
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Copper-catalyzed synthesis of allenylboronic acids. Access to sterically encumbered homopropargylic alcohols and amines by propargylboration†
Jian
Zhao‡
a,
Sybrand J. T.
Jonker‡
 a,
Denise N.
Meyer
a,
Göran
Schulz
a,
C. Duc
Tran
a,
Lars
Eriksson
b and
Kálmán J.
Szabó
*a
a,
Denise N.
Meyer
a,
Göran
Schulz
a,
C. Duc
Tran
a,
Lars
Eriksson
b and
Kálmán J.
Szabó
*a
aDepartment of Organic Chemistry, Stockholm University, SE-106 91 Stockholm, Sweden. E-mail: kalman.j.szabo@su.se
bDepartment of Materials and Environmental Chemistry, Stockholm University, SE-106 91 Stockholm, Sweden
First published on 19th February 2018
Abstract
Tri- and tetrasubstituted allenylboronic acids were prepared via a new versatile copper-catalyzed methodology. The densely functionalized allenylboronic acids readily undergo propargylboration reactions with ketones and imines without any additives. Catalytic asymmetric propargylborylation of ketones is demonstrated with high stereoselectivity allowing for the synthesis of highly enantioenriched tertiary homopropargyl alcohols. The reaction is suitable for kinetic resolution of racemic allenylboronic acids affording alkynes with adjacent quaternary stereocenters.
Introduction
Allylation and propargylation of carbonyl compounds and imines is an attractive method for the highly selective synthesis of homoallylic and propargylic alcohols and amines.1 Development of new methodologies for synthesis of enantioenriched compounds with allyl and propargyl groups is particularly important, as these motifs often occur in natural products.2 Accordingly, a number of excellent methods have been reported for asymmetric propargylation.1c,3 The most recent trends in this field involve synthesis of sterically encumbered propargylic alcohols and imines occurring in natural products.2a Asymmetric catalysis for construction of acyclic small-molecules with adjacent quaternary carbon centers is one of the most challenging synthetic transformations.4 This structural motif is abundant in terpenoid natural products, such as in tertiary prenyl derivatives.2b,c Formation of a carbon–carbon single bond between sterically encumbered quaternary carbons is a particularly difficult synthetic problem. The strong steric repulsion between the bulky substituents (none of them is a hydrogen) leads to an elongated and very weak carbon–carbon σ-bond, which is difficult to create and easy to cleave.5Stereoselective propargylboration with allenylboron species became one of the most important transformations for creation of sterically crowded propargylic alcohols and imines.6 One of the important synthetic strategies for the formation of a tertiary stereocenter is based on the reaction of allenyl or propargylic boron reagents with ketones in the presence of a chiral catalyst (Fig. 1a). Schaus,6b Shibasaki6a and Fandrick6v reported these types of propargylboration reactions using mono- or disubstituted allenyl- and propargylboronates. Another very efficient method involves synthesis of secondary and tertiary propargylic alcohols without application of organometallic reagents using asymmetric metal catalysis (Fig. 1b).7 Krische reported an efficient method for tert-prenylation for terpenoid construction (Fig. 1c).6w By this method a propargylic all-carbon quaternary center could be created adjacent to a secondary alcohol. However, as far as we know, formation of vicinal quaternary carbon centers has never been reported for asymmetric propargylation reactions. In this paper we report our results for achievement of this goal via asymmetric propargylboration of ketones with tetrasubstituted allenylboronic acids (Fig. 1d).
 | ||
| Fig. 1 Asymmetric catalysis toward sterically encumbered homopropargylic alcohols. | ||
Results and discussion
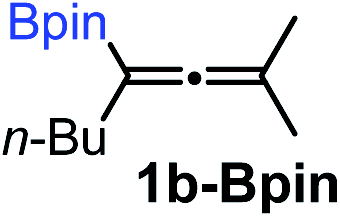
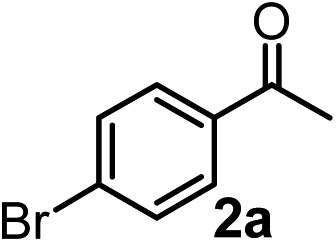
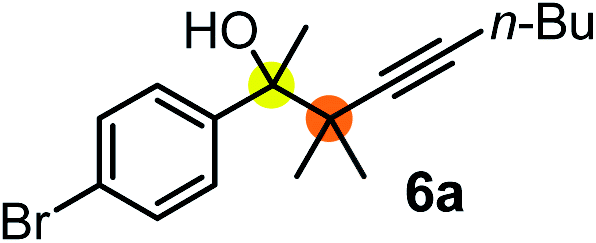
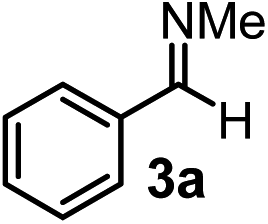
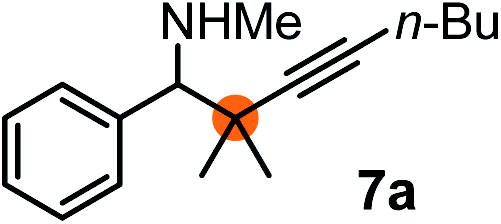
Relatively few methods are available for the synthesis of allenylboronates and their analogs. The parent (unsubstituted) allenylboronate and a few alkyl-substituted derivatives can be prepared via Grignard (or other allenyl- or propargyl-metal mediated) reactions.6e,p,q,8 More recently, Cu- and Pd-catalyzed methods have been reported by the group of Ito/Sawamura and subsequently our and other groups for the synthesis of densely (tri- and tetra-) substituted allenyl-Bpin (Bpin = pinacolborane) derivatives.9However, densely functionalized, easily accessible tetrasubstituted allenyl-Bpin compounds have a relatively low reactivity profile. These compounds, such as 1a-Bpin, react directly (without additives) with aldehydes6k,9a,b but according to our studies they are completely unreactive (Fig. 2) with ketones (2a) and imines (3a) at ambient temperature. Thus, homopropargyl alcohols and amines with adjacent quaternary carbons cannot be accessed using allenyl-Bpin reagents under mild conditions, which is required for a highly selective carbon–carbon bond formation.
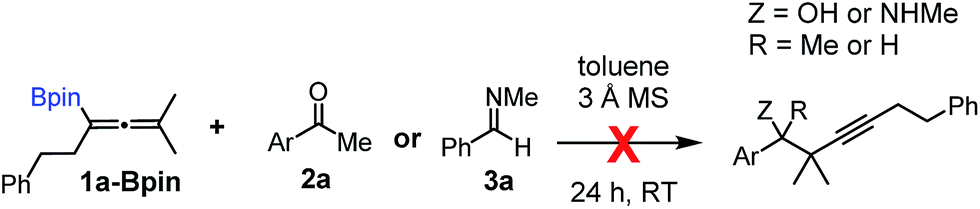 | ||
| Fig. 2 Pinacol protected allenylboronic acids are reluctant to react with ketones and imines. | ||
The structural analogy between allyl-10 and allenylboron species suggests11 that allenylboronic acids are expected to be much more reactive than allenyl-Bpin (or other diol protected boron) reagents utilized to access crowded homopropargyl alcohols and amines. However, the lack of efficient methodologies for the preparation of densely functionalized allenylboronic acids is a fundamental problem for the implementation of this concept. Therefore, we undertook development of the first transition metal catalyzed synthesis of tri- and tetra-substituted allenylboronic acids, and subsequently we have exploited the synthetically useful properties of the unprotected B(OH)2 group for synthesis of enantiomerically enriched propargylic alcohols with vicinal quaternary carbons.
Synthesis of allenylboronic acids
Copper-catalyzed transformation of propargylic carbonates under basic conditions was used for the borylation process.9a–c Similar to the catalytic synthesis of allylboronic acids, we employed diboronic acid12 (5a) as the B(OH)2 source (Table 1).10a Application of the acid form 5a instead of B2pin2 (ref. 9a–c) (5b) under basic reaction conditions for synthesis of 1 required three important synthetic innovations: (1) the catalyst (CuOMe) was generated, in situ from mesitylcopper(I) (MesCu) and MeOH; (2) ethylene glycol was used for in situ protection of the boronic acid functionality in the product;13 and (3) we used weakly coordinating and easily removable P(OMe)3 as the ligand.|
|
||
|---|---|---|
| Entry | Conditions | Yieldb [%] |
| a General procedure: 4a (0.10 mmol), 5 (0.15 mmol), mesitylcopper(I) (0.01 mmol), P(OMe)3 (0.02 mmol), ethylene glycol (0.30 mmol), and 3 Å MS were stirred in MeOH (1 mL) at −10 °C for 24 h. b 1H NMR-yields. c 10 mol%. d 20 mol%. e Protodeborylation occurs. | ||
| 1 | No change | 76 |
| 2 | CuClc and KOMed instead of MesCu | 52 |
| 3 | CuClc and NaOMed instead of MesCu | 49 |
| 4 | CuClc and LiOMed instead of MesCu | 65 |
| 5 | Culc and LiOMed instead of MesCu | 29 |
| 6 | PPh3 instead of P(OMe)3 | 46 |
| 7 | PCy3 or P(O-iPr)3 instead of P(OMe)3 | 0e |
| 8 | 1,3-Propanediol instead of ethylene glycol | 49 |
| 9 | Without ethylene glycol | 66 |
| 10 | Without 3 Å MS | 75 |
| 11 | At 0 °C | 16e |
| 12 | THF or toluene instead of MeOH | 0 |
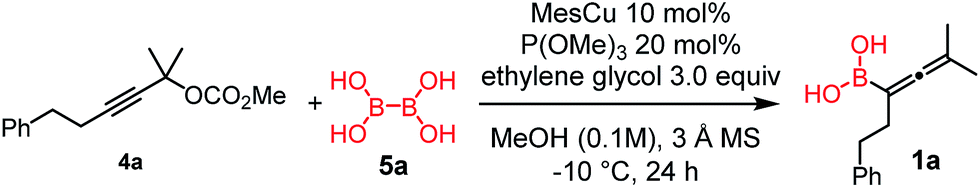
Using the optimized conditions, allenylboronic acid 1a could be obtained with 76% NMR yield (Table 1, entry 1). When the Cu-catalyst was generated from CuCl and alkali methoxides instead of MesCu/MeOH, the yields decreased to 49–65% (entries 2–4). Using CuI instead of CuCl led to a sharp decrease in the yield to 29% (entry 5). Phosphite P(OMe)3 could be replaced by PPh3, albeit in a diminished yield of 46% (entry 6). Application of bulky phosphorus based ligands, such as PCy3 and P(O-iPr)3, led to 0% yield of 1a (entry 7). In these reactions, large amounts of protodeborylated allene formed, which indicates that 1a is probably generated but likely undergoes a Cu-catalyzed protodeborylation. In the presence of ethylene glycol (cf. entries 9 and 1), the yield increased and only traces of protodeborylation products were observed. We found that in this in situ protection step, ethylene glycol is more efficient than its homolog, 1,3-propanediol (cf. entries 1 with 8–9). The addition of molecular sieves had a relatively weak effect on the yield (entry 10). When the reaction was conducted at 0 °C instead of −10 °C a large amount of protodeborylated product formed and the yield dropped substantially. Changing the solvent from methanol to toluene or THF prevented the formation of 1a (entry 12). Allenylboronic acid 1a is oxygen sensitive and resistant to crystallization (similar to analogs 1b–j). Therefore, the purification was done by quenching the reaction mixture with 0.5 M HCl solution (to remove the glycol protecting group) and subsequent toluene extraction of the allenylboronic acid product. The resultant toluene solution of 1a (and 1b–j) can be stored under Ar (at −18 °C for several weeks) and used for all synthetic applications presented below (Tables 3–5). Compound 1a can easily be converted to allenylboronates by adding the corresponding alcohols. For example 1a and pinacol readily give 1a-Bpin (see ESI† page 6). Reaction of 1a with diethanolamine leads to 1a-ean (Fig. 3). Easy formation of 1a-ean can be exploited for further purification of 1a by implementation of a methodology reported by Santos and co-workers (Fig. 3).14
 | ||
| Fig. 3 (a) and (b) Purification of allenylboronic acids 1a and 1b. (c) Attempted transformation of the pinacolate analogue 1a-Bpin with diethanolamine. | ||
This method is based on reaction of the (extracted) toluene solution of 1a with diethanolamine (Fig. 3a). The esterification of the B(OH)2 group was very fast12d and the diethanolamine ester of 1a (1a-ean) is precipitated from toluene. Allenylboronate 1a-ean was an air- and moisture stable solid, which could be stored for several months. We attempted purification of 1a-ean by silica gel chromatography but this purification method led to decomposition of 1a-ean. Thus, after washing of 1a-ean with ether, degassed toluene and 0.5 M HCl solution were added and the pure 1a was extracted to the toluene phase. The purification process includes a slight loss of 1a (yield 73%).
The 1H-NMR spectrum (in toluene-d8) of purified 1a (Fig. 4) clearly shows a peak (“d”) at 4.27 ppm, which belongs to the unprotected B(OH)2 group. From this spectrum it appears that the sample does not contain any glycol or other esters of the B(OH)2 group. The sample obtained by toluene extraction of the aqueous reaction mixture of the borylation is very similar (see the ESI†) to the purified one.
 | ||
| Fig. 4 NMR spectra of allenylboronic acid 1a with the characteristic B(OH)2 peak “d”. | ||
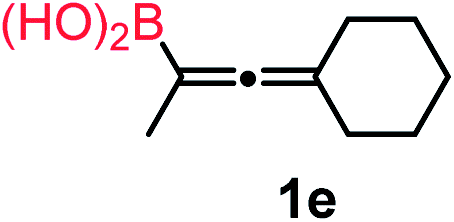
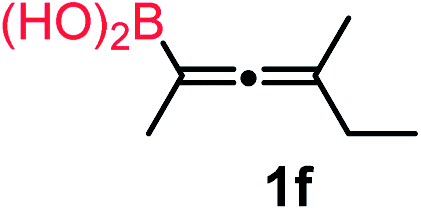
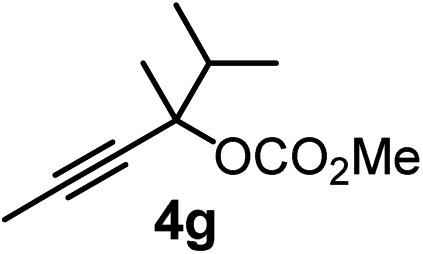
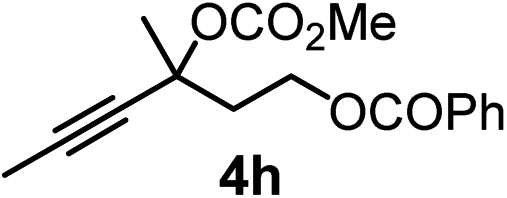
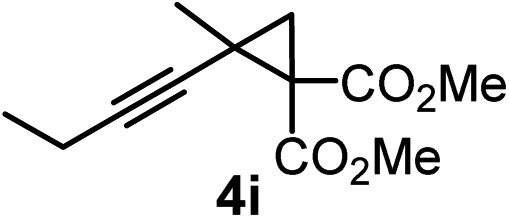
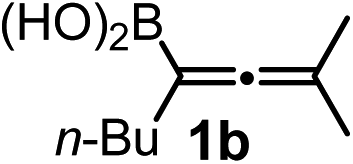
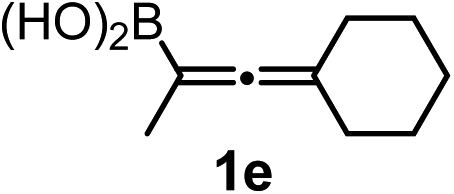
We have explored the synthetic scope of the above borylation reaction (Table 2) using various propargylic carbonates (4b–h, 4j) and 5a under the above (Table 1) optimized conditions. Compound 4b, a close analog of 4a reacted with excellent yield (94%) affording allenylboronic acid 1b (entry 1). The butyl substituent can be replaced with more (4c) or less (4d) sterically demanding groups to provide the corresponding products in 61–67% yield (entries 2–3). The substituent can be varied at the propargylic carbon as well. Compound 4e gives 1e with 59% yield and substrates with two different propargylic substituents (4f-g) also react readily (entries 5 and 6) providing racemic allenylboronic acids 1f-g. The borylation reaction also tolerates various types of substituents. In 4h only the propargylic carbonate is transformed, while the aliphatic carboxylate remains unchanged (entry 7). We succeeded in performing the borylation of propargyl cyclopropane 4i (entry 8). In this reaction, the cyclopropane ring opens instead of displacement of the carbonate leaving group. Even secondary propargylic carbonates (4j) could be borylated, albeit with a lower yield (34%) than their tertiary analogs (entry 9). The presented borylation method was scaled up thirty-fold with some drop of the yield to 62% (entry 1). Compound 1b could also be further purified by the Santos method (Fig. 3b), as 1b-ean precipitated as a solid. However, diethanolamine esters of the other allenylboronic acids (1c–j) were not precipitated from the toluene solution, and therefore these compounds cannot be purified further by this method.
| Entry | Substrate | Product | Yieldb [%] |
|---|---|---|---|
| a General procedure: 4a (0.10 mmol), 5 (0.15 mmol), mesitylcopper(I) (0.01 mmol), P(OMe)3 (0.02 mmol), ethylene glycol (0.30 mmol), and 3 Å MS were stirred in MeOH (1 mL) at −10 °C for 24 h. b 1H NMR-yield. c Yield at 3 mmol scale. | |||
| 1 |
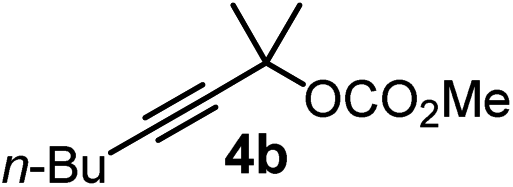
|

|
94 |
| 62c | |||
| 2 |

|
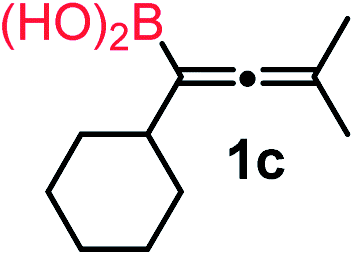
|
67 |
| 3 |
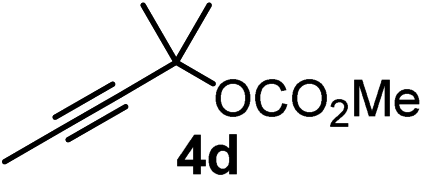
|

|
61 |
| 4 |
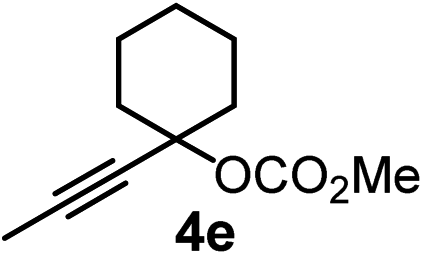
|
|
59 |
| 5 |
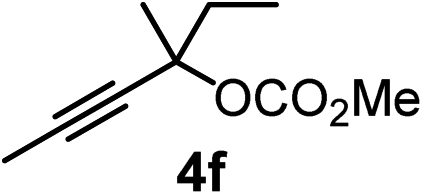
|
|
80 |
| 6 |
|

|
63 |
| 7 |
|

|
83 |
| 8 |
|

|
59 |
| 9 |
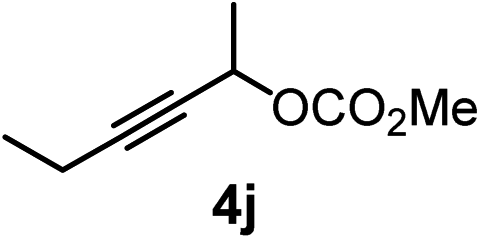
|

|
34 |
Extension of the borylation to synthesis of other allenyl boronates
To our delight, the above described procedure for synthesis of allenylboronic acids 1a–j was suitable for synthesis of other allenylboronates by replacement of the diboron reagent from diboronic acid 5a to other species, such as B2pin2 (5b), neopentyl ester 5c or chiral pinane ester 5d (Fig. 5). The first method (Fig. 5a) is a useful complement to the previously reported copper and palladium catalyzed methods for synthesis of allenyl-Bpin compounds,9a–c such as 1a-Bpin. Compound 1a-Bnep is less stable than 1a-Bpin, for example it decomposes under silica gel chromatography (Fig. 5b). However, it is air- and moisture stable, i.e. it is easier to handle than its unprotected counterpart 1a. Compound 1a-Bpne is a stable easily accessible chiral allenylboronate that can be purified by silica gel chromatography (Fig. 5c) and it can be stored in a refrigerator for several weeks. Similar to 1a-Bpin (Fig. 2) 1a-Bpne was unreactive toward ketones (such as 2a and 2c) at ambient temperature in 24 hours.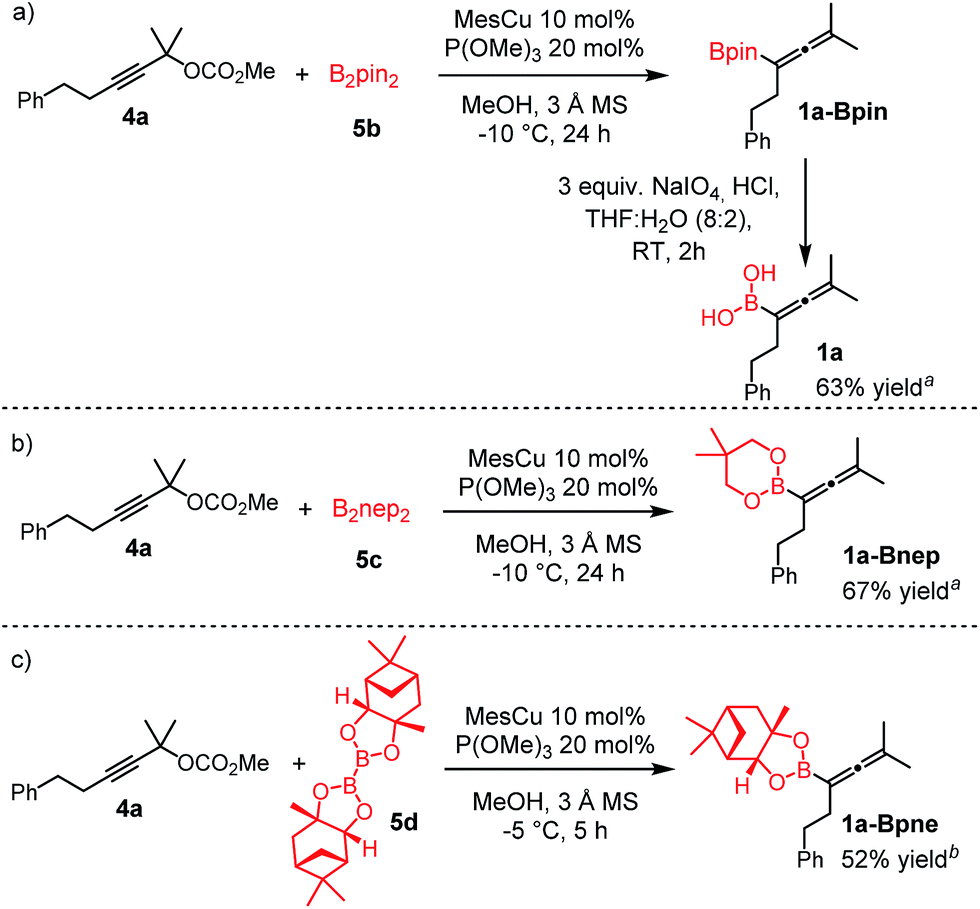 | ||
| Fig. 5 Extension of the methodology for synthesis of allenyl boronates. a1H NMR yield. bIsolated yield. | ||
Attempts for preparation of allenylboronic acids from allenyl boronates
Of course, the most efficient route to unprotected allenylboronic acids is using the above synthesis with application of unprotected diboronic acid 5a (Tables 1 and 2). However, in order to extend the scope of the synthesis of allenylboronic acids, we studied the possibilities of hydrolysis of protected allenyl-Bpin compounds, such as 1a-Bpin. The Santos method14 can be used for deprotection of various alkyl-Bpin derivatives, even aryl-Bpin compounds affording the corresponding organoboronic acids. However, the reaction of 1a-Bpin with diethanolamine did not result in any formation of 1a-ean at 40 °C in 48 h (Fig. 3c), and thus this method cannot be used for accessing allenylboronic acids from pinacol esters.We tested another method based on oxidative hydrolysis of the pinacolborane functionality in the presence of NaIO4 reported by Falck and co-workers.15 This method was also used by Petasis and co-workers6u for oxidative hydrolysis of mono- and disubstituted allenyl-Bpin compounds. The oxidative hydrolysis of 1a-Bpin was successful. Using this method after the borylation procedure, we were able to obtain 1a with 63% overall yield (Fig. 5a). This yield was somewhat lower than the analog process using B2(OH)4 (5a) as the boron source (76%) but it is still viable for obtaining 1a. We note that the level of purity of 1a obtained by this multi-step procedure (Fig. 5a) is lower than by using 5a as the boronate source (Table 1, entry 1) because of the use of more chemicals (e.g. 3 equiv. NaIO4) and pinacol as the protecting group.
Propargylboration of ketones and imines with allenylboronic acids
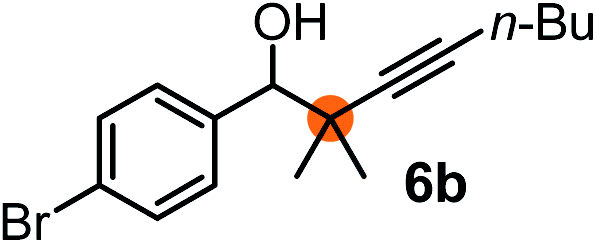
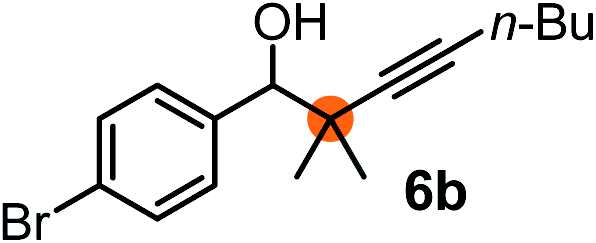
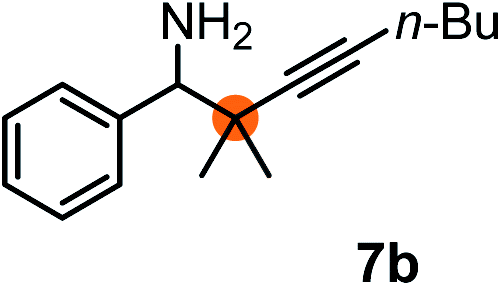
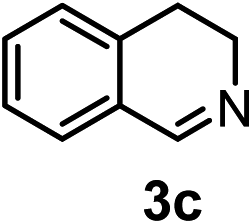
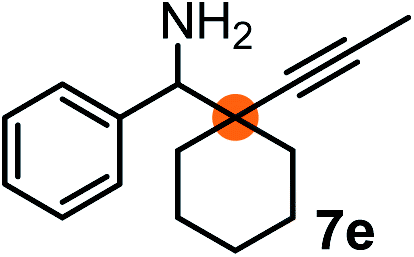
Subsequently, we have studied the reactivity of these allenylboronic acids (Table 3). Compound 1b reacted rapidly with aldehyde 2b in toluene in the presence of molecular sieves without any further additives. The reaction was complete in 10 minutes at room temperature affording 6b in 87% yield (entry 1). Under the same conditions (in 10 min) its Bpin analogue (1b-Bpin) did not provide any 6b (entry 2) demonstrating that under identical reaction conditions 1b is much more reactive than 1b-Bpin. As mentioned above (Fig. 2) a densely functionalized allenyl-Bpin compound 1a-Bpin did not react with ketones and imines in 24 h at room temperature. In contrast, ketones (such as 2a) and aldimines 3a-b reacted readily under these reaction conditions affording the corresponding homopropargylic alcohol (6a) and amine (7a-b) products in high yields (entries 3–5) confirming the superior reactivity of allenylboronic acids over their allenyl-Bpin counterparts. Dihydroisoquinoline 3c also reacted smoothly affording homopropargyl derivative 7c in 96% yield (entry 6). Even indole1h3d underwent homopropargylation with 1a at the C2 position affording 7d in 82% yield (entry 7). Finally, we were able to react 1e with imine 3b affording compound 7e. Remarkable features of the above reactions are the facile formation of one (entries 1, 4–8) or two (entry 3) quaternary centers connecting the newly formed C–C bond. The very high reactivity of an unprotected allenylboronic acid is the consequence of its ability to form the highly reactive boroxine (with the aid of molecular sieves), which is the anhydride of the boronic acid.16 Our DFT studies have shown that imines react with a much lower activation energy with allylboroxines than with allylboronic acids.10c,16 The reason is that the boroxines are much stronger Lewis acids than the corresponding boronic acids (or boronates).16





Asymmetric propargylboration towards congested homopropargyl alcohols
Schaus and Barnett6b studied the reaction of the glycol ester of the parent and disubstituted allenylboronates with ketones. These authors found that in the presence of BINOL catalysts under the influence of microwave heating, useful levels of enantioselectivity could be achieved. We have found that tetra-substituted allenylboronic acid 1b reacted readily with ketone 2a at room temperature without the need for microwave heating (Table 4, entry 1). In the presence of catalytic amounts of (S)-dibromo-BINOL 8a and EtOH, sterically encumbered homopropargylic alcohol 6a with vicinal quaternary carbons was obtained with remarkably high enantioselectivity (94% ee) and in excellent yield (95%). Interestingly, the yield was higher in this reaction than in the racemic reaction (Table 3, entry 3) indicating that the reaction was accelerated in the presence of (S)-dibromo-BINOL 8a. Application of an aliphatic alcohol, such as EtOH, in stoichiometric amount was essential to get a high enantioselectivity, in particular, when a catalytic amount of dibromo-BINOL 8a was applied (entries 2 and 3). This observation is also in agreement with our previous experience with asymmetric allylation of ketones10e and imines10f and with the related DFT modeling study.16 When a stoichiometric amount of 8a in the absence of EtOH was used, the ee dropped to 77% from 94% (Table 4, entry 2). When the amount of 8a was reduced to 15 mol% and the reaction was conducted without any aliphatic alcohols the reaction proceeds with poor ee of 44%. The beneficial effects of addition of an aliphatic alcohol to increase the selectivity of the asymmetric allyl- and allenylboration by organoboronic acids are now a well-understood feature of these reactions (see the section below on the mechanism). However, the efficiency of a certain aliphatic alcohol on the improvement of the enantioselectivity depends on both the organoboronic acid and the employed electrophile. For example, for asymmetric allylboration of ketones,10e addition of tBuOH had the most favourable effect on the enantioselectivity. However, when we replaced EtOH with tBuOH in the asymmetric propargylboration reaction the ee dropped substantially from 94% to 55% (entry 4). Notably, finding the most efficient aliphatic alcohol improving the enantioselectivity of the allyl- and propargylboration reactions is inherently easier for allyl- and allenylboronic acids than their organoboronate counterparts. In organoboronates the B(OH)2 group is usually protected with an aliphatic alcohol (such as pinacol, glycol, isopropanol etc.), which may have detrimental effects on the enantioselectivity of the allyl- or propargylboration process. An additionally useful synthetic feature of the above asymmetric propargylboration reaction is that using (R)-dibromo-BINOL (the enantiomer of 8a) as the catalyst the enantiomeric form of 6a can be obtained with high ee and yield (entry 5).
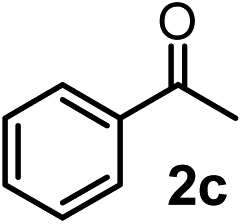
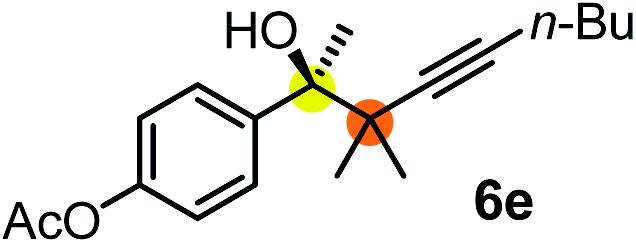
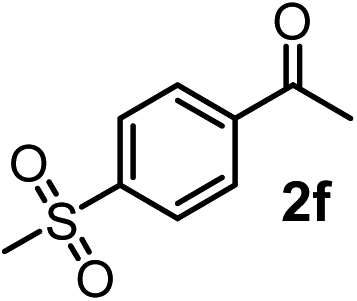
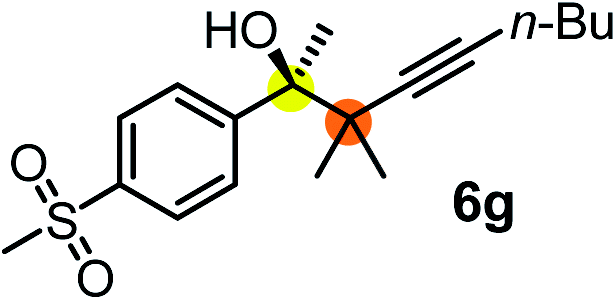
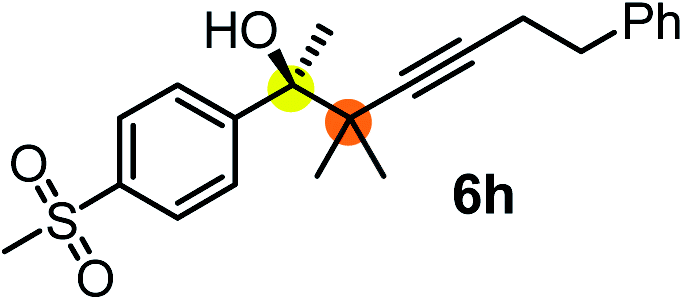
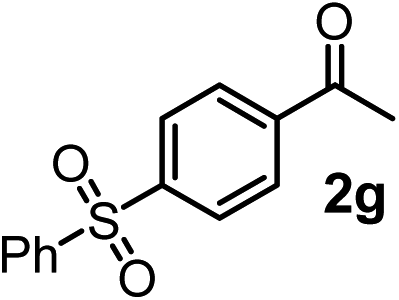
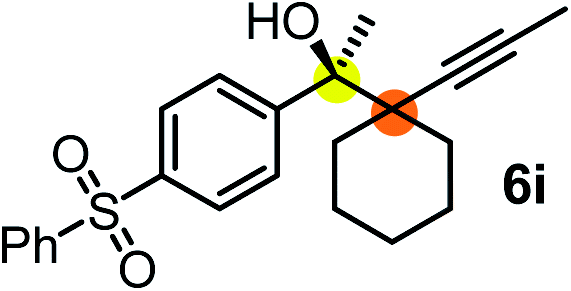
We have briefly studied the synthetic scope of the above described asymmetric propargylation reactions for synthesis of encumbered homopropargylic alcohols with vicinal quaternary centers including a reversed prenyl motif (Table 5). Using the above described achiral allenylboronic acids 1a–e and ketones 2c–g the expected homopropargylic alcohols formed with 90–99% ee and 62–90% yields. The studied reactions involved the parent acetophenone 2c (entry 1) and analogs with cyano 2d (entry 2), acetate 2e and bromo 2a (entry 4) functionalities (entry 3). We made several derivatives (6g–i) containing a sulfone group in order to obtain crystalline products for determination of the absolute configuration of the products (entries 5–7) via X-ray diffraction. Unfortunately, all these products (6g–i) were oils resistant to crystallization. Finally, we succeeded in obtaining crystals of the ester of 6g (ESI†), which were suitable for X-ray analysis. The absolute configuration of the stereogenic carbon in 6g-ester was R, and thus we assigned all products arising from the (S)-dibromo-BINOL 8a catalyzed reactions as the R-enantiomers. The reaction can be easily scaled up by five-fold without a significant change in yield or ee (entry 5).
| Entry | Substrates | Product | Yieldb [%] | ee | |
|---|---|---|---|---|---|
| a EtOH (0.2 mmol) and 8a were added to 1 (0.1 mmol) in toluene (0.2 M) with 3 Å MS, then 3 h later 2 (0.15 mmol) was added and this mixture was stirred for 48 h at RT. b Isolated yields. c Reaction time 72 h. d Reaction time 90 h. Conc. was 0.1 M. 20 mol% 8a. e 0.5 mmol scale, using 30 mol% 8a and the reaction time was 90 h. | |||||
| 1 | 1b |
|
|
75 | 97 |
| 2 | 1b |

|

|
67 | 91 |
| 3 | 1b |
|
|
90 | 96 |
| 4c | 1c | 2a |

|
77 | 90 |
| 5d | 1b |
|
|
62 (70e) | 94 (96e) |
| 6c | 1a | 2f |
|
63 | 96 |
| 7c | 1e |
|
|
64 | 99 |
Racemic allenylboronic acids are not supposed to react with high selectivity in conventional asymmetric catalysis. However, Schaus and co-workers6b have shown that disubstituted (racemic) allylboronates undergo kinetic resolution with benzophenone (2c) in the presence of 8a. We have also found that racemic 1g reacted with ketone 2a and a stoichiometric amount of 8a, affording 9 in very high enantio- (96% ee) and diastereoselectivity (Fig. 6). The reaction of 1g with 2a was slower than with the dimethyl substituted achiral allenylboronic acids most probably because the steric congestion was further increased by the presence of an isopropyl group. Therefore, the reaction temperature was increased to 45 °C, and 100 mol% of BINOL had to be employed for full conversion of ketone 2a. Interestingly, in the absence of BINOL 8a boronic acid 1g did not react with ketone 2a under the otherwise identical conditions. This is another indication that the reactivity of allenylboronic acid is increased in the presence of BINOL 8a (Fig. 6).
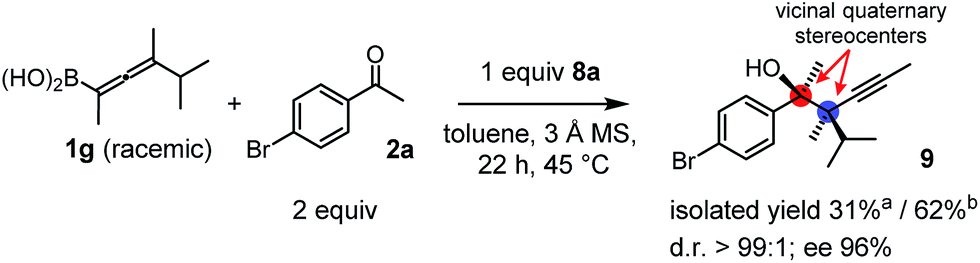 | ||
| Fig. 6 Kinetic resolution of 1g affording a single enantiomeric product with adjacent quaternary stereocenters. aYield is based on racemic 1g. bYield is based on the reactive enantiomer of 1g. | ||
As far as we know compound 9 is the first reported enantioenriched homopropargylic compound, which was synthesized by asymmetric catalysis forming adjacent quaternary stereogenic centers. Compound 9 was obtained as an oil, which resisted functionalization. The tertiary alcohol center was assigned on the basis of the X-ray structure of 6g-ester. The relative configuration was tentatively assigned as syn based on the similarities of the spectral data and optical rotation reported by Schaus for a related product.6b
Suggested mechanism
The suggested mechanism for the stereoselection, exemplified by the reaction of 1b with 2c in the presence of (S)-dibromo-BINOL 8a (Table 5, entry 1), is shown in Fig. 7. We assume that BINOL 8a esterifies the B(OH)2 group of 1b to give chiral allenylboronate 10a and the reaction proceeds via a Zimmerman-Traxler (Z.-T.) TS (Fig. 7).6d,e,k The assumption of the diesterification of 1b with 8a is based on our16 and others'17 DFT studies on asymmetric allylboration of ketones. These studies show that diesters of BINOL, such as 10a, react with a much lower activation barrier with ketones than mono-esters or esters of aliphatic alcohols. In the case of the Re-face arrangement the Me group of ketone 2c clashes with one of the bromo substituents of the BINOL moiety, therefore this TS is disfavoured. However, in the Si-face TS this steric repulsion does not occur, and therefore, formation of the R-product is favoured (Fig. 7).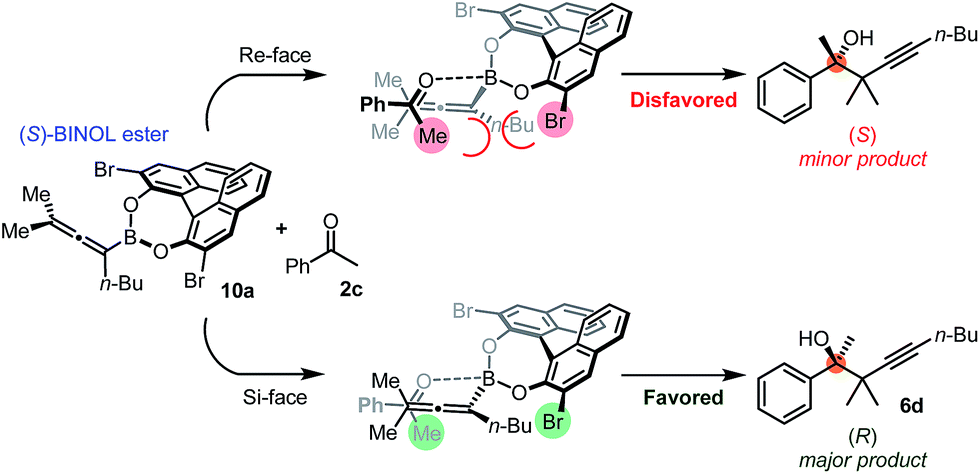 | ||
| Fig. 7 Plausible mechanism of the stereoselection exemplified with the propargylation of 2c. | ||
The proposed catalytic cycle for the propargylboration is given in Fig. 8. Accordingly, the enantioselective version of the reaction starts with mono- or diesterification of allenyl boronic acid 1b with EtOH affording 1b-OR. This esterification process is very fast and it can be observed by 1H NMR spectroscopy (see ESI† page 27). As mentioned above,16 boronic esters of aliphatic alcohols (such as EtOH) react much slower (if at all) than allylboronic acids/boroxines with ketones.10b,11 Thus 1b-OR does not react directly with ketone 2c, effectively shutting down the racemic background reaction (Table 3, entry 3). Therefore, when the reaction was performed without EtOH (Table 4, entry 3), the yield remained high but the ee dropped considerably indicating that considerable amount of racemic product was formed by the reaction of 1b or its boroxine and 2a. Compound 1b-OR may undergo transesterification with BINOL 8a forming a highly reactive chiral allenylboronate 11a. The transesterification of aliphatic esters (such as ethyl-ester) of 1b is probably much faster than for pinacol ester. The difficult transesterification of 1a-Bpin with diethanolamine is mentioned above (Fig. 3c). The very high reactivity of BINOL esterified allylboronic acids towards electrophiles was demonstrated by our recent DFT studies.16 The high reactivity of 11a is due to the presence of phenolic oxygen atoms, which conjugate less efficiently with the empty B(pπ) orbital than the oxygen atoms of aliphatic alcohols (e.g. EtOH). Therefore, the B(pπ) orbital of 11a will be an efficient electron acceptor for the O(nπ) orbital of ketone 2c in the cyclic TS (Fig. 7) of the reaction. The stereoselectivity of the reaction is determined in the 11a + 2c → 11b process (Fig. 7). Formation of 11b involves trapping of the BINOL catalyst. The added EtOH probably mediates decomposition of 11b, effectively releasing the BINOL catalyst 8a back into the catalytic cycle. Thus, the uncatalyzed racemic propargylation can be suppressed by the “dual action” of EtOH (i.e.1b → 1b-OR and 11b → 8a processes) increasing the ee of the reaction.
 | ||
| Fig. 8 Proposed mechanism for the asymmetric propargylation exemplified with the reaction of 1b with 2c. | ||
Conclusion
We have presented a new approach for the propargylboration of ketones and imines, which is suitable for synthesis of sterically encumbered homopropargylic alcohols and amines. The key reagent in this process is unprotected allenylboronic acid 1. In this study, we report the development of the first transition metal catalysed procedure for the synthesis of allenylboronic acids. This process is based on the application of diboronic acid as the commercially available B(OH)2 source and utilizes easily accessible propargylic carbonates. The diboron reagent scope of the reaction is also broad. The newly developed process is suitable for synthesis of allenylboronates as well, including chiral ones. The broad application of allenylboronic acids with various electrophiles is based on the high reactivity of the unprotected B(OH)2 group. The addition of molecular sieves leads to the formation of allenylboroxine, which is highly reactive towards carbonyls and imines. Therefore, tetra-substituted allenylboronic acids readily react with aldehydes, ketones, imines, and indoles at room temperature. Convenient in situ esterification of the B(OH)2 group with the diboromo-BINOL/EtOH system allows for the catalytic asymmetric propargylboration of ketones at room temperature affording sterically encumbered homopropargyl alcohols with neighbouring quaternary carbons. In addition, kinetic resolution of racemic allenylboronic acid could be performed in the presence of (S)-dibromo-BINOL (8a) catalyst. This reaction is suitable for creation of vicinal quaternary stereocenters with high enantio- and diastereoselectivity. We hope that similar to allylboronic acids,10g–j access to allenylboronic acids will also inspire new synthetic applications towards complex organic molecules and natural products.2Conflicts of interest
There are no conflicts to declare.Acknowledgements
Support from VR, the Wenner-Gren Foundation (fellowship for J. Z.), and ERASMUS stipends for G. S. and C. D. T. are gratefully acknowledged. Generous gifts of B2(OH)4 and bis[(+)-pinanediolato]diboron from Allychem are also appreciated.Notes and references
- (a) D. G. Hall, Boronic Acids, Wiley, Weinheim, 2011 Search PubMed; (b) C. Diner and K. J. Szabó, J. Am. Chem. Soc., 2017, 139, 2 CrossRef CAS PubMed; (c) C.-H. Ding and X.-L. Hou, Chem. Rev., 2011, 111, 1914 CrossRef CAS PubMed; (d) H. M. Wisniewska and E. R. Jarvo, J. Org. Chem., 2013, 78, 11629 CrossRef CAS PubMed; (e) J. Cid, H. Gulyas, J. J. Carbo and E. Fernández, Chem. Soc. Rev., 2012, 41, 3558 RSC; (f) D. Leonori and V. K. Aggarwal, Acc. Chem. Res., 2014, 47, 3174 CrossRef CAS PubMed; (g) M. Yus, J. C. González-Gómez and F. Foubelo, Chem. Rev., 2011, 111, 7774 CrossRef CAS PubMed; (h) F. Nowrouzi and R. A. Batey, Angew. Chem., Int. Ed., 2013, 52, 892 CrossRef CAS PubMed.
- (a) C. I. Keeling, J. Bohlmann and T. P. Begley, in Wiley Encyclopedia of Chemical Biology, John Wiley & Sons, Inc., 2007 Search PubMed; (b) S. E. O'Connor and J. J. Maresh, Nat. Prod. Rep., 2006, 23, 532 RSC; (c) A. J. Kochanowska-Karamyan and M. T. Hamann, Chem. Rev., 2010, 110, 4489 CrossRef CAS PubMed.
- (a) J. A. Marshall, Chem. Rev., 2000, 100, 3163 CrossRef CAS PubMed; (b) J. Feng, M. Holmes and M. J. Krische, Chem. Rev., 2017, 117, 12564 CrossRef CAS PubMed; (c) S. W. Kim, W. Zhang and M. J. Krische, Acc. Chem. Res., 2017, 50, 2371 CrossRef CAS PubMed.
- (a) I. Marek, Y. Minko, M. Pasco, T. Mejuch, N. Gilboa, H. Chechik and J. P. Das, J. Am. Chem. Soc., 2014, 136, 2682 CrossRef CAS PubMed; (b) K. W. Quasdorf and L. E. Overman, Nature, 2014, 516, 181 CrossRef CAS PubMed.
- C. G. Watson, A. Balanta, T. G. Elford, S. Essafi, J. N. Harvey and V. K. Aggarwal, J. Am. Chem. Soc., 2014, 136, 17370 CrossRef CAS PubMed.
- (a) S.-L. Shi, L.-W. Xu, K. Oisaki, M. Kanai and M. Shibasaki, J. Am. Chem. Soc., 2010, 132, 6638 CrossRef CAS PubMed; (b) D. S. Barnett and S. E. Schaus, Org. Lett., 2011, 13, 4020 CrossRef CAS PubMed; (c) B. L. Kohn, N. Ichiishi and E. R. Jarvo, Angew. Chem., Int. Ed., 2013, 52, 4414 CrossRef CAS PubMed; (d) M. Chen and W. R. Roush, J. Am. Chem. Soc., 2012, 134, 10947 CrossRef CAS PubMed; (e) A. S. Tsai, M. Chen and W. R. Roush, Org. Lett., 2013, 15, 1568 CrossRef CAS PubMed; (f) P. Jain, H. Wang, K. N. Houk and J. C. Antilla, Angew. Chem., Int. Ed., 2012, 51, 1391 CrossRef CAS PubMed; (g) L. R. Reddy, Org. Lett., 2012, 14, 1142 CrossRef CAS PubMed; (h) B. M. Partridge, L. Chausset-Boissarie, M. Burns, A. P. Pulis and V. K. Aggarwal, Angew. Chem., Int. Ed., 2012, 51, 11795 CrossRef CAS PubMed; (i) B. Jung and A. H. Hoveyda, J. Am. Chem. Soc., 2012, 134, 1490 CrossRef CAS PubMed; (j) E. Alicea-Matías and J. A. Soderquist, Org. Lett., 2017, 19, 336 CrossRef PubMed; (k) Y. Sasaki, M. Sawamura and H. Ito, Chem. Lett., 2011, 40, 1044 CrossRef CAS; (l) L. C. Hirayama, T. D. Haddad, A. G. Oliver and B. Singaram, J. Org. Chem., 2012, 77, 4342 CrossRef CAS PubMed; (m) E. M. Vieira, F. Haeffner, M. L. Snapper and A. H. Hoveyda, Angew. Chem., Int. Ed., 2012, 51, 6618 CrossRef CAS PubMed; (n) D. R. Fandrick, K. R. Fandrick, J. T. Reeves, Z. Tan, W. Tang, A. G. Capacci, S. Rodriguez, J. J. Song, H. Lee, N. K. Yee and C. H. Senanayake, J. Am. Chem. Soc., 2010, 132, 7600 CrossRef CAS PubMed; (o) Y. Yamashita, Y. Cui, P. Xie and S. Kobayashi, Org. Lett., 2015, 17, 6042 CrossRef CAS PubMed; (p) R. Haruta, M. Ishiguro, N. Ikeda and H. Yamamoto, J. Am. Chem. Soc., 1982, 104, 7667 CrossRef CAS; (q) E. J. Corey, C. M. Yu and D. H. Lee, J. Am. Chem. Soc., 1990, 112, 878 CrossRef CAS; (r) M. Chen and W. R. Roush, J. Am. Chem. Soc., 2013, 135, 9512 CrossRef CAS PubMed; (s) C. Allais and W. R. Roush, Org. Lett., 2017, 19, 2646 CrossRef CAS PubMed; (t) X.-D. Hu, C.-A. Fan, F.-M. Zhang and Y. Q. Tu, Angew. Chem., Int. Ed., 2004, 43, 1702 CrossRef CAS PubMed; (u) F. Liepouri, G. Bernasconi and N. A. Petasis, Org. Lett., 2015, 17, 1628 CrossRef CAS PubMed; (v) K. R. Fandrick, D. R. Fandrick, J. T. Reeves, J. Gao, S. Ma, W. Li, H. Lee, N. Grinberg, B. Lu and C. H. Senanayake, J. Am. Chem. Soc., 2011, 133, 10332 CrossRef CAS PubMed; (w) K. D. Nguyen, D. Herkommer and M. J. Krische, J. Am. Chem. Soc., 2016, 138, 5238 CrossRef CAS PubMed.
- (a) Y. Yang, I. B. Perry, G. Lu, P. Liu and S. L. Buchwald, Science, 2016, 353, 144 CrossRef CAS PubMed; (b) F. Meng, F. Haeffner and A. H. Hoveyda, J. Am. Chem. Soc., 2014, 136, 11304 CrossRef CAS PubMed; (c) T. Liang, S. K. Woo and M. J. Krische, Angew. Chem., Int. Ed., 2016, 55, 9207 CrossRef CAS PubMed; (d) K. C. Harper and M. S. Sigman, Science, 2011, 333, 1875 CrossRef CAS PubMed.
- H. C. Brown, U. R. Khire and U. S. Racherla, Tetrahedron Lett., 1993, 34, 15 CrossRef CAS.
- (a) H. Ito, Y. Sasaki and M. Sawamura, J. Am. Chem. Soc., 2008, 130, 15774 CrossRef CAS PubMed; (b) T. S. N. Zhao, Y. Yang, T. Lessing and K. J. Szabó, J. Am. Chem. Soc., 2014, 136, 7563 CrossRef CAS PubMed; (c) J. Zhao and K. J. Szabó, Angew. Chem., Int. Ed., 2016, 56, 1502 CrossRef PubMed; (d) L. Mao, K. J. Szabó and T. B. Marder, Org. Lett., 2017, 19, 1204 CrossRef CAS PubMed.
- (a) M. Raducan, R. Alam and K. J. Szabó, Angew. Chem., Int. Ed., 2012, 51, 13050 CrossRef CAS PubMed; (b) R. Alam, M. Raducan, L. Eriksson and K. J. Szabó, Org. Lett., 2013, 15, 2546 CrossRef CAS PubMed; (c) R. Alam, A. Das, G. Huang, L. Eriksson, F. Himo and K. J. Szabó, Chem. Sci., 2014, 5, 2732 RSC; (d) A. Das, R. Alam, L. Eriksson and K. J. Szabó, Org. Lett., 2014, 16, 3808 CrossRef CAS PubMed; (e) R. Alam, T. Vollgraff, L. Eriksson and K. J. Szabó, J. Am. Chem. Soc., 2015, 137, 11262 CrossRef CAS PubMed; (f) R. Alam, C. Diner, S. Jonker, L. Eriksson and K. J. Szabó, Angew. Chem., Int. Ed., 2016, 55, 14417 CrossRef CAS PubMed; (g) Z. Lu, M. Yang, P. Chen, X. Xiong and A. Li, Angew. Chem., Int. Ed., 2014, 53, 13840 CrossRef CAS PubMed; (h) K. J. Bruemmer, R. R. Walvoord, T. F. Brewer, G. Burgos-Barragan, N. Wit, L. B. Pontel, K. J. Patel and C. J. Chang, J. Am. Chem. Soc., 2017, 139, 5338 CrossRef CAS PubMed; (i) Q. Tan, X. Wang, Y. Xiong, Z. Zhao, L. Li, P. Tang and M. Zhang, Angew. Chem., Int. Ed., 2017, 56, 4829 CrossRef CAS PubMed; (j) D. Böse, P. Niesobski, M. Lübcke and J. Pietruszka, J. Org. Chem., 2014, 79, 4699 CrossRef PubMed.
- H. C. Brown, U. S. Racherla and P. J. Pellechia, J. Org. Chem., 1990, 55, 1868 CrossRef CAS.
- (a) L. T. Pilarski and K. J. Szabó, Angew. Chem., Int. Ed., 2011, 50, 8230 CrossRef CAS PubMed; (b) G. A. Molander, S. L. J. Trice and S. D. Dreher, J. Am. Chem. Soc., 2010, 132, 17701 CrossRef CAS PubMed; (c) V. J. Olsson, S. Sebelius, N. Selander and K. J. Szabó, J. Am. Chem. Soc., 2006, 128, 4588 CrossRef CAS PubMed; (d) S. Sebelius, V. J. Olsson and K. J. Szabó, J. Am. Chem. Soc., 2005, 127, 10478 CrossRef CAS PubMed.
- G. A. Molander, S. L. J. Trice and B. Tschaen, Tetrahedron, 2015, 71, 5758 CrossRef CAS PubMed.
- J. Sun, M. T. Perfetti and W. L. Santos, J. Org. Chem., 2011, 76, 3571 CrossRef CAS PubMed.
- J. R. Falck, M. Bondlela, S. K. Venkataraman and D. Srinivas, J. Org. Chem., 2001, 66, 7148 CrossRef CAS PubMed.
- G. Huang, C. Diner, K. J. Szabó and F. Himo, Org. Lett., 2017, 19, 5904 CrossRef CAS PubMed.
- R. S. Paton, J. M. Goodman and S. C. Pellegrinet, Org. Lett., 2008, 11, 37 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures and compound characterization data are given. CCDC 1550097. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7sc05123a |
| ‡ J. Z. and S. J. T. J. contributed equally. |
| This journal is © The Royal Society of Chemistry 2018 |