
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Protein stabilization by tuning the steric restraint at the reverse turn†
Priyanka
Lahiri‡
 ,
Hitesh
Verma‡
,
Ashraya
Ravikumar
and
Jayanta
Chatterjee
*
,
Hitesh
Verma‡
,
Ashraya
Ravikumar
and
Jayanta
Chatterjee
*
Molecular Biophysics Unit, Indian Institute of Science, Bangalore 560012, India. E-mail: jayanta@iisc.ac.in
First published on 24th April 2018
Abstract
Reverse turns are solvent-exposed motifs in proteins that are crucial in nucleating β-sheets and drive the protein folding. The solvent-exposed nature makes reverse turns more amenable to chemical modifications than α-helices or β-sheets towards modulating the stability of re-engineered proteins. Here, we utilize van der Waals repulsive forces in tuning the steric restraint at the reverse turn. The steric restraint induced upon N-methylation of the i+1–i+2 amide bond at the reverse turn results in well-folded and stable β-sheets in aqueous solution at room temperature. The developed superactive turn inducing motif is tolerant to a wide variety of functional groups present on coded amino acids making the designed turn fully compatible with bioactive loops in proteins. We demonstrate that the steric restraint and the functional groups at the reverse turn act in synergy to modulate the folding of re-engineered β-sheets. Introduction of the turn motifs onto a three-stranded β-sheet protein, Pin 1 WW domain, resulted in various analogs showing a cooperative two-state transition with thermal stability (TM) ranging from 62 °C to 82 °C. Despite modulating the stability of Pin 1 variants by ∼2.8 kcal mol−1 (ΔΔGf), the native fold in all the protein variants was found to be unperturbed. This structural stability is brought about by conformational preorganization at the engineered reverse turn that results in strong intramolecular hydrogen bonds along the three dimensional structure of the protein. Thus, this simple loop engineering strategy via two amino acid substitution provides us a “toolkit” to modulate the stability of β-sheet containing peptides and proteins in aqueous solution that will greatly expand the scope of de novo protein and foldamer design.
Introduction
An elegant combination of various secondary structure motifs, i.e., α-helices, β-strands and reverse turns acts as a framework for any protein structure.1 Recapitulation of such secondary structure elements in isolation from the protein environment is a challenging task and often requires covalent/non-covalent constraints to recover their native conformational and functional properties.2 Nevertheless, this approach has found tremendous utility in delivering small to medium sized constrained peptides and proteins and has revolutionized the development of peptide based therapeutics2c and the generation of novel non-antibody based scaffolds.3Utilizing such covalent and non-covalent constraints to engineer proteins and enzymes with improved thermodynamic stability has been another desirable area of interest. Tight packing in the hydrophobic core of a protein by optimizing the van der Waals interaction via substitution with non-polar amino acids is one of the most common strategies to enhance the protein stability.4 However, it was observed that if the volume of amino acid substitution is above the tolerance limit, the local increase in strain energy leads to de-stabilization of re-engineered proteins.5 Likewise, covalent tethering of protein secondary structures by disulfide bridge formation between canonical and noncanonical amino acid pairs and recently by thioether stapling has also found great success in enhancing the protein and enzyme stability.6
Free peptide bond rotation is a major driving force for the unfolding of a protein.7 Thus, the restriction of such free bond rotation at a site that is important for the folding of a protein would have a significant contribution towards reducing the conformational entropy of the unfolded state. β-Turns are such regions in a protein which are known to drive the protein folding process by nucleating the formation of β-sheets.8 Moreover, unlike α-helices and β-sheets the distinguishing feature of β-turns is their occurrence at solvent-exposed sites in globular proteins. Thus, in the context of protein engineering, β-turns are more amenable to chemical modifications, which would minimally perturb the native structure of a protein. A few turn-mimetics like R-nipecotic acid-S-nipecotic acid,9 charged dibenzofuran,10 Hot = Tap (derived from fusion of γ-hydroxythreonine and 4-thiaproline)11 and tetrasubstituted alkenes12 have been successfully utilized to re-engineer the conformational stability of (semi)synthetic proteins. However, these turn mimetics require elaborate chemical synthesis and lack the functional group diversity found in reverse turns of bioactive proteins. Therefore, we are particularly keen to develop super-active turn-inducing motifs8e bearing a diverse set of amino acids with different functional groups that can be easily and site-selectively incorporated into proteins via synthetic and semi-synthetic methods.
Unlike most earlier reported studies that utilize a covalent restraint to minimize the conformational entropy at the reverse turn, we chose to adopt a strategy based on the non-covalent van der Waals repulsion to tune the steric interactions at the β-turn. Inspired by our earlier study, we minimally modified the i+1–i+2 amide bond at the turn by N-methylation.13 Surprisingly, a single N-methylation was found to introduce a favorable pseudoallylic strain between various atoms of the i+1 and i+2 residues in the β-turn (Fig. 1), thereby minimizing its conformational entropy and imparting thermodynamic stability to the re-engineered proteins. We observed that the pseudoallylic pA1,3 strain is critical in nucleating the formation of a β-sheet in aqueous solution than the pseudoallylic pA1,2 strain. However, the presence of both pA1,3 and pA1,2 strain enhances the foldedness of the peptide. Since this pseudoallylic strain is common to any α-amino acid bearing a Cβ, it allowed us to functionalize the reverse turn with any desired amino acid. Furthermore, our screening led to the identification of several super-active turn inducing motifs that show remarkable thermodynamic stability of the re-engineered three-stranded β-sheet protein, Pin 1 WW domain, without altering its native conformation.
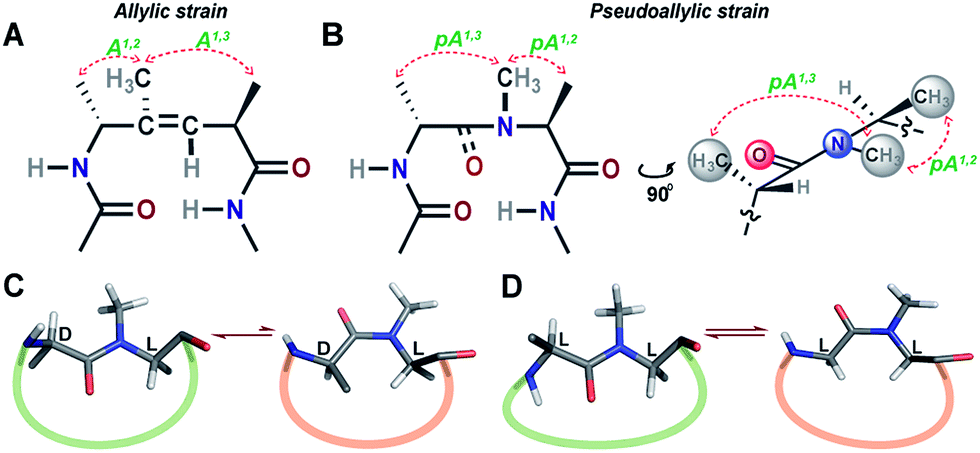 | ||
| Fig. 1 (A) The trisubstituted alkene isostere with the A1,2 and A1,3 allylic strain; note the hydrophobic nature of the reverse turn. (B) N-Methylated i+2 residue at the reverse turn displaying the pseudoallylic strain pA1,3 and pA1,2 between heterochiral residues. A side view representation of pseudoallylic strain pA1,3 and pA1,2. (C) In cyclic peptides with N-methylated heterochiral residues, the trans conformation of the N-methylated amide bond is energetically more favorable than the cis conformation. (D) While, in cyclic peptides with N-methylated homochiral residues, both cis and trans conformations are energetically favorable, as observed from the solution NMR studies (ESI†). | ||
Results and discussion
Differential control of cis/trans isomerism in peptide bonds by pseudoallylic strain
Towards re-engineering the conformational stability of proteins while maintaining the polar functional group diversity observed at the reverse turn, we sought to minimally modify the i+1–i+2 amide bond. We were inspired by the turn-inducing properties of trisubstituted alkene isosteres, in which the allylic A1,3 and A1,2 strain (Fig. 1A) plays a critical role in folding the polypeptide into a β-sheet conformation.12,14 However, the loss of electrostatic potential associated with the substitution of the amide bond with E-alkene that is devoid of any amino acid functional groups at the i+1 and i+2 sites15 could severely compromise the bioactivity of the re-engineered protein, wherein the molecular recognition is mediated by the amino acid sequence at the reverse turn.N-Methylation of an amide bond induces conformational restriction in a peptide backbone due to the pseudoallylic strain pA1,3 and pA1,2 between (i)CNMe/(i−1)Cβ and (i)CNMe/(i)Cβ, respectively (Fig. 1B).16 By analyzing the conformations of various literature reported N-methylated model and bioactive cyclic peptides (ESI†), we identified that the N-methylation-induced pseudoallylic strain between two heterochiral residues (L-/D- or D-/L-) strictly favors a trans peptide conformation, whereas when present between two homochiral residues (L-/L- or D-/D-), there is an equal probability for the N-methylated peptide bond to adopt a cis or a trans conformation (Fig. 1C and D).17
This finding encouraged us to evaluate the role of pseudoallylic strain in governing the cis/trans equilibrium of the N-methylated peptide bond in unrestrained linear peptides (Table 1) using NMR in aqueous buffer. We observed that in addition to the pA1,2 strain (L1), the introduction of pA1,3 strain increases the Ktrans/cis in the heterochiral system (L3) and significantly decreases the Ktrans/cis in the homochiral system (L2). The reduced Ktrans/cis in the homochiral system is a result of the unfavorable 1,3-diaxial strain in the trans conformation, which is absent in the heterochiral system (ESI†). Furthermore, the pA1,3 strain (L4) alone does not provide sufficient conformational restriction to favor the trans conformation; however, in combination with the pA1,2 strain (L3) it increases the Ktrans/cis. These results clearly indicate that the differential behavior of an N-methylated peptide bond depends on the neighboring substituents with the highest propensity to adopt a trans conformation when the flanking residues are heterochiral.
| Peptide | Strain | Sequence | K trans/cis |
|---|---|---|---|
| L1 | pA 1,2 | Ac-VG(NMe)AF-NH2 | 4.7 |
| L2 | pA 1,3 , pA1,2 | Ac-VA(NMe)AF-NH2 | 2.2 |
| L3 | pA 1,3 , pA1,2 | Ac-Va(NMe)AF-NH2 | 5.9 |
| L4 | pA 1,3 | Ac-Va(NMe)GF-NH2 | 1.8 |
Evidence of conformational restriction imposed by N-methylation
In order to understand the conformational restraint imposed by N-methylation of the peptide bond, we analyzed the Ramachandran plot of N-methylated linear tetrapeptides with varying pseudoallylic strain. We observed that irrespective of the chirality of N-methylated residue, N-methylation restrains Ψ of the preceding residue (compare Ψi+1 in Fig. 2A with that in Fig. 2C and D). On the other hand, N-methylation constrains both Φ and Ψ of the N-methylated residue (Fig. 2B–D). We clearly noted that the introduction of pA1,3 strain (e.g., i+1 residue in Fig. 2D as opposed to i+1 residue in Fig. 2B) or pA1,2 strain (e.g., i+2 residue in Fig. 2D as opposed to i+2 residue in Fig. 2C) significantly restricts the allowed conformational space about the N-methylated peptide bond. This finding correlates well with the restricted conformational freedom of N-methylated residues obtained from conformational energy calculations in N-methylated alanine and phenylalanine dipeptide models by Momany.18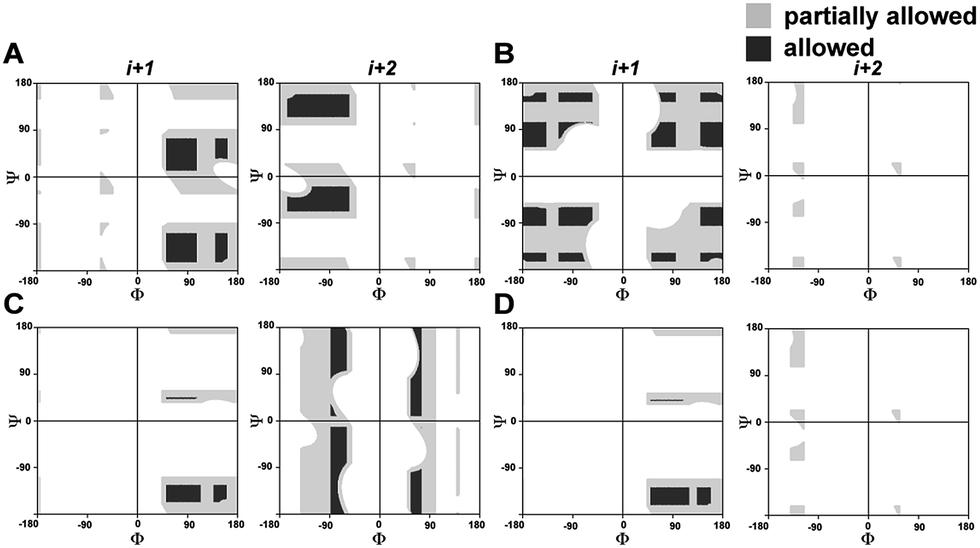 | ||
| Fig. 2 Partially allowed (grey) and allowed (black) torsion angles of the flanking amino acid residues (i+1 and i+2) about non-N-methylated and singly N-methylated peptide bonds. (A) Ac-AaAA-NH2 (absence of pA1,3 and pA1,2 strain); (B) Ac-AG(NMe)AA-NH2 (only pA1,2 strain); (C) Ac-Aa(NMe)GA-NH2 (only pA1,3 strain); (D) Ac-Aa(NMe)AA-NH2 (both pA1,3 and pA1,2 strain). | ||
Incorporation of pseudoallylic strain yields stable β-hairpins in aqueous solution
To demonstrate the applicability of pseudoallylic strain in inducing stable β-turns, we engineered the i+1 and i+2 residues in the reverse turn of a water-soluble β-sheet peptide, pG from the Gellman laboratory (Table 2).19| Peptide | Strain | Sequence |
|---|---|---|
| 1 | pA 1,2 | Ac-RYVEVG(NMe)AKKILQ-NH2 |
| 2 | pA 1,3 | Ac-RYVEVa(NMe)GKKILQ-NH2 |
| 3 | pA 1,3 , pA1,2 | Ac-RYVEVa(NMe)AKKILQ-NH2 |
| 4 | — | Ac-RYVEVaAKKILQ-NH2 |
| pG | — | Ac-RYVEVpGKKILQ-NH2 |
Initially, we synthesized four molecules with varying pseudoallylic strain in the reverse turn to analyze the critical role of N-methylation in inducing the β-sheet stability (Table 2). We acquired the CD (circular dichroism) spectrum at room temperature (25 °C) and pH 3.8 to obtain qualitative information on the foldedness of each peptide. Comparing the CD spectrum of 1, 4 and 2, 3 (Fig. 3B), we observed that only the latter showed the signature of a β-sheet peptide with a broad minimum centered at about 215 nm. Next, to quantitate the folding of each peptide, we resorted to 2D NMR studies at 25 °C (pH 3.8). The 1H NMR clearly indicated enhanced dispersion of HN resonance and upfield shift of Leu11 Hδ1/2 in the order 3 > 2 > 4 > 1 (Fig. 3C) which is not a consequence of the peptide aggregation as determined by the 1H NMR dilution experiments (ESI†). This order corroborates well with the Hα secondary chemical shifts of the strand residues in the individual peptides, as residues in a β-strand undergo a downfield shift as compared to a random coil (Fig. 3D).20 We also determined the summation of Hα secondary chemical shifts, which indicates the overall foldedness of the β-sheet peptide (Fig. 3E).21 The best folding is observed in 3 with both pA1,2 and pA1,3 followed by 2 with only pA1,3. Thus, pA1,3 is crucial in nucleating a stable reverse turn which is further strengthened by pA1,2, verifying our theoretical findings from the Ramachandran plot. Although, the control peptide 4 indicates that the i+1 and i+2 Cβ induces a certain amount of conformational restriction in the reverse turn, the introduction of an additional CH3 moiety in the amide bond (3), due to van der Waals repulsion, provides added stability to the β-sheet.
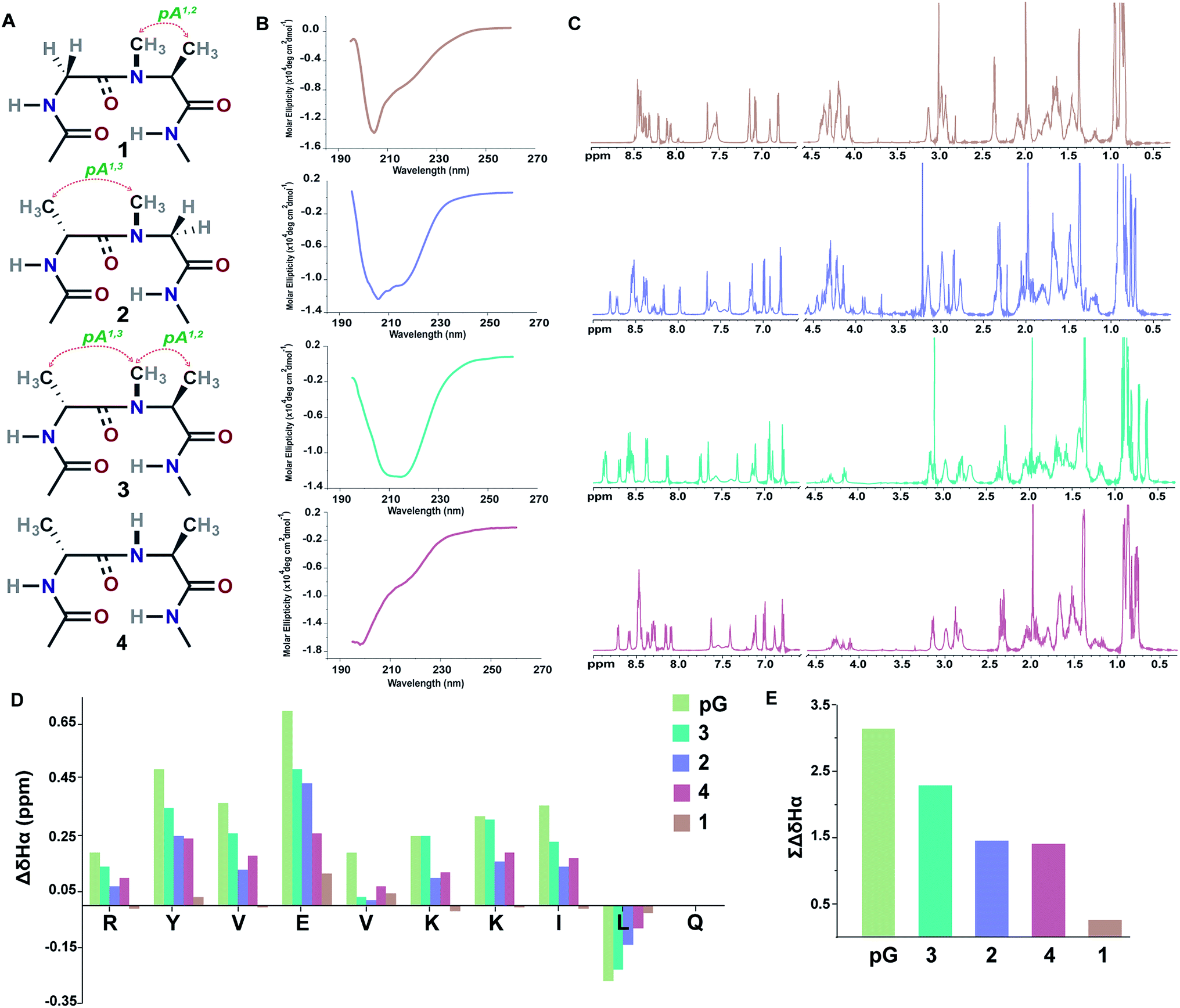 | ||
| Fig. 3 (A) Interplay of pseudoallylic strain (pA1,2 and pA1,3) in the reverse turn to induce the β-sheet folding. Note the spatial positioning of the methyl groups between i+1 Cα and i+2 Cα which modulates the folding of the polypeptide in aqueous solution at 25 °C. (B) CD spectrum of the peptides in acetate buffer (pH 3.8). (C) 1H NMR in acetate buffer (pH 3.8), clearly indicating the enhanced dispersion of the HN resonance and upfield shifted, distinguishable Leu11 Hδ1/2 in the well-folded peptides. (D) Secondary chemical shifts obtained from the respective random coil control peptides, where the D-residue is substituted with the L-residue. Leu11 Hα shows a negative deviation due to its proximity to the phenyl ring of Tyr2 in the folded analogs. In 1, the secondary chemical shifts were determined by subtracting the Hα chemical shifts from the mean random coil chemical shifts of the respective residues obtained from the random coils of 2 and 3. The ring constrained reverse turn motif, pG, was taken as a reference.19 (E) Summation of the secondary chemical shifts; only the absolute values were considered. | ||
Encouraged by this finding, we set out to systematically scan the i+1 and i+2 residues of the reverse turn in the β-sheet peptide with diverse functional groups, but fixed N-methylation at the i+1–i+2 amide bond.
Functional group diversity in the reverse turn of β-hairpin peptides
Our primary library was based on 3 that showed the maximum foldedness. Since the pseudoallylic strain pA1,3 is critical in folding the peptide, keeping the i+1 substitution constant, we initially sought to substitute the i+2 residue with various L-amino acid side chains and assess their influence on the β-sheet stability using ∑CSD (Fig. 4A).21 The characteristic NOEs observed in these compounds (Fig. 4B) indicated the occurrence of a βII′ turn, where the i+2 N-methyl and the i+2 Cβ are syn-periplanar. Most amino acids were tolerated at the i+2 site; however, the unfavorable steric clash (pA1,2 strain) between i+2 N-methyl and i+2 Cγ (due to the unrestricted rotation about χ1 in valine and isoleucine) leads to destabilization of 3b and 3a. In contrast, in 3e the β-branch demonstrates almost a two-fold increase in the ∑CSD resulting from the restricted χ1 rotation due to a probable intramolecular hydrogen bond between Thr–OH and its C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O (ESI†). Moreover, when the distance between the isopropyl group and the N-methyl group increases, e.g., in the γ-branched amino acid leucine (3k), we obtained a more than two-fold increase in the ∑CSD compared to that of 3b. Another notable observation was the increase in the foldedness of the β-sheet by incorporating positive charges, e.g., in 3l and 3n at the termini of an unbranched amino acid side chain 3f. These results show how modulation of local steric interactions and charges at a single site in the β-turn can alter the overall foldedness of the β-sheet.
O (ESI†). Moreover, when the distance between the isopropyl group and the N-methyl group increases, e.g., in the γ-branched amino acid leucine (3k), we obtained a more than two-fold increase in the ∑CSD compared to that of 3b. Another notable observation was the increase in the foldedness of the β-sheet by incorporating positive charges, e.g., in 3l and 3n at the termini of an unbranched amino acid side chain 3f. These results show how modulation of local steric interactions and charges at a single site in the β-turn can alter the overall foldedness of the β-sheet.
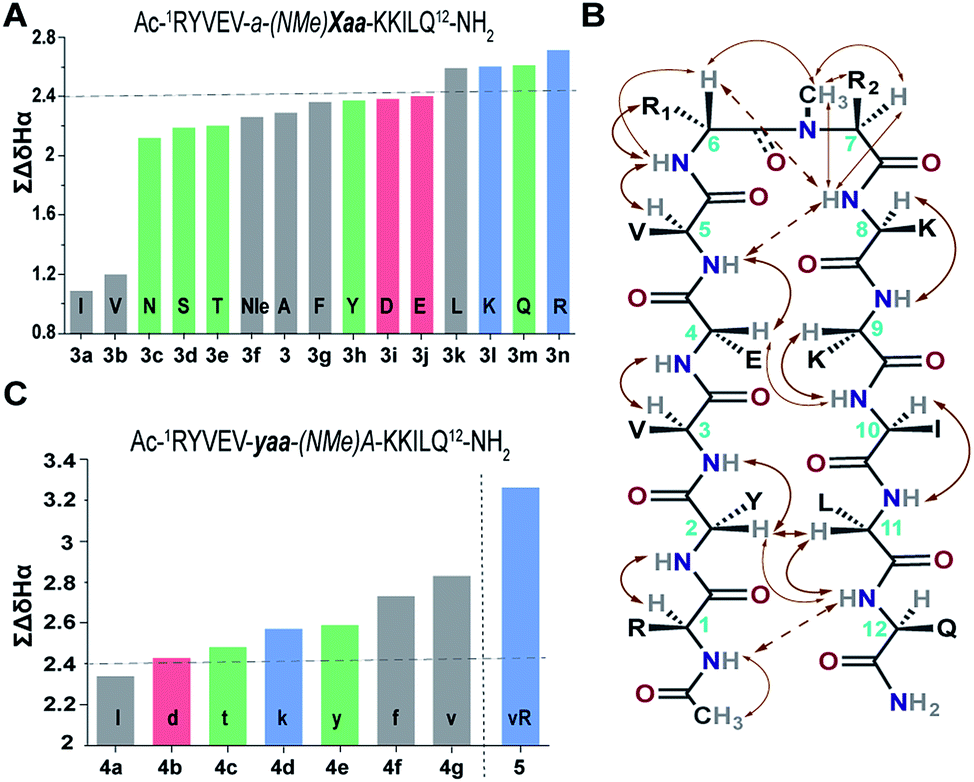 | ||
| Fig. 4 (A) ∑CSD of the primary library with L-amino acid substitutions (Xaa) at the i+2 site. (B) The characteristic NOEs, where the thick arrows denote strong (1.8–2.1 Å), the thin arrows denote medium (2.2–2.5 Å) and the broken arrows denote weak (>2.6 Å) NOEs. (C) ∑CSD of the secondary library with i+1 substitutions (yaa) along with 5 derived from the combination of 4g and 3n. All the NMR measurements were done in acetate buffer (pH 3.8) at 25 °C. | ||
Next, towards the thermodynamic analysis of i+2 substituted linear peptides, we estimated the folded population of selected β-sheets and their free energy of folding (ΔGfold) in water at 25 °C (Table 3) utilizing the respective fully folded disulfide bridged cyclic variant and unfolded random coil peptides by following the established methods.19,22 Overall, we observed that the increase in ∑CSD correlates well with the folded population of the β-sheets. We note that the introduction of pA1,3 alone is not sufficient to form an isolated stable β-sheet in water as reflected by the ΔGf of 0.17 ± 0.2 kcal mol−1 in 2. However, with the additional incorporation of pA1,2, the equilibrium shifts towards the folded side as observed in 3 (ΔGf of −0.48 ± 0.1 kcal mol−1). The presence of a hydrophobic β-branch (3a and 3b) is detrimental to the folding of the peptide, whereas a –CH3 to –OH substitution dramatically alters the folded population (ΔΔGf ∼ −0.6 kcal mol−1 between 3b and 3e). Thus, we observe that a combination of pA1,2 and polar functional groups enhances the stability of the β-sheet by −1.0 kcal mol−1 (ΔΔGf between 3n and 2).
| Fraction foldeda (%) | % folded population | ΔGfold (kcal mol−1) | ||||
|---|---|---|---|---|---|---|
| Val3 | Val5 | Lys8 | Ile10 | |||
| a Fraction folded and ΔGfold were calculated from the respective fully folded disulfide bridged cyclic variant and an unfolded random coil by established methods (ESI). | ||||||
| 2 | 39 | 60 | 36 | 38 | 43 ± 10 | 0.17 ± 0.2 |
| 3 | 68 | 80 | 68 | 59 | 69 ± 7 | −0.48 ± 0.1 |
| 3b | 38 | 63 | 32 | 30 | 41 ± 13 | 0.22 ± 0.2 |
| 3e | 64 | 80 | 67 | 59 | 67 ± 8 | −0.42 ± 0.2 |
| 3n | 78 | 99 | 76 | 70 | 81 ± 11 | −0.86 ± 0.3 |
| 4g | 84 | 99 | 81 | 75 | 85 ± 9 | −1.03 ± 0.3 |
| 5 | 95 | 99 | 89 | 85 | 92 ± 5 | −1.44 ± 0.4 |
| pG | 81 | 87 | 83 | 76 | 82 ± 3 | −0.90 ± 0.1 |
Subsequently, we constructed a secondary library with selected D-amino acid substitutions at the i+1 site with N-methylated alanine at the i+2 site. To our satisfaction, we observed the strong tolerance of various functional groups at the reverse turn (Fig. 4C). Of note, the β-branched amino acid valine, when present at the i+2 site (3b), by virtue of the unfavorable pA1,2 strain destabilizes the β-sheet, whereas when present at the i+1 site, the favorable pA1,3 strain enhances the stability of the β-sheet (4g) which is comparable to the stability of the ring constrained reverse turn motif pG (Table 3). Thus, the presence of an isopropyl group results in a 0.5–0.7 kcal mol−1 destabilization (ΔΔGf between 3 and 3b) or stabilization (ΔΔGf between 3 and 4g) when present at the i+2 or i+1 site, respectively (Table 3).
i+1 and i+2 residues induce a concerted effect on β-sheet stability
The enhanced thermodynamic stability of the β-sheet resulting from the conformational restriction in the reverse turn by the isopropyl group at the i+1 site led us to re-engineer the most folded analog obtained from the i+2 substitution library (3n). To our amazement, the introduction of the i+1 isopropyl group into 3n resulted in an additional 0.58 kcal mol−1 stabilization (ΔΔGf between 5 and 3n) yielding a super stable monomeric β-sheet 5. This result suggests that the i+1 and i+2 substituents act synergistically to enhance the foldedness of the engineered β-sheet. Additionally, although almost all functional groups are tolerated at the reverse turn, certain amino acids, leucine, lysine, glutamine, and arginine at the i+2 site and D-aspartate, D-threonine, D-lysine, D-tyrosine, D-phenylalanine and D-valine at the i+1 site, display high propensity to stabilize the β-sheet (demarcated by the dotted line at ∑CSD of 2.4 in Fig. 4A and C).To gain molecular-level insight into the factors responsible for the enhanced thermodynamic stability of the β-sheets, we determined the high-resolution NMR structures of 3n, 4g and 5, which show an increasing order of stability. The conformation of the three β-sheets revealed that the presence of β-branched D-valine at the i+1 residue in 4g (Fig. 5B) and 5 (Fig. 5C) leads to conformational restriction at the β-turn as opposed to non-β-branched residues as observed in 3n (Fig. 5A). This conformational restriction was also demonstrated by the narrow conformational space of the β-branched i+1 residue as opposed to the non-β-branched i+2 residue of 4g and 5 in the Ramachandran plots of the conformational ensemble (ESI†). We also noted that the Φ and Ψ values of the turn residues in 3n, 4g, and 5 (Table 4) represent a βII′ type turn and beautifully overlap with the theoretically predicted values of these torsion angles by the introduction of pA1,2 and pA1,3 strain in linear tetrapeptides (Fig. 2D).
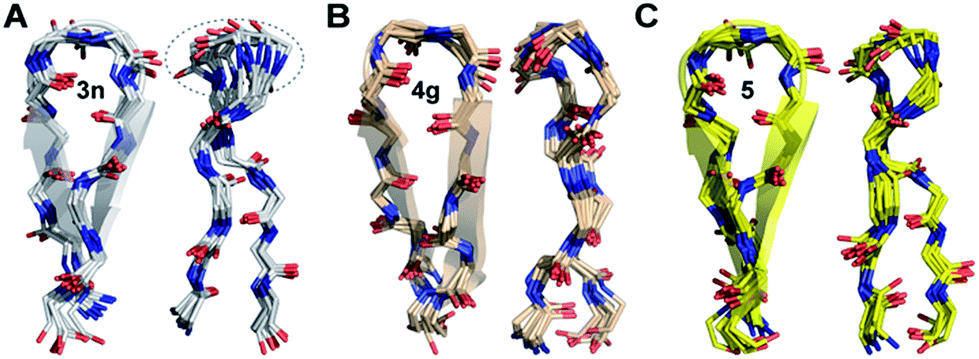 | ||
| Fig. 5 The backbone overlay of an ensemble of ten minimum energy conformations obtained from 10 ns restrained molecular dynamics simulation of (A) 3n, (B) 4g and (C) 5 in water. Note the flexibility of the β-turn with a shorter β-strand registry in 3n. The 90° rotation of the β-sheets clearly depicts the enhanced right-handed twist in 3n and 5. The dotted lines indicate the flexibility of the turn in 3n as opposed to 5 and 4g. The side chains are omitted for the sake of clarity (ESI†). The NMR spectra of 3n, 4g and 5 were acquired in acetate buffer (pH 3.8) at 25 °C. | ||
| i+1 | i+2 | |||
|---|---|---|---|---|
| Φ | Ψ | Φ | Ψ | |
| 3n | 59 ± 12 | −115 ± 11 | −115 ± 10 | 8 ± 10 |
| 4g | 62 ± 10 | −123 ± 9 | −101 ± 13 | −1 ± 11 |
| 5 | 70 ± 9 | −131 ± 9 | −111 ± 11 | 13 ± 11 |
| βII′ | 60 | −120 | −80 | 0 |
Thus, this conformational restriction results in a well-formed β-turn with a strong hydrogen bond between Lys8HN and Val5CO, which plays a critical role in the folding transition of a β-hairpin23 and eventually dictates the elongated registry of the β-strands in 4g and 5. The temperature coefficient of all the amide protons except for Lys8HN in these β-hairpins shows very high temperature dependence indicating the β-turn to be the most persistent structure in the hairpin.24 Curiously, the extent of foldedness in these hairpin peptides measured in terms of ΔGfold (Table 3) corroborates with the temperature coefficient25 of Lys8HN (Δδ/ΔT of −3.6 ppb K−1 (3b) < −2.3 ppb K−1 (2) < −1.6 ppb K−1 (3) < −1.0 ppb K−1 (3n) ≅ −1.0 ppb K−1 (4g) < −0.6 ppb K−1 (5)). Thus, it is tempting to speculate that the increased conformational restriction in the β-turn possibly results in an enhanced strength of the intramolecular hydrogen bond stabilizing the β-turn.
Another remarkable feature was the stark right-handed twist observed in 3n and 5 typical of β-sheet rich proteins26 owing to the presence of arginine at the i+2 site in the reverse turn. The observed right-handed twist was also indicated by the presence of strong NOEs between Lys9Hβ/γ and Tyr2Hδ in the ROESY spectrum27 and a slight red-shift of the CD minimum in 3n and 5 as opposed to 4g (ESI†).28 Although we failed to fully understand the mechanistic origin of this right-handed twist, the conformation of these two peptides determined in vacuo indicates that the guanidinium group in arginine might be involved in hydrogen bonding with the peptide backbone, playing a critical role in governing the right-handed twist. Interestingly, protegrin-1, a potent antimicrobial peptide displaying a right-handed twisted β-hairpin conformation29 (PDB ID: 1PG1), also shows the involvement of the guanidinium group of arginine at the i+2 site in side chain-backbone hydrogen bonding.
Re-engineering a three-stranded β-sheet protein using the developed turn motifs
The enhanced thermodynamic stability of the monomeric β-hairpins in water is fundamentally attributed to the conformational preorganization of the β-turn attained by different i+1 and i+2 side-chain functional groups which imposes a favorable steric constraint on the β-turn. Such preorganization of the main chain torsion angles by side-chain substitution confers extraordinary conformational stability to proteins and peptides as was observed in collagen triple helices by substituting proline with 4-fluoroproline and 4-methylproline.32This encouraged us to utilize the designed turn motifs with myriad side-chain functional groups to engineer the conformational stability of proteins. Since antiparallel β-sheets are quite prevalent in proteins and have found great utility towards the development of non-antibody based- and hyperstable peptide scaffolds,33 we chose to incorporate our β-turn motifs into the three-stranded β-sheet protein, Pin 1 WW domain.30,34 The folding of the Pin 1 WW domain is rate-limited by the formation of loop 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 35 and it was shown by Kelly et al. that the introduction of a type II′ β-turn affords the fastest folding of the WW domain.12,36 Thus, we re-engineered the loop 1 in the Pin 1 WW domain to demonstrate the role of pseudoallylic strain in modulating the thermodynamic stability of proteins (Fig. 6A and B).
35 and it was shown by Kelly et al. that the introduction of a type II′ β-turn affords the fastest folding of the WW domain.12,36 Thus, we re-engineered the loop 1 in the Pin 1 WW domain to demonstrate the role of pseudoallylic strain in modulating the thermodynamic stability of proteins (Fig. 6A and B).
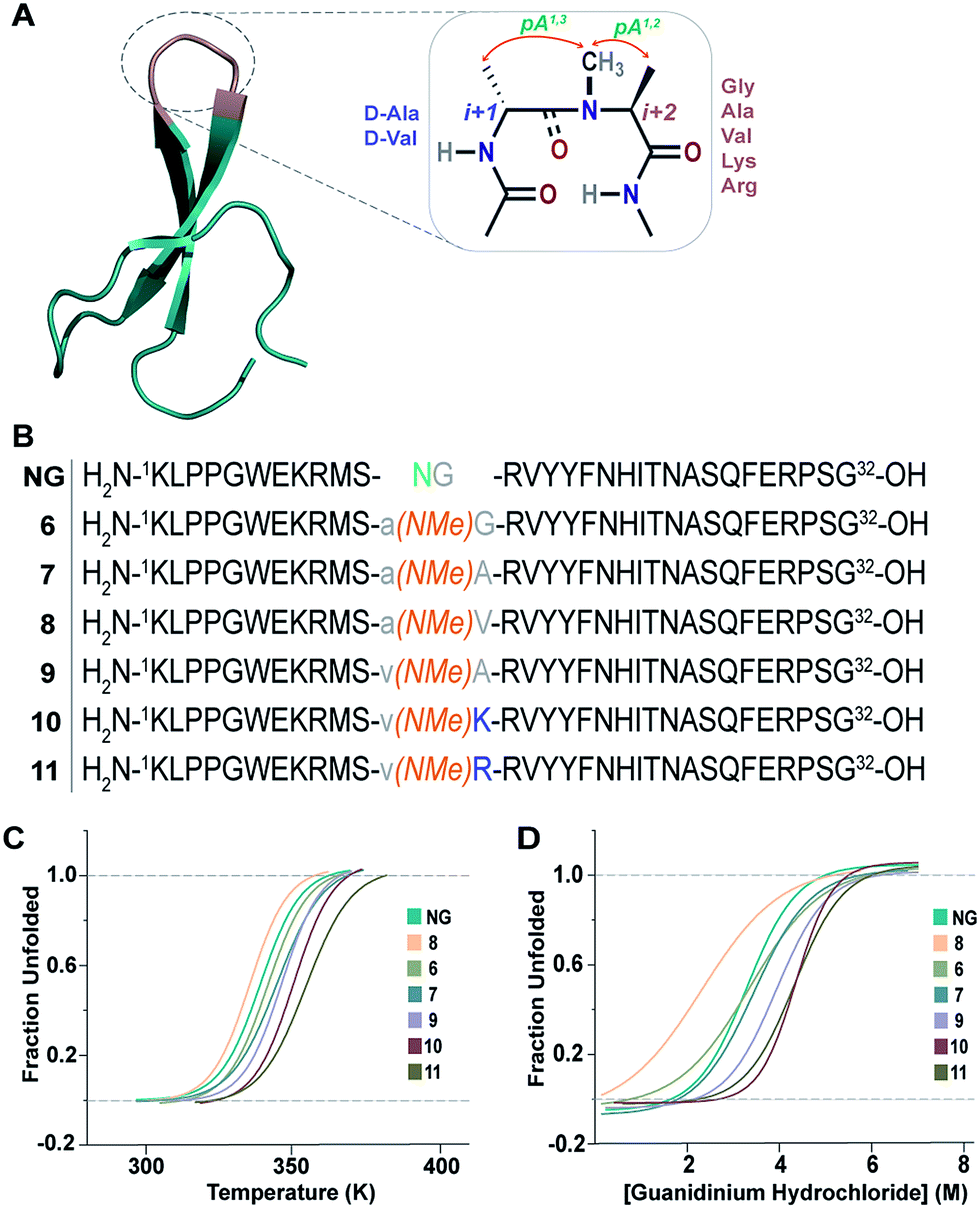 | ||
| Fig. 6 (A) Pin 1 WW domain with 2 residues (–NG–) in loop 1 (NG) (PDB ID: 1ZCN), which is re-engineered by introducing the pseudoallylic strain by incorporating different amino acids. (B) NG and the six-synthesized reverse-turn engineered Pin 1 variants. (C) Thermal unfolding of the Pin 1 variants monitored at 227 nm by variable temperature CD with 10 μM protein concentration in 20 mM sodium phosphate buffer, pH 7.0. The lines represent fits of the data to a two-state folding model.30 (D) Fluorescence-monitored guanidine hydrochloride denaturation curve of the Pin 1 variants with 2.5 μM protein concentration. The lines represent fits of the data to a two-state folding model.31 | ||
All the Pin 1 variants were chemically synthesized, which showed the characteristic maximum at 227 nm in CD spectra indicating the presence of a folded protein (ESI†). Their thermodynamic stability (Table 5) was assessed by thermal (Fig. 6C) and chemical (Fig. 6D) denaturation studies to obtain the TM and free energy of folding (ΔGf), respectively. Unlike the model 12-mer peptide, the incorporation of pA1,3 strain at the reverse turn in 6 was sufficient to impart reasonable thermodynamic stability (TM of 68.7 ± 0.2 °C) to the Pin 1 protein due to multiple hydrophobic interactions along the β-stands in the entire protein. The TM values of all the Pin 1 variants studied were independent of the protein concentration over a range of 5–50 μM indicating their monomeric nature (ESI†). Furthermore, the incorporation of both pA1,3 and pA1,2 strain results in additional stabilization of the Pin 1 by ∼1.0 kcal mol−1 (ΔΔGf) in 7 (Table 5). The introduction of a β-branched amino acid, valine, at the i+2 site (8) by virtue of the unfavorable pA1,2 strain destabilizes the Pin 1 by ∼0.9 kcal mol−1 (ΔΔGf). However, when incorporated at the i+1 site (9), it stabilizes the protein by ∼1.2 kcal mol−1 (ΔΔGf), further strengthening the critical role of steric interaction at the reverse turn in governing the thermodynamic stability of proteins. Nevertheless, the most exciting observation was the synergism between i+1 and i+2 residues at the reverse turn in enhancing the thermodynamic stability of the Pin 1 protein as observed in 10 (ΔΔGf of ∼1.6 kcal mol−1) and 11 (ΔΔGf of ∼1.9 kcal mol−1) with the incorporation of lysine and arginine at the i+2 site, respectively, which corroborated our findings in the 12-mer model peptide.
| T M (°C) | ΔGf (kcal mol−1) | ΔΔGf (kcal mol−1) | |
|---|---|---|---|
| 6 | 68.7 ± 0.2 | 1.87 ± 0.04 | 0 |
| 7 | 72.0 ± 0.4 | 2.93 ± 0.06 | −1.06 |
| 8 | 62.5 ± 0.4 | 0.97 ± 0.2 | 0.90 |
| 9 | 74.3 ± 0.3 | 3.10 ± 0.02 | −1.23 |
| 10 | 78.4 ± 0.6 | 3.45 ± 0.01 | −1.58 |
| 11 | 81.8 ± 0.4 | 3.77 ± 0.01 | −1.90 |
| NG | 66.9 ± 0.3 | 1.75 ± 0.2 | — |
Next, we wondered whether this large change in thermodynamic stability between 8 and 11 (ΔTM of ∼20 °C) corresponding to about 2.8 kcal mol−1 (ΔΔGf) results from significant perturbation in the secondary structure of the re-engineered Pin 1. To identify the structural alterations in the solution conformation of Pin 1 variants, we acquired the 1H, TOCSY, and NOESY spectra of each Pin 1 variant (40–60 μM) at 15 °C. On comparing the backbone Hα CSD of the re-engineered Pin 1 variants with the wild-type apo-Pin 1 WW domain34b and the βI′ turn containing Pin 1 (NG)34c (Fig. 7), quite unexpectedly we observed that despite such a large change in thermodynamic stability, the native fold in all the re-engineered variants stays intact (ESI†). This was also validated by the characteristic NOEs and the stark upfield-shifted 1H resonances (ESI†). Collectively, these observations indicate the potential of the developed turn-inducing motifs in modulating the thermodynamic stability of proteins and enzymes without perturbing their native structure.38
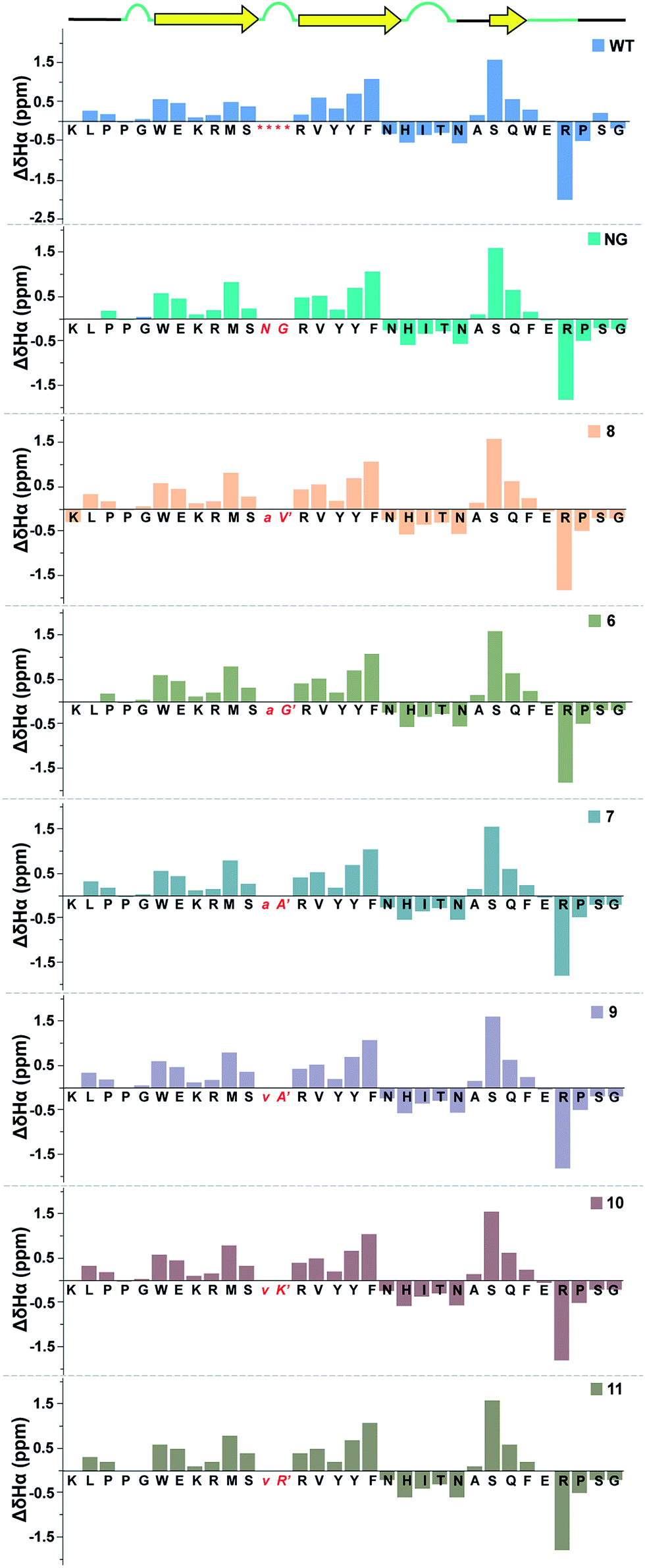 | ||
| Fig. 7 Comparison of backbone Hα chemical shift deviation of the apo-Pin 1 WW domain (WT) with the βI′ turn (NG) and the re-engineered Pin 1 variants deduced from the predicted random coil values,37 indicating minimal perturbation in the secondary structure. The –RSSG– motif in loop 1 of WT is indicated by an asterisk. | ||
Enhanced global stability correlates with the increased local stability of the intramolecularly hydrogen bonded structure
Finally, we sought to investigate whether the conformational preorganization at the reverse turn eventually affects the global stability of the Pin 1 protein by stabilizing the folded conformation of the protein involved in intramolecular hydrogen bonds. Therefore, to qualitatively estimate the strength of the hydrogen bonds, we measured the amide temperature coefficients of the least and most stable Pin 1 variants 8 and 11, respectively. It was quite satisfying to note that even at 35 °C, there was minimal change in the Hα chemical shift deviation in these variants indicating that the native fold was unperturbed (ESI†). This observation suggests the potential of this loop modification in engineering therapeutic proteins. Moreover, as there were no temperature induced chemical shift changes associated with the loss of the secondary structure of the protein, the amide temperature coefficients in these Pin 1 variants are good indicators of intramolecular hydrogen bonds.25,39Overall, we observed more positive amide temperature coefficient values in 11 than 8 (Fig. 8A); however, we chose to analyze the amide protons that are involved in intramolecular hydrogen bonding.40 Interestingly, the most notable changes (difference in Δδ/ΔT ≥ 1 ppb K−1 between the identical amino acids in 8 and 11) were observed in Arg14 and Ser11 that are directly involved in stabilizing the reverse turn (loop 1) along with Tyr16 and Tyr,17 which are hydrogen bonded to the residues in N-term and C-term strands, respectively (Fig. 8B). Likewise, Asn19 and His20 that are hydrogen bonded to C-term and N-term strand residues, respectively, also displayed a stark increase. Thus, we note that most residues of the Pin 1 central strand in 11 show strong hydrogen bonding as compared to 8 to the amino acids of the flanking N-term and C-term strands indicating higher stability of the intramolecularly hydrogen bonded region in 11.
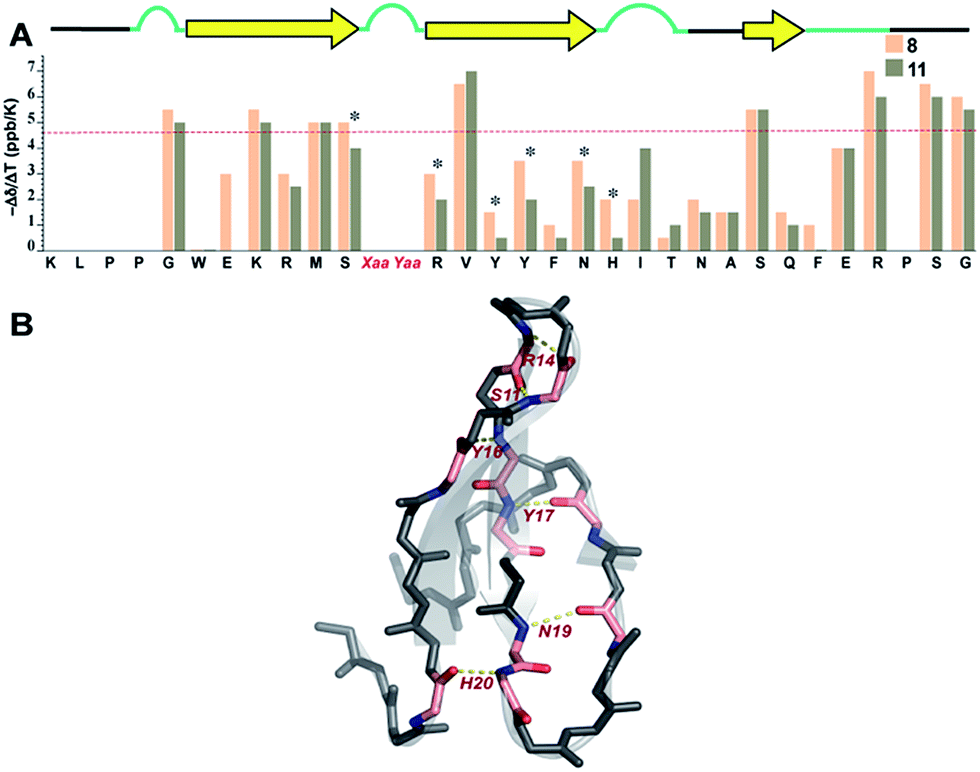 | ||
| Fig. 8 (A) Amide temperature coefficients of the least and most stable Pin 1 analogs 8 and 11, respectively, obtained by analysing a series of TOCSY spectra acquired from 288 K to 308 K. The asterisks indicate the residues that show a change greater than or equal to 1 ppb K−1. The red dotted line marks the barrier at −4.6 ppb K−1, above which the values indicate the absence of intramolecular hydrogen bonding. (B) Residues showing a change greater than or equal to 1 ppb K−1 are highlighted in pink, which are mapped on Pin 1-NG (PDB ID: 1ZCN). | ||
Conclusion
In summary, we demonstrate that the non-covalent constraint induced by N-methylation of a peptide bond differentially modulates the population of cis/trans isomers about the tertiary amide bond. This property of an N-methylated peptide bond along with its heavily restricted conformational space allowed us to probe the strength of pseudoallylic strain pA1,2 and pA1,3 in engineering reverse turns with different functional groups present on coded amino acids. Utilizing the combination of pseudoallylic strain and the functional groups on coded amino acids, we could impart remarkable conformational stability to isolated β-sheets and proteins in aqueous solution at room temperature. This enhanced conformational stability is presumably achieved by preorganization of the β-turn by the formation of stable hydrogen bonds at the reverse turn in the transition state.23,40b Such preorganization is achieved by incorporating diverse side-chain substituents at the i+1 and i+2 sites of a β-turn, which induces favorable steric constraints upon the main chain. Thus, by modifying the side-chain substituents at the turn, we could successfully tune the stability of β-sheets. Interestingly, a single amino acid substitution at the reverse turn also contributes towards modulating the right-handed twist in isolated β-sheets.Since reverse turns are solvent exposed, and the pseudoallylic strain provided a basic conformational stability to the reverse turn, it allowed us to probe the effect of various amino acid substitution on the stability of a mini-protein, Pin 1 WW domain. We show that re-engineering the non-repetitive motif like β-turns with non-natural amino acids can have a large contribution to protein stability (ΔΔGf of ∼2.8 kcal mol−1 between the least and the most stable Pin 1 analogs). Hence, with the remarkable advancement in tools to generate semi-synthetic proteins41 and genetic techniques to incorporate noncanonical amino acids into proteins,42 we strongly believe that this protein engineering strategy will find its use in enhancing the thermodynamic stability of proteins,6c,43 enzymes44 and foldamers.45
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors acknowledge DBT through the project BT/COE/34/SP15219/2015 to JC, DBT-IISc partnership program and IISc (MHRD) for funding this work. We also acknowledge the DST-FIST program, DST & DBT funded NMR and proteomics facility at IISc. We thank Prof. Debnath Pal for the access to the computational facility and Prof. Siddhartha P Sarma for help with the NMR experiments. PL and AR acknowledge MHRD and HV acknowledges UGC for research fellowships.References
- J. S. Richardson, Adv. Protein Chem., 1981, 34, 167 CrossRef CAS PubMed.
- (a) Y. J. Chung, L. A. Christianson, H. E. Stanger, D. R. Powell and S. H. Gellman, J. Am. Chem. Soc., 1998, 120, 10555 CrossRef CAS; (b) A. G. Cochran, R. T. Tong, M. A. Starovasnik, E. J. Park, R. S. McDowell, J. E. Theaker and N. J. Skelton, J. Am. Chem. Soc., 2001, 123, 625 CrossRef CAS PubMed; (c) L. D. Walensky and G. H. Bird, J. Med. Chem., 2014, 57, 6275 CrossRef CAS PubMed; (d) S. J. Maynard, A. M. Almeida, Y. Yoshimi and S. H. Gellman, J. Am. Chem. Soc., 2014, 136, 16683 CrossRef CAS PubMed; (e) T. A. Hill, N. E. Shepherd, F. Diness and D. P. Fairlie, Angew. Chem., Int. Ed., 2014, 53, 13020 CrossRef CAS PubMed; (f) B. L. Kier, J. M. Anderson and N. H. Andersen, J. Am. Chem. Soc., 2015, 137, 5363 CrossRef CAS PubMed.
- R. Vazquez-Lombardi, T. G. Phan, C. Zimmermann, D. Lowe, L. Jermutus and D. Christ, Drug Discovery Today, 2015, 20, 1271 CrossRef CAS PubMed.
- (a) A. E. Eriksson, W. A. Baase, J. A. Wozniak and B. W. Matthews, Nature, 1992, 355, 371 CrossRef CAS PubMed; (b) D. Mendel, J. A. Ellman, Z. Y. Chang, D. L. Veenstra, P. A. Kollman and P. G. Schultz, Science, 1992, 256, 1798 CAS; (c) B. I. Dahiyat and S. L. Mayo, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 10172 CrossRef CAS PubMed.
- (a) K. Yutani, K. Ogasahara, T. Tsujita and Y. Sugino, Proc. Natl. Acad. Sci. U. S. A., 1987, 84, 4441 CrossRef CAS PubMed; (b) M. Karpusas, W. A. Baase, M. Matsumura and B. W. Matthews, Proc. Natl. Acad. Sci. U. S. A., 1989, 86, 8237 CrossRef CAS PubMed; (c) S. Ventura, M. C. Vega, E. Lacroix, I. Angrand, L. Spagnolo and L. Serrano, Nat. Struct. Biol., 2002, 9, 485 CrossRef CAS PubMed.
- (a) M. Matsumura, G. Signor and B. W. Matthews, Nature, 1989, 342, 291 CrossRef CAS PubMed; (b) C. R. Robinson and R. T. Sauer, Biochemistry, 2000, 39, 12494 CrossRef CAS PubMed; (c) T. Liu, Y. Wang, X. Z. Luo, J. Li, S. A. Reed, H. Xiao, T. S. Young and P. G. Schultz, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 5910 CrossRef CAS PubMed; (d) E. J. Moore, D. Zorine, W. A. Hansen, S. D. Khare and R. Fasan, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 12472 CrossRef CAS PubMed.
- C. N. Pace, J. M. Scholtz and G. R. Grimsley, FEBS Lett., 2014, 588, 2177 CrossRef PubMed.
- (a) P. N. Lewis, F. A. Momany and H. A. Scheraga, Proc. Natl. Acad. Sci. U. S. A., 1971, 68, 2293 CrossRef CAS PubMed; (b) I. D. Kuntz, J. Am. Chem. Soc., 1972, 94, 4009 CrossRef CAS PubMed; (c) S. S. Zimmerman and H. A. Scheraga, Proc. Natl. Acad. Sci. U. S. A., 1977, 74, 4126 CrossRef CAS PubMed; (d) V. Munoz, P. A. Thompson, J. Hofrichter and W. A. Eaton, Nature, 1997, 390, 196 CrossRef CAS PubMed; (e) A. M. Marcelino and L. M. Gierasch, Biopolymers, 2008, 89, 380 CrossRef CAS PubMed.
- U. Arnold, M. P. Hinderaker, B. L. Nilsson, B. R. Huck, S. H. Gellman and R. T. Raines, J. Am. Chem. Soc., 2002, 124, 8522 CrossRef CAS PubMed.
- R. Kaul, S. Deechongkit and J. W. Kelly, J. Am. Chem. Soc., 2002, 124, 11900 CrossRef CAS PubMed.
- B. Eckhardt, W. Grosse, L. O. Essen and A. Geyer, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 18336 CrossRef CAS PubMed.
- A. A. Fuller, D. Du, F. Liu, J. E. Davoren, G. Bhabha, G. Kroon, D. A. Case, H. J. Dyson, E. T. Powers, P. Wipf, M. Gruebele and J. W. Kelly, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 11067 CrossRef CAS PubMed.
- D. Ghosh, P. Lahiri, H. Verma, S. Mukherjee and J. Chatterjee, Chem. Sci., 2016, 7, 5212 RSC.
- (a) M. Sukopp, L. Marinelli, M. Heller, T. Brandl, S. L. Goodman, R. W. Hoffman and H. Kessler, Helv. Chim. Acta, 2002, 85, 4442 CrossRef CAS; (b) J. B. Xiao, B. Weisblum and P. Wipf, Org. Lett., 2006, 8, 4731 CrossRef CAS PubMed.
- P. Wipf, T. C. Henninger and S. J. Geib, J. Org. Chem., 1998, 63, 6088 CrossRef CAS PubMed.
- J. Chatterjee, C. Gilon, A. Hoffman and H. Kessler, Acc. Chem. Res., 2008, 41, 1331 CrossRef CAS PubMed.
- J. Chatterjee, F. Rechenmacher and H. Kessler, Angew. Chem., Int. Ed., 2013, 52, 254 CrossRef CAS PubMed.
- P. Manavalan and F. A. Momany, Biopolymers, 1980, 19, 1943 CrossRef CAS PubMed.
- F. A. Syud, J. F. Espinosa and S. H. Gellman, J. Am. Chem. Soc., 1999, 121, 11577 CrossRef CAS.
- (a) D. S. Wishart, B. D. Sykes and F. M. Richards, J. Mol. Biol., 1991, 222, 311 CrossRef CAS PubMed; (b) D. S. Wishart, B. D. Sykes and F. M. Richards, Biochemistry, 1992, 31, 1647 CrossRef CAS PubMed.
- G. A. Lengyel, R. C. Frank and W. S. Horne, J. Am. Chem. Soc., 2011, 133, 4246 CrossRef CAS PubMed.
- C. D. Tatko and M. L. Waters, J. Am. Chem. Soc., 2004, 126, 2028 CrossRef CAS PubMed.
- R. M. Culik, H. Jo, W. F. DeGrado and F. Gai, J. Am. Chem. Soc., 2012, 134, 8026 CrossRef CAS PubMed.
- M. S. Searle, D. H. Williams and L. C. Packman, Nat. Struct. Biol., 1995, 2, 999 CrossRef CAS PubMed.
- T. Cierpicki and J. Otlewski, J. Biomol. NMR, 2001, 21, 249 CrossRef CAS PubMed.
- F. R. Salemme, Prog. Biophys. Mol. Biol., 1983, 42, 95 CrossRef CAS PubMed.
- F. A. Syud, H. E. Stanger and S. H. Gellman, J. Am. Chem. Soc., 2001, 123, 8667 CrossRef CAS PubMed.
- (a) M. C. Manning, M. Illangasekare and R. W. Woody, Biophys. Chem., 1988, 31, 77 CrossRef CAS PubMed; (b) A. Iyer, S. J. Roeters, V. Kogan, S. Woutersen, M. Claessens and V. Subramaniam, J. Am. Chem. Soc., 2017, 139, 15392 CrossRef CAS PubMed.
- R. L. Fahrner, T. Dieckmann, S. S. L. Harwig, R. I. Lehrer, D. Eisenberg and J. Feigon, Chem. Biol., 1996, 3, 543 CrossRef CAS PubMed.
- R. Kaul, A. R. Angeles, M. Jager, E. T. Powers and J. W. Kelly, J. Am. Chem. Soc., 2001, 123, 5206 CrossRef CAS PubMed.
- R. F. Greene and C. N. Pace, J. Biol. Chem., 1974, 249, 5388 CAS.
- M. D. Shoulders, K. A. Satyshur, K. T. Forest and R. T. Raines, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 559 CrossRef CAS PubMed.
- G. Bhardwaj, V. K. Mulligan, C. D. Bahl, J. M. Gilmore, P. J. Harvey, O. Cheneval, G. W. Buchko, S. V. S. R. K. Pulavarti, Q. Kaas, A. Eletsky, P. S. Huang, W. A. Johnsen, P. J. Greisen, G. J. Rocklin, Y. F. Song, T. W. Linsky, A. Watkins, S. A. Rettie, X. Z. Xu, L. P. Carter, R. Bonneau, J. M. Olson, E. Coutsias, C. E. Correnti, T. Szyperski, D. J. Craik and D. Baker, Nature, 2016, 538, 329 CrossRef CAS PubMed.
- (a) M. Jager, H. Nguyen, J. C. Crane, J. W. Kelly and M. Gruebele, J. Mol. Biol., 2001, 311, 373 CrossRef CAS PubMed; (b) J. A. Kowalski, K. Liu and J. W. Kelly, Biopolymers, 2002, 63, 111 CrossRef CAS PubMed; (c) M. Jager, Y. Zhang, J. Bieschke, H. Nguyen, M. Dendle, M. E. Bowman, J. P. Noel, M. Gruebele and J. W. Kelly, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 10648 CrossRef CAS PubMed; (d) M. S. Ardejani, E. T. Powers and J. W. Kelly, Acc. Chem. Res., 2017, 50, 1875 CrossRef CAS PubMed.
- T. Sharpe, A. L. Jonsson, T. J. Rutherford, V. Daggett and A. R. Fersht, Protein Sci., 2007, 16, 2233 CrossRef CAS PubMed.
- F. Liu, D. G. Du, A. A. Fuller, J. E. Davoren, P. Wipf, J. W. Kelly and M. Gruebele, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 2369 CrossRef CAS PubMed.
- D. S. Wishart, C. G. Bigam, A. Holm, R. S. Hodges and B. D. Sykes, J. Biomol. NMR, 1995, 5, 67 CrossRef CAS PubMed.
- E. K. Culyba, J. L. Price, S. R. Hanson, A. Dhar, C. H. Wong, M. Gruebele, E. T. Powers and J. W. Kelly, Science, 2011, 331, 571 CrossRef CAS PubMed.
- (a) N. H. Andersen, J. W. Neidigh, S. M. Harris, G. M. Lee, Z. H. Liu and H. Tong, J. Am. Chem. Soc., 1997, 119, 8547 CrossRef CAS; (b) N. J. Baxter and M. P. Williamson, J. Biomol. NMR, 1997, 9, 359 CrossRef CAS PubMed.
- (a) S. Deechongkit, P. E. Dawson and J. W. Kelly, J. Am. Chem. Soc., 2004, 126, 16762 CrossRef CAS PubMed; (b) S. Deechongkit, H. Nguyen, E. T. Powers, P. E. Dawson, M. Gruebele and J. W. Kelly, Nature, 2004, 430, 101 CrossRef CAS PubMed.
- (a) P. E. Dawson, T. W. Muir, I. Clarklewis and S. B. H. Kent, Science, 1994, 266, 776 CAS; (b) T. W. Muir, D. Sondhi and P. A. Cole, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 6705 CrossRef CAS PubMed; (c) M. B. Soellner, K. A. Dickson, B. L. Nilsson and R. T. Raines, J. Am. Chem. Soc., 2003, 125, 11790 CrossRef CAS PubMed.
- (a) J. A. Johnson, Y. Y. Lu, J. A. Van Deventer and D. A. Tirrell, Curr. Opin. Chem. Biol., 2010, 14, 774 CrossRef CAS PubMed; (b) C. C. Liu and P. G. Schultz, Annu. Rev. Biochem., 2010, 79, 413 CrossRef CAS PubMed.
- T. W. Craven, M. K. Cho, N. J. Traaseth, R. Bonneau and K. Kirshenbaum, J. Am. Chem. Soc., 2016, 138, 1543 CrossRef CAS PubMed.
- (a) U. Arnold, M. P. Hinderaker, J. Koditz, R. Golbik, R. Ulbrich-Hofmann and R. T. Raines, J. Am. Chem. Soc., 2003, 125, 7500 CrossRef CAS PubMed; (b) A. Tam, U. Arnold, M. B. Soellner and R. T. Raines, J. Am. Chem. Soc., 2007, 129, 12670 CrossRef CAS PubMed; (c) C. Mayer, M. M. Muller, S. H. Gellman and D. Hilvert, Angew. Chem., Int. Ed., 2014, 53, 6978 CrossRef CAS PubMed.
- (a) C. M. Goodman, S. Choi, S. Shandler and W. F. DeGrado, Nat. Chem. Biol., 2007, 3, 252 CrossRef CAS PubMed; (b) W. S. Horne and S. H. Gellman, Acc. Chem. Res., 2008, 41, 1399 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7sc05163h |
| ‡ Equal contribution. |
| This journal is © The Royal Society of Chemistry 2018 |