSC; (b) C.-I. Lin, R. M. McCarty and H.-w. Liu, Angew. Chem., Int. Ed., 2017, 56, 3446 CrossRef CAS PubMed.
- (a) N. Steffan, I. A. Unsöld and S.-M. Li, ChemBioChem, 2007, 8, 1298 CrossRef CAS PubMed; (b) I. A. Unsöld and S. M. Li, Microbiology, 2005, 151, 1499 CrossRef PubMed; (c) J. C. Gebler, A. B. Woodside and C. D. Poulter, J. Am. Chem. Soc., 1992, 114, 7354 CrossRef CAS.
- (a) D. Jakubczyk, J. Z. Cheng and S. E. O'Connor, Nat. Prod. Rep., 2014, 31, 1328 RSC; (b) O. Rigbers and S. M. Li, J. Biol. Chem., 2008, 283, 26859 CrossRef CAS PubMed.
- L. Y. Luk, Q. Qian and M. E. Tanner, J. Am. Chem. Soc., 2011, 133, 12342 CrossRef CAS PubMed.
- D. D. Schwarzer, P. J. Gritsch and T. Gaich, Angew. Chem., Int. Ed., 2012, 51, 11514 CrossRef CAS PubMed.
- X. Liu and C. T. Walsh, Biochemistry, 2009, 48, 11032 CrossRef CAS PubMed.
- S. M. Barry, J. A. Kers, E. G. Johnson, L. Song, P. R. Aston, B. Patel, S. B. Krasnoff, B. R. Crane, D. M. Gibson, R. Loria and G. L. Challis, Nat. Chem. Biol., 2012, 8, 814 CrossRef CAS PubMed.
- E. A. Merritt and B. Olofsson, Angew. Chem., Int. Ed., 2009, 48, 9052 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2018 |