
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFluorescent dyes and probes for super-resolution microscopy of microtubules and tracheoles in living cells and tissues†
Gražvydas
Lukinavičius
 *,
Gyuzel Y.
Mitronova
,
Sebastian
Schnorrenberg
,
Alexey N.
Butkevich
,
Hannah
Barthel
,
Vladimir N.
Belov
and
Stefan W.
Hell
*,
Gyuzel Y.
Mitronova
,
Sebastian
Schnorrenberg
,
Alexey N.
Butkevich
,
Hannah
Barthel
,
Vladimir N.
Belov
and
Stefan W.
Hell
Max Planck Institute for Biophysical Chemistry, Department of NanoBiophotonics, Am Fassberg 11, 37077 Göttingen, Germany. E-mail: grazvydas.lukinavicius@mpibpc.mpg.de
First published on 26th February 2018
Abstract
We introduce fluorogenic tubulin probes based on the recently reported fluorescent dyes (510R, 580CP, GeR and SiR) and chemotherapy agents – taxanes (docetaxel, cabazitaxel and larotaxel). The cytotoxicity of the final probe, its staining performance and specificity strongly depend on both components. We found correlation between the aggregation efficiency (related to the spirolactonization of fluorophore) and cytotoxicity. Probe optimization allowed us to reach 29 ± 11 nm resolution in stimulated emission depletion (STED) microscopy images of the microtubule network in living human fibroblasts. Application to living fruit fly (Drosophila melanogaster) tissues highlighted two distinct structures: microtubules and tracheoles. We identified 6-carboxy isomers of 580CP and SiR dyes as markers for chitin-containing taenidia, a component of tracheoles. STED microscopy revealed correlation between the taenidia periodicity and the diameter of the tracheole. Combined tubulin and taenidia STED imaging showed close interaction between the microtubules and respiratory networks in living tissues of the insect larvae.
Introduction
The fast development of super-resolution microscopy (nanoscopy) is accompanied by the emergence of new fluorescent dyes serving as molecular probes.1 Particular attention is paid to the compatibility of these probes with living cells.2 Key features of good live-cell imaging probes are high cell permeability, excitation/emission in the far-red or near-infrared region, acceptable photostability, brightness, lowest possible off-target binding and little or no toxicity. Ideally, the probes are fluorogenic.3 The most straightforward strategy towards obtaining such highly desirable probes is the linking of a small-molecule ligand to the desired fluorophore.4 In the majority of cases, the fluorescent dye and the targeting moiety are of similar size, so that both parts contribute to the properties of the final probe.Paclitaxel (Fig. 1a) is one of the most successful anti-cancer drugs used for the treatment of ovarian, breast, and lung cancers.5 It is a natural product first isolated from the bark of the yew tree Taxus brevifolia in 1971.6 Since then, a large variety of paclitaxel analogues was reported.5,7 Flutax-2 is the most extensively used compound for fluorescent imaging of tubulin. It comprises of paclitaxel linked at position 7 via a beta-alanine linker to the fluorophore Oregon Green (OG).8 Other tubulin probes reported in the literature are the conjugates of taxanes with boron-dipyrromethene (BODIPY) dyes, rhodamines, coumarins, carbo-, germano- or silicon-rhodamines.2b,c,8a,9 However, no systematic studies have been published describing the influence of the fluorescent dye on cytotoxicity, staining performance or fluorescence intensity change upon target binding. On the other hand, a large set of taxane drugs, designed to avoid multiple drug resistance (MDR) conferring efflux pumps, offers a perspective to apply them for the generation of new fluorescent probes with improved cell-permeability.10 For example, cabazitaxel and larotaxel are poorly recognized by MDR proteins, and they are able to cross the blood–brain barrier.11 In our study, we chose and combined recently reported fluorescent dyes compatible with stimulated emission depletion (STED) microscopy (510R, 580CP, GeR and SiR),12 with targeting moieties based on docetaxel,13 cabazitaxel11a,14 and larotaxel11b (Fig. 1a–c). Next, we treated living cells with these synthetic probes and examined their cytotoxicity. We found a correlation between the fluorogenic properties of the probe (related to spirolactonization or aggregation of the dye) and toxicity. This observation enabled us to select the optimal fluorescent tubulin probes (featuring minimal cytotoxicity, high fluorogenicity and cell permeability) for three well separated excitation wavelengths. Recently, live-cell reversible saturable optical linear fluorescence transition (RESOLFT) nanoscopy on fluorescent protein labelled microtubule filaments of Drosophila melanogaster larvae was reported.15 To analyse the performance of the generated tubulin probes, we stained living tissues of dissected D. melanogaster larvae. The acquired images displayed high contrast and provided detailed characteristic arrangement of the microtubule network in living tissues – including brain, intestine and body wall muscle. Further analysis showed that the tubulin probes display off-target staining of the insect tracheoles. We demonstrated that the ligand-free fluorescent dyes could be applied for specific staining of taenidia in the tracheoles of D. melanogaster. These results not only emphasize that both the fluorophore and the targeting moiety contribute to the specificity of the final probe, but also provide a toolbox for further design of advanced fluorescent probes.
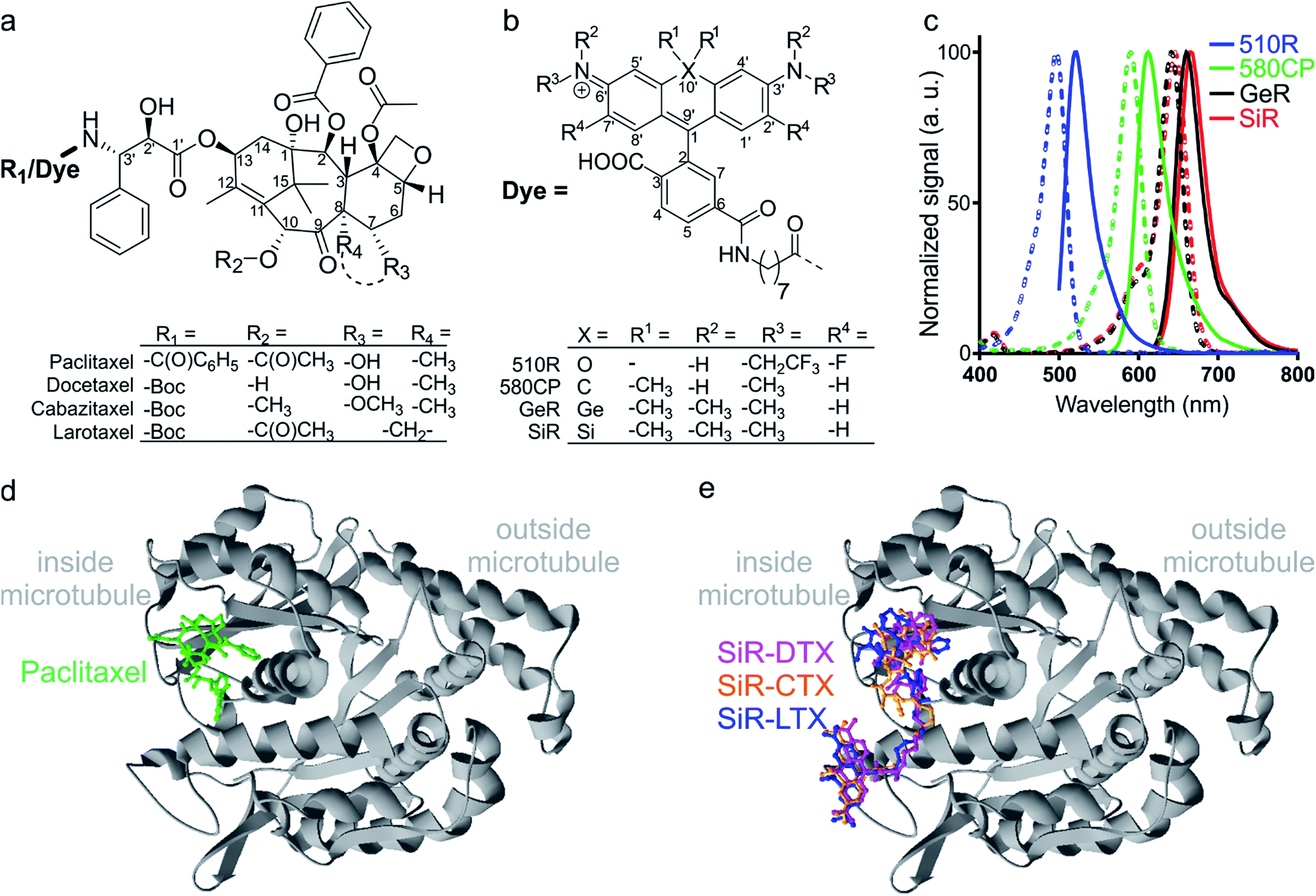 | ||
| Fig. 1 Chemical structures and spectra of fluorescent tubulin-binding probes. (a) Structures of paclitaxel and taxanes used for probe generation: docetaxel (Boc-DTX), cabazitaxel (Boc-CTX) and larotaxel (Boc-LTX). (b) Structures of fluorescent dyes together with the C8-linker used for the synthesis of the tubulin probes. (c) Absorption (dashed line) and emission (solid line) spectra of dyes used for the generation of the tubulin probes. (d) High-resolution cryo-electron microscopy reconstruction of β-tubulin (grey) bound to paclitaxel (green). For clarity, α-tubulin molecule, GTP/GDP and Mg2+ were omitted from the original PDB file (PDB ID: 5SYF). (e) Model of three different SiR probes (SiR-DTX – magenta, SiR-CTX – orange and SiR-LTX – blue) docked to β-tubulin. The same β-tubulin structure as in (d) is used. | ||
Results and discussion
Chemical synthesis of fluorescent tubulin probes
The design of new tubulin probes is based on the replacement of the tert-butyloxycarbonyl (Boc) group in taxanes with a fluorophore attached via a linear C8-alkyl chain at the 3′-position (Fig. 1a and b). This approach proved to be optimal for the generation of SiR-tubulin, GeR-tubulin and SiR700-tubulin probes based on docetaxel.2c,9b,16 In this study, we designed and systematically characterized these tubulin probes for their application in super-resolution STED microscopy. First, we performed the docking of the model compounds – SiR-based ligands – to the paclitaxel binding site of β-tubulin, by using the AutoDock Vina17 program and a recently published cryo-EM structure (Fig. 1d and e).18 In all cases, the fluorophore is positioned in the binding pocket located on the surface of the protein and formed by the first alpha helix (11–28 aa) followed by the loop (29–41 aa). This suggests that the fluorophores should be located inside the microtubule lumen and under ideal conditions, the labelled structure should appear as a tube of ∼20 nm diameter in the microscopy images (Fig. 1d and e and S1†).We synthesized all possible 12 probes containing 510R, 580CP, GeR and SiR fluorophores conjugated with three tubulin binding moieties – docetaxel (Boc-DTX), cabazitaxel (Boc-CTX) and larotaxel (Boc-LTX) (Fig. 1a–c and Table S1†). Importantly, the selected fluorophores can form the non-fluorescent spironolactones depending on the dielectric constant of the environment (D).12a,bD0.5 parameter allows to estimate the sensitivity of the dye to surrounding polarity and is equal to the dielectric constant at which dye has half maximum absorbance. This advantageous property is required for fluorogenicity and therefore, dyes with sufficiently high D0.5 values (580CP, GeR and SiR)2c,12a,b were chosen (Table S1†). We assumed that our new probes were fluorogenic as well.12a,b,19 As the D0.5 value of the 510R dye has not been reported, we measured it to be equal to 53, predicting the possibility of desirable fluorogenic behaviour (Table S1†).12a
Fluorogenic properties of the tubulin probes
First, we characterized the spectroscopic properties of the new probes by measuring their absorbance and fluorescence spectra as well as quantum yields (QY) (Table S2 and Fig. S3†). Conjugation of the fluorophores to the targeting moieties induced ∼5 nm bathochromic shift of the absorbance and emission maxima compared to the unconjugated dyes (Tables S1 and S2†). The absorbance and QY of the dyes are considerably decreased upon conjugation and this effect is especially significant for the larotaxel derivatives (Table S2†). The addition of an anionic detergent sodium dodecyl sulfate (SDS, 0.1%) recovers both the absorbance and the fluorescence intensities (Fig. S3, Tables S2 and S3†). Based on our previous work,2c,12b we hypothesized that the fluorescence intensity of the new probes is modulated by both spirolactonization and aggregate formation (Fig. 2a). If only spirolactonization occurred, the changes in absorbance and fluorescence should be proportional, resulting in a constant fluorescence QY. However, we observed significant decrease in QY of the probes compared to the free dyes (Tables S1 and S2†). This phenomenon directly indicated possible aggregation leading to fluorescence quenching (e.g. by homo FRET, energy transfer from the excited state of the dye to the neighbouring ligand or the closed-ring form of another dye residue). The loss of charges due to spirolactonization of the fluorophore might induce self-aggregation of the probe and fluorescence quenching as reflected by the drop in QY (Fig. 2a and Table S2†). To further test this, we plotted the changes of fluorescence versus absorbance upon SDS addition and observed significant deviation from the linear equation going through the axis origin (y = x) (Fig. S4a†). All data points are above the direct proportionality line supporting our model based on aggregation and spirolactonization (Fig. 2a). The larotaxel derivatives demonstrated the largest deviations from the line of equality suggesting the strongest aggregate formation characteristic of these probes among the tested fluorescent taxanes.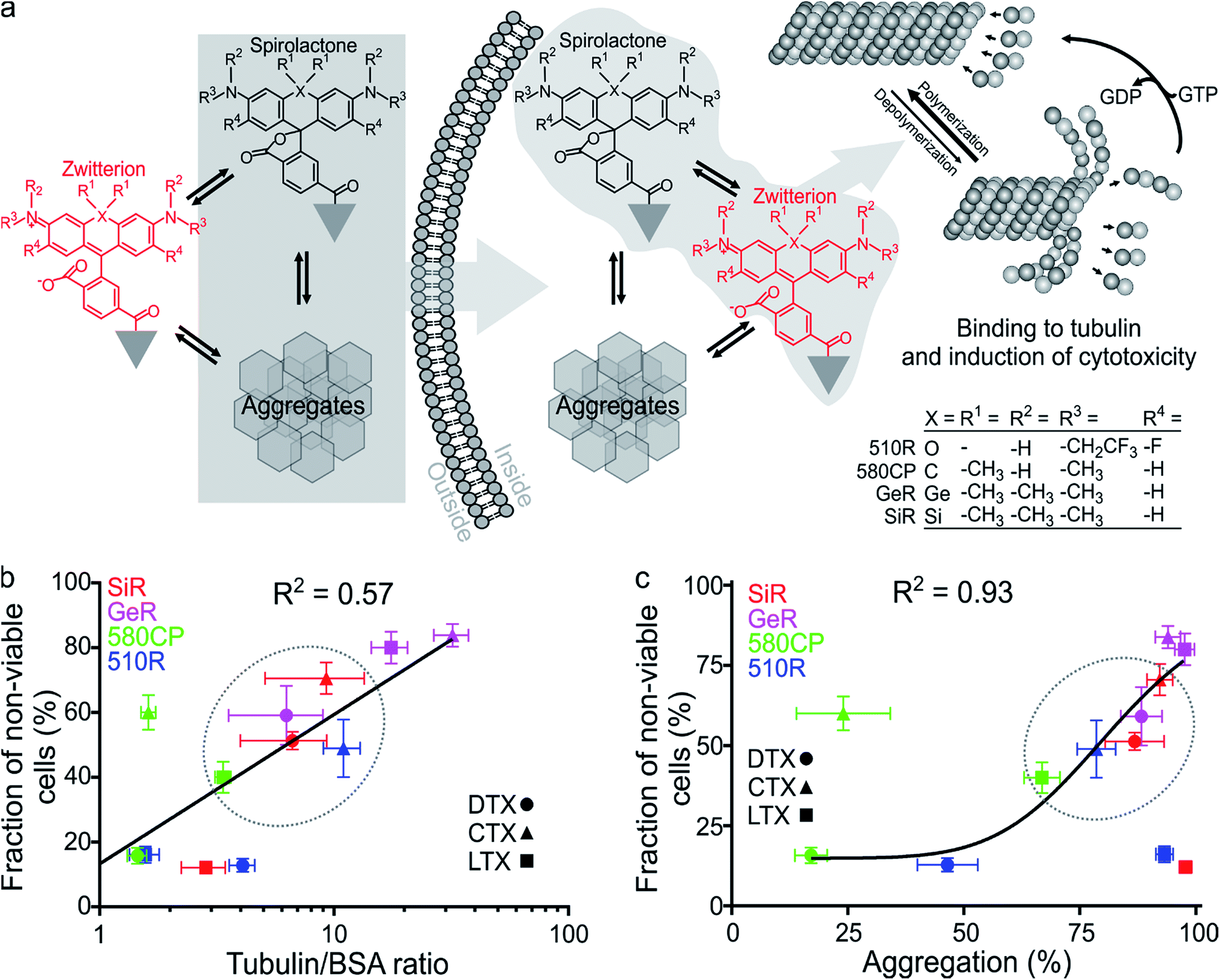 | ||
| Fig. 2 Properties of fluorescent tubulin probes. (a) Proposed mechanism of cell permeability and cytotoxicity in living cells. Targeting moiety (grey triangle) of these probes contains taxanes (DTX – docetaxel, CTX – cabazitaxel or LTX – larotaxel), which are able to stabilize polymerised tubulin resulting in increased apparent polymerization rate. Black structure shows the spirolactone form, which is non-absorbing and non-fluorescent in the visible light spectral region, red – the absorbing and fluorescent zwitterion form. Grey hexagons indicate aggregates present in the equilibrium. The spirolactone form and aggregates (grey background on the left) are efficiently entering the cell. Inside, free spirolactone and zwitterion (grey background on the right) can stabilize tubulin and induce cytotoxicity. GTP – guanosine-5′-triphosphate, GDP – guanosine-5′-diphosphate. (b) Correlation between the fluorescence increase upon binding of 2 μM probes to 1 mg ml−1 tubulin and measured cytotoxicity in cells incubated in growth medium containing 1 μM probe after 24 h. Blue symbols correspond to 510R derivatives, green – 580CP derivatives, purple – GeR derivatives and red – SiR derivatives. Black line represents fitting to a linear equation. (c) Correlation between the probe aggregation efficiency in vitro and cytotoxicity after 24 h. In both cases probe concentration is 1 μM. Black line represents fitting to a dose–response equation in GraphPad Prism 6 (EC50 = 83 ± 22%). Data are presented as mean values with standard deviations, N ≥ 3. Dashed ellipse shows the region corresponding to the optimal probes showing moderate toxicity. | ||
We further investigated the formation of aggregates and imaged them by phase contrast light microscopy (Fig. S4b†). The formed aggregates were clearly detected in the images corresponding to PBS (phosphate buffered saline) solutions of 510R-docetaxel (510R-DTX), 510R-larotaxel (510R-LTX), 580CP-cabazitaxel (580CP-CTX) and SiR-larotaxel (SiR-LTX) probes. In contrast, less pronounced aggregation was observed in case of 580CP-LTX and GeR-DTX. Encouraged by these observations, we developed an assay which permits estimation of the aggregation extent (Fig. S5a†). It is based on the separation of the aggregated and soluble fractions of the probes by centrifugation. In addition, the collected pellet can be completely dissolved in the PBS containing 0.1% SDS. This allows measurement of the fluorescence in the supernatant and in the dissolved pellet fractions after the aggregates were spun down. If the centrifugation step is omitted, the fluorescence was mainly present in solution (Fig. S5b†). The distribution of fluorescence between the two fractions correlated well with the fluorogenicity of the probes (Fig. S5c†).
Finally, we measured the fluorescence intensity changes upon addition of the target protein – tubulin. The most fluorogenic probes are based on GeR and SiR fluorophores, which have the highest D0.5 value (Fig. S6, Tables S1 and S3†). Interestingly, the fluorescence intensity change was lower for larotaxel derivatives, as compared to cabazitaxel or docetaxel-derived probes. This suggests that these aggregates formed in aqueous buffers are very stable and cannot dissociate upon binding to tubulin. In contrast, the 580CP-larotaxel probe (580CP-LTX) displayed the largest fluorescence increase among the 580CP-based probes indicating the dissociation of the aggregates after binding to tubulin. These observations prompted us to verify whether the synthetic probes induce tubulin stabilization as efficiently as the corresponding parent compounds. We found that the replacement of an N-Boc group with a C8-alkyl-dye functionality decreases the apparent tubulin polymerization rate up to 5-fold (Fig. S7 and Table S3†). The smallest reduction was observed for 510R dye derivatives, while 580CP-, GeR- and SiR-containing probes were significantly less potent in stabilizing tubulin.
Cytotoxicity of the fluorescent tubulin probes
Taxanes are well known cytotoxic chemotherapy agents, but derivatization of these drugs may reduce their impact on cell cycle because of the changes in the cell membrane permeability and/or the target binding efficiency. Goodwin et al. demonstrated that transcellular permeability is a function of the molecule's hydrophobicity.20 On the other hand, the hydrophobicity of SiR probes, as it has been shown recently, is proportional to their fluorogenicity and vice versa.2c,16 We assumed that this might also be true for 510R-, 580CP- and GeR-based probes. We also assumed that the probe cytotoxicity is a superposition of the ability to enter the cell and the target binding efficiency. The spirolactone form is largely responsible for the cell permeability of the probe, as it is significantly more hydrophobic than the zwitterionic form (Table S2†). In addition, if aggregates are formed, they can be internalized into the cell via endocytosis pathways employing similar mechanisms described for nanoparticles (Fig. 2a).21 Indeed, nanoparticles have been extensively exploited for improving the permeability of paclitaxel.22To test this hypothesis, we examined the toxicity of our probes by incubating them with living Hela cells for 24 h and measuring the distribution of the cell population over the cell cycle (Fig. S8†). Paclitaxel derivatives are known to halt the cell cycle at the mitosis (M) phase leading to apoptosis and fragmentation of the cell DNA.23 We measured the accumulation of cells containing less DNA than cells at the G1 phase. The highest toxicity was observed for the probes derived from cabazitaxel, while larotaxel- and docetaxel-based probes were considerably less toxic (Fig. S8†). In the fluorophore series, the cytotoxicity decreased in the order GeR > SiR > 580CP ≈ 510R. In contrast, non-conjugated dyes did not change the population distribution over the cell cycle histograms (Fig. S8†).
Finally, we correlated the probe-induced toxicity with the fluorescence increase upon binding to tubulin and the aggregation extent. We assumed that if the concentration of free tubulin largely exceeds the probe concentration, the binding efficiency of the fluorogenic ligands will also be proportional to the fluorescence intensity increase, which is likely to occur in living cells due to high tubulin abundancy (2.4 mg ml−1 or 24 μM dimer).24 In agreement, we observed a significant correlation (R2 = 0.57) between the probe cytotoxicity and fluorescence increase upon tubulin binding (Fig. 2b). Furthermore, we observed a strong correlation between the cytotoxicity and the aggregation extent (R2 = 0.93, Fig. 2c), but no significant correlation between apparent tubulin polymerization rate and cytotoxicity (Fig. S9†). This behaviour can be explained if the cell permeability of probes is enhanced by the formation of aggregates. Similarly, nanoparticles, a higher order structures of small molecules, are known to demonstrate improved cellular uptake profiles.21,25 This supports our suggestion that the probe cytotoxicity is a superposition of the ability to enter the cell and the target binding efficiency. However, if we assume that the deviations from the linear correlation observed for 510R-DTX, 510R-LTX, 580CP-CTX and SiR-LTX probes are significant (Fig. 2b and c), we may conclude that a certain degree of off-targeting, specific to the particular combination of a dye, a linker and a targeting moiety, is taking place. These results highlights the importance of a full experimental study of newly generated probes regarding their staining performance and cytotoxicity.
Staining of tubulin in living cells
The most critical parameter of all fluorescent probes is the target staining efficiency. We imaged living human fibroblast cells labelled with our tubulin probes using confocal microscopy. The staining intensity of microtubules varied significantly among the probes (Fig. S10†). In general, docetaxel-based probes showed a lower staining intensity compared to the probes based on the other two ligands. Cabazitaxel-derived probes stained tubulin efficiently in combination with all fluorophores (Fig. S10†). This behaviour correlates with the observed cytotoxicity, suggesting that the better binding to tubulin leads to a higher microtubule stabilization resulting in higher cytotoxicity of the probes. However, larotaxel-derived probes demonstrated the highest variation: 580CP-LTX showed low toxicity and excellent microtubules staining, GeR-LTX was highly toxic and stained microtubules poorly, 510R-LTX demonstrated low cytotoxicity, but highlighted the cytosol without microtubule structures, and SiR-LTX was neither toxic nor staining microtubules (Fig. S10 and Table S3†). This complex behaviour might stem from the strong aggregation of larotaxel probes (and higher content of the closed-ring isomer in the equilibrium) affecting both permeability and the binding efficiency to tubulin in vitro and in vivo.Taking into account the known mechanisms of fluorogenicity, cell permeability, as well as cytotoxicity, we attempted to identify the probes that efficiently stain microtubules and induce minimal perturbation to the cell cycle. Our results clearly show that the best combinations of the targeting moiety and fluorescent dye are the following: cabazitaxel in combination with 510R and SiR dyes (510R-CTX and SiR-CTX), larotaxel-580CP (580CP-LTX), and docetaxel-GeR (GeR-DTX). For the first time, we demonstrate that larotaxel derivative 580CP-LTX can be used for the staining of microtubules in living cells. It should be noted that most of the probes showed cytotoxicity at high concentrations (1 μM) after 24 h of incubation. We addressed this by determining the concentration of the probe at which it can be used for tubulin staining without substantial toxic effects over a 24 h period: < 250 nM for 580CP-LTX and GeR–DTX; ≤62.5 nM for 510R-CTX and SiR-CTX (Fig. S11 and Table S3†). In addition, we recorded time-lapse movies of the microtubule dynamics at these concentrations (Videos S1–S4†).
Super-resolution imaging of tubulin in living mammalian cells
After the characterization of the probes, we turned our attention towards their performance in live-cell STED nanoscopy. We stained and imaged living human fibroblasts with optimal probes: 510R-CTX, 580CP-LTX, SiR-CTX and GeR-DTX (Fig. 3a). The optical resolution in STED microscopy strongly depends on the de-excitation beam power and the fluorophore-specific saturation intensity Isat, defined as the intensity needed to deplete half of the excited molecules.26 For 580CP-, GeR- and SiR-based probes, we used a 775 nm STED laser, and for the 510R dye – a 587 nm STED laser. We measured the full width at half maximum (FWHM) of microtubule filaments at different de-excitation laser powers and calculated the Isat values (Fig. 3b and c and Table S3†). The highest optical resolution corresponding to the smallest microtubule FWHM could be achieved using the 510R-CTX and SiR-CTX probes (Table S3†). The apparent diameter of the microtubules is 35 ± 13 nm corresponding to 29 ± 11 nm optical resolution in living cells after taking into account the real microtubule diameter of ∼20 nm (Fig. 3b and c, S1 and S12a, Table S3†).18 To the best of our knowledge, this is the highest STED image resolution as yet reported in living cells. In addition, excellent performance of the probes was confirmed by acquiring high quality STED images at the sub-cytotoxic concentrations (Fig. S13†).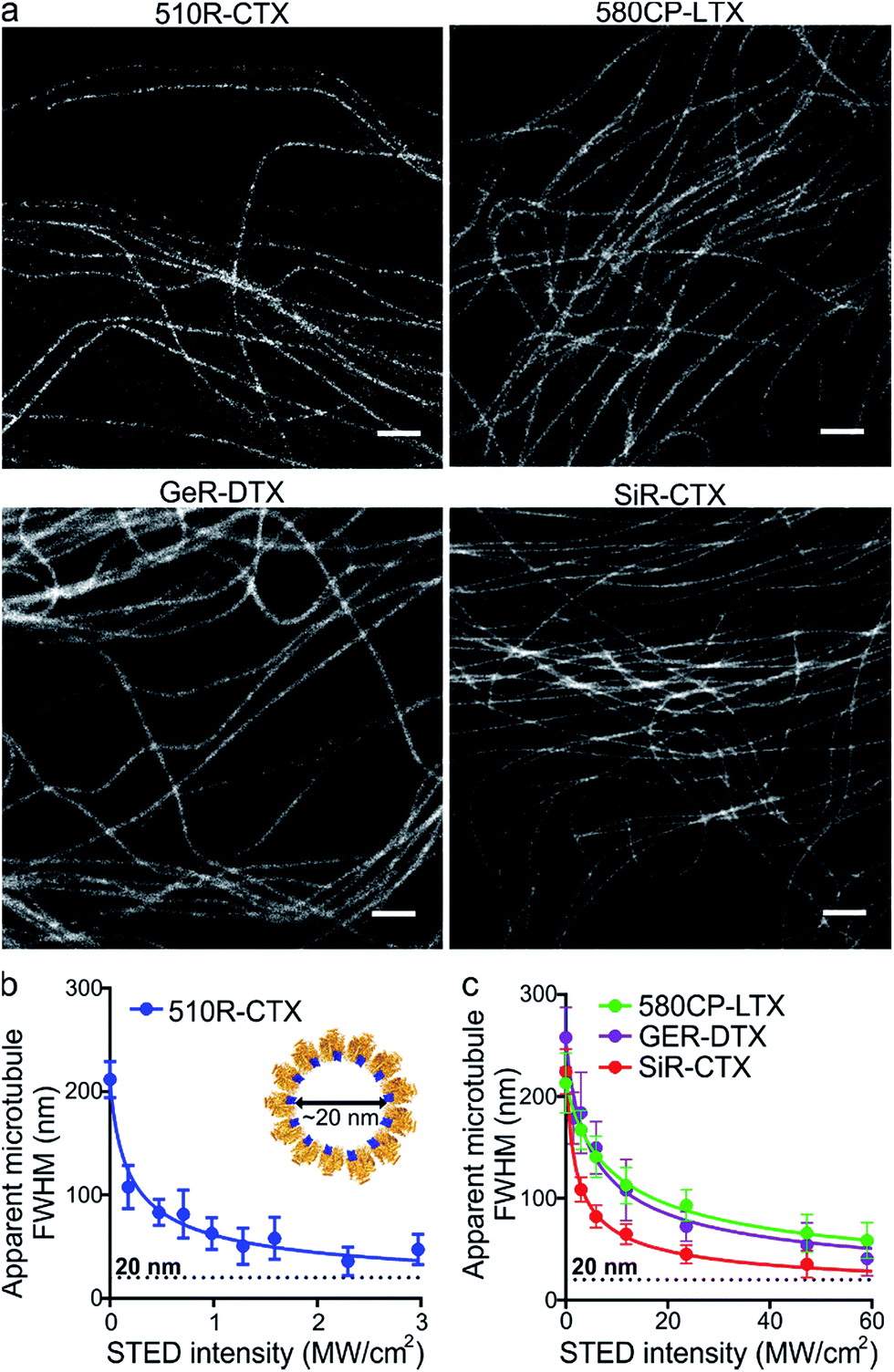 | ||
| Fig. 3 STED nanoscopy of living cells stained with fluorescent tubulin probes. (a) Selected images of tubulin filaments chosen for each fluorescent dye. 510R-CTX images were acquired on a custom built STED microscope equipped with a 587 nm STED laser. 580CP-LTX, GeR-DTX and SiR-CTX images were recorded on an Abberior STED 775 QUAD scanning microscope. Scale bars: 1 μm. (b) The apparent microtubule FWHM as a function of the intensity of the 587 nm STED laser used for imaging of specimens stained with 510R-CTX. Data are presented as mean values with standard deviations, N ≥ 10 microtubules in at least 3 different fields of view. Insert shows Cryo-electron microscopy model of 13 reconstructed tubulin (orange) protofilaments in the microtubule cross-section bound to paclitaxel (blue). The model predicts the apparent diameter of the microtubule stained with taxane derivatives to be ∼20 nm. (c) The apparent microtubule FWHM as a function of the 775 nm STED laser intensity used for imaging of specimens stained with 580CP-LTX (green), GeR-DTX (purple) and SiR-CTX (red). Data are presented as mean values with standard deviations, N ≥ 10 microtubules in at least 3 different fields of view. | ||
Super-resolution imaging of tubulin in living fruit fly tissues
Finally, we examined our probes for tubulin staining in living tissues of D. melanogaster and embarked on the probes based on the red-emitting dyes 580CP and SiR to minimize phototoxicity and light scattering in the tissue. Further, this dye combination allows two-colour STED nanoscopy.12a,27 To this end, we stained tissues from D. melanogaster larvae with 1 μM of 580CP–LTX or SiR-CTX probes. We observed bright tubulin staining of all analysed cell types, including cells of intestine, body wall muscles and brain of dissected third instar larvae. SiR-CTX staining of the microtubules allowed us to image their dynamics in the body wall muscles (Video S5†). Next to this, we noticed strong tubulin staining in the tracheole cells. STED images of the labelled microtubule network demonstrated significantly better resolution (∼60 nm) in various cell types and structures when compared with confocal images (Fig. S12b and c†).Staining of taenidia in insect tracheole with unconjugated fluorescent dyes
Besides the specific staining of microtubule filaments in the tracheole cells, we also observed a periodic pattern (Fig. S14†). Most likely, our probes could stain tracheoles themselves, which were previously observed and reported by Yalgin et al. as being surrounded with the tubulin network.28 This study used an anti-tubulin antibody to label the microtubule network in tracheole cells but observed neither periodical structures nor pattern. In a control experiment, we performed a competitive displacement of the fluorescent probes with non-fluorescent parent compounds in order to test if the periodic structures were indeed tubulin-specific (Fig. S15a and c†). In the images obtained, we could not detect the stained microtubules, however, the tracheolar network and its periodic structures were clearly highlighted. We therefore tested whether the tracheoles could be efficiently stained by simply adding non-conjugated carboxyrhodamine dyes. Indeed, 6-carboxy isomers of 580CP–COOH and SiR–COOH showed clearly visible enrichment in the respiratory system of D. melanogaster larvae living tissues (Fig. S15b and d†). We thought that such staining was arising from the affinity of non-conjugated dye to the chitin present in these insects. Accordingly, in addition to a diffuse background staining we observed an excellent co-localization of SiR–COOH fluorescence signal with the chitin-specific dye Calcofluor White M2R (Fig. S16a†).29 Furthermore, the chitin-rich larva cuticle showed bright staining with both 580CP–COOH and SiR–COOH (Fig. S16b and c†). Importantly, we could obtain STED images of the labelled taenidia of tracheoles and measure their periodicity, which correlated with their diameter (Fig. 4d and e, S15e and f†). The observed direct correlation is not surprising since the main function of taenidia is to keep the tracheole open for air flow and bigger diameter tracheoles require thicker taenidia with larger spacing.30 For the first time, it allowed us to identify 6-carboxy isomers of 580CP and SiR dyes as chitin-specific stains compatible with STED nanoscopy. We took advantage of this knowledge and recorded two-colour STED images of tracheole taenidia surrounded by the tubulin network of tracheole cells stained with 580CP–LTX and SiR–COOH (Fig. 4f). Both structures are of size below the diffraction barrier, and STED nanoscopy images enabled us to see the details in living tissues and with high specificity that were previously only visible using (cryo-)electron microscopy.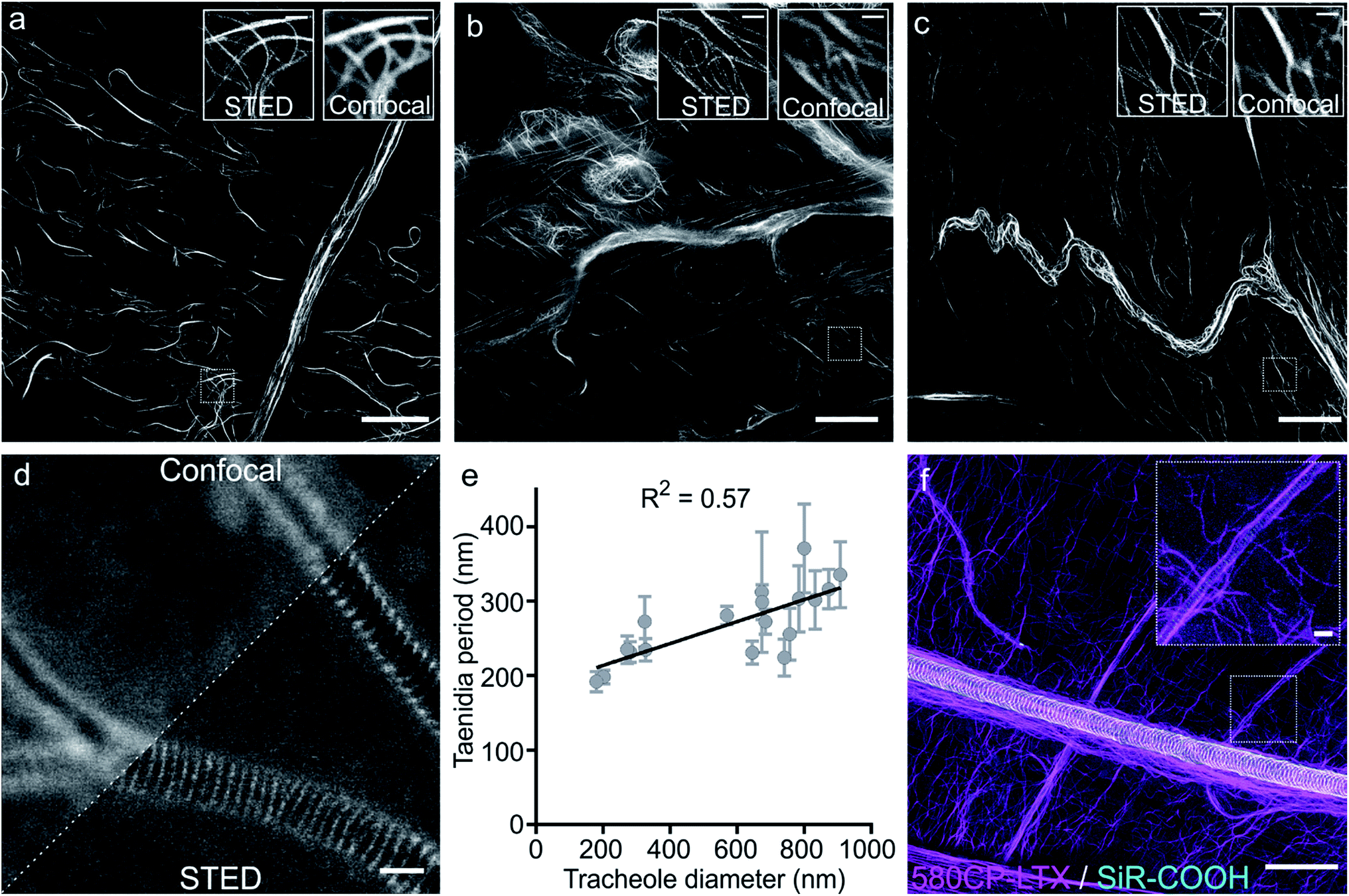 | ||
| Fig. 4 STED nanoscopy images of living tissues of Drosophila melanogaster larvae. (a) Larval brain stained with 1 μM SiR-CTX. (b) Intestine stained with 1 μM SiR-CTX. (c) Body wall muscle stained with 1 μM 580CP-LTX. White rectangles show locations of the zoom-in regions shown in the inserts. Scale bars: 10 μm in the large field of view and 1 μm in the zoom-in images. (d) Confocal and STED images of tracheoles stained with 1 μM SiR–COOH for 1 h. Scale bar: 1 μm. (e) Correlation between the tracheole diameter and periodicity of its taenidia. Periodicity is presented as mean with standard deviation, N ≥ 7. Tracheole diameter is presented as a single measurement in the region where periodicity was measured. (f) Two-color image of body wall muscle microtubule network stained with 1 μM 580CP-LTX and tracheoles stained with 1 μM SiR–COOH. Insert shows zoom-in image of the region shown in panel as a white rectangle. Image shows microtubule network following tracheole. Images were acquired on an Abberior STED 775 QUAD scanning microscope. Scale bars: 5 μm in the large field of view and 1 μm in the zoom-in image. | ||
Conclusions
In conclusion, we designed and prepared a set of fluorescent probes targeting tubulin by linking fluorescent dyes and taxanes. These probes are suitable for super-resolution STED microscopy of living cells and tissues and allow observation of tubulin structures with optical resolution below the diffraction limit (<250 nm). We provide further evidences supporting the mechanism of fluorogenicity proposed earlier and demonstrate the correlation between the aggregation efficiency in vitro and cytotoxicity in cellulo. We have reached 29 ± 11 nm resolution in STED microscopy images using our optimal probes. To the best of our knowledge, this is the highest STED image resolution reported in living cells. We envision that the optimization of both the fluorophore and the ligand part of a fluorescent probe will be extended to the probes targeting other cellular structures. For the first time, we identified the 6-carboxy isomers of 580CP and SiR dyes as efficient stains for chitin and successfully used them for labelling of taenidia in tracheoles of living insect tissues. This allowed us to reveal the periodic taenidia pattern present in tracheoles. The combination of a free carboxyrhodamine fluorescent dye with a specific tubulin probe allowed us to acquire two-colour STED images of both tracheolar and microtubular networks in living insect tissues. We anticipate that our new specific tubulin and chitin probes will enable further studies of these structures using STED nanoscopy in living cells and tissues.Experimental
Molecular docking
A Tubulin structure was downloaded from PDB database (PDB ID: 5SYF) and only β-tubulin molecule was used for the docking experiment. Ligands were drawn using ChemDraw Professional 15.1 and prepared for docking with AutoDock Tools version 1.5.6 (ref. 31). The docking simulation was performed using the Vina Autodock version 1.1.2 (ref. 17). Twenty binding modes were generated starting from random configurations of ligand that had fully flexible torsional degrees of freedom. The dynamics of β-tubulin was not taken into account. Only the conformation with lowest binding energy was selected.Evaluation of the fluorogenic properties of 510R
The absorption spectra of 510R free carboxylic acid were recorded on a Varian Cary 4000 UV-Vis spectrophotometer in quartz cuvettes (3 ml) in aqueous dioxane solutions (4 μM) with water content varying from 10 to 100% and dielectric constant values (D) from 5.6 to 78.3 respectively. The maximal extinction (εmax) at 496 nm was detected in water solutions. The normalized ε/εmax ratio at 496 nm were taken for D0.5 determination.Measurements of quantum yields
Fluorescence quantum yields (absolute values) were obtained on a Quantaurus-QY absolute PL quantum yield spectrometer (model C11347-12, Hamamatsu) at ambient temperature (25 °C), excitation wavelengths were 20 nm shorter than the absorption maxima of the measured compounds; all measurements were performed in triplicates.Estimation of probe fluorescence increase upon SDS addition and target binding
Fluorescence increase of the probes upon SDS addition was measured by preparing 2 μM probe solution in PBS buffer (Lonza, Cat. no. BE17-516F) with or without 0.1% SDS (Acros Organics). The samples prepared in 1.5 ml tubes (Eppendorf) were incubated at room temperature for 1 h before measurements.Fluorescence increase of taxane-based probes binding to tubulin was estimated using the following procedure: the substrate from 1 mM DMSO stock solution was diluted to the final concentration of 3 μM in 1 mg ml−1 tubulin (Cytoskeleton, Cat. no. T240-C) solution in the general tubulin buffer (80 mM PIPES pH 6.9, 2 mM MgCl2 and 0.5 mM EGTA, Cytoskeleton, Cat. no. BST01-010) supplemented with 1 mM GTP (Thermo Fisher Scientific, Cat. no. R0461). Each sample of the final volume of 0.5 ml prepared in 1.5 ml tubes (Eppendorf) was incubated at 37 °C for 5 h before measurements. During this incubation tubulin polymerization occurs which can be detected by significant increase of the solution viscosity.
Absorption and fluorescence were measured with a multiwell plate reader Spark® 20M (Tecan) in glass bottom 96-well plates (MatTek, Cat. No. PBK96G-1.5-5-F) at room temperature (25 °C). Absorption of solutions was recorded from 320 nm to 850 nm with wavelength step size of 1 nm. The background absorption of a glass bottom plate was measured in wells containing only buffer and subtracted from the spectra of the samples. Fluorescence emission of the free 510R dye or its probes was recorded from 480 nm to 850 nm with 5 nm bandwidth and 2 nm step size while exciting at 470 nm with a 20 nm bandwidth. Fluorescence emission of the free 580CP or its probes was recorded from 550 nm to 850 nm with 5 nm bandwidth and 2 nm step size while exciting at 525 nm with a 15 nm bandwidth. Fluorescence emission of the free SiR/GeR or their probes was recorded from 620 nm to 850 nm with 5 nm bandwidth and 2 nm step size while exciting at 595 nm with a 15 nm bandwidth.
All samples were prepared in technical duplicates, which were repeated three times as three independent experiments performed on different days. Ratios F(PBS+SDS)/F(PBS) or F(PBS+tubulin)/F(PBS+BSA) of fluorescence signals at emission maxima were calculated.
Estimation of the probe aggregation
The estimation of aggregation extent was performed using a centrifugation-based assay. The 1 μM solutions of probes were prepared by adding 1000× DMSO stock solution to PBS (Lonza, Cat. no. BE17-516F). After 1 h incubation at RT, the solutions were centrifuged for 1 h at 20 °C in a centrifuge set at 16000g. The pellet and solution fractions were separated, the pellet was suspended in PBS + 0.1% SDS and the supernatant was supplemented with SDS to 0.1% concentration. After 1 h incubation at RT, the pellet was fully dissolved, and fluorescence was measured for both supernatant and pellet fractions with a multiwell plate reader Spark® 20M (Tecan) in a glass bottom 96-well plate (MatTek, Cat. no. PBK96G-1.5-5-F) at room temperature (25 °C). The following settings were used for excitation and emission: 510R – 500 and 525 nm, 580CP – 585 and 610 nm, GeR – 640 and 660 nm, SiR – 650 and 670 nm, respectively. In all cases, the bandwidth was set to 5 nm. Aggregation percentage was calculated using the following formula: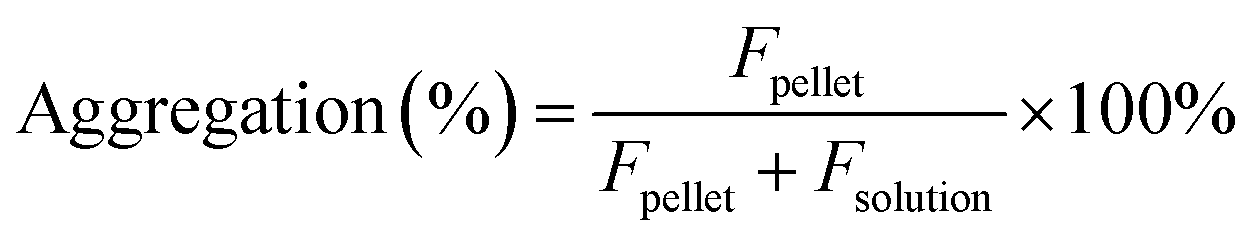 | (1) |
In vitro tubulin polymerization assay
Measurements of the polymeric tubulin stabilization enhancement upon addition of the probes were performed using the commercial tubulin polymerization fluorescence assay kit available from Cytoskeleton, Inc. (Cat. BK011P). It is based on DAPI (4′,6-diamidino-2-phenylindole) fluorescence change upon tubulin polymerization.32 Taxanes are known to stabilize tubulin in the polymerized state resulting in an increased apparent polymerization rate. All measurements were carried out according to the manufacturer's standard protocol. Samples were measured with a multiwell plate reader Spark® 20M (Tecan) in a half-area 96-well plate (Greiner Bio-One Cat. 675076) at 37 °C. Fluorescence emission was detected at 430 nm while exciting at 350 nm. The excitation and emission bandwidths for all measurements were set to 20 and 10 nm, respectively. All samples were prepared in duplicates and fluorescence was measured over 45 min (one measurement per minute) until a stable signal was achieved. The data points were normalized to the highest value of each set. The obtained polymerization curves were analysed using GraphPad Prism version 6.0 and fitted to a model describing plateau followed by one-phase exponential association:| y = y0 + (plateau − y0) × (1 − e−K×(x−x0)), | (2) |
Maintenance and preparation of cells
Human primary dermal fibroblasts and HeLa cells were cultured in high-glucose DMEM (Life Technologies, Cat. no. 31053-028) supplemented with GlutaMAX-1 (Life Technologies, Cat. no. 35050-038) and 10% fetal bovine serum (FBS, Life Technologies, Cat. no. 10270-106) in a humidified 5% CO2 incubator at 37 °C. The cells were split every 3–4 days or at confluence. Cells were seeded in glass bottom 12-well plates (MatTek Corporation, Cat. no. P12G-1.0-14-F).Cells were stained with the fluorescent probes in DMEM (Thermo Fisher Scientific, Cat. no. 31053-028) supplemented with 10% FBS (Thermo Fisher Scientific, Cat. no. 10082139) at 37 °C and 5% CO2. Afterwards, the cells were washed 2 times with HBSS (Hanks' balanced salt solution, Lonza, Cat. no. BE10-527F). Imaging was performed in DMEM with 10% FBS.
Maintenance and preparation of Drosophila melanogaster
Wildtype OregonR Drosophila melanogaster were raised on standard cornmeal–yeast–agar medium at 25 °C and used for all experiments. For staining of living Drosophila melanogaster tissues, wandering third instar larvae were dissected in 1× PBS (Phosphate Buffered Saline) pH 7.4 (Life Technologies, Carlsbad, California USA) and the inverted front half of the larva was incubated with probes of 1 μM in 1× PBS for 1 h at room temperature. After single washing step with 1× PBS, isolated tissues were mounted in 1× PBS under a coverslip and sealed with nontoxic duplicating silicone (picodent, Wipperfuerth, Germany) to prevent evaporation of the medium during imaging.Cell cycle analysis by imaging flow cytometry
HeLa cells were grown in 6-well plates (250.000 cells per well) for 24 h in the presence of the fluorescent probe in variable concentrations. The probes were dissolved in DMSO to 1 mM concentration, thus control samples were prepared by adding only DMSO. We found that HeLa cells do not adhere strongly to the plastic bottom of the 6-well plate and thus the trypsinisation step could be omitted. The cells were simply washed off and suspended in 1 ml of the growth medium by intensively pipetting up and down. Next, the cells were processed according to the NucleoCounter® NC-3000™ two-step cell cycle analysis protocol. Specifically, ∼500.000 cells were harvested by centrifuging at room temperature for 5 min at 400g. Afterwards, the cells were resuspended in 250 μl lysis solution (Solution 10, Chemometec Cat. no. 910-3010) supplemented with 10 μg ml−1 DAPI (Solution 12, Chemometec Cat. no. 910-3012), incubated at 37 °C for 5 min. Then 250 μl of stabilization solution (Solution 11, Chemometec Cat. no. 910-3011) was added. Cells were counted on a NucleoCounter® NC-3000™ in NC-Slide A2™ slides (Chemometec, Cat. no. 942-0001) loaded with ∼30 μl of each of the cell suspensions into the chambers of the slide. Each time, ∼10.000 cells in total were measured, and the obtained cell cycle histograms were analysed with ChemoMetec NucleoView NC-3000 software, version 2.1.25.8. All experiments were repeated three times and the results are presented as means with standard deviations. The obtained mean values were compared by running multiple t-tests on GraphPad Prism version 6.0 software.Confocal microscope
Confocal imaging was performed on a Leica SP8 (Leica Microsystems, Mannheim, Germany) inverted confocal microscope equipped with an HC PL APO CS2 63×/1.40 OIL objective. Images were acquired using a 700 Hz bidirectional scanner, a pixel size of 70 nm × 70 nm, a pinhole of 95.6 μm (1 AU) and frame averaging of 3. Hoechst 33342 and Calcofluor White M2R were excited with a 405 nm laser and detected with a regular PMT in the 415–470 nm range. Fluorescent tubulin probes were excited and detected using the following parameters: 510R probes – excited with 488 nm laser and detected with Leica HyD detector set within the spectral range of 520–570 nm, 580CP probes – excited with 561 nm laser and detected in the range of 600–650 nm, GeR or SiR probes – excited with 633 nm laser and detected in the range of 670–720 nm.STED microscope with 775 nm depletion laser
Confocal and STED images were acquired on an Abberior STED 775 QUAD scanning microscope (Abberior Instruments GmbH, Germany) equipped with 485 nm, 561 nm and 640 nm 40 MHz pulsed excitation lasers, a pulsed 775 nm 40 MHz STED laser, and an UPlanSApo 100×/1.40 Oil objective. The following detection windows were used: GFP/A488 channel 525/50 nm, TMR/Cy3 channel 615/20 nm and Cy5 channel 685/70 nm. Pixel size was 20–50 nm for all confocal and STED images acquired on this setup. Laser powers were optimized for each sample. Estimation of STED effect was performed by varying STED laser power from 0 to 100% while measuring cells stained with tubulin probes. The given STED laser intensities were calculated from the power entering the back aperture of the objective and the donut area in the sample plane. We estimated the size of the donut based on the reflection of the STED laser beam from gold nanoparticles (Sdonut,775 = 4.23 × 10−9 cm2, delimited by the FWHM of the donut crest). The obtained data were fitted to the following equation using GraphPad Prism 6: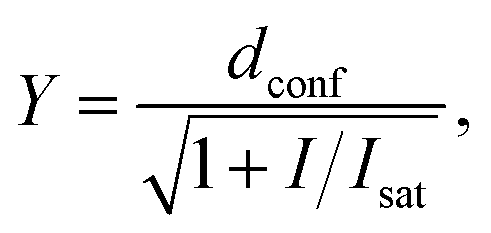 | (3) |
STED microscope with 587 nm depletion laser
STED pulses were delivered by a Raman-shifted fiber laser (Rainbow prototype system, IPG Photonics, Mountain View, CA, USA), which was operated at a wavelength of 587 nm with a repetition rate of 20 MHz and a pulse duration of 1.2 ns. For spectral clean-up, we used a BrightLine 591/6 filter (Semrock, Inc., Rochester, NY, USA). To create the helical phase pattern for lateral resolution enhancement, we applied the 582 nm vortex of a VPP-1b vortex phase plate (RPC Photonics, Inc., Rochester, NY, USA). For excitation at 488 nm, we used a PicoTA laser (TOPTICA Photonics AG, Gräfelfing, Germany and PicoQuant, Berlin, Germany) with a pulse duration of ∼100 ps, filtered by a BrightLine 488/6 clean-up filter (Semrock, Inc.). The output of the lasers was passed through appropriate acousto-optical modulators (MT110-1.5-VIS and MT110-A1-VIS, AA Opto-electronics, Orsay, France) for intensity modulation and switching, and delivered to the setup via polarization-maintaining single-mode fibers.Laser beams were overlaid using the following dichroic mirrors: 460DCXRU (angle-tuned) and 565DCXRU (both Chroma Technology Corp., Bellows Falls, VT, USA). Beam scanning was accomplished with a home-built scanner (“Quad-Scanner”) consisting of four galvanometric mirrors. Furthermore, we used a tube lens from Leica Microsystems GmbH (Wetzlar, Germany) in combination with a 100 × NA 1.4 plan-apo oil-immersion objective lens (HCX-PL-APO 100×/1.4–0.7 OIL CS, Leica Microsystems GmbH). A piezo translator (Mipos 100 PL CAP, piezosystem jena GmbH, Jena, Germany) moved the objective lens along the optical axis for precise focus control.
Fluorescence was collected with the same objective lens, descanned and imaged onto a pinhole of variable size (MPH16, Thorlabs, Inc., Newton, NJ, USA). For the present experiments, it was set to correspond to 1.0 × the diameter of the Airy disc at the location of the pinhole. Fluorescence was filtered with a BrightLine 525/50 emission filter (Semrock, Inc.) and detected with an avalanche photodiode (SPCM-AQRH-13, Excelitas Technologies Corp., Waltham, MA, USA).
STED laser pulses were sampled to trigger excitation pulses and to synchronize both lasers on a pulse-to-pulse basis using home-built electronics. For STED imaging, fluorescence detection was time-gated on a nanosecond timescale using home-built electronics. For both experiment control and image acquisition, we used the ImSpector software (Abberior Instruments GmbH, Göttingen, Germany). We realized hardware control via a field-programmable gate array board (PCIe-7852R, National Instruments Corp., Austin, TX, USA).
For STED imaging with a 587 nm de-excitation laser, we performed 4 line scans with 5 μs pixel dwell time and 23 nm pixel size. We calculated the STED laser intensities in the same manner as described for the 775 nm depletion laser, based on the power values and the measured donut area (Sdonut,587 = 2.96 × 10−9 cm2). Obtained data were fitted to eqn (3) using GraphPad Prism 6.
Processing and visualization of acquired images
All acquired or reconstructed images were processed and visualized using Fiji.33 Line profiles were measured using the “straight line” tool with the line width set to 3 pixels. All profiles were fitted to Gaussian or Lorentz distributions using appropriate tools available on GraphPad Prism 6.The optical resolution of STED microscopy images was calculated using the equation:
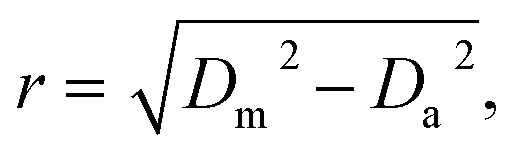 | (4) |
Conflicts of interest
G. L. has filed a patent application on SiR derivatives. V. N. B. and S. W. H. own shares in Abberior GmbH which markets the 510R dye as Abberior LIVE 510. S.W.H. owns shares in Abberior Instruments GmbH manufacturing Abberior STED 775 QUAD scanning microscope.Acknowledgements
The authors acknowledge the financial support provided by the German Bundesministerium für Bildung und Forschung (grant FKZ 13N14122). The authors are grateful to Prof. Dr Stefan Jakobs for providing equipment and reagents for the Drosophila melanogaster experiments. The authors acknowledge Jan Seikowski, Jens Schimpfhauser and Jürgen Bienert for the synthesis and characterization of numerous tubulin probes. G.L. is grateful to the Max Planck Society for a Nobel Laureate Fellowship. The authors thank Dr Dirk Kamin, Tanja Gilat and Dr Ellen Rothermel for technical assistance, and acknowledge Jaydev Jethwa for proofreading the manuscript.Notes and references
- (a) S. J. Sahl, S. W. Hell and S. Jakobs, Nat. Rev. Mol. Cell Biol., 2017, 18, 685–701 CrossRef CAS PubMed; (b) S. W. Hell, Angew. Chem., Int. Ed. Engl., 2015, 54, 8054–8066 CrossRef CAS PubMed.
- (a) J. B. Grimm, A. K. Muthusamy, Y. Liang, T. A. Brown, W. C. Lemon, R. Patel, R. Lu, J. J. Macklin, P. J. Keller, N. Ji and L. D. Lavis, Nat. Methods, 2017, 14, 987–994 CrossRef CAS PubMed; (b) A. N. Butkevich, V. N. Belov, K. Kolmakov, V. V. Sokolov, H. Shojaei, S. C. Sidenstein, D. Kamin, J. Matthias, R. Vlijm, J. Engelhardt and S. W. Hell, Chem.–Eur. J., 2017, 23, 12114–12119 CrossRef CAS PubMed; (c) G. Lukinavičius, L. Reymond, E. D'Este, A. Masharina, F. Gottfert, H. Ta, A. Guther, M. Fournier, S. Rizzo, H. Waldmann, C. Blaukopf, C. Sommer, D. W. Gerlich, H. D. Arndt, S. W. Hell and K. Johnsson, Nat. Methods, 2014, 11, 731–733 CrossRef PubMed; (d) K. Umezawa, M. Yoshida, M. Kamiya, T. Yamasoba and Y. Urano, Nat. Chem., 2017, 9, 279–286 CrossRef CAS PubMed; (e) G. Zhang, S. Zheng, H. Liu and P. R. Chen, Chem. Soc. Rev., 2015, 44, 3405–3417 RSC.
- (a) L. D. Lavis, Biochemistry, 2017, 56, 5165–5170 CrossRef CAS PubMed; (b) G. Lukinavičius and K. Johnsson, Nat. Chem., 2014, 6, 663–664 CrossRef PubMed.
- T. Terai and T. Nagano, Pflugers Arch., 2013, 465, 347–359 CrossRef CAS PubMed.
- C. C. Rohena and S. L. Mooberry, Nat. Prod. Rep., 2014, 31, 335–355 RSC.
- M. C. Wani, H. L. Taylor, M. E. Wall, P. Coggon and A. T. McPhail, J. Am. Chem. Soc., 1971, 93, 2325–2327 CrossRef CAS PubMed.
- Y. F. Wang, Q. W. Shi, M. Dong, H. Kiyota, Y. C. Gu and B. Cong, Chem. Rev., 2011, 111, 7652–7709 CrossRef CAS PubMed.
- (a) I. Barasoain, J. F. Diaz and J. M. Andreu, Methods Cell Biol., 2010, 95, 353–372 CAS; (b) J. F. Diaz, R. Strobe, Y. Engelborghs, A. A. Souto and J. M. Andreu, J. Biol. Chem., 2000, 275, 26265–26276 CrossRef CAS PubMed.
- (a) J. A. Evangelio, M. Abal, I. Barasoain, A. A. Souto, M. P. Lillo, A. U. Acuna, F. Amat-Guerri and J. M. Andreu, Cell Motil. Cytoskeleton, 1998, 39, 73–90 CrossRef CAS PubMed; (b) A. N. Butkevich, H. Ta, M. Ratz, S. Stoldt, S. Jakobs, V. N. Belov and S. W. Hell, ACS Chem. Biol., 2018, 13, 475–480 CrossRef CAS PubMed; (c) M. M. Lee, Z. Gao and B. R. Peterson, Angew. Chem., Int. Ed. Engl., 2017, 56, 6927–6931 CrossRef CAS PubMed.
- S. Nobili, I. Landini, T. Mazzei and E. Mini, Med. Res. Rev., 2012, 32, 1220–1262 CrossRef CAS PubMed.
- (a) A. Abidi, J. Pharmacol. Pharmacother., 2013, 4, 230–237 CrossRef CAS PubMed; (b) O. Metzger-Filho, C. Moulin, E. de Azambuja and A. Ahmad, Expert Opin. Invest. Drugs, 2009, 18, 1183–1189 CrossRef CAS PubMed.
- (a) A. N. Butkevich, G. Y. Mitronova, S. C. Sidenstein, J. L. Klocke, D. Kamin, D. N. Meineke, E. D'Este, P. T. Kraemer, J. G. Danzl, V. N. Belov and S. W. Hell, Angew. Chem., Int. Ed. Engl., 2016, 55, 3290–3294 CrossRef CAS PubMed; (b) G. Lukinavičius, K. Umezawa, N. Olivier, A. Honigmann, G. Yang, T. Plass, V. Mueller, L. Reymond, I. R. Correa, Jr., Z. G. Luo, C. Schultz, E. A. Lemke, P. Heppenstall, C. Eggeling, S. Manley and K. Johnsson, Nat. Chem., 2013, 5, 132–139 CrossRef PubMed; (c) G. Y. Mitronova, V. N. Belov, M. L. Bossi, C. A. Wurm, L. Meyer, R. Medda, G. Moneron, S. Bretschneider, C. Eggeling, S. Jakobs and S. W. Hell, Chem.–Eur. J., 2010, 16, 4477–4488 CrossRef CAS PubMed.
- (a) J. E. Cortes and R. Pazdur, J. Clin. Oncol., 1995, 13, 2643–2655 CrossRef CAS PubMed; (b) R. Pazdur, A. P. Kudelka, J. J. Kavanagh, P. R. Cohen and M. N. Raber, Cancer Treat. Rev., 1993, 19, 351–386 CrossRef CAS PubMed.
- M. D. Galsky, A. Dritselis, P. Kirkpatrick and W. K. Oh, Nat. Rev. Drug Discovery, 2010, 9, 677–678 CrossRef CAS PubMed.
- S. Schnorrenberg, T. Grotjohann, G. Vorbruggen, A. Herzig, S. W. Hell and S. Jakobs, eLife, 2016, 5 Search PubMed.
- G. Lukinavičius, L. Reymond, K. Umezawa, O. Sallin, E. D'Este, F. Gottfert, H. Ta, S. W. Hell, Y. Urano and K. Johnsson, J. Am. Chem. Soc., 2016, 138, 9365–9368 CrossRef PubMed.
- O. Trott and A. J. Olson, J. Comput. Chem., 2010, 31, 455–461 CAS.
- E. H. Kellogg, N. M. A. Hejab, S. Howes, P. Northcote, J. H. Miller, J. F. Diaz, K. H. Downing and E. Nogales, J. Mol. Biol., 2017, 429, 633–646 CrossRef CAS PubMed.
- H. N. Kim, M. H. Lee, H. J. Kim, J. S. Kim and J. Yoon, Chem. Soc. Rev., 2008, 37, 1465–1472 RSC.
- J. T. Goodwin, R. A. Conradi, N. F. Ho and P. S. Burton, J. Med. Chem., 2001, 44, 3721–3729 CrossRef CAS PubMed.
- V. Mailander and K. Landfester, Biomacromolecules, 2009, 10, 2379–2400 CrossRef PubMed.
- P. Ma and R. J. Mumper, J. Nanomed. Nanotechnol., 2013, 4, 1000164 Search PubMed.
- T. H. Wang, H. S. Wang and Y. K. Soong, Cancer, 2000, 88, 2619–2628 CrossRef CAS PubMed.
- D. L. Gard and M. W. Kirschner, J. Cell Biol., 1987, 105, 2191–2201 CrossRef CAS PubMed.
- (a) E. Frohlich, Int. J. Nanomed., 2012, 7, 5577–5591 CrossRef PubMed; (b) K. Cho, X. Wang, S. Nie, Z. G. Chen and D. M. Shin, Clin. Cancer Res., 2008, 14, 1310–1316 CrossRef CAS PubMed.
- B. Harke, J. Keller, C. K. Ullal, V. Westphal, A. Schonle and S. W. Hell, Opt. Express, 2008, 16, 4154–4162 Search PubMed.
- A. N. Butkevich, G. Lukinavičius, E. D'Este and S. W. Hell, J. Am. Chem. Soc., 2017, 139, 12378–12381 CrossRef CAS PubMed.
- C. Yalgin, M. R. Karim and A. W. Moore, J. Visualized Exp., 2011, 57, e3662 Search PubMed.
- M. V. Elorza, H. Rico and R. Sentandreu, J. Gen. Microbiol., 1983, 129, 1577–1582 CAS.
- M. R. Webster, J. J. Socha, L. Teresi, P. Nardinocchi and R. De Vita, Bioinspiration Biomimetics, 2015, 10, 066011 CrossRef PubMed.
- G. M. Morris, R. Huey, W. Lindstrom, M. F. Sanner, R. K. Belew, D. S. Goodsell and A. J. Olson, J. Comput. Chem., 2009, 30, 2785–2791 CrossRef CAS PubMed.
- D. Bonne, C. Heusele, C. Simon and D. Pantaloni, J. Biol. Chem., 1985, 260, 2819–2825 CAS.
- J. Schindelin, I. Arganda-Carreras, E. Frise, V. Kaynig, M. Longair, T. Pietzsch, S. Preibisch, C. Rueden, S. Saalfeld, B. Schmid, J. Y. Tinevez, D. J. White, V. Hartenstein, K. Eliceiri, P. Tomancak and A. Cardona, Nat. Methods, 2012, 9, 676–682 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7sc05334g |
| This journal is © The Royal Society of Chemistry 2018 |