
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMicrohydration of PAH+ cations: evolution of hydration network in naphthalene+-(H2O)n clusters (n ≤ 5)†
Kuntal
Chatterjee
and
Otto
Dopfer
 *
*
Institut für Optik und Atomare Physik, Technische Universität Berlin, Hardenbergstr. 36, 10623 Berlin, Germany. E-mail: dopfer@physik.tu-berlin.de; Tel: +49 30 31423018
First published on 24th January 2018
Abstract
The interaction of polycyclic aromatic hydrocarbon molecules with water (H2O = W) is of fundamental importance in chemistry and biology. Herein, size-selected microhydrated naphthalene cation nanoclusters, Np+-Wn (n ≤ 5), are characterized by infrared photodissociation (IRPD) spectroscopy in the C–H and O–H stretch range to follow the stepwise evolution of the hydration network around this prototypical PAH+ cation. The IRPD spectra are highly sensitive to the hydration structure and are analyzed by dispersion-corrected density functional theory calculations (B3LYP-D3/aug-cc-pVTZ) to determine the predominant structural isomers. For n = 1, W forms a bifurcated CH⋯O ionic hydrogen bond (H-bond) to two acidic CH protons of the bicyclic ring. For n ≥ 2, the formation of H-bonded solvent networks dominates over interior ion solvation, because of strong cooperativity in the former case. For n ≥ 3, cyclic Wn solvent structures are attached to the CH protons of Np+. However, while for n = 3 the W3 ring binds in the CH⋯O plane to Np+, for n ≥ 4 the cyclic Wn clusters are additionally stabilized by stacking interactions, leading to sandwich-type configurations. No intracluster proton transfer from Np+ to the Wn solvent is observed in the studied size range (n ≤ 5), because of the high proton affinity of the naphthyl radical compared to Wn. This is different from microhydrated benzene+ clusters, (Bz-Wn)+, for which proton transfer is energetically favorable for n ≥ 4 due to the much lower proton affinity of the phenyl radical. Hence, because of the presence of polycyclic rings, the interaction of PAH+ cations with W is qualitatively different from that of monocyclic Bz+ with respect to interaction strength, structure of the hydration shell, and chemical reactivity. These differences are rationalized and quantified by quantum chemical analysis using the natural bond orbital (NBO) and noncovalent interaction (NCI) approaches.
1. Introduction
The interactions and chemical reactions of aromatic molecules and their cations with surrounding solvent molecules are of fundamental importance for many phenomena in chemistry and biology.1–4 In particular, the interaction of benzene and polycyclic aromatic hydrocarbon molecules (PAH) and their cations (PAH+) with water (H2O = W) plays a crucial role in the areas of combustion,5 organic chemistry,6–14 astrochemistry,15–26 and biomolecular recognition.3,4,27–29 For example, intermolecular interactions involving aromatic hydrocarbons and water are the driving force for many biochemical processes, such as the conformation and folding of proteins, base pair stacking in DNA, drug design, macromolecular assemblies, and biological membranes. The relative strengths of the various interaction types (e.g., OH⋯π, CH⋯O, cation⋯π)4,28,30–32 strongly depend on the charge state of the PAH. In the context of astrochemistry, PAH molecules, their radical cations (PAH+), and their protonated species (H+PAH) are suggested to be carriers of the unidentified infrared emission bands as well as the diffuse interstellar bands.33–38 Concerning ions of the most simple PAH (naphthalene, C10H8, Np), the postulation39 and tentative identification of the Np+ cation as a DIB carrier40,41 provided a great stimulus for the plethora of laboratory spectroscopy of both Np+ (ref. 39 and 42–51) and H+Np52–58 in the gas phase and isolated in rare gas matrices in both the infrared and optical range of the electromagnetic spectrum. In addition, the spectroscopy and photochemistry of Np and Np+ deposited in ice or on ice grains as well as in aqueous solution have been studied.6,18,19,23,59,60For a deeper understanding of the interaction between water and PAH molecules in various charge states at the molecular level, the accurate knowledge of the involved interaction potential is required. To this end, the combination of spectroscopy of molecular clusters isolated in the gas phase with quantum chemical calculations provides the most direct access to this potential. Herein, we combine infrared photodissociation (IRPD) spectroscopy with dispersion-corrected density functional theory (DFT) calculations to probe the initial microhydration steps in size-selected cold Np+-Wn clusters produced in a supersonic plasma expansion using electron ionization. This approach has recently been applied in our laboratory to a variety of microhydrated aromatic ions.61–72 Significantly, the employed cluster ion source generates predominantly the most stable isomer of a given cluster ion, because of the sequential cluster growth realized in the high-pressure region in the supersonic expansion.73,74
Surprisingly, despite their importance only very scarce information is available for the spectroscopy of PAH(±)-Wn clusters, and this is even true for the most simple Np(±)-Wn clusters. For example, no spectral data have been reported yet for neutral Np-Wn clusters, and thus all information about their structure and interaction potential has to barely rely on rather limited computational studies available only for n ≤ 2.75 The latter predict π H-bonded structures, in which the W ligands bind via OH⋯π stacking to the aromatic π-electron system of Np either as single W ligands or as a H-bonded W2 dimer. The situation is more favorable for Np−-Wn anion clusters, which were characterized by photoelectron76–78 and IR79 spectroscopy up to the size range n = 6. While Np− is unstable with respect to electron detachment because of the negative electron affinity of Np, microhydrated Np−-Wn clusters with n ≥ 1 are stable and can be produced in supersonic expansions. Computational analysis of the IR spectra of Np−-Wn reveals that also the anion clusters prefer π H-bonded structures, in which W (n = 1), a H-bonded W2 dimer (n = 2), or cyclic Wn clusters (n = 3 and 4) bind to the π cloud of Np−via (multiple) OH⋯π interactions.79 The first and only information about Np+-Wn cation clusters prior to our work came from a very recent mass spectrometric study,14 in which Np+-Wn clusters up to n = 6 were generated by injecting Np+ cations produced by electron ionization into a drift cell of an ion mobility mass spectrometer containing water vapor. For the n = 1 dimer, the signal was strong enough to measure clustering equilibria, which yield a binding enthalpy of −ΔH = 7.8 ± 1 kcal mol−1 (2730 ± 350 cm−1) for Np+-W. Out of the four isomers calculated for Np+-W, the three lowest-energy structures have a bifurcated CH⋯O H-bond, while the π-bonded structure is least stable (Fig. 1). Although the calculated binding energy of the global minimum (7.7 kcal mol−1) agrees with the measured enthalpy, the other two H-bonded isomers are also quite low in energy (6.8 and 6.5 kcal mol−1) and thus no reliable conclusion about the cluster structures can be drawn from the mass spectra.14 For n > 1, only very weak mass peaks were reported, and the most stable computed structures determined for n = 2–6 have a linear H-bonded Wn chain attached to Np+.14
 | ||
| Fig. 1 Structures of W, Np+ (with numbering of carbon atoms), and the (18), (12), (23), and (π) isomers of Np+-W calculated at the B3LYP-D3/aug-cc-pVTZ level. Binding energies (D0) and bond lengths are given in cm−1 and Å, respectively. Numbers in parenthesis correspond to relative energies and free energies (E0, G). | ||
In a recent spectroscopic study,72 we analysed the IRPD spectrum of Np+-W recorded in the C–H and O–H stretch (νCH/OH) range by dispersion-corrected DFT calculations (B3LYP-D3/aug-cc-pVTZ) to obtain the first spectroscopic information about the interaction of a PAH+ cation with W. This spectral range is highly sensitive to the details of the H-bonded structure. The analysis of the observed IR spectrum confirms that the bifurcated CH⋯O binding motif involving two CH groups of two different rings (denoted Np+-W(18) in Fig. 1) corresponds indeed to the global minimum of Np+-W predicted by the calculations, and no other isomer is identified in the cold molecular beam. In addition to the reliable structure determination, frequency-dependent photofragmentation branching ratios monitored for IRPD of cold Np+-W-Ar clusters yield a spectroscopic determination of the Np+-W binding energy (D0 = 2800 ± 300 cm−1), in excellent agreement with the calculated value (D0 = 2773 cm−1) and the enthalpy derived from mass spectrometry (−ΔH = 2730 ± 350 cm−1).14 Detailed analysis using the natural bond orbital (NBO) approach elucidates the binding mechanisms and interaction strengths of the various possible bifurcated CH⋯O H-bonding motifs,72 as well as differences in structure and binding energy between the most stable Np+-W isomer and that of the monohydrated benzene cation (Bz+-W).
Herein, we extend our combined IRPD and DFT approach to larger Np+-Wn clusters up to n ≤ 5 to reliably determine the structure of the microhydration network formed around this most simple PAH+ cation. Surprisingly, our results yield cyclic solvent structures, in disagreement with the recent computational study predicting linear Wn structures.14 Comparison between neutral,72,75 anionic,76–79 and cationic14,72 polyhydrated Np(±)-Wn clusters elucidates the drastic effects of the charge state on the hydration network and the interaction strength. Comparison of Np+-Wn with the related and well-studied (Bz-Wn)+ clusters10,12,61,62,73,80–88 reveals the qualitative difference in structure and chemical reactivity of both cluster systems. The Np+-W interaction is quite different from that of Bz+-W with respect to structure, interaction strength, and binding mechanism.14,72 In addition, the proton affinity of the naphthyl radical is much larger than that of the phenyl radical. Hence, the structure of the hydration shell and also the chemical reactivity with respect to intracluster proton transfer are expected to be very different for (Np-Wn)+ and (Bz-Wn)+.
2. Experimental and computational techniques
2.1 Experimental methods
IRPD spectra of mass-selected Np+-Wn clusters with n ≤ 5 are recorded in a tandem quadrupole mass spectrometer coupled to an electron-ionization cluster ion source described in detail elsewhere.73,89 Briefly, Np+-Wn clusters are produced in a pulsed supersonic plasma expansion by electron and/or chemical ionization of Np and subsequent clustering reactions in the high-pressure regime of the expansion.72 The expanding gas mixture is generated by passing Ar carrier gas (8–10 bar) through a reservoir containing solid Np (Sigma-Aldrich, >99%, heated to 70 °C). To produce hydrated clusters, 10 μl of distilled water are added to the gas line before the sample reservoir. The desired Np+-Wn parent clusters are mass-selected in the first quadrupole mass filter and irradiated in an adjacent octopole ion guide with a tunable IR laser pulse (νIR) of an optical parametric oscillator (IR-OPO), which is pumped by a nanosecond Q-switched Nd:YAG laser. The IR-OPO laser pulses are characterized by an energy of 2–5 mJ in the C–H/O–H stretch range, a repetition rate of 10 Hz, and a bandwidth of 1 cm−1. Calibration of νIR to better than 1 cm−1 is accomplished by a wavemeter. Resonant vibrational excitation upon single-photon absorption leads to evaporation of a single W ligand corresponding to following photodissociation reaction:| Np+-Wn + νIR → Np+-Wn−1 + W |
2.2 Computational techniques
Quantum chemical calculations are carried out for Np+-Wn at the B3LYP-D3 level to determine their structural, vibrational, and energetic properties.90 Initial extensive manual screening of the potential energy surface is conducted using the cc-pVTZ basis set and final optimization is performed with the larger aug-cc-pVTZ basis set. The dispersion-corrected B3LYP-D3 functional describes the electrostatic, induction, and dispersion forces of aromatic clusters rather well, and reproduces their experimental IR spectra and binding energies to satisfactory accuracy using the aug-cc-pVTZ basis set.67,68,71,91 This is particularly true for the properties of Np, Np+, W, and the Np+-W interaction, as was demonstrated in our recent report.72 In addition, spin contamination of the radical cation clusters is negligible at this computational level.72 All coordinates are relaxed in the search for stationary points, and their nature as minima or transition states is verified by harmonic frequency analysis. Harmonic intramolecular vibrational frequencies are linearly scaled by the factor 0.96266, derived from fitting calculated harmonic O–H and C–H stretch frequencies of W and Np to experimental values.72 Harmonic scaled IR stick spectra are convoluted with a Gaussian line shape (fwhm = 10 cm−1) for facile comparison to the experimental IRPD spectra. All reported binding energies (D0), relative energies (E0), and relative free energies (G) are corrected for harmonic vibrational zero-point energies. Energies are not corrected for basis set superposition error, because it was found to be very small (<1%) for Np+-W and related microhydrated aromatic cations at this computational level.71 Natural bond orbital (NBO) analysis is employed to evaluate charge distribution and charge transfer in Np+-Wn, as well as second-order perturbation energies (E(2)) of donor–acceptor orbital interactions involved in the H-bonds.92,93 Interaction types and strengths are determined using the noncovalent interaction (NCI) approach.94,95 To this end, the reduced gradient of the electron density, s(ρ) ∼ |grad(ρ)|/ρ4/3, is evaluated as a function of the electron density ρ oriented by the sign of the second eigenvalue λ2 of the Hessian, ρ* = ρsign(λ2). The ρ* values (given in a.u.) provide a measure of the strengths of the intermolecular bonds.3. Results and discussion
Fig. 2 compares the IRPD spectra of Np+-Wn in the 2950–3800 cm−1 spectral range recorded in the Np+-Wn−1 fragment channel. This range covers the aromatic C–H modes of Np+ (νCH) and the O–H stretch modes of the W ligands (νOH), and thus provides a sensitive probe of the evolution of the structure of the H-bonded hydration network around Np+ in its 2Au electronic ground state. The positions and widths of the transitions observed are listed in Table 1, along with their suggested vibrational and isomer assignments. In general, the bands labeled A–C occurring in the 3600–3800 cm−1 range are attributed to free O–H stretch modes of the W ligands, whereas transitions D in the 3200–3600 cm−1 range arise from H-bonded O–H stretch vibrations of the Wn hydration network. Finally, band E near 3080 cm−1 is assigned to aromatic C–H stretch modes of Np+. In the following, we discuss the structural evolution of the hydration network of Np+-Wn by comparing the measured IRPD spectra to computed linear IR absorption spectra of the most stable isomers as a function of the cluster size (n). Clearly, the IRPD spectra of Np+-Wn in Fig. 2 show a large variation in appearance as the number of W molecules in the cluster increases, thereby illustrating the qualitative changes in the hydration network structure induced by sequential addition of W ligands in this size regime.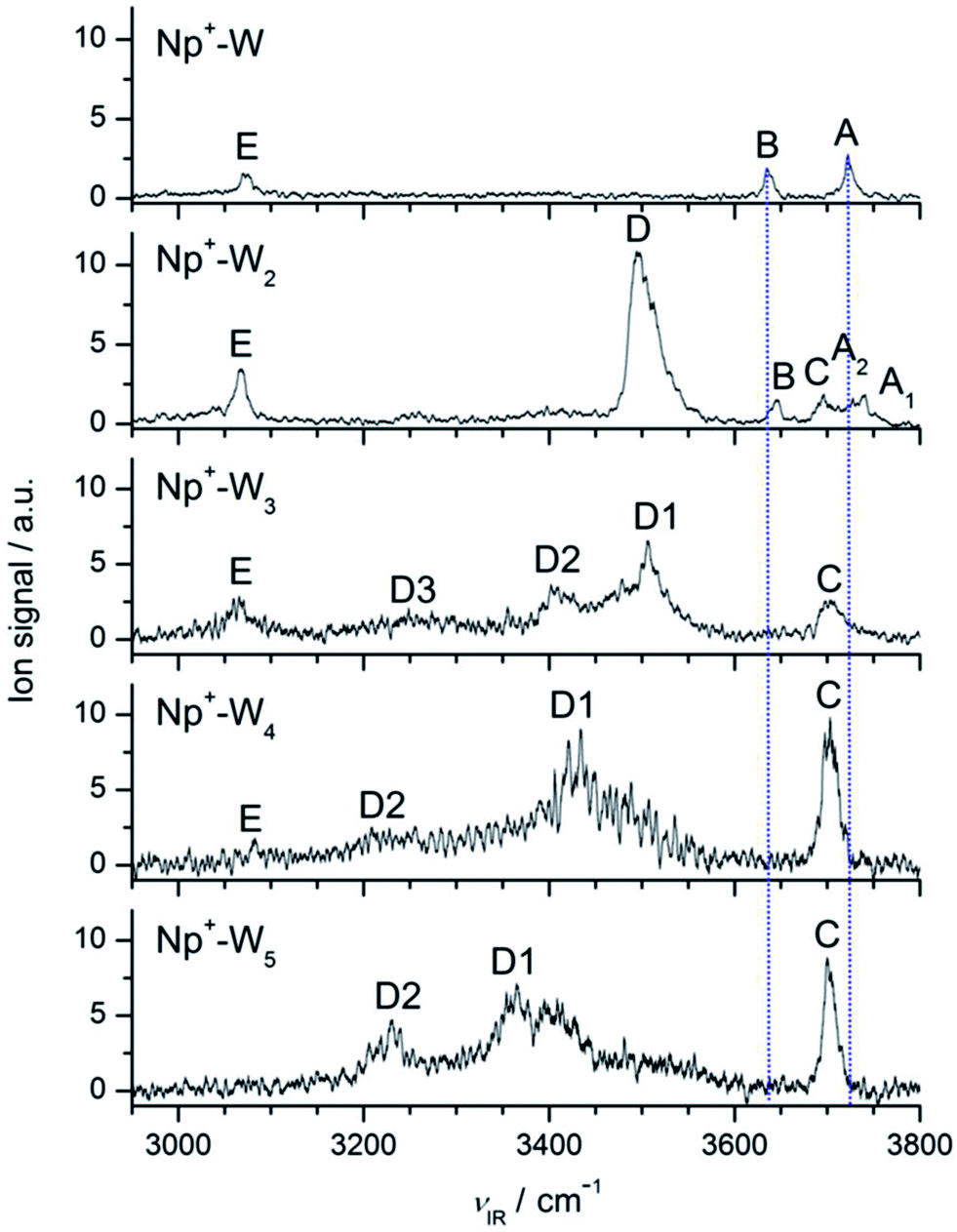 | ||
| Fig. 2 IRPD spectra of Np+-Wn (n = 1–5) recorded in the Np+-Wn−1 fragment channel in the O–H and C–H stretch range. The positions, widths, and vibrational and isomer assignments of the transitions observed (A–E) are listed in Table 1. The dashed lines are included to guide the eye for illustrating relative positions of the free O–H stretch bands (A–C). | ||
| Cluster | Exp (cm−1) | Vibration | Calca (cm−1) | Isomer |
|---|---|---|---|---|
| a IR intensity (in km mol−1) and vibrational symmetry are listed in parentheses. For the νCH modes, only the by far most intense calculated vibration is listed. b Ref. 97, 107, 127 and 128. | ||||
| W | 3657b | ν 1 | 3656 (5, a1) | |
| 3756b | ν 3 | 3755 (63, b2) | ||
| W2 | 3601b | ν b | 3540 (341, a′) | |
| 3654b | ν 1 | 3650 (9, a′) | ||
| 3735b | ν f | 3727 (87, a′) | ||
| 3746b | ν 3 | 3745 (84, a′′) | ||
| Np+-W | E 3072 (13) | ν CH | 3062 (59, a1) | Np+-W(18) |
| B 3635 (10) | ν 1 | 3641 (34, a1) | Np+-W(18) | |
| A 3722 (9) | ν 3 | 3729 (95, b1) | Np+-W(18) | |
| Np+-W2 | E 3068 (10) | ν CH | 3051 (143, a′) | Np+-W2(18) |
| ν CH | 3063 (108, b3u) | Np+-W2(1845) | ||
| D 3496 (33) | ν b | 3434 (696, a′) | Np+-W2(18) | |
| B 3646 (13) | ν 1 | 3649 (22, a′) | Np+-W2(18) | |
| ν 1 | 3642 (65, ag) | Np+-W2(1845) | ||
| C 3696 (14) | ν f | 3708 (84, a′) | Np+-W2(18) | |
| A2 3728 (10) | ν 3 | 3731 (186, b1u) | Np+-W2(1845) | |
| A1 3740 (9) | ν 3 | 3741 (99, a′′) | Np+-W2(18) | |
| Np+-W3 | E 3065 (24) | ν CH | 3123 (18) | Np+-W3(c1) |
| ν CH | 3079 (113) | Np+-W3(c2) | ||
| D3 3248 (broad) | 2βOH | |||
| D2 3402 (broad) | ν b | 3415 (162) | Np+-W3(c1) | |
| ν b | 3417 (85) | Np+-W3(c2) | ||
| D1 3507 (broad) | ν b | 3485 (313) | Np+-W3(c1) | |
| 3528 (197) | ||||
| ν b | 3472 (380) | Np+-W3(c2) | ||
| 3502 (335) | ||||
| C 3703 (28) | ν f | 3713 (79) | Np+-W3(c1) | |
| 3711 (137) | ||||
| 3708 (120) | ||||
| ν f | 3717 (68) | Np+-W3(c2) | ||
| 3715 (143) | ||||
| 3714 (126) | ||||
| Np+-W4 | E 3082 (4) | ν CH | 3087 (4) | Np+-W4(c) |
| D2 3210 (broad) | ν b | 3240 (56) | Np+-W4(c) | |
| 2βOH | ||||
| D1 3433 (broad) | ν b | 3319 (752) | Np+-W4(c) | |
| 3332 (778) | ||||
| 3375 (170) | ||||
| C 3703 (16) | ν f | 3710 (94) | Np+-W4(c) | |
| 3708 (122) | ||||
| 3708 (153) | ||||
| 3706 (18) | ||||
| Np+-W5 | D2 3230 (49) | ν b | 3180 (182) | Np+-W5(c1) |
| 2βOH | ||||
| D1 3365 (broad) | ν b | 3256 (1327) | Np+-W5(c1) | |
| 3281 (816) | ||||
| 3317 (429) | ||||
| 3356 (134) | ||||
| C 3700 (13) | ν f | 3712 (117) | Np+-W5(c1) | |
| 3710 (146) | ||||
| 3708 (84) | ||||
| 3707 (74) | ||||
| 3706 (29) | ||||
3.1 Np+ and W monomers
The geometric, vibrational, and electronic structures of W, Np, and Np+ obtained at the B3LYP-D3/aug-cc-pVTZ level were discussed in detail in our earlier report on Np+-W.72 Briefly, the O–H bond parameters of W in its 1A1 ground state (ROH = 0.9617 Å, ν1 = 3656 cm−1, ν3 = 3755 cm−1) agree well with the experimental data (0.9578 Å, 3657 and 3756 cm−1).96,97 The optimized planar structures of both Np and Np+ have D2h symmetry. The calculated geometry and vibrational frequencies of neutral Np in its 1Ag electronic ground state are in excellent agreement with the experimental data.98–100 The 2Au ground electronic state of Np+ is derived by removal of a bonding π(au) electron from the highest occupied molecular orbital of Np, which is delocalized over the aromatic ring.48,50,51,72 The NBO analysis reveals that the excess positive charge is mostly distributed over the peripheral hydrogen atoms of the bicyclic aromatic ring.72 In contrast to large C–C bond length changes, ionization induces only minor contractions of the C–H bonds (≤1 mÅ). Importantly, the IR activities of all C–H stretch fundamentals is very weak (<2 km mol−1),72 and hence νCH fundamentals of Np+ have never been detected, neither in the gas phase72 nor in rare gas matrices.43,1013.2 Np+-W
The possible structural isomers of Np+-W and the assignment of its IRPD spectrum have been discussed in detail in the previous communication.72 They shall be briefly summarized herein for completeness, because they provide the basis for the discussion of the larger clusters. The four nonequivalent minima on the Np+-W potential14,72 are shown in Fig. 1, and their predicted IR spectra are compared in Fig. 3 to the measured IRPD spectrum. For comparison, also the spectra calculated for bare Np+ and W are shown (although at a different intensity scale). The attraction in all four Np+-W isomers is largely given by the electrostatic charge–dipole interaction between the positive charge on Np+ and the dipole moment of W. As a consequence, the dipole of W points away from the Np+ cation in all isomers. The Np+-W(18) global minimum with C2v symmetry features a symmetric bifurcated CH⋯O ionic H-bond, in which two neighboring CH groups of the two different aromatic rings act as proton donors for two intermolecular H-bonds to the two in-plane lone pairs of W. Its calculated dissociation energy of D0 = 2773 cm−1 is in good agreement with the values extracted from IRPD spectra of Np+-W-Ar recorded in various fragment channels (D0 = 2800 ± 300 cm−1),72 and mass spectrometric experiments (−ΔH = 2730 ± 350 cm−1).14 The other two planar Np+-W(12) and Np+-W(23) isomers with Cs and C2v symmetry have a similar bifurcated CH⋯O H-bond motif, with the main difference that both CH proton donor groups belong to the same aromatic ring. As a result, the CH⋯O H-bonds deviate substantially more from linearity than those in the (18) isomer. Therefore, they are significantly weaker and longer, leading to correspondingly smaller binding energies of D0 = 2458 and 2340 cm−1, respectively. This view is fully supported by the NBO analysis of the donor–acceptor energies of the orbital interactions involved in the CH⋯O H-bond motif.72 The three in-plane Np+-W isomers are separated by calculated barriers of Vb = 99–485 cm−1. The fourth Np+-W(π) isomer with a π-bonded structure and D0 = 1947 cm−1 is less stable than the in-plane isomers. | ||
| Fig. 3 Comparison of experimental IRPD spectrum of Np+-W to linear IR absorption spectra of all four nonequivalent isomers calculated at the B3LYP-D3/aug-cc-pVTZ level (Fig. 1, Table 1). For comparison, also the spectra calculated for Np+ and W are shown (at a different intensity scale). | ||
The consequences of the formation of the Np+-W dimer on the appearance of its IR spectrum in the O–H and C–H stretch range are visualized in Fig. 3 for the four ligand binding sites. The large disparity in the ionization energies of Np and W (IE = 65![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 687 and 101787 cm−1)51,102 implies that the positive charge in (Np–W)+ mainly remains on the Np moiety, justifying the notation of Np+-W. Nonetheless, formation of the Np+-W dimer is accompanied by modest charge transfer from Np+ to the W ligand, which scales with the interaction strength. For example, Δq = 12, 7, and 8 me for the (18), (12), and (23) isomers. As a result of this charge transfer, the O–H bonds of W become weaker, leading to minor red shifts in the symmetric and antisymmetric O–H stretch modes from the frequencies of bare W, ν1/3 = 3656/3755 cm−1. For the (18) global minimum, the effects are largest, with ΔrOH = 1.7 mÅ and Δν1/3 = −15/−26 cm−1. In addition to the Δν1/3 shifts, the relative IR intensity of ν1 is strongly enhanced compared to that of bare W, which is typical for cation-W dimers.61,73,80 Concerning the aromatic C–H bonds of Np+, the largest impact is observed for the CH proton donor groups. The elongation of the C–H bonds upon formation of the CH⋯O ionic H-bond is rather minor, and the corresponding red shift in νCH is small. For example, for the (18) global minimum, the values are ΔrCH = 0.8 mÅ and ΔνCH = −5 cm−1. However, the IR intensity enhancement is substantial with up to two orders of magnitude. As a consequence, the nearly IR inactive νCH modes of bare Np+ (ICH ≤ 2 km mol−1) become visible in the Np+-W spectrum of the in-plane isomers, and reach intensities comparable to the νOH fundamentals (e.g., ICH = 59 km mol−1 for (18)). Of course, for Np+-W(π) there is essentially no effect on the C–H bond properties upon monohydration.
687 and 101787 cm−1)51,102 implies that the positive charge in (Np–W)+ mainly remains on the Np moiety, justifying the notation of Np+-W. Nonetheless, formation of the Np+-W dimer is accompanied by modest charge transfer from Np+ to the W ligand, which scales with the interaction strength. For example, Δq = 12, 7, and 8 me for the (18), (12), and (23) isomers. As a result of this charge transfer, the O–H bonds of W become weaker, leading to minor red shifts in the symmetric and antisymmetric O–H stretch modes from the frequencies of bare W, ν1/3 = 3656/3755 cm−1. For the (18) global minimum, the effects are largest, with ΔrOH = 1.7 mÅ and Δν1/3 = −15/−26 cm−1. In addition to the Δν1/3 shifts, the relative IR intensity of ν1 is strongly enhanced compared to that of bare W, which is typical for cation-W dimers.61,73,80 Concerning the aromatic C–H bonds of Np+, the largest impact is observed for the CH proton donor groups. The elongation of the C–H bonds upon formation of the CH⋯O ionic H-bond is rather minor, and the corresponding red shift in νCH is small. For example, for the (18) global minimum, the values are ΔrCH = 0.8 mÅ and ΔνCH = −5 cm−1. However, the IR intensity enhancement is substantial with up to two orders of magnitude. As a consequence, the nearly IR inactive νCH modes of bare Np+ (ICH ≤ 2 km mol−1) become visible in the Np+-W spectrum of the in-plane isomers, and reach intensities comparable to the νOH fundamentals (e.g., ICH = 59 km mol−1 for (18)). Of course, for Np+-W(π) there is essentially no effect on the C–H bond properties upon monohydration.
The comparison of the IRPD spectrum measured for Np+-W with the linear IR spectra calculated for the four Np+-W isomers suggests an immediate assignment of the experimental spectrum to the most stable (18) isomer, because of the good agreement with respect to both the positions and relative intensities of the transitions.72 The bands A, B, and E observed at 3722, 3635, and 3072 cm−1 are attributed to ν3, ν1, and νCH of (18) calculated at 3729, 3641, and 3062 cm−1, with systematic deviations of 7–10 cm−1, respectively. The single intense νCH normal coordinate of (18) corresponds mostly to a symmetric elongation of the C1–H and C8–H bonds, which is in phase with a minor elongation of the opposite C4–H and C5–H bonds. The detection of three single narrow transitions in the IRPD spectra of Np+-W (and also the colder Np+-W-Ar cluster not shown here) indicates that the experimental spectrum is mainly produced by the (18) isomer.72 The predicted IR spectra of the other three isomer have νOH and νCH frequencies, which are clearly different from those of (18) when considering the achieved spectral resolution. Thus, if any of the less stable local minima would be present in the expansion, the spectral features measured in the νOH and νCH range should exhibit splittings and/or shoulders, in disagreement with the experimental observation. Hence, the population of the (12), (23), and (π) isomers of Np+-W is below the detection limit. This result is in line with the relative (free) energies of the four isomers (Fig. 1).
3.3 Np+-W2
Fig. 4 summarizes the low-energy Np+-W2 structures considered in this work, along with the geometry of bare W2. Np+-W2 clusters with π-bonded W ligands are no longer discussed here, because of their low binding energies. There are two principal ways to add a second W ligand to the H-bonded Np+-W dimer to form Np+-W2. In the first category, which corresponds to the formation of a H-bonded solvent network, an H-bonded W2 dimer is attached to the Np+ cation via an in-plane bifurcated CH⋯O ionic H-bond. Depending on the three nonequivalent binding sites, these are denoted Np+-W2(18), Np+-W2(12), and Np+-W2(23). Their total binding energies of D0 = 5529, 5019, and 4851 cm−1 follow the same trend as found for the Np+-W dimers (Fig. 1), and their relative free energies (G) are similar to the relative energies (E0). The formation of the H-bonded solvent network is strongly cooperative because of the nonadditive induction forces, as will be illustrated in some detail quantitatively for the (18) isomers of n = 1 and n = 2. Attachment of the second W ligand strongly strengthens the ionic CH⋯O bond, which contracts from 2.327 Å (n = 1) to 2.242 Å (n = 2). This result is in line with the larger proton affinity of W2 as compared to W (PA = 808 and 691 kJ mol−1).103,104 As a consequence of the stronger CH⋯O bond, the C–H proton donor bonds are elongated from 1.0828 to 1.0836 Å, and the corresponding C–H stretch frequency decreases from 3062 to 3051 cm−1 along with an enhancement of the IR intensity from 59 to 143 km mol−1. On the other hand, the H-bond of the W2 dimer becomes much stronger by the presence of the Np+ cation. The nearby positive charge polarizes the first W ligand and the additional induced dipole strengthens the W–W H-bond. The isolated W2 dimer has a calculated dissociation energy of D0 = 1108 cm−1, in good agreement with the experimental value of 1105 ± 10 cm−1,105 illustrating that the chosen B3LYP-D3/aug-cc-pVTZ level reliably describes the W–W interaction. The W–W OH⋯O H-bond contracts from 1.947 Å in free W2 to 1.826 Å in Np+-W2(18). The total binding energy of D0 = 5529 cm−1 for Np+-W2(18) exceeds the sum of the dissociation energies of W2 (1108 cm−1) and the Np+-W(18) dimer (2773 cm−1) by 42%, providing a quantitative energetic measure for the strong cooperativity. The formation of the W2 dimer in Np+-W2 has unique signatures in the νOH range of the IR spectrum (Fig. 5). The localized bound O–H stretch of the proton donor is largely red shifted to νb = 3434 cm−1 with high IR intensity (696 km mol−1), because of the elongated O–H bond (0.9759 Å), while the uncoupled free O–H stretch of the dangling OH group (0.9617 Å) of this W molecule occurs at νf = 3708 cm−1. The νb mode in Np+-W2(18) is shifted to lower frequency by 106 cm−1 compared to bare W2 (3540 cm−1), as a result of the stronger OH⋯O H-bond. The coupled ν1 and ν3 frequencies of the proton acceptor OH groups (0.9627 Å) are predicted at 3649 and 3741 cm−1, i.e. they occur 8 and 11 cm−1 blueshifted from those of the n = 1 cluster, because the W molecule is further away from the Np+ charge and thus their O–H bonds are shorter than in n = 1 (0.9634 Å). Further measures of the degree of noncooperativity provided by the NBO and NCI analyses are detailed in ESI (Fig. S1–S3, Table S1†). Both the NBO and NCI indicators suggest that the neutral OH⋯O H-bond in Np+-W2(18) is stronger than the CH⋯O ionic H-bond! Similar cooperative effects as found for the (18) structure of Np+-W2 are also observed for the (12) and (23) isomers, although both the OH⋯O and CH⋯O H-bonds are weaker in these less stable local minima as compared to the (18) isomer. | ||
| Fig. 4 Optimized structures of W2 and most stable isomers of Np+-W2 calculated at the B3LYP-D3/aug-cc-pVTZ level. The (18), (12), and (23) isomers correspond to formation of a H-bonded solvent network, while the (18/45), (18/12), and (12/23) isomers are examples of interior ion solvation. Binding energies (D0) and bond lengths are given in cm−1 and Å, respectively. Numbers in parenthesis correspond to relative energies and free energies (E0, G). | ||
 | ||
| Fig. 5 Comparison of experimental IRPD spectrum of Np+-W2 to linear IR absorption spectra of the most stable (18) and (18/45) isomers of Np+-W2 calculated at the B3LYP-D3/aug-cc-pVTZ level (Fig. 4, Table 1). For comparison, also the spectrum calculated for bare W2 is shown (at a different intensity scale). Comparison of the IRPD spectrum to linear IR spectra of less stable isomers is available in Fig. S4 in ESI.† | ||
In the second category of Np+-W2 structures, which corresponds to interior ion solvation, both W ligands bind separately to the Np+ cation via two individual CH⋯O ionic H-bonds. The three most stable examples of this category are Np+-W2(18/45), Np+-W2(18/12), and Np+-W2(12/23), with binding energies of D0 = 5395, 4817, and 4512 cm−1, respectively (Fig. 4). Other possible isomers such as (18/23), (18/34), etc. are less stable. In contrast to clusters with an H-bonded solvent network, the nonadditive polarization forces are slightly noncooperative for interior ion solvation, mainly because of enhanced charge delocalization. This effect shall be illustrated in some detail quantitatively for the (18) and (18/45) isomers of n = 1 and n = 2, respectively, which are both characterized by bifurcated CH⋯O ionic H-bonds to the CαH groups of Np+. The (18/45) isomer with D2h symmetry has two equivalent CH⋯O H-bonds with R = 2.341 Å and D0 = 2698 cm−1, while the corresponding bond in the n = 1 cluster (C2v) is slightly stronger (R = 2.327 Å, D0 = 2773 cm−1). Energetically, the noncooperativity thus is evaluated as 3%. As a consequence of the slightly weaker CH⋯O H-bond, the C–H proton donor bonds contract from 1.0828 Å (n = 1) to 1.0826 Å (n = 2), and the corresponding (averaged) C–H stretch frequency increases from 3064 to 3070 cm−1. In addition, the O–H bonds of the individual W ligands are less affected by the interaction with Np+ in n = 2 as compared to n = 1 (ΔrOH = 1.5 vs. 1.7 mÅ, Δν1/3 = −14/−24 vs. −15/−26 cm−1). Similar noncooperative effects are observed for other isomers with interior ion solvation.
According to the calculations, the most stable Np+-W2 isomer is the (18) isomer with W2 attached to C1H and C8H. The next stable isomer is (18/45) with E0 = 134 cm−1 and G = 521 cm−1, i.e. the formation of an H-bonded network is favored over interior ion solvation (Fig. 4). The other considered isomers of both categories are substantially higher in energy, and from experience with the analysis of the n = 1 spectrum, they are not expected to substantially contribute to the experimental IRPD spectrum of Np+-W2. To this end, the IR spectra calculated for the (18) and (18/45) isomers are compared in Fig. 5 to the measured IRPD spectrum. The latter exhibits six transitions at 3740 (A1), 3728 (A2), 3696 (C) 3646 (B), 3496 (D), and 3068 (E) cm−1. The bands D and C are newly observed in the n = 2 spectrum (Fig. 2) and are a clear signature of a Np+-W2 isomer with a H-bonded W2 dimer. They correspond to the bound and free O–H stretch modes of the proton donor W molecule (νb/f) in the W2 dimer. As a consequence, transitions D and C at 3496 and 3696 cm−1 are assigned to νb and νf of the (18) isomer, in good agreement with the calculated frequencies of 3434 and 3708 cm−1. The remaining free O–H stretch modes of the terminal W acceptor molecule are predicted at ν1 = 3649 and ν3 = 3741 cm−1, and thus are assigned to transitions B and A1 observed at 3646 and 3740 cm−1, respectively. Transition E at 3068 cm−1 is then attributed to νCH of the (18) isomer predicted at 3051 cm−1. The experimental red shift of νCH upon attachment of the second W ligand (−4 cm−1) is somewhat smaller than the predicted shift (−11 cm−1). In addition, the enhanced relative intensity of νCH compared to the free νOH bands observed experimentally is nicely reproduced by the spectrum calculated for (18). While for n = 1, νCH is less intense than νOH, the opposite is true for n = 2. Overall, most of the major transitions of the IRPD spectrum of Np+-W2 agree well with those predicted for the (18) global minimum with respect to position and relative intensity. The other less stable Np+-W2 local minima with a W2 structure, (12) and (23), have a qualitatively similar IR spectrum (Fig. S4 in ESI†) but may be excluded for several reasons. First, the calculated relative energies are rather high (E0 > 500 cm−1), and thus their presence may be excluded for thermodynamic reasons. For the n = 1 cluster, all local minima with E0 > 300 cm−1 are below the detection limit. In addition, since the OH⋯O H-bonds are weaker in the (12) and (23) isomers as compared to the (18) isomer (R = 1.841 and 1.846 vs. 1.862 Å), their bound O–H stretch frequencies occur at significantly higher energies (νb = 3456 and 3462 vs. 3434 cm−1). However, band D assigned to νb occurs as a single peak and does not show any splitting. The asymmetric blueshaded band contour of band D with a sharp rise on the red side and a long monotonic decreasing tail on the blue side is typical for excitation of proton donor stretch vibrations73 and not an indication for the presence of further isomers. Similarly, band E assigned to νCH does not exhibit any splitting or shoulder. While for the (12) isomer the νCH intensity is predicted to be much weaker than for (18) (ICH = 23 vs. 143 km mol−1) and thus difficult to detect, the most intense νCH fundamental of (23) is predicted to occur with substantial intensity at 3094 cm−1 (52 km mol−1). This frequency is far from the measured νCH band E at 3068 cm−1, and thus the population of (23) is clearly below the detection limit.
Although at first glance the IRPD spectrum of Np+-W2 can mostly be assigned to the dominating (18) global minimum, closer inspection reveals subtle hints for the minor presence of a second isomer, which belongs to the class of interior ion solvation. The clearest signature is transition A2 at 3728 cm−1, which occurs close to band A of Np+-W at 3722 cm−1. This band thus is attributed to the free ν3 mode of a W ligand directly attached to Np+. The small observed blue shift of 6 cm−1 from n = 1 to n = 2 is consistent with the noncooperative effect when attaching the second W ligand. Hence, band A2 of Np+-W2 is assigned to ν3 of the by far most stable (18/45) isomer with interior ion solvation predicted at 3731 cm−1. The other transitions calculated for this isomer (ν3 = 3642 cm−1, νCH = 3063 cm−1) overlap with bands B and E at 3646 and 3068 cm−1 mainly assigned to the (18) global minimum. This overlap may explain the somewhat higher relative intensity of band B in the IRPD spectrum, which cannot be explained by the calculated spectrum of (18) alone. Comparing the ratio of the integrated band intensities of bands A1 and A2 (>3:1) with the IR cross sections calculated for ν3 of the (18) and (18/45) isomers (1:2), we estimate the maximum abundance of the (18/45) local minimum to be below 20%. This result is consistent with its relative (free) energy of E0 = 134 (521) cm−1. An alternative explanation for band A2 may be unresolved structure of (hindered) internal rotation of the terminal W ligand of the (18) isomer of Np+-W2. Although we cannot exclude this interpretation, we favor currently the scenario with the coexistence of the two isomers. Other isomers with interior ion solvation may safely be excluded for thermodynamic reasons (E0 > 700 cm−1). In addition, their spectra predicted in the νCH range do not match well with band E, because of their weaker CH⋯O H-bonds (Fig. S4 in ESI†).
3.4 Np+-W3
Interestingly, the IRPD spectrum of Np+-W3 in Fig. 2 is dominated in the free O–H stretch range by band C at νf = 3703 cm−1. Unlike the spectra for the n = 1 and n = 2 clusters, the n = 3 spectrum does not show anymore pronounced peaks A and B assigned to coupled ν3 and ν1 transitions of W ligands acting barely as proton acceptors in H-bonding. While there is definitively no signal around 3650 cm−1 (B, ν1), some weak signal intensity in the blue shoulder of band C near 3730 cm−1 may be attributed to ν3 transitions (A). On the other hand, there are intense and broad transitions D1 and D2 peaking at 3507 and 3402 cm−1, which clearly come from H-bonded O–H stretch modes. These observations are taken as evidence that the predominant Np+-W3 structures contributing to the IRPD spectrum have only W ligands, which act as proton donor in H-bonds, either as single donor or double donor. In such structures, a cyclic H-bonded W3 trimer is attached to the Np+ cation. The still high intensity of transition E at 3065 cm−1 indicates that at least in some of these Np+-W3 clusters, the cyclic W3 unit is connected to the Np+ cation via CH⋯O ionic H-bonding.The calculations yield a large number of stable structures for Np+-W3. Similar to the case of Np+-W2, the possible Np+-W3 isomers may be classified into those with interior ion solvation and those forming a single hydration network. The latter may further be divided into those having a linear, branched, or cyclic W3 unit. Several of the low-energy structures representing prototypes for these classes of Np+-W3 clusters are shown in Fig. 6 and S5 in ESI.† Amongst all optimized structures, Np+-W3(c1) with D0 = 8355 cm−1 is the most stable isomer (E0 = 0). In this structure, a cyclic neutral distorted H-bonded W3 trimer is attached to the Np+ cation via two nearly linear CH⋯O ionic H-bonds involving the C1H and C8H proton donor groups. For comparison, the structure and IR spectrum calculated for bare W3 are shown in Fig. S6 and S7 in ESI.† Its dissociation energy for W loss (D0 = 2809 cm−1) is in good agreement with the experimental value of 2650 ± 150 cm−1.106 The three intermolecular OH⋯O bonds in Np+-W3(c1) are rather different (R = 1.880, 1.904, 2.059 Å), giving rise to three isolated bound O–H stretch oscillators (νb = 3415, 3485, 3528 cm−1, Fig. 7) due to rather different O–H bond lengths (0.9762, 0.9733, 0.9712 Å). In contrast, the free O–H bonds have essentially the same length (0.9619 ± 0.0001 Å), producing three free O–H stretch bands with similar frequency (3708–3713 cm−1). A similar cyclic structure Np+-W3(c2) shown in Fig. 6 with D0 = 7846 cm−1 is slightly higher in energy (E0 = 509 cm−1). In the most stable linear (18) isomer with D0 = 8140 cm−1 (E0 = 215 cm−1), a linear W3 chain is attached to the Np+ cation via a bifurcated CH⋯O ionic H-bond between the acidic C1H and C8H protons and the first W ligand of the chain (Fig. S5 in ESI†). Such linear chains may also start between the less acidic C1H and C2H protons or the C2H and C3H protons, yielding the less stable (12) and (23) isomers with D0 = 7524 and 7262 cm−1 (E0 = 831 and 1093 cm−1). Np+-W3 clusters with a branched W3 network are less stable than those with a linear W3 chain (e.g., D0 = 7787 versus 8140 cm−1 for attachment of W3 at the C8H and C1H protons). Amongst all the isomers with interior ion hydration, the most stable (18/45) isomer with D0 = 8059 cm−1 (E0 = 296 cm−1) binds a W2 unit via bifurcated CH⋯O H-bonds to C1H and C8H and a single W ligand to the C4H and C5H protons. As expected, the most stable Np+-W3 isomer, in which all three W ligands bind separately to Np+via individual ionic CH⋯O H-bonds, is the (18/45/12) isomer with D0 = 7300 cm−1 (E0 = 1055 cm−1). When considering entropic effects at room temperature, the linear and branched structures as well as structures with interior ion solvation become more favorable than cyclic structures because the latter ones are more rigid (see G values in Fig. 6 and S5 in ESI†).
 | ||
| Fig. 6 Optimized structures of the two most stable cyclic Np+-W3 isomers obtained at the B3LYP-D3/aug-cc-pVTZ level. Binding energies (D0) and bond lengths are given in cm−1 and Å, respectively. Numbers in parenthesis correspond to relative energies and free energies (E0, G). | ||
 | ||
| Fig. 7 Comparison of experimental IRPD spectrum of Np+-W3 to linear IR absorption spectra of the most stable cyclic isomers of Np+-W3 calculated at the B3LYP-D3/aug-cc-pVTZ level (Fig. 6, Table 1). For comparison, also the spectrum calculated for bare W3 is shown (at a different intensity scale). | ||
The IRPD spectrum of Np+-W3 is compared in Fig. 7 to the IR spectra calculated for the two considered cyclic isomers (c1) and (c2), while comparison to IR spectra of the other isomers is available in Fig. S8 in ESI.† As mentioned above, the lack of bands A and B in the Np+-W3 spectrum provides a strong indication that the experimental spectrum is largely dominated by cyclic isomers, and indeed it can fully be rationalized by the two lowest-energy cyclic isomers (Fig. 7). The resulting assignments are summarized in Table 1. While the (c1) global minimum can account well for free and bound O–H stretch bands (C, D1, D2), it fails to reproduce the intense νCH band E. In contrast, the (c2) isomer can explain the latter transition and, in addition, also the O–H stretch part of its spectrum is compatible with the experimental one. The minor signal in the blue wing of band C near 3730 cm−1 may provide weak evidence for the presence of the most stable linear (18) isomer (Fig. S8 in ESI†), which for energetic reasons may also have a significant population. Because its terminal W is far away from the Np+ charge, its ν1/3 bands have low IR activity, and thus may not readily be detected here. The broad transition D3 near 3250 cm−1 cannot be rationalized by any of the calculated Np+-W3 isomers. This band thus is tentatively attributed to the first overtone of the bending vibrations of the W ligands (2βOH), which is expected in this spectral range and not included in the harmonic calculations. Clearly, the IR spectrum predicted for cyclic W3 (Fig. 7) is quite different from that of Np+-W3(c1). The perturbation by the nearby Np+ cation arises from both charge-induced and steric effects.
3.5 Np+-W4
The evolution of the IRPD spectra of Np+-Wn in Fig. 2 yields the following major conclusions for the n = 4 cluster. Bands A and B assigned to ν3 and ν1 modes of W ligands not acting as H-bond donors, which are strong for n ≤ 2 and very weak and/or absent for n = 3, definitively disappear for n = 4. This result indicates that the n = 4 clusters are dominated by isomers with a cyclic W4 unit. Second, compared to the spectra of the n = 1–3 clusters, also the relative intensity of band E attributed to νCH of Np+ becomes very weak relative to the νOH transitions for n = 4. This observation indicates that the population of Np+-W4 isomers with CH⋯O ionic H-bonds of Np+ to the W4 unit is small, and the cyclic W4 ring may preferentially bind above or below the Np+ ring. The O–H stretch spectral range is now completely dominated by the sharp band C and a broader band D1 characteristic for νf and νb of single-donor W ligands.Out of the many stable structures computed for Np+-W4, the cyclic Np+-W4(c) isomer shown in Fig. 8 is the most stable one, with a total binding energy of D0 = 12557 cm−1. In this structure, a distorted neutral H-bonded W4 unit is located above the aromatic Np+ ring, with a strongly nonplanar bifurcated CH⋯O H-bond motif to one W molecule of the W4 cycle. As a result, the C1H⋯O and C8H⋯O H-bonds are rather weak (2.685 and 2.491 Å) compared to the planar bifurcated CH⋯O H-bonds found for n ≤ 3. Like for n = 3, also for n = 4 all free O–H bonds of the cyclic W4 ring point away from the Np+ cation to maximize the electrostatic charge–dipole attraction. For comparison, also a low-energy linear Np+-W4(l) isomer is shown in Fig. 8 (D0 = 11103 cm−1), although its relative energy is already rather high compared to Np+-W4(c), E0 = 1454 cm−1. Note that this linear isomer differs from that reported in ref. 14 such that the W ligands are closer to the Np+ cation. The global minimum reported in ref. 14 could not be optimized at the current theoretical level.
 | ||
| Fig. 8 Optimized structures of the most stable Np+-W4 isomers with a cyclic (c) and linear (l) W4 solvent cluster obtained at the B3LYP-D3/aug-cc-pVTZ level. Binding energies (D0) and bond lengths are given in cm−1 and Å, respectively. Numbers in parenthesis correspond to relative energies and free energies (E0, G). | ||
The IRPD spectrum of Np+-W4 is compared in Fig. 9 to the IR spectra calculated for the cyclic and linear isomers shown in Fig. 8. In general, the overall appearance of the Np+-W4(c) spectrum agrees well with the measured spectrum, in particular in view of the fact that the computations somewhat overestimate the red shift of the bound O–H stretch bands (see Fig. 5 for n = 2). This is true for the overall appearance, the positions and relative intensities of the transitions, and the simplicity of the spectrum. Hence, bands C and D1 at 3703 and 3433 cm−1 in the IRPD spectrum are assigned to νf and νb modes of Np+-W4(c), calculated in the narrow 3706–3710 cm−1 range for νf and more widely spread out at 3240, 3319, 3332, and 3375 cm−1 for νb (Table 1). The transition D2 at 3210 cm−1 is again tentatively attributed to 2βOH, and may also cover the lowest-frequency νb mode (3240 cm−1). Consistent with the experimental spectrum, the νCH intensity of this isomer is very small (4 km mol−1). In fact, the visible band E at 3082 cm−1 is probably not (only) produced by the Np+-W4(c) global minimum but may be an indicator for the presence of less stable cyclic and/or linear/branched Np+-W4 isomers with stronger CH⋯O contacts. One such example may be the computed Np+-W4(l) isomer. On the other hand, this isomer has the ν1 and ν3 transitions typical of the terminal W ligand, which are not detected in the experimental spectrum, although their IR activity is predicted to be higher than that of νCH. Interestingly, the spectrum calculated for cyclic W4 (Fig. S6 in ESI†) is not too different from that of Np+-W4(c1) (Fig. 9). The strongly IR active ring stretch mode of W4 is measured at νb = 3416 cm−1,107i.e. somewhat lower than the maximum of band D1 of Np+-W4 at 3433 cm−1. In addition, it is substantially higher than the predicted frequency of νb = 3297 cm−1, confirming that the DFT calculations overestimate the red shift of the bound O–H stretch frequencies.
 | ||
| Fig. 9 Comparison of experimental IRPD spectrum of Np+-W4 to linear IR absorption spectra of the most stable cyclic and linear isomers of Np+-W4 calculated at the B3LYP-D3/aug-cc-pVTZ level (Fig. 8, Table 1). For comparison, also the spectrum calculated for bare W4 is shown (at a different intensity scale). | ||
3.6 Np+-W5
The following conclusions are evident from the comparison of the IRPD spectrum of Np+-W5 with those of the smaller hydrates in Fig. 2. First, band C near 3700 cm−1 gains further in relative intensity, because the number of single-donor W ligands in the clusters increases and their νf bands occur all at the same position. In addition, its width becomes narrower, because the binding energy for W elimination becomes smaller as the cluster size increases. Second, like for the other n ≥ 3 clusters, bands A and B are absent, indicating that clusters with cyclic W5 units dominate the cluster population. Third, band E disappears completely for n = 5, revealing that the observed cluster structures have no (or only a weak) CH⋯O ionic H-bond. Thus, the trend observed already for n = 4 continues for n = 5, and the spectrum points toward Np+-W5 clusters with cyclic W5 rings above the Np+ plane. Fourth, the transition D1 associated for νb transitions becomes broader and shifted to lower frequency, indicating that the OH⋯O H-bonds of the W5 solvent network become stronger. Finally, similar to band C, the band D2 assigned to the 2βOH overtone increases in relative intensity, because the number of W ligands in the cluster becomes larger and possibly the anharmonic interaction (Fermi resonances) with the nearby νb fundamentals becomes stronger. Part of the D2 intensity may also arise from νb transitions.The most stable structure out of the plethora of minima found for Np+-W5 is the Np+-W5(c1) isomer shown in Fig. 10, with a total binding energy of D0 = 15927 cm−1. In this structure, a distorted cyclic neutral W5 pentamer is connected to the most acidic C1H and C8H protons by a strongly nonplanar bifurcated CH⋯O ionic H-bond to a single W molecule. This structure is similar to the most stable Np+-W4(c) structure of the n = 4 cluster, with the simple insertion of one W ligand into the cyclic water cluster ring. All W molecules act as single-donor single-acceptor, except for the W molecule connecting the W5 cycle to Np+. For comparison, a related cyclic Np+-W5(c2) isomer is also shown in Fig. 10, in which one W ligand is a double acceptor in the cyclic W5 pentamer. This less stable structure is substantially higher in energy (E0 = 2718 cm−1), illustrating that W5 rings with only single-donor single-acceptor molecules are energetically very favorable. Other linear or branched structures or cyclic structures with smaller Wn<5 rings (ring-tail structures) are not considered in detail because they are less stable and, similar to Np+-W4, do not fit the experimental spectrum in the free O–H stretch range. Similar to the n = 4 cluster, the global minimum reported in ref. 14 with a linear W5 chain pointing away from Np+ could not be found at the current theoretical level.
 | ||
| Fig. 10 Optimized structures of the two most stable cyclic Np+-W5 isomers obtained at the B3LYP-D3/aug-cc-pVTZ level. Binding energies (D0) and bond lengths are given in cm−1 and Å, respectively. Numbers in parenthesis correspond to relative energies and free energies (E0, G). For comparison, also the spectrum calculated for bare W5 is shown (at a different intensity scale). | ||
In Fig. 11 the IRPD spectrum measured for Np+-W5 is compared to the IR spectra calculated for the (c1) and (c2) isomers of Np+-W5 and bare W5. The spectrum of the most stable Np+-W5(c1) isomer agrees well with the measured spectrum, when we recall that the calculations underestimate the frequencies of the bound O–H stretch bands (νb). The resulting assignments are listed in Table 1. All νf modes of Np+-W5(c1) are predicted in the narrow range of 3709 ± 3 cm−1, in good agreement with band C at 3700 cm−1 (which has a width of 13 cm−1). The highly red shifted νb modes assigned to the OH⋯O H-bonds at 3180, 3256, 3281, 3317, and 3356 cm−1 correlate with the broad bands D2 and D1 peaking at 3230 and 3365 cm−1, respectively. As for the other cluster sizes, band D2 may also contain contributions from 2βOH overtone transitions. Although the Np+-W5(c2) isomer is quite high in energy, clusters of this type may account for the signal observed in the 3400–3600 cm−1 range because of their weaker OH⋯O bonds and thus higher νb frequencies. Comparison of the IR spectra calculated for Np+-W5(c1) and bare W5 reveals that the presence of Np+ causes a smaller perturbation than for the n = 4 case. Again, the strongly IR active νb modes of W5 calculated at 3248 cm−1 (averaged value) are around 100 cm−1 lower than the measured band (3360 cm−1),107 confirming that the calculations overestimate the red shifts of the bound O–H stretch frequencies of the cyclic Wn ring.
 | ||
| Fig. 11 Comparison of experimental IRPD spectrum of Np+-W5 to linear IR absorption spectra of the cyclic Np+-W5 isomers calculated at the B3LYP-D3/aug-cc-pVTZ level (Fig. 10, Table 1). | ||
4. Further discussion
4.1 Cluster growth
The analysis of the IRPD spectra utilizing the DFT calculations provides a detailed and consistent picture of the preferential sequential cluster growth in microhydrated Np+-Wn clusters. The observed IRPD spectra can readily be explained by the most stable clusters predicted by the calculations. For the most probable cluster growth, we obtain the following scenario. As discussed in detail in our previous report,72 the population of the Np+-W dimer (n = 1) is dominated by the Np+-W(18) isomer, in which W binds in a planar bifurcated CH⋯O H-bond to the most acidic C1H and C8H protons of Np+. For n = 2, the formation of a H-bonded solvent network is more favorable than interior ion solvation, even though the cation–dipole interaction in Np+-W (D0 = 2800 ± 300 cm−1)72 is nearly three times stronger than the neutral W–W H-bond bond (D0 = 1105 ± 10 cm−1).105 This at first glance surprising result is attributed to the massive effects of the nonadditive three-body induction forces. For the formation of the H-bonded solvent network, these are highly cooperative, while for interior ion solvation they are slightly noncooperative. For Np+-W2, these can be quantified as +42% for the H-bonded Np+-W2(18) global minimum and −3% for the Np+-W2(18/45) local minimum, respectively. For Np+-W2, the cooperative effects involved in H-bonded networks eventually override the large difference between the Np+-W and W–W binding energies. Thus, the detected Np+-W2 clusters are mostly attributed to the Np+-W2(18) global minimum, in which a W2 dimer is attached to the acidic C1H and C8H protons via a planar bifurcated CH⋯O ionic H-bond (D0 = 5529 cm−1). A minor part of the population (<20%) is tentatively assigned to the less stable Np+-W2(18/45) isomer, in which two individual W ligands separately bind to Np+via equivalent planar bifurcated CH⋯O H-bonds (D0 = 5395 cm−1). For n ≥ 3, isomers with interior ion solvation are not observed anymore, and only the more stable Np+-Wn structures with cyclic H-bonded Wn rings are detected. However, while for n = 3 these rings bind in the Np+ plane to the acidic C–H protons of Np+via two linear CH⋯O H-bonds, for n = 4 and 5 the cyclic Wn rings are located above the aromatic plane and are connected to Np+via strongly nonplanar bifurcated CH⋯O ionic H-bonds. In such sandwich-type structures, the W ligands are closer to the positive charge of the Np+ cation, and thus can maximize the strong electrostatic charge–dipole and inductive charge-induced dipole interactions. The gain in electrostatic and induction energy is larger than the energy penalty arising from the deviation from linearity and planarity of the CH⋯O ionic H-bonding. Some additional stabilization may also come from increased dispersion between the cyclic Wn ring and the π-electron system of the aromatic Np+ ring in the sandwich-type configuration. Thus, the Np+-Wn structures with n = 4 and 5 strongly benefit from the cooperative induction forces arising from the cyclic H-bonded ring, and the sandwich configuration maximizes both dispersion and electrostatic forces. In addition, the Wn rings in Np+-Wn are all more or less distorted structures derived from those of isolated neutral cyclic Wn rings (Fig. S6 in ESI†). Although all of them have only single-donor single-acceptor solvent molecules in the most stable Np+-Wn isomers, the free dangling OH groups are all pointing away from the Np+ cation to optimize the cation–dipole attraction. When considering the predicted Np+-Wn global minima, the incremental dissociation energies, ΔD0(n) = D0(n) − D0(n − 1), are 2773, 2756, 2826, 4202, and 3370 cm−1 for n = 1–5, respectively. Although there is no monotonic trend in the whole considered size range, ΔD0 increases for small n and peaks at n = 4, illustrating the strong cooperativity effects for the hydration network above the Np+ cation. The magnitude of these incremental binding energies is also in line with the experimental observation of single W loss under the single-photon IRPD conditions employed in the present work (with hνIR ≤ 4000 cm−1). The structure of the cyclic neutral Wn clusters are distorted by the presence of the Np+ cation, and their IR spectroscopic signatures change as well, although their coarse geometries remain unaffected (Fig. S6 and S7 in ESI†). Also, their total and incremental binding energies, which vary as ΔD0(n) = 1108, 2810, 3539, and 2521 cm−1 for n = 2–5, respectively, are smaller than those of the corresponding Np+-Wn clusters, illustrating the cooperative effect of the nearby positive charge on the cyclic H-bonded network. As expected, the averaged OH⋯O bond length in bare Wn clusters decreases monotonically as 1.947 > 1.903 > 1.764 > 1.733 Å for n = 2–5. Interestingly, the corresponding variation in Np+-Wn is strongly nonmonotonic (1.826 < 1.972 > 1.789 > 1.753 Å for n = 2–5). In addition, for n ≥ 3 the H-bond lengths in Np+-Wn is even larger than in bare Wn although the incremental binding energies are smaller. This effect is attributed to the steric effects of the Np+ cation on the structure of the H-bonded Wn network. Interestingly, also the interaction of the Wn cluster with the CH groups of Np+ changes nonmonotonically in the most stable Np+-Wn clusters, with averaged CH⋯O bond lengths of 2.327 > 2.242 < 2.467 < 2.588 > 2.571 Å. The drop from n = 1 to n = 2 is due to cooperativity, while the increase for n ≥ 3 arises from the fact that the CH⋯O bond becomes weaker because of the formation of the sandwich structures with the Wn rings lying above the Np+ plane. The destabilization of the CH⋯O interaction with increasing cluster size is clearly visible in the decreasing intensity of νCH in the IR spectra (band E in Fig. 2). Interestingly, the evolution of the microhydration network around Np+ is similar to that inferred for the heterocyclic aromatic imidazolium ion, in which the two acidic NH groups of the monocyclic five-membered ring are protected by substitution with bulky alkyl groups.108 Finally, the solvation of the Np+ cation by nonpolar ligands (such as Ar) is preferentially via π-stacking, because for these clusters dispersion and polarization forces dominate the attraction.48,49,72,109Significantly, the cluster growth scenario derived herein from IRPD spectroscopy and DFT calculations is very different from the one reported previously from mass spectrometry and computations.14 In the latter study, Np+-Wn clusters are generated by electron ionization of Np and injecting these ions into a drift cell filled with He buffer gas and water vapor with partial cooling down to 249 K. In the resulting mass spectra, Np+-Wn clusters can be detected up to n = 6, although for cluster sizes with n ≥ 2 the ion signal is barely above the noise level. This is in marked contrast to our much more efficient electron-impact supersonic plasma cluster ion source, in which via the same sequential cluster growth mechanism much higher yields of Np+-Wn clusters can be generated under cold molecular beam conditions and at concentrations sufficient to record IRPD action spectra. The low Np+-Wn yield in the previous study14 allowed to perform cluster aggregation equilibria to be measured only for n = 1, and the obtained binding enthalpy of −ΔH = 2730 ± 350 cm−1 is in good agreement with our recent spectroscopic value of D0 = 2800 ± 300 cm−1.72 In addition to mass spectra, the previous study also reported results of DFT calculations at the B3LYP and M06-2X levels for Np+-Wn clusters up to n = 6.14 For n = 1 and 2, the reported minima14 are in good agreement with our structures. However, for n ≥ 3 there is a qualitative discrepancy between the previous study14 and the most stable structures derived herein from both IR spectroscopy and DFT computations. No cyclic structures were reported (and possibly also not considered) previously,14 and it was claimed that in the most stable Np+-Wn clusters, a linear H-bonded Wn chain is attached to Np+via CH⋯O bonding. This is in contrast to our computations, which clearly show that cyclic structures are by far more stable than linear ones for n ≥ 3, a conclusion strongly supported by our IRPD spectra. If linear H-bonded chains were attached to Np+ at the C1H and C8H protons, transitions A, B, and E should be detected in the Np+-Wn spectra with substantial intensity (in disagreement with the experimental observation). Since our electron-impact cluster ion source is known to produce the most stable isomers of a given cluster size,73,74 we are confident that the cyclic structures are indeed the global minima for Np+-Wn with n ≥ 3. The failure of finding the global minima in the previous study14 illustrates the rather limitated capabilities of mass spectrometry and incomplete structural survey by computations for finding the most stable cluster structures. At the same time, it highlights the high sensitivity of vibrational spectroscopy in the X–H stretch range for the determination of the H-bonded solvation networks in polyhydrated clusters.
The ionization energy of Np (IE = 8.144 eV)51 is by far lower than that of Wn clusters (>10 eV for n ≤ 20).110,111 Consequently, the excess positive charge is mostly localized on the aromatic molecule even for large n, in line with the notation Np+-Wn. There is some partial charge transfer from Np+ to the Wn network, which for the most stable Np+-Wn isomers amounts to Δq = 12, 14, 22, 28, and 32 me for n = 1–5 according to the NBO analysis (Fig. S3 in ESI†). Interestingly, the magnitude of the charge transfer for the cyclic solvent structures is slightly larger than that predicted for linear chain structures (e.g., Δq = 23 me for n = 6).14 Similarly, the proton affinity of the naphthyl radical (PA = 234.5 kcal mol−1)11 is by far larger than those of Wn clusters in this size range (PA = 167–216 kcal mol−1 for n = 1–6).112 Thus, no proton transfer to the solvent cluster occurs in (Np-Wn)+, because C10H8+-Wn clusters are much more stable than C10H7–H+Wn. Test calculations for (Np-Wn)+ clusters with a protonated H+Wn solvent cluster with n = 4 and n = 5 reveal that they are more than 1 eV less stable than corresponding Np+-W4 clusters (Fig. S9 in ESI†).
It is instructive to compare the properties of Np+-Wn clusters characterized herein with those of neutral72,75 and anionic clusters76–79 to extract the considerable impact of the charge on the hydration network of PAHs. So far, no experimental data are available for neutral Np-Wn clusters, and thus all information for this complex has to rely on computations carried out for n ≤ 2.72,75 According to the calculations, the most stable Np–W and Np–W2 clusters have π H-bonded structures, in which either a single W ligand or a H-bonded W2 forms two intermolecular OH⋯π H-bonds with the aromatic π electron system of Np. In such π H-bonds, the OH protons can favourably interact with the negative π electron cloud.72,75,113 Because of the lack of the positive charge, the interaction is much weaker in neutral Np-Wn than in the Np+-Wn cation. Ionization of the n = 1 and n = 2 clusters thus causes a drastic change in the potential energy surface with respect to both the structure and interaction strength of the aromatic molecule with Wn from the OH⋯π H-bonded neutral clusters to CH⋯O H-bonded cation clusters. Such ionization-induced changes in the hydration motif give rise to solvent rearrangement reactions occurring on the picosecond timescale.87,114 These may be probed in the future for Np-Wn clusters by time-resolved pump-probe IR spectroscopy,114,115 a technique recently applied to monitor solvent rearrangement reactions in related aromatic clusters in real time.116–120 Although Np− is an unstable anion because of its negative electron affinity, stable microhydrated Np−-Wn clusters can be produced in supersonic expansions and have been characterized by photoelectron (n ≤ 8)76–78 and IR (n ≤ 6)79 spectroscopy. The computational analysis of these IR spectra79 yields multiple π H-bonded structures for Np−-Wn for n ≤ 4, in which H-bonded Wn clusters are attached to a single side of Np. For n = 3 and 4, cyclic solvent structures with single-donor single-acceptor W molecules are most favourable. However, in contrast to the cation clusters, in Np−-Wn the H atoms not involved in the OH⋯O H-bonded Wn network point toward the Np− anion to form OH⋯π H-bonds. For Np+-Wn cations, this configuration is repulsive and the free OH groups point away from Np+. In all three charge states, the formation of Wn networks strongly benefit from cooperativity.
4.2 Comparison to (Bz-Wn)+ clusters
Comparison between Np+-Wn and (Bz-Wn)+ clusters reveals the effects of the second aromatic ring present in Np on the interaction between PAH+ cations and W. The geometric and electronic structure of (Bz-Wn)+ clusters are well characterized by IR (n ≤ 23)61,62,73,80–83 and electronic (n ≤ 4)84 spectroscopy, photoionization spectroscopy (n = 1),85 mass spectrometry (n ≤ 40),10,12,86 and quantum chemistry.87,88 A detailed comparison between the Np+-W and Bz+-W dimers (n = 1) has been presented in our previous report.72 Briefly, the structures and H-bonding motifs in Bz+-W and Np+-W are principally different. While in the Np+-W(18) global minimum, the W ligands forms a bifurcated CH⋯O H-bond to two adjacent CαH groups of two different rings, in the most stable Bz+-W(12) structure W is attached to two CH groups obviously from the same ring. Because in Bz+ the excess charge is delocalized over only a single ring, the electrostatic interaction in Bz+-W is stronger than in Np+-W. This view is consistent with the NBO72 and NCI (Table S1 in ESI†) analyses of the charge distribution in both ions, which show that the CH protons in Np+ carry a lower positive partial charge. This electrostatic effect is partly compensated for by the more linear H-bonds in Np+-W(18) enabled by simultaneous binding of W to the two fused rings. As a net result, the dissociation energy of Bz+-W (D0 = 3290 ± 120 cm−1)83 inferred from spectroscopic measurements is still substantially larger than that of Np+-W (D0 = 2800 ± 300 cm−1),72 in good agreement with the values calculated at the B3LYP-D3/aug-cc-pVTZ level (D0 = 3209 and 2773 cm−1) and binding enthalpies extracted from mass spectrometry (−ΔH = 3150 ± 350 and 2730 ± 350 cm−1).12,14 For both aromatic monohydrates, there is a large geometry change from the neutral cluster (π H-bonded)113,121–126 to the cation cluster (CH⋯O H-bonded).61,73,80,87Similar to Np+-W2, also for the Bz+-W2 trimer the isomer with a W2 dimer attached to Bz+ is found to be more stable than isomers with individual W ligands and both isomers are observed in the IR spectra.62,81 Previous computations and IR spectra indicate that the low-energy isomers of Bz+-W3 have linear and branched W3 networks attached to Bz+, while cyclic isomers have not been identified as minima on the potential.13,62,81 This is in contrast to Np+-W3, for which our calculations indicate that cyclic structures are not only stable but also yield the global minimum. This difference is rationalized by the stronger Bz+-W bond with larger electrostatic interactions, which are considerably stronger than the W–W bonds and thus favor branched and linear structures via directional charge–dipole forces. In Np+-Wn, the Np+-W bond is weaker and less dominant. Therefore, in Np+-W3 the W–W H-bond interactions become more competitive and can maximize the number of H-bonds by forming a cyclic ring.
Starting from n ≥ 4, in (Bz-Wn)+ clusters one proton from Bz+ is transferred to the Wn solvent cluster, i.e. the ground state structure has the form C6H5–H+Wn, in which a phenyl radical binds to the surface of a protonated water cluster. This intracluster ion–molecule reaction is inferred from IR62,81,82 and electronic84 spectroscopy and is consistent with mass spectrometry10,12,13 and calculations.13,81,88 The proton affinity of Wn clusters increases with cluster size n (PA = 691, 808, 862, 900, 904, 908 kJ mol−1 for n = 1–6),10,103,112 and starting from n = 4 it becomes larger than the one of the phenyl radical (884 kJ mol−1).103 Thus, for (Bz-Wn)+ with n ≥ 4, the proton-transferred structure C6H5–H+Wn is thermodynamically more stable than the Bz+-Wn form. In contrast, the proton affinity of the naphthyl radical is rather high (981 kJ mol−1),11 so that proton transfer from Np+ to Wn in (Np-Wn)+ is thermodynamically not feasible in the cluster size range studied here.
5. Concluding remarks
In summary, microhydrated clusters of Np+ are characterized by IR spectroscopy and dispersion-corrected DFT calculations to probe the stepwise evolution of the structure of the microhydration network around this prototypical PAH+ cation. Significantly, the presented data provide the first spectroscopic impression of the Np+-Wn (and thus the PAH+-Wn) interaction and the stepwise hydration process at the molecular level. The IR spectra recorded in the C–H and O–H stretch range are highly sensitive to the solvation network and change substantially with cluster size. Hence, their computational analysis provides unprecedented insight into the preferential cluster growth. Notably, the cold Np+-Wn clusters are produced in a supersonic plasma beam expansion. Thus, the predominant population corresponds to the global minima on the respective potential energy surfaces, which is supported by the analysis of the IR spectra. The salient results can be summarized as follows.(1) The most stable Np+-W dimer (n = 1) is stabilized by a strong, planar, and symmetric bifurcated CH⋯O ionic H-bond, in which the two lone pairs of W bind to two CH protons belonging to different rings of the bicyclic Np+ ion. The CH⋯O H-bond in Np+-W differs qualitatively in both structure and interaction type from the corresponding CH⋯O H-bond in Bz+-W. The H-bond in Bz+-W is stronger than in Np+-W (3290 ± 120 versus 2800 ± 300 cm−1), because the positive charge density is more localized in the monocyclic Bz+ cation leading to larger charge–dipole attraction. However, because the bifurcated H-bonds in Np+-W deviate much less from linearity, enabled by the simultaneous interaction with two fused aromatic rings, the molecular orbital interaction stabilizing the H-bond is stronger than in Bz+-W. These qualitative differences in bonding between Bz+-W and Np+-W will also hold for larger PAH+-W clusters, because they arise from the presence of fused rings in Np+ and other PAH+ ions. The quantum chemical NBO and NCI analyses quantitatively explain these differences.
(2) For Np+-Wn clusters with n ≥ 2, two types of clusters can compete, namely those with interior ion solvation and those with a H-bonded hydration network. The former are slightly less stabilized by small noncooperative nonadditive induction forces due to charge delocalization. In contrast, the latter strongly benefit from cooperative nonadditive polarization forces due to enhanced H-bond strengths in the multiple H-bonded network. Consequently, Np+-W2 clusters (n = 2) with a H-bonded W2 dimer attached to the Np+ cation via a bifurcated CH⋯O H-bond dominate the population (>80%), because they are more stable than structures in which two individual W ligands bind separately to Np+. The degree of cooperativity and noncooperativity can be quantified by considering the binding energies, the vibrational frequency shifts upon H-bonding, and the NCI and NBO analyses (charge transfer and molecular orbital interactions). Concerning the binding energy, the cooperativity is quantified as +42% for the most stable Np+-W2 isomer with a W2 dimer, while the noncooperativity is −3% for the most stable Np+-W2 isomer with two single W ligands.
(3) Np+-Wn clusters with n ≥ 3 prefer a cyclic Wn network anchored to the Np+ cation via CH⋯O ionic H-bonds. In all these Np+-Wn clusters, the cyclic Wn network is composed of single-donor single-acceptor W ligands. In the most stable Np+-W3 cluster, cyclic W3 is attached to Np+ in the aromatic plane via two single linear CH⋯O H-bonds, while in Np+-Wn with n = 4 and 5 the cyclic Wn clusters bind to Np+via a single nonplanar bifurcated CH⋯O H-bond to form sandwich-like structures. This subtle change in structural motifs arises from the optimization of the sum of various competing contributions, such as steric hindrance (repulsion), strengths and number of the different H-bonds (OH⋯O, CH⋯O), charge–dipole (electrostatics) and charge-induced dipole (polarization) attractions, and dispersion forces. In the larger n = 4 and 5 sandwich structures, electrostatic, polarization, and dispersion forces as well as the OH⋯O H-bonds of the Wn subclusters are enhanced, while the CH⋯O H-bond is less favored but also less important for the total interaction energy. For the smaller n = 3 cluster, the CH⋯O H-bonds still provide an important contribution.
(4) The presence of the Np+ cation in Np+-Wn has an important impact on the structure and IR spectrum of the Wn unit, although the coarse structure of Wn remains unaffected (e.g., monocyclic for n = 3–5). In particular, the free OH groups of the single-donor single-acceptor cyclic network in Np+-Wn with n = 3–5 all point away from the positive charge.
(5) The cyclic Np+-Wn clusters (n = 3–5) obtained here by IR spectroscopy and DFT calculations deviate substantially from the ones with linear Wn chains predicted recently by mass spectrometry and DFT calculations,14 indicating that the former combined approach is much more sensitive in predicting reliable cluster structures.
(6) Comparison of cationic Np+-Wn with neutral Np-Wn and anionic Np−-Wn illustrates the important impact of the charge state on the PAH(±)-Wn interaction. While anionic and neutral Np(−)-Wn clusters benefit from (multiple) OH⋯π H-bonds and thus have H-bonded Wn clusters attached to the aromatic π-electron system, these configurations are repulsive for cationic Np+-Wn, because the cation–dipole forces turn the free OH groups of Wn away from Np+.
(7) Comparison of (Np-Wn)+ with (Bz-Wn)+ reveals the drastic differences in intermolecular interaction, microhydration, and chemical reactivity upon attachment of the second ring. First, for n ≤ 2, the H-bonds in Bz+-Wn are stronger than in Np+-Wn, because the charge is more localized in the former ion, leading to increasing electrostatic and polarization forces. Second, for the same reason, Bz+-W3 prefers a linear/branched W3 structure over a cyclic W3 ring observed in Np+-W3. Third, (Bz-Wn)+ clusters with n ≥ 4 exhibit proton transfer to solvent, because the proton affinity of Wn≥4 exceeds the one of the phenyl radical. No such intracluster proton transfer is observed for (Np-Wn)+ because of the substantially higher proton affinity of the naphthyl radical. The same will be true for (PAH-Wn)+ clusters with larger PAH molecules because the proton affinity of their radicals will be even higher.
In future work, this combined spectroscopic and computational strategy may be employed to extend these cluster studies to larger degree of hydration, larger PAH+ cations, different solvents (polar/nonpolar, protic/nonprotic), and protonated PAH. Exploration of larger (PAH-Wn)+ cations eventually converges to the limit of PAH+ cations embedded in ice or located on ice surfaces, which are particularly relevant for astrochemical applications. Because the charge remains on Np+ even in large polyhydrated clusters, the highly reactive Np+ radical cation generated in ice grains or on their surfaces by radiation or particle impact is readily available for ion–molecule reactions involving other organic molecules deposited in and/or on the grains.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by Deutsche Forschungsgemeinschaft (DFG, DO 729/3).References
- E. A. Meyer, R. K. Castellano and F. Diederich, Angew. Chem., Int. Ed., 2003, 42, 1210–1250 CrossRef CAS PubMed.
- L. M. Salonen, M. Ellermann and F. Diederich, Angew. Chem., Int. Ed., 2011, 50, 4808–4842 CrossRef CAS PubMed.
- J. P. Schermann, Spectroscopy and Modelling of Biomolecular Building Blocks, Elsevier, Amsterdam, 2008 Search PubMed.
- P. Hobza and K. Müller-Dethlefs, Non-covalent Interactions, The Royal Society of Chemistry, Cambridge, 2009 Search PubMed.
- P. Weilmünster, A. Keller and K. H. Homann, Combust. Flame, 1999, 116, 62–83 CrossRef.
- S. Steenken, C. J. Warren and B. C. Gilbert, J. Chem. Soc., Perkin Trans. 2, 1990, 335–342 RSC.
- M. Mons, I. Dimicoli and F. Piuzzi, Int. Rev. Phys. Chem., 2002, 21, 101–135 CrossRef CAS.
- B. Brutschy, Chem. Rev., 1992, 92, 1567–1587 CrossRef CAS.
- H. Mohan and J. P. Mittal, J. Phys. Chem. A, 1999, 103, 379–383 CrossRef CAS.
- A. Courty, M. Mons, J. Le Calve, F. Piuzzi and I. Dimicoli, J. Phys. Chem. A, 1997, 101, 1445–1450 CrossRef CAS.
- W. Y. Feng and C. Lifshitz, Int. J. Mass Spectrom. Ion Process., 1996, 152, 157–168 CrossRef CAS.
- Y. Ibrahim, E. Alsharaeh, K. Dias, M. Meot-Ner and M. S. El-Shall, J. Am. Chem. Soc., 2004, 126, 12766–12767 CrossRef CAS PubMed.
- Y. M. Ibrahim, M. Meot-Ner, E. H. Alshraeh, M. S. El-Shall and S. Scheiner, J. Am. Chem. Soc., 2005, 127, 7053–7064 CrossRef CAS PubMed.
- I. K. Attah, S. P. Platt, M. Meot-Ner, M. S. El-Shall, S. G. Aziz and A. O. Alyoubi, Chem. Phys. Lett., 2014, 613, 45–53 CrossRef CAS.
- L. J. Allamandola, M. P. Bernstein, S. A. Sandford and R. L. Walker, Space Sci. Rev., 1999, 90, 219–232 CrossRef CAS PubMed.
- T. Henning and F. Salama, Science, 1998, 282, 2204–2210 CrossRef CAS PubMed.
- K. Sellgren, Spectrochim. Acta, Part A, 2001, 57, 627–642 CrossRef CAS.
- M. S. Gudipati, J. Phys. Chem. A, 2004, 108, 4412–4419 CrossRef CAS.
- M. P. Bernstein, S. A. Sandford, A. L. Mattioda and L. J. Allamandola, Astrophys. J., 2007, 664, 1264–1272 CrossRef CAS.
- M. P. Bernstein, S. A. Sandford, L. J. Allamandola, J. S. Gillette, S. J. Clemett and R. N. Zare, Science, 1999, 283, 1135–1138 CrossRef CAS PubMed.
- S. G. Murthy and J. A. Louis, Astrophys. J., Lett., 2003, 596, L195–L198 CrossRef.
- A. M. Cook, A. Ricca, A. L. Mattioda, J. Bouwman, J. Roser, H. Linnartz, J. Bregman and L. J. Allamandola, Astrophys. J., 2015, 799, 14 CrossRef.
- J. Krausko, J. K'Ekuboni Malongwe, G. Bicanova, P. Klan, D. Nachtigallova and D. Heger, J. Phys. Chem. A, 2015, 119, 8565–8578 CrossRef CAS PubMed.
- A. Simon, J. A. Noble, G. Rouaut, A. Moudens, C. Aupetit, C. Iftner and J. Mascetti, Phys. Chem. Chem. Phys., 2017, 19, 8516–8529 RSC.
- J. A. Noble, C. Jouvet, C. Aupetit, A. Moudens and J. Mascetti, Astron. Astrophys., 2017, 599, A124 CrossRef.
- A. L. F. de Barros, A. L. Mattioda, A. Ricca, G. A. Cruz-Diaz and L. J. Allamandola, Astrophys. J., 2017, 848, 112 CrossRef PubMed.
- P. Ball, Chem. Rev., 2008, 108, 74–108 CrossRef CAS PubMed.
- G. A. Jeffrey and W. Saenger, Hydrogen Bonding in Biological Systems, Springer, Heidelberg, 1991 Search PubMed.
- D. P. Zhong, S. K. Pal and A. H. Zewail, Chem. Phys. Lett., 2011, 503, 1–11 CrossRef CAS.
- P. Hobza and R. Zahradnik, Intermolecular Complexes: The Role of van der Waals Systems in Physical Chemistry and in the Biodisciplines, Elsevier, Amsterdam, 1988 Search PubMed.
- P. Hobza and Z. Havlas, Chem. Rev., 2000, 100, 4253–4264 CrossRef CAS PubMed.
- D. A. Dougherty, Science, 1996, 271, 163–168 CAS.
- L. J. Allamandola, A. G. G. M. Tielens and J. R. Barker, Astrophys. J., 1985, 290, L25–L28 CrossRef CAS.
- A. Leger and L. d'Hendecourt, Astron. Astrophys., 1985, 146, 81–85 CAS.
- A. Leger and J. L. Puget, Astron. Astrophys., 1984, 137, L5–L8 CAS.
- G. H. Herbig, Annu. Rev. Astron. Astrophys., 1995, 33, 19–73 CrossRef CAS.
- A. G. G. M. Tielens, Annu. Rev. Astron. Astrophys., 2008, 46, 289–337 CrossRef CAS.
- T. Snow, L. V. Page, Y. Keheyan and V. M. Bierbaum, Nature, 1998, 391, 259–260 CrossRef CAS PubMed.
- F. Salama and L. J. Allamandola, Astrophys. J., 1992, 395, 301–306 CrossRef CAS.
- S. Iglesias-Groth, A. Manchado, D. A. García-Hernández, J. I. G. Hernández and D. L. Lambert, Astrophys. J., Lett., 2008, 685, L55–L58 CrossRef CAS.
- T. P. Snow, Astrophys. J., 1992, 401, 775–777 CrossRef CAS.
- F. Salama and L. J. Allamandola, J. Chem. Phys., 1991, 94, 6964–6977 CrossRef CAS.
- D. M. Hudgins, S. A. Sandford and L. J. Allamandola, J. Phys. Chem., 1994, 98, 4243–4253 CrossRef CAS PubMed.
- D. Romanini, L. Biennier, F. Salama, A. Kachanov, L. J. Allamandola and F. Stoeckel, Chem. Phys. Lett., 1999, 303, 165–170 CrossRef CAS PubMed.
- L. Biennier, F. Salama, L. J. Allamandola and J. J. Scherer, J. Chem. Phys., 2003, 118, 7863–7872 CrossRef CAS.
- L. Biennier, F. Salama, M. Gupta and A. O'Keefe, Chem. Phys. Lett., 2004, 387, 287–294 CrossRef CAS.
- O. Sukhorukov, A. Staicu, E. Diegel, G. Rouillé, T. Henning and F. Huisken, Chem. Phys. Lett., 2004, 386, 259–264 CrossRef CAS.
- H. Piest, G. von Helden and G. Meijer, Astrophys. J., Lett., 1999, 520, L75–L78 CrossRef CAS.
- T. Pino, N. Boudin and P. Brechignac, J. Chem. Phys., 1999, 111, 7337–7347 CrossRef CAS.
- J. Oomens, A. J. A. van Roij, G. Meijer and G. von Helden, Astrophys. J., 2000, 542, 404–410 CrossRef CAS.
- M. C. R. Cockett, H. Ozeki, K. Okuyama and K. Kimura, J. Chem. Phys., 1993, 98, 7763–7772 CrossRef CAS.
- U. J. Lorenz, N. Solca, J. Lemaire, P. Maitre and O. Dopfer, Angew. Chem., Int. Ed., 2007, 46, 6714–6716 CrossRef CAS PubMed.
- H. Knorke, J. Langer, J. Oomens and O. Dopfer, Astrophys. J., Lett., 2009, 706, L66–L70 CrossRef CAS.
- A. M. Ricks, G. E. Douberly and M. A. Duncan, Astrophys. J., 2009, 702, 301–306 CrossRef CAS.
- I. Alata, R. Omidyan, M. Broquier, C. Dedonder, O. Dopfer and C. Jouvet, Phys. Chem. Chem. Phys., 2010, 12, 14456–14458 RSC.
- I. Alata, M. Broquier, C. Dedonder-Lardeux, C. Jouvet, M. Kim, W. Y. Sohn, S. Kim, H. Kang, M. Schütz, A. Patzer and O. Dopfer, J. Chem. Phys., 2011, 134, 074307 CrossRef PubMed.
- A. Patzer, M. Schütz, C. Jouvet and O. Dopfer, J. Phys. Chem. A, 2013, 117, 9785–9793 CrossRef CAS PubMed.
- O. Krechkivska, Y. Liu, K. L. K. Lee, K. Nauta, S. H. Kable and T. W. Schmidt, J. Phys. Chem. Lett., 2013, 4, 3728–3732 CrossRef CAS.
- M. P. Bernstein, J. P. Dworkin, S. A. Sandford and L. J. Allamandola, Meteorit. Planet. Sci., 2001, 36, 351–358 CrossRef CAS.
- M. P. Bernstein, S. A. Sandford and L. J. Allamandola, Astrophys. J., Lett., 1996, 472, L127–L130 CrossRef CAS PubMed.
- N. Solcà and O. Dopfer, Chem. Phys. Lett., 2001, 347, 59–64 CrossRef.
- N. Solcà and O. Dopfer, J. Phys. Chem. A, 2003, 107, 4046–4055 CrossRef.
- M. Schmies, M. Miyazaki, M. Fujii and O. Dopfer, J. Chem. Phys., 2014, 141, 214301 CrossRef PubMed.
- J. Klyne, M. Schmies, M. Fujii and O. Dopfer, J. Phys. Chem. B, 2015, 119, 1388–1406 CrossRef CAS PubMed.
- J. Klyne, M. Schmies, M. Miyazaki, M. Fujii and O. Dopfer, Phys. Chem. Chem. Phys., 2018 10.1039/c1037cp04659f , in press.
- H. S. Andrei, N. Solca and O. Dopfer, J. Phys. Chem. A, 2005, 109, 3598–3607 CrossRef CAS PubMed.
- M. Schütz, Y. Matsumoto, A. Bouchet, M. Öztürk and O. Dopfer, Phys. Chem. Chem. Phys., 2017, 19, 3970–3986 RSC.
- A. Bouchet, M. Schütz and O. Dopfer, ChemPhysChem, 2016, 17, 232–243 CrossRef CAS PubMed.
- O. Dopfer, A. Patzer, S. Chakraborty, I. Alata, R. Omidyan, M. Broquier, C. Dedonder and C. Jouvet, J. Chem. Phys., 2014, 140, 124314 CrossRef PubMed.
- M. Schütz, K. Sakota, R. Moritz, M. Schmies, T. Ikeda, H. Sekiya and O. Dopfer, J. Phys. Chem. A, 2015, 119, 10035–10051 CrossRef PubMed.
- J. Klyne, M. Miyazaki, M. Fujii and O. Dopfer, Phys. Chem. Chem. Phys., 2018 10.1039/c1037cp06132c , in press.
- K. Chatterjee and O. Dopfer, Phys. Chem. Chem. Phys., 2017, 19, 32262–32271 RSC.
- O. Dopfer, Z. Phys. Chem., 2005, 219, 125–168 CrossRef CAS.
- N. Solcà and O. Dopfer, J. Phys. Chem. A, 2001, 105, 5637–5645 CrossRef.
- E. M. Cabaleiro-Lago, J. Rodríguez-Otero and Á. Peña-Gallego, J. Phys. Chem. A, 2008, 112, 6344–6350 CrossRef CAS PubMed.
- J. Schiedt, W. J. Knott, K. Le Barbu, E. W. Schlag and R. Weinkauf, J. Chem. Phys., 2000, 113, 9470–9478 CrossRef CAS.
- S. A. Lyapustina, S. Xu, J. N. Nilles and K. H. Bowen Jr, J. Chem. Phys., 2000, 112, 6643–6648 CrossRef CAS.
- S. H. Lee, J. H. Kim, I. Chu and J. K. Song, Phys. Chem. Chem. Phys., 2009, 11, 9468–9473 RSC.
- B. J. Knurr, C. L. Adams and J. M. Weber, J. Chem. Phys., 2012, 137, 104303 CrossRef PubMed.
- M. Miyazaki, A. Fujii, T. Ebata and N. Mikami, Chem. Phys. Lett., 2001, 349, 431–436 CrossRef CAS.
- M. Miyazaki, A. Fujii, T. Ebata and N. Mikami, Phys. Chem. Chem. Phys., 2003, 5, 1137–1148 RSC.
- M. Miyazaki, A. Fujii, T. Ebata and N. Mikami, J. Phys. Chem. A, 2004, 108, 10656–10660 CrossRef CAS.
- M. Miyazaki, A. Fujii and N. Mikami, J. Phys. Chem. A, 2004, 108, 8269–8272 CrossRef CAS.
- M. Miyazaki, A. Fujii, T. Ebata and N. Mikami, Chem. Phys. Lett., 2004, 399, 412–416 CrossRef CAS.
- B.-M. Cheng, J. R. Grover and E. A. Walters, Chem. Phys. Lett., 1995, 232, 364–369 CrossRef CAS.
- Y. Ibrahim, E. Alsharaeh, M. Rusyniak, S. Watson, M. M. N. Mautner and M. S. El-Shall, Chem. Phys. Lett., 2003, 380, 21–28 CrossRef CAS.
- H. Tachikawa and M. Igarashi, J. Phys. Chem. A, 1998, 102, 8648–8656 CrossRef CAS.
- M. Shimizu, E. Yamashita, M. Mitani and Y. Yoshioka, Chem. Phys. Lett., 2006, 432, 22–26 CrossRef CAS.
- O. Dopfer, Int. Rev. Phys. Chem., 2003, 22, 437–495 CrossRef CAS.
- M. J. Frisch, et al., GAUSSIAN09, D.01, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- J. Klyne and O. Dopfer, J. Mol. Spectrosc., 2017, 337, 124–136 CrossRef CAS.
- E. D. Glendening, J. K. Badenhoop, A. E. Reed, J. E. Carpenter, J. A. Bohmann, C. M. Morales, C. R. Landis and F. Weinhold, NBO 6.0, Theoretical Chemistry, University of Wisconsin, Madison, 2013 Search PubMed.
- A. E. Reed, L. A. Curtiss and F. Weinhold, Chem. Rev., 1988, 88, 899–926 CrossRef CAS.
- E. R. Johnson, S. Keinan, P. Mori-Sanchez, J. Contreras-Garcia, A. J. Cohen and W. T. Yang, J. Am. Chem. Soc., 2010, 132, 6498–6506 CrossRef CAS PubMed.
- J. Contreras-Garcia, E. R. Johnson, S. Keinan, R. Chaudret, J. P. Piquemal, D. N. Beratan and W. T. Yang, J. Chem. Theor. Comput., 2011, 7, 625–632 CrossRef CAS PubMed.
- A. G. Csaszar, G. Czako, T. Furtenbacher, J. Tennyson, V. Szalay, S. V. Shirin, N. F. Zobov and O. L. Polyansky, J. Chem. Phys., 2005, 122, 214305 CrossRef PubMed.
- G. Herzberg, Molecular Spectra and Molecular Structure. II. Infrared and Raman Spectra of Polyatomic Molecules, Krieger Publishing Company, Malabar, Florida, 1991 Search PubMed.
- S. N. Ketkar and M. Fink, J. Mol. Struct., 1981, 77, 139–147 CrossRef CAS.
- V. I. Ponomarev, O. S. Filipenko and L. O. Atovmyan, Kristallografiya, 1976, 21, 392–394 CAS.
- K. B. Hewett, M. H. Shen, C. L. Brummel and L. A. Philips, J. Chem. Phys., 1994, 100, 4077–4086 CrossRef CAS.
- J. Szczepanski, D. Roser, W. Personette, M. Eyring, R. Pellow and M. Vala, J. Phys. Chem., 1992, 96, 7876–7881 CrossRef CAS.
- A. W. Potts and W. C. Price, Proc. R. Soc. London, Ser. A, 1972, 326, 181–197 CrossRef.
- P. J. Linstrom and W. G. Mallard, NIST Chemistry WebBook, NIST Standards and Technology, Gaithersburg MD, 20899, 2017, http://webbook.nist.gov Search PubMed.
- D. J. Goebbert and P. G. Wenthold, Eur. J. Mass Spectrom., 2004, 10, 837–845 CrossRef CAS PubMed.
- B. E. Rocher-Casterline, L. C. Ch'ng, A. K. Mollner and H. Reisler, J. Chem. Phys., 2011, 134, 211101 CrossRef PubMed.
- L. C. Ch'ng, A. K. Samanta, Y. Wang, J. M. Bowman and H. Reisler, J. Phys. Chem. A, 2013, 117, 7207–7216 CrossRef PubMed.
- F. Huisken, M. Kaloudis and A. Kulcke, J. Chem. Phys., 1996, 104, 17–25 CrossRef CAS.
- J. M. Voss, B. M. Marsh, J. Zhou and E. Garand, Phys. Chem. Chem. Phys., 2016, 18, 18905–18913 RSC.
- T. Vondrák, S. Sato and K. Kimura, Chem. Phys. Lett., 1996, 261, 481–485 CrossRef.
- S. Tomoda and K. Kimura, Chem. Phys. Lett., 1983, 102, 560–564 CrossRef CAS.
- L. Belau, K. R. Wilson, S. R. Leone and M. Ahmed, J. Phys. Chem. A, 2007, 111, 10075–10083 CrossRef CAS PubMed.
- R. Knochenmuss, O. Cheshnovsky and S. Leutwyler, Chem. Phys. Lett., 1988, 144, 317–323 CrossRef CAS.
- S. Suzuki, P. G. Green, R. E. Bumgarner, S. Dasgupta, W. A. Goddard III and G. A. Blake, Science, 1992, 257, 942–945 CAS.
- O. Dopfer and M. Fujii, Chem. Rev., 2016, 116, 5432–5463 CrossRef CAS PubMed.
- M. Fujii and O. Dopfer, Int. Rev. Phys. Chem., 2012, 31, 131–173 CrossRef CAS.
- K. Tanabe, M. Miyazaki, M. Schmies, A. Patzer, M. Schütz, H. Sekiya, M. Sakai, O. Dopfer and M. Fujii, Angew. Chem., Int. Ed., 2012, 51, 6604–6607 CrossRef PubMed.
- M. Wohlgemuth, M. Miyazaki, M. Weiler, M. Sakai, O. Dopfer, M. Fujii and R. Mitric, Angew. Chem., Int. Ed., 2014, 53, 14601–14604 CrossRef CAS PubMed.
- M. Miyazaki, T. Nakamura, M. Wohlgemuth, R. Mitric, O. Dopfer and M. Fujii, Phys. Chem. Chem. Phys., 2015, 17, 29969–29977 RSC.
- M. Wohlgemuth, M. Miyazaki, K. Tsukada, M. Weiler, O. Dopfer, M. Fujii and R. Mitric, Phys. Chem. Chem. Phys., 2017, 19, 22564–22572 RSC.
- M. Miyazaki, A. Naito, T. Ikeda, J. Klyne, K. Sakota, H. Sekiya, O. Dopfer and M. Fujii, Phys. Chem. Chem. Phys., 2018 10.1039/c1037cp06127g , in press.
- H. S. Gutowski, T. Emilsson and E. Arunan, J. Chem. Phys., 1993, 99, 4883–4893 CrossRef.
- R. N. Pribble and T. S. Zwier, Science, 1994, 265, 75–79 CAS.
- T. S. Zwier, Annu. Rev. Phys. Chem., 1996, 47, 205–241 CrossRef CAS.
- D. Feller, J. Phys. Chem. A, 1999, 103, 7558–7561 CrossRef CAS.
- B. Brutschy, Chem. Rev., 2000, 100, 3891–3920 CrossRef CAS PubMed.
- K. S. Kim, P. Tarakeshwar and J. Y. Lee, Chem. Rev., 2000, 100, 4145–4185 CrossRef CAS PubMed.
- Z. S. Huang and R. E. Miller, J. Chem. Phys., 1989, 91, 6613–6631 CrossRef CAS.
- K. Kuyanov-Prozument, M. Y. Choi and A. F. Vilesov, J. Chem. Phys., 2010, 132, 014304 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed NBO and NCI analyses, structures and IR spectra of less stable Np+-Wn isomers and Wn clusters, Cartesian coordinates of all optimized structures. See DOI: 10.1039/c7sc05124g |
| This journal is © The Royal Society of Chemistry 2018 |