
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTranslating molecular detections into a simple temperature test using a target-responsive smart thermometer†
Jingjing
Zhang
 ,
Hang
Xing
and
Yi
Lu
*
,
Hang
Xing
and
Yi
Lu
*
Department of Chemistry, University of Illinois at Urbana-Champaign, Urbana, IL 61801, USA. E-mail: yi-lu@illinois.edu
First published on 7th March 2018
Abstract
While it has been well recognized that affordable and pocket-size devices play a major role in environmental monitoring, food safety and medical diagnostics, it often takes a tremendous amount of resources to develop such devices. Devices that have been developed are often dedicated devices that can detect only one or a few targets. To overcome these limitations, we herein report a novel target-responsive smart thermometer for translating molecular detection into a temperature test. The sensor system consists of a functional DNA–phospholipase A2 (PLA2) enzyme conjugate, a liposome-encapsulated NIR dye, and a thermometer interfaced with a NIR-laser device. The sensing principle is based on the target-induced release of PLA2 from the DNA–enzyme conjugate, which catalyzes the hydrolysis of liposome to release the NIR dye inside the liposome. Upon NIR-laser irradiation, the released dye can convert excitation energy into heat, producing a temperature increase in solution, which is detectable using a thermometer. Considering the low cost and facile incorporation of the system with suitable functional DNAs to recognize many targets, the system demonstrated here makes the thermometer an affordable and pocket-size meter for the detection and quantification of a wide range of targets.
Introduction
The ability to detect and quantify a broad range of targets at home or in the field is very important for applications such as environmental monitoring, food safety and medical diagnostics.1–5 In order to carry out such measurements quantitatively, portable devices such as fluorimeters are often required, and, given the wide availability of smartphones, devices that can interface with smartphones are also preferred.6,7 Despite the progress made so far, most portable devices still require a fair amount of resources for their development and manufacture, making the devices unaffordable to most users at home. In addition, most of these devices are not small enough to be placed in pockets for convenient and frequent use. To overcome these limitations, we and others have taken advantage of the widely available pocket-sized personal glucose meters (PGMs) and developed novel methods to convert binding of non-glucose targets by functional DNAs (e.g., aptamers and DNAzymes) into glucose so that anyone can use the PGM for the quantification of a wide range of targets from heavy metal ions to organic toxins, cancers and pathogens.8–12 Even though millions of diabetics have PGMs, most other people do not possess PGMs at home. Therefore, there is a need to search for other portable devices that are even more widely available and cheaper.13,14The thermometer is arguably the most widely available, smallest and cheapest quantitative device for measurements. The determination of the temperature is one of the most important analytical methods in environmental monitoring and medical diagnostics.15–17 As a result, various thermometers were developed for scientific research,18,19 environmental monitoring,20,21 and personal healthcare.22 However, most of the commercialized thermometers are still limited in that they can only detect a single “target”, temperature.
A major challenge in using a thermometer to measure a broad range of targets beyond temperatures is to find a method that can link the binding of any targets with a change of temperature so that one can use any thermometer to detect the target and measure its concentrations. Conventional calorimetric biosensors utilize enzymatic reactions to generate heat and correlate it with the concentration of the substrate.23–28 Most of the methods, however, are either time-consuming, labor-intensive, high cost, or require highly technical expertise and sophisticated instrumentation, which limits their application for field work or POC testing.29,30 To overcome the limitation, integrated sensors based on the photo-thermal effect of hemoglobin have been described, but their sensitivity is low, due to the relatively low thermal generation efficiency of hemoglobin.31–33 While the exothermic chip sensor, based on an aptamer-modified hydrogel to modulate the capillary fluid flow rate to control the heat produced by the exothermic reagent (e.g. NaOH) dissolution, can provide high sensitivity,34 its performance is often affected by the endogenous fluid viscosity, making it unsuitable for the analysis of biological samples (e.g. blood or serum). Recently, a nanoparticle-mediated photothermal bioassay using a thermometer as the signal reader has been developed for detecting biomolecules (e.g. Prostate-specific antigen) using antibodies.35 However, antibodies don't have good sensitivity and selectivity when applied for detecting small molecule targets (e.g. metal ions and small molecule drugs). Therefore, it is still a challenge to integrate thermometers with a convenient and highly sensitive sensing system for quantitative bioanalysis of a wide range of targets.
In this paper, we take advantage of the excellent photo-thermal properties of indocyanine green (ICG) that is encapsulated in liposomes and target-responsive functional DNA (aptamer or DNAzyme) conjugated to phospholipase A2 (PLA2) to provide a quantitative link between the target and the temperature readout of a thermometer for on-site and real-time detections. Our sensor system includes several key features: first, a biotin-DNA immobilized onto magnetic beads (MBs) by means of streptavidin–biotin binding, and the DNA–PLA2 conjugate synthesized via the maleimide–thiol reaction, both of which hybridize with the target-responsive functional DNAs (e.g. structure-switching aptamer or RNA-cleaving DNAzyme) to form a DNA sandwich structure; this design can provide a general platform for the recognition of a wide range of targets. Second, ICG is the only near-infrared (NIR) photosensitizer dye approved by the United States Food and Drug Administration (FDA) for clinical use and has a wide range of applications in imaging and photo-thermal therapy,36,37 because it can effectively convert NIR laser light into heat. Third, liposomes are spherical lipid vesicles having a phospholipid bi-layered membrane structure,38 which have been widely used as carriers of different signaling tracers with high carrying capacity.39–42 Finally, the encapsulated ICG can be easily released by PLA2, which is a superfamily of enzymes that degrade phospholipids by cleaving the sn-2 acyl ester bond of glycerol phospholipids to produce free fatty acids and lysolipids.43,44 For this reason, PLA2 could provide a link between the target concentration and the temperature readout.
Results and discussion
The sensor reported in this work, called target-responsive smart thermometer (TRESTM), is comprised of three components (Scheme 1): a functional DNA conjugated to the PLA2 enzyme at its 5′ end and immobilized onto magnetic beads (MBs) at its 3′ end, which establishes the relationship between target concentration and enzyme concentration based on the target-induced release of the DNA–PLA2 conjugate; liposome-encapsulated ICG, which acts as a signal amplifier; and a NIR-laser integrated thermometer, which is used as a downstream transducer for temperature signal output. The binding of a target by the functional DNA can result in the release of PLA2 from the functional DNA–PLA2 conjugates that are immobilized on the MBs, and the released PLA2 then catalyzes the hydrolysis of the liposome to release the ICG from its core. Upon NIR-laser light irradiation in a photo-thermal chamber (Scheme 1b), the released ICG can convert the light excitation energy into heat, producing a temperature increase in solution, which is detectable using a super-fast Thermapen (Scheme 1c and d). Since the output temperature signal is related to the presence and concentration of the released ICG in solution, which in turn depends on the presence and concentration of the released PLA2 by the target, the presence and concentration of the target can be determined by monitoring the temperature change in the system. Furthermore, by taking advantage of the enormous amplification afforded by liposome-encapsulated ICG, the system can greatly enhance the high sensitivity of the novel thermometer transducer system, and hence lead to an ultrasensitive thermosensor.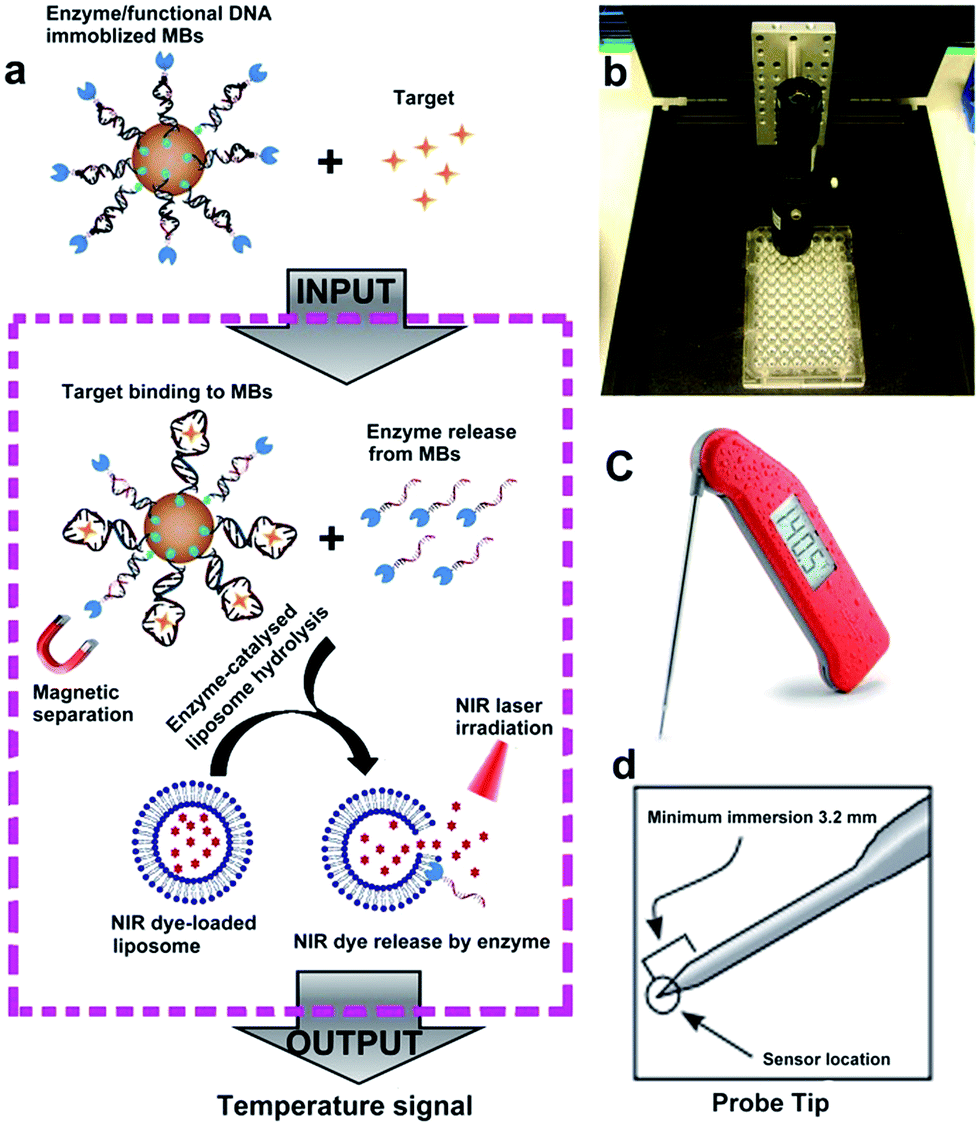 | ||
| Scheme 1 (a) Schematic view of the design and working mechanism of the sensor based on a functional DNA and thermometer. (b) Photo of the photothermal chamber including a NIR laser pointer, and a 96-well colorless plate. (c) Photo of the super-fast Thermapen with 3 second readings that were used to detect the temperature change during laser irradiation. (d) Probe tip with a minimum immersion of 3.2 mm, which could measure the solution temperature in a small volume (180 μL). | ||
Fig. S1a† illustrates the synthetic approach to prepare the DNA–PLA2 conjugate using the maleimide–thiol reaction.45 The UV-vis absorption spectrum of the purified DNA–PLA2 conjugate overlays well with the sum of the spectra of its two components, DNA and PLA2 (Fig. S1b†), suggesting successful conjugation. The successful conjugation has also been confirmed by SDS-PAGE, followed by fluorescence and photographic imaging. In this experiment, the thiolated DNA was modified with fluorescein so that the free DNA and DNA–PLA2 conjugate could be fluorescently imaged (Fig. S1c†), while the free PLA2 was stained with Coomassie Brilliant Blue (Fig. S1d†). Another important component of our thermometer system is the liposome that contains ICG, which can be prepared by the film hydration and extrusion method to sequester a large amount of the ICG dye for signal amplification.46–48 The UV-vis absorption spectrum of liposome-ICG shows a significant red shift (by 22 nm) from that of free ICG (Fig. 1a), suggesting that ICG has been integrated into the lipid bilayers of the liposome after encapsulation, as reported previously.49 The dynamic light scattering (DLS) spectrum suggests an average hydration diameter of ∼155 nm with a uniform distribution (Fig. S2†). After incubating the liposome-ICG with PLA2 for 1 h, the average hydration diameter increased to about 221 nm, indicating the hydrolysis of liposome-ICG by PLA2. The enzymatic hydrolysis reaction was further analyzed by monitoring the released ICG in the supernatant using UV absorption spectra (Fig. 1b), demonstrating that even subtle changes in PLA2 concentration (e.g. 5 nM) could release a large amount of the ICG dye, which can be subsequently transduced into an enhanced temperature signal.
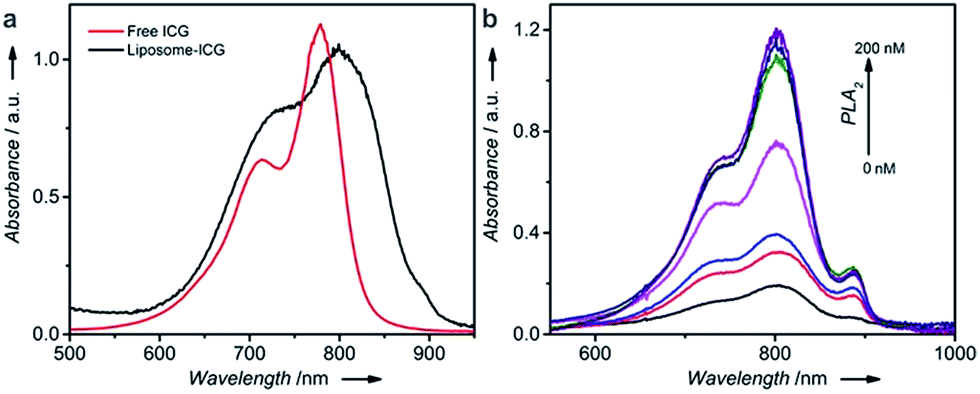 | ||
| Fig. 1 (a) UV-vis spectra of free ICG and liposome-ICG. (b) UV-vis spectra of the supernatant collected after incubating liposome-ICG with different concentrations of PLA2 for 2 h. Bottom to top: 0, 5, 10, 20, 50, 100, and 200 nM. | ||
To test the ability of the above TRESTM to detect a broad range of targets, we first explored quantitative detection of cocaine using a cocaine aptamer. Cocaine is an addictive drug, and serves as a representative model target for testing new aptamer-based analytical techniques due to unmet diagnostic needs.50,51 As shown in Fig. 2a, a DNA sandwich structure was assembled on MBs by connecting the DNA–PLA2 conjugate to a biotinylated capture DNA (biotin-DNA) through simultaneous hybridization with the cocaine aptamer. In the presence of cocaine, a target-specific structure switching of the aptamer could cause the disassembly of the DNA sandwich structure.52 This disassembly process was verified by fluorescence assays using FAM-labeled DNA–PLA2 conjugates (Fig. 2b). The fluorescence intensity increased as a function of increasing cocaine concentrations, establishing a quantitative relationship between the concentration of the target cocaine and the amount of DNA–PLA2 released into the solution. The released DNA–PLA2 conjugates were allowed to catalyze the hydrolysis of the liposome to release the ICG from its core, which converted the light excitation energy into heat, producing a temperature increase in solution that can be measured with a thermometer. To release as much ICG as possible for maximal signal output, the concentration of liposome-ICG, determined using a calibration curve of UV absorbance versus ICG concentration (Fig. S3†), was chosen as 0.25 mg mL−1 in subsequent experiments. In addition, to obtain high analytical performance, some important detection parameters, including the time of enzymatic hydrolysis and NIR irradiation, were also optimized (Fig. S4†).
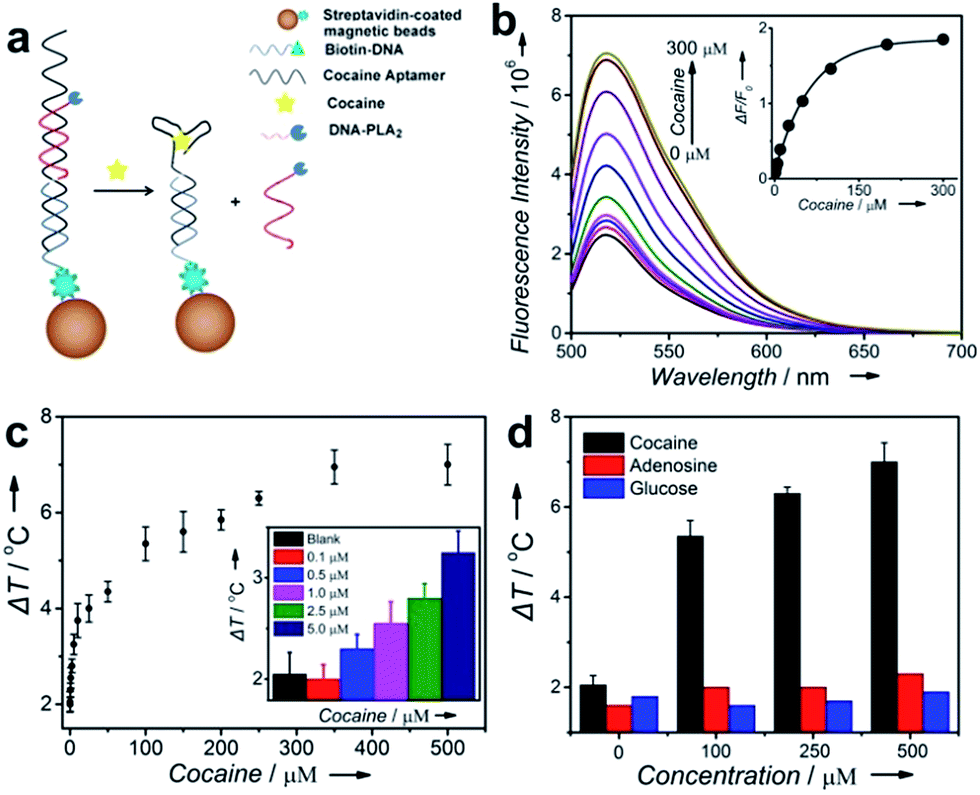 | ||
| Fig. 2 Design and performance of cocaine detection using the TRESTM system. (a) Cocaine-induced release of immobilized DNA–PLA2 conjugates. (b) Fluorescence spectra of the release of FAM-labeled DNA–PLA2 conjugates upon the addition of cocaine at different concentrations. Inset: calibration curve for cocaine detection. (c) Cocaine detection in buffer using the TRESTM. Inset: comparison of the temperature increase at low concentrations of cocaine. (d) Selectivity of cocaine detection using the TRESTM. | ||
In the absence of cocaine, the temperature increase (ΔT), defined as the temperature change of the supernatant solution before and after the NIR laser irradiation, was ∼2 °C, which may be due to the nonspecific release of the DNA–PLA2 conjugate or ICG leakage during centrifugation. In contrast, under the same incubation conditions, ΔT increased with the increasing input of cocaine and reached a plateau at 300 μM with temperature increased up to 7 °C (Fig. 2c). This observation is in agreement with the fluorescence results, which indicated that all the releasable DNA–PLA2 conjugates were released at this high concentration of cocaine. This TRESTM exhibited a wide dynamic range from 0 to 500 μM and showed a limit of detection of 3.8 μM. This sensitivity is comparable to that of commercial cocaine test kits and other portable meter-based cocaine sensors.10
To demonstrate the selectivity of the TRESTM, we further carried out a control experiment with two common biological compounds (adenosine and glucose). Continuous addition of up to 0.5 mM adenosine or glucose showed a negligible signal change. In contrast, the temperature response of cocaine at 100 μM was much higher (Fig. 2d). These results indicate that the TRESTM retained the aptamer selectivity with a highly specific response to the target, cocaine.
To test the feasibility of applying the TRESTM sensor in real samples, we explored the detection of cocaine in human serum, one of the most challenging media that contain a variety of proteins and other interfering molecules.53 By testing 18 serum samples spiked with different concentrations of cocaine with our TRESTM sensor, we found that the calibration curve in the presence of 20% serum (Fig. S5†) was similar to that in PBS buffer. The detection limit was 15.1 μM, which was comparable with that obtained in the buffer, suggesting that the other components of the serum did not interfere significantly with sensor performance. The results revealed that the TRESTM system showed promising applicability in complex body fluids.
Another key issue that needs to be addressed, especially for in-home or in-field diagnostics, is the acceptable shelf life of the TRESTM system. Previous research has shown that aqueous-instability of ICG limits its application as a fluorescence agent for imaging and sensing purposes. To address the question of whether such an instability affects the temperature signal in target detection, we monitored the output temperature signal to assess the stability of liposome-ICG as a signal reporter species by using 250 μM cocaine as the input. As shown in Fig. S6,† after refrigerated storage for one month, our TRESTM system retained more than 80% of its initial sensitivity, demonstrating that the liposome-ICG is stable and has a suitable shelf life for practical applications.
To demonstrate the generality of our TRESTM sensor, we extended our methodology from aptamers to DNAzymes for the on-site detection of uranyl (UO22+), which is a radioactive and potentially carcinogenic species. To achieve the detection, the DNA–PLA2 conjugates were connected to biotin-DNA immobilized MBs by the UO22+-specific DNAzyme (39E) and its substrate (39S) via 12-base-pair hybridization (Fig. 3a).54 Upon the addition of UO22+, the DNAzymes catalyzed the cleavage of their substrate at the 3′-phosphoester bond of the ribonucleotide A (rA) and subsequent release of the DNA–PLA2 conjugate, which was verified by fluorescence assays (Fig. 3b). A dose–response fluorescence curve for UO22+ detection is shown in the inset of Fig. 3b, indicating the possibility to further connect the UO22+ detection with the TRESTM system. As expected, our TRESTM sensor allowed the quantitative analysis of UO22+ in a dynamic range from 0 to 1 mM (Fig. 3c). The detection limit was calculated to be 25.7 nM (6.8 μg L−1), which is comparable to that of our previous PGM-based detection system (9.1 nM) and much lower than the US Environmental Protection Agency (EPA) regulated level (30 μg L−1 or 113 nM) for drinking water. In addition, the sensor maintained excellent selectivity for UO22+ over various other biologically relevant metal ions at physiologically relevant concentrations (Fig. 3d).
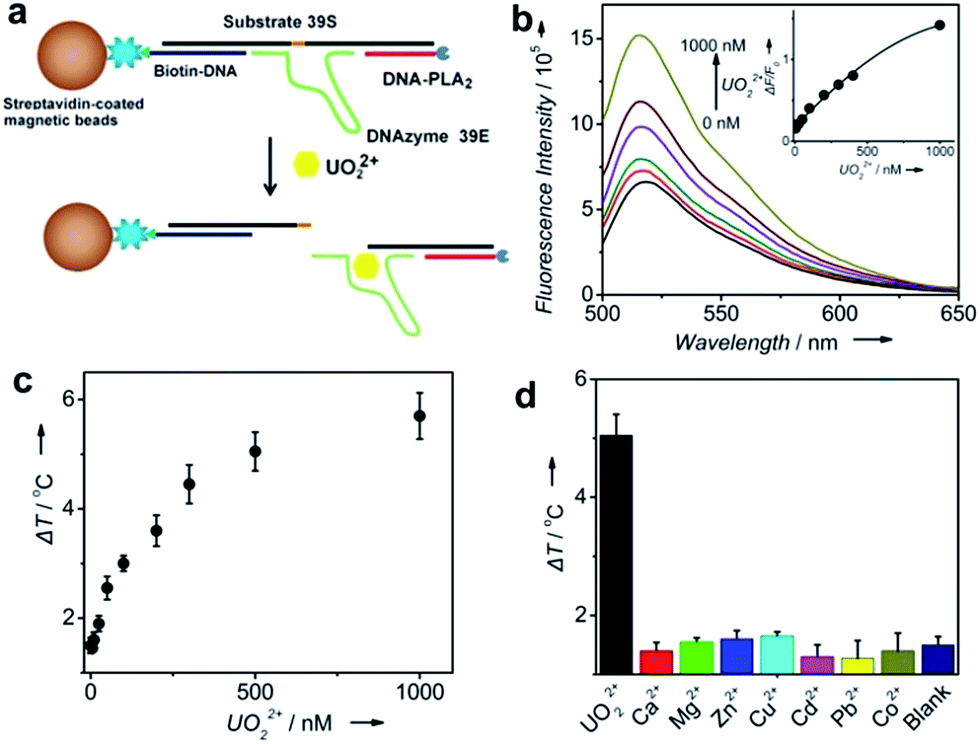 | ||
| Fig. 3 Design and performance of UO22+ detection using the TRESTM system. (a) UO22+-induced release of immobilized DNA–PLA2 conjugates. (b) Fluorescence spectra of the release of FAM-labeled DNA–PLA2 conjugates upon the addition of UO22+ at different concentrations. Inset: calibration curve for UO22+ detection. (c) UO22+ detection in buffer using the PTM. (d) Selectivity of UO22+ detection using the TRESTM system (UO22+, 500 nM; Ca2+ and Mg2+, 100 μM; other metal ions, 1 μM). | ||
Conclusions
In summary, we have demonstrated a new sensor system in which the presence and concentration of a target can be transformed to a detectable temperature change using a portable thermometer. The sensor system is based on the target-induced release of DNA–PLA2 conjugates from a functional DNA duplex immobilized on MBs, and subsequent hydrolysis of liposome-ICG and NIR-laser irradiation to produce a temperature signal. Using this system, cocaine and uranium have been detected quantitatively through a direct temperature reading. As more functional DNAs are available to bind a broad range of targets, our TRESTM will provide a generic general approach for quantitative detection of a wide range of targets at home or in the field using a portable thermometer, which is among the cheapest and most widely used analytical devices. Although the sensing system was not fully integrated into a single device, as a proof-of-concept, the sensing platform was shown to hold considerable promise for the future in terms of portability by taking advantage of well-developed microfluidic or lateral flow technology.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We wish to thank the U.S. National Institutes of Health (GM124316) for financial support.Notes and references
- M. H. Lin, J. J. Wang, G. B. Zhou, J. B. Wang, N. Wu, J. X. Lu, J. M. Gao, X. Q. Chen, J. Y. Shi, X. L. Zuo and C. H. Fan, Angew. Chem., Int. Ed. Engl., 2015, 54, 2151–2155 CrossRef CAS PubMed.
- M. Balcioglu, B. Z. Buyukbekar, M. S. Yavuz and M. V. Yigit, J. Biomol. Struct. Dyn., 2015, 33, 47–48 Search PubMed.
- H. Q. Zhang, M. D. Lai, A. Zuehlke, H. Y. Peng, X. F. Li and X. C. Le, Angew. Chem., Int. Ed. Engl., 2015, 54, 14326–14330 CrossRef CAS PubMed.
- Y. J. Song, Y. Y. Huang, X. W. Liu, X. J. Zhang, M. Ferrari and L. D. Qin, Trends Biotechnol., 2014, 32, 132–139 CrossRef CAS PubMed.
- J. S. Sun, Y. L. Xianyu and X. Y. Jiang, Chem. Soc. Rev., 2014, 43, 6239–6253 RSC.
- S. Ryu, I. Yoo, S. Song, B. Yoon and J. M. Kim, J. Am. Chem. Soc., 2009, 131, 3800–3801 CrossRef CAS PubMed.
- S. Freddi, L. Sironi, R. D'Antuono, D. Morone, A. Dona, E. Cabrini, L. D'Alfonso, M. Collini, P. Pallavicini, G. Baldi, D. Maggioni and G. Chirico, Nano Lett., 2013, 13, 2004–2010 CrossRef CAS PubMed.
- Y. Xiang and Y. Lu, Nat. Chem., 2011, 3, 697–703 CrossRef CAS PubMed.
- J. J. Zhang, Y. Xiang, M. Wang, A. Basu and Y. Lu, Angew. Chem., Int. Ed. Engl., 2016, 55, 732–736 CrossRef PubMed.
- L. Yan, Z. Zhu, Y. Zou, Y. S. Huang, D. W. Liu, S. S. Jia, D. M. Xu, M. Wu, Y. Zhou, S. Zhou and C. J. Yang, J. Am. Chem. Soc., 2013, 135, 3748–3751 CrossRef CAS PubMed.
- Z. Z. Wang, Z. W. Chen, N. Gao, J. S. Ren and X. G. Qu, Small, 2015, 11, 4970–4975 CrossRef CAS PubMed.
- J. Su, J. Xu, Y. Chen, Y. Xiang, R. Yuan and Y. Q. Chai, Chem. Commun., 2012, 48, 6909–6911 RSC.
- K. Tram, P. Kanda, B. J. Salena, S. Y. Huan and Y. F. Li, Angew. Chem., Int. Ed. Engl., 2014, 53, 12799–12802 CrossRef CAS PubMed.
- Z. Zhu, Z. C. Guan, D. Liu, S. S. Jia, J. X. Li, Z. C. Lei, S. C. Lin, T. H. Ji, Z. Q. Tian and C. Y. J. Yang, Angew. Chem., Int. Ed. Engl., 2015, 54, 10448–10453 CrossRef CAS PubMed.
- C. H. Marton, G. S. Haldeman and K. F. Jensen, Ind. Eng. Chem. Res., 2011, 50, 8468–8475 CrossRef CAS.
- H. F. Arata, Y. Rondelez, H. Noji and H. Fujita, Anal. Chem., 2005, 77, 4810–4814 CrossRef CAS PubMed.
- C. Gota, K. Okabe, T. Funatsu, Y. Harada and S. Uchiyama, J. Am. Chem. Soc., 2009, 131, 2766–2767 CrossRef CAS PubMed.
- D. Zhou, M. Lin, X. Liu, J. Li, Z. L. Chen, D. Yao, H. Z. Sun, H. Zhang and B. Yang, ACS Nano, 2013, 7, 2273–2283 CrossRef PubMed.
- Y. J. Cui, H. Xu, Y. F. Yue, Z. Y. Guo, J. C. Yu, Z. X. Chen, J. K. Gao, Y. Yang, G. D. Qian and B. L. Chen, J. Am. Chem. Soc., 2012, 134, 3979–3982 CrossRef CAS PubMed.
- S. Lee, J. H. Lee, M. Kim, J. Kim, M. J. Song, H. I. Jung and W. Lee, Appl. Phys. Lett., 2013, 103, 143114 CrossRef.
- S. Vermeir, B. M. Nicolai, P. Verboven, P. Van Gerwen, B. Baeten, L. Hoflack, V. Vulsteke and J. Lammertyn, Anal. Chem., 2007, 79, 6119–6127 CrossRef CAS PubMed.
- G. L. Ke, C. M. Wang, Y. Ge, N. F. Zheng, Z. Zhu and C. J. Yang, J. Am. Chem. Soc., 2012, 134, 18908–18911 CrossRef CAS PubMed.
- M. Yakovleva, S. Bhand and B. Danielsson, Anal. Chim. Acta, 2013, 766, 1–12 CrossRef CAS PubMed.
- F. E. Torres, M. I. Recht, J. E. Coyle, R. H. Bruce and G. Williams, Curr. Opin. Struct. Biol., 2010, 20, 598–605 CrossRef CAS PubMed.
- P. Paul, M. Hossain and G. S. Kumar, J. Chem. Thermodyn., 2011, 43, 1036–1043 CrossRef CAS.
- L. Zhang and T. Dong, J. Micromech. Microeng., 2013, 23, 045011 CrossRef.
- M. Yakovleva, O. Buzas, H. Matsumura, M. Samejima, K. Igarashi, P. O. Larsson, L. Gorton and B. Danielsson, Biosens. Bioelectron., 2012, 31, 251–256 CrossRef CAS PubMed.
- S. Zhou, Y. F. Zhao, M. Mecklenburg, D. J. Yang and B. Xie, Biosens. Bioelectron., 2013, 49, 99–104 CrossRef CAS PubMed.
- Y. H. Choi, M. G. Kim, D. H. Kang, J. Sim, J. Kim and Y. J. Kim, J. Micromech. Microeng., 2012, 22, 045022 CrossRef.
- L. Zhang, T. Dong, X. Y. Zhao, Z. C. Yang and N. M. M. Pires, IEEE Eng. Med. Biol., 2012, 523–526 CAS.
- B. S. Kwak, H. J. Kim, H. O. Kim and H. I. Jung, Biosens. Bioelectron., 2010, 26, 1679–1683 CrossRef CAS PubMed.
- B. S. Kwak, H. O. Kim, J. H. Kim, S. Lee and H. I. Jung, Biosens. Bioelectron., 2012, 35, 484–488 CrossRef CAS PubMed.
- C. Yi, J. H. Lee, B. S. Kwak, M. X. Lin, H. O. Kim and H. I. Jung, Sens. Actuators, B, 2014, 191, 305–312 CrossRef CAS.
- B. B. Gao, H. Liu and Z. Z. Gu, Lab Chip, 2016, 16, 525–531 RSC.
- G. Fu, S. T. Sanjay, M. Dou and X. Li, Nanoscale, 2016, 8, 5422–5427 RSC.
- A. Hannah, G. Luke, K. Wilson, K. Homan and S. Emelianov, ACS Nano, 2014, 8, 250–259 CrossRef CAS PubMed.
- L. Wu, S. T. Fang, S. Shi, J. Z. Deng, B. Liu and L. T. Cai, Biomacromolecules, 2013, 14, 3027–3033 CrossRef CAS PubMed.
- D. C. Turner, D. Moshkelani, C. S. Shemesh, D. Luc and H. L. Zhang, Pharm. Res., 2012, 29, 2092–2103 CrossRef CAS PubMed.
- K. Y. Chumbimuni-Torres, J. Wu, C. Clawson, M. Galik, A. Walter, G. U. Flechsig, E. Bakker, L. F. Zhang and J. Wang, Analyst, 2010, 135, 1618–1623 RSC.
- C. K. Kim and S. J. Lim, Liposomes, 2003, 373, 260–277 CAS.
- S. J. Lim and C. K. Kim, Anal. Biochem., 1997, 247, 89–95 CrossRef CAS PubMed.
- H. G. Nie, S. J. Liu, R. Q. Yu and J. H. Jiang, Angew. Chem., Int. Ed. Engl., 2009, 48, 9862–9866 CrossRef CAS PubMed.
- K. Jorgensen, J. Davidsen and O. G. Mouritsen, FEBS Lett., 2002, 531, 23–27 CrossRef CAS PubMed.
- H. Xing, C. L. Zhang, G. Ruan, J. J. Zhang, K. Hwang and Y. Liu, Anal. Chem., 2016, 88, 1506–1510 CrossRef CAS PubMed.
- A. S. D. S. Indrasekara, B. J. Paladini, D. J. Naczynski, V. Starovoytov, P. V. Moghe and L. Fabris, Adv. Healthcare Mater., 2013, 2, 1370–1376 CrossRef CAS PubMed.
- S. T. Proulx, P. Luciani, S. Derzsi, M. Rinderknecht, V. Mumprecht, J. C. Leroux and M. Detmar, Cancer Res., 2010, 70, 7053–7062 CrossRef CAS PubMed.
- S. K. Cool, B. Geers, S. Roels, S. Stremersch, K. Vanderperren, J. H. Saunders, S. C. De Smedt, J. Demeester and N. N. Sanders, J. Controlled Release, 2013, 172, 885–893 CrossRef CAS PubMed.
- F. L. Tansi, R. Ruger, M. Rabenhold, F. Steiniger, A. Fahr, W. A. Kaiser and I. Hilger, Small, 2013, 9, 3659–3669 CrossRef CAS PubMed.
- N. Beziere, N. Lozano, A. Nunes, J. Salichs, D. Queiros, K. Kostarelos and V. Ntziachristos, Biomaterials, 2015, 37, 415–424 CrossRef CAS PubMed.
- J. S. Swensen, Y. Xiao, B. S. Ferguson, A. A. Lubin, R. Y. Lai, A. J. Heeger, K. W. Plaxco and H. T. Soh, J. Am. Chem. Soc., 2009, 131, 4262–4266 CrossRef CAS PubMed.
- B. R. Baker, R. Y. Lai, M. S. Wood, E. H. Doctor, A. J. Heeger and K. W. Plaxco, J. Am. Chem. Soc., 2006, 128, 3138–3139 CrossRef CAS PubMed.
- J. L. He, Z. S. Wu, H. Zhou, H. Q. Wang, J. H. Jiang, G. L. Shen and R. Q. Yu, Anal. Chem., 2010, 82, 1358–1364 CrossRef CAS PubMed.
- A. X. Zheng, J. Li, J. R. Wang, X. R. Song, G. N. Chen and H. H. Yang, Chem. Commun., 2012, 48, 3112–3114 RSC.
- J. W. Liu, A. K. Brown, X. L. Meng, D. M. Cropek, J. D. Istok, D. B. Watson and Y. Lu, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 2056–2061 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Details of the experimental procedures and other figures. See DOI: 10.1039/c7sc05325h |
| This journal is © The Royal Society of Chemistry 2018 |