
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Second-generation CK2α inhibitors targeting the αD pocket†
Jessica
Iegre‡
 a,
Paul
Brear‡
b,
Claudia
De Fusco‡
ac,
Masao
Yoshida
ad,
Sophie L.
Mitchell
a,
Maxim
Rossmann
b,
Laura
Carro
a,
Hannah F.
Sore
a,
Marko
Hyvönen
*b and
David R.
Spring
*a
a,
Paul
Brear‡
b,
Claudia
De Fusco‡
ac,
Masao
Yoshida
ad,
Sophie L.
Mitchell
a,
Maxim
Rossmann
b,
Laura
Carro
a,
Hannah F.
Sore
a,
Marko
Hyvönen
*b and
David R.
Spring
*a
aDepartment of Chemistry, University of Cambridge, CB2 1EW, Cambridge, UK. E-mail: spring@ch.cam.ac.uk
bDepartment of Biochemistry, University of Cambridge, CB2 1GA, Cambridge, UK. E-mail: mh256@cam.ac.uk
cStructure Biophysics & FBLG, Discovery Sciences, IMED Biotech Unit, AstraZeneca, Cambridge, UK
dR&D Division, Daiichi Sankyo Co., Ltd., 1-2-58, Hiromachi, Shinagawa-ku, Tokyo 140-8710, Japan
First published on 20th February 2018
Abstract
CK2 is a critical cell cycle regulator that also promotes various anti-apoptotic mechanisms. Development of ATP-non-competitive inhibitors of CK2 is a very attractive strategy considering that the ATP binding site is highly conserved among other kinases. We have previously utilised a pocket outside the active site to develop a novel CK2 inhibitor, CAM4066. Whilst CAM4066 bound to this new pocket it was also interacting with the ATP site: herein, we describe an example of a CK2α inhibitor that binds completely outside the active site. This second generation αD-site binding inhibitor, compound CAM4712 (IC50 = 7 μM, GI50 = 10.0 ± 3.6 μM), has numerous advantages over the previously reported CAM4066, including a reduction in the number of rotatable bonds, the absence of amide groups susceptible to the action of proteases and improved cellular permeability. Unlike with CAM4066, there was no need to facilitate cellular uptake by making a prodrug. Moreover, CAM4712 displayed no drop off between its ability to inhibit the kinase in vitro (IC50) and the ability to inhibit cell proliferation (GI50).
Introduction
CK2 is a serine/threonine kinase that is a key regulator of many cellular processes and is involved in cellular proliferation and anti-apoptotic mechanisms.1In vivo it exists mainly as a holoenzyme composed of two catalytic (α and/or α′) and a dimer of regulatory (β) subunits, but it can also be found as the isolated subunits.2 Unlike most other kinases it is constitutively active and more than 300 proteins have been identified as CK2 substrates, making it probably one of the most pleiotropic proteins in eukaryotic systems.3 Elevated levels of CK2 have been found in a variety of cancers, including leukaemia, breast, lung, prostate, colorectal, renal and glioblastoma brain tumours.4,5 It has been shown that cancer cells are particularly susceptible to CK2 inhibition because they rely on high levels of the kinase to survive.6 CK2 overexpression has been associated with multi-drug resistance phenotypes and it has been demonstrated that CK2α inhibition leads to an increased uptake of known drugs in multidrug resistant cells.7,8 It has been shown that CK2 inhibitors are able to limit the growth of a range of cancer cell lines.9,10 Hence, CK2 has been recognised as a highly promising target for anti-cancer therapies.Like the majority of kinase inhibitors, most of the known CK2 inhibitors target the ATP binding site, presenting the issue of poor selectivity over other kinases.11–13 This is the case for CX4945 (known as silmitasertib), the first in class CK2 inhibitor currently in phase II clinical trials.14,15 The IC50 of CX4945 against CK2 is 1 nM but it also inhibits 12 other kinases with nanomolar affinity and it is more potent against Clk2 than against CK2.16,17
Previous work from our groups led to the discovery of a new binding pocket on CK2α, which is located adjacent to the ATP binding site. This pocket was revealed in a X-ray crystallographic screen, in which several weakly binding fragments where found to occupy this novel site formed through a movement of the αD helix, hence the name of αD pocket.18,19 Through fragment growing and linking, we generated a novel selective CK2 inhibitor: CAM4066 (Fig. 1).
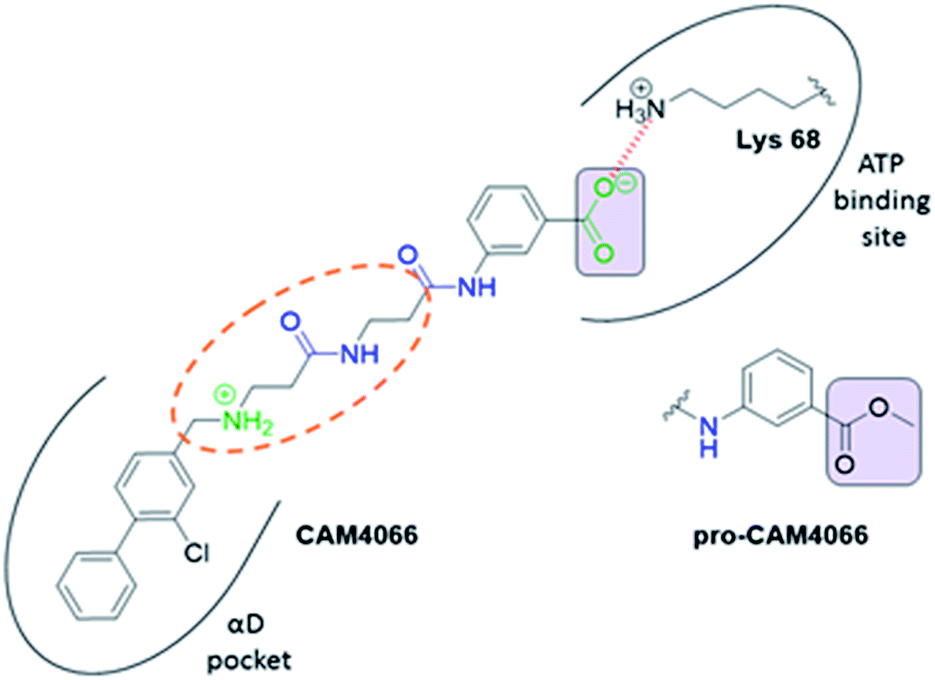 | ||
| Fig. 1 Structure of CAM4066 and pro-CAM4066. Zwitterionic elements are coloured in dark green, amide bonds in blue and the difference between CAM4066 and its prodrug highlighted in purple. The interaction between CAM4066 and the highly conserved Lys68 is shown as red dashed line. The flexible linker is circled in orange. The αD pocket and ATP binding site are reported as black curves. | ||
CAM4066 was a valuable tool for validating the concept of using the αD site to develop selective inhibitors of CK2α; however, it has several structural features that are undesirable in a lead molecule or chemical tool. These features, shown in Fig. 1, include a long flexible linker (circled in orange), a zwitterionic nature (the amine and the carboxylate are highlighted in green), amide bonds (coloured blue) and a high MW, which is often associated with poor oral bioavailability (Fig. 1). Moreover, the carboxylate forms a salt bridge with the conserved Lys68 in the ATP binding site. As expected due to its physicochemical properties, CAM4066 suffers from poor cellular permeability and therefore the methyl ester derivative, pro-CAM4066, was used as a pro-drug to improve cellular activity and target engagement.18 The aim of this work was to develop enhanced CK2α inhibitors that have improved physico-chemical properties and bind to the αD pocket without reaching deep into the ATP pocket. Our ideal lead-like candidate would have a smaller number of rotatable bonds (<10), not be susceptible to protease action (absence of amide groups), and be cell permeable without resorting to the use of a pro-drug. In addition, we aimed to develop inhibitors that do not rely on any of the conserved interactions within the ATP binding site.
The strategy (shown in Fig. 2) involved a fragment optimisation and a fragment-growing stage, followed by merging of the best compounds. Firstly, we would optimise the αD site fragment further to gain higher affinity and secondly, we would grow the fragment into the upper part of the αD pocket in order to gain inhibition.
 | ||
| Fig. 2 Optimisation strategy adopted in this project using 1 as the fragment starting point. | ||
Finally, the compounds with the most promising substitution patterns would be combined to provide the final inhibitor that would show inhibition of the kinase activity, good cell permeability and efficacy in cellular assays.
Results and discussion
Previously we have reported our preliminary studies on the exploration of the αD pocket, based on the development of primary hits from the crystallographic screen, which led to the identification of compound 1 (Kd = 267 μM), shown in Fig. 2. Our strategy to optimise the αD site fragment was to concentrate on ring A of the biaryl structure; however, a brief investigation of ring B was also performed. In parallel, optimisation of the amine substituent was carried out growing in the channel that connects the αD site and the ATP binding pocket.Optimization of the αD site fragment
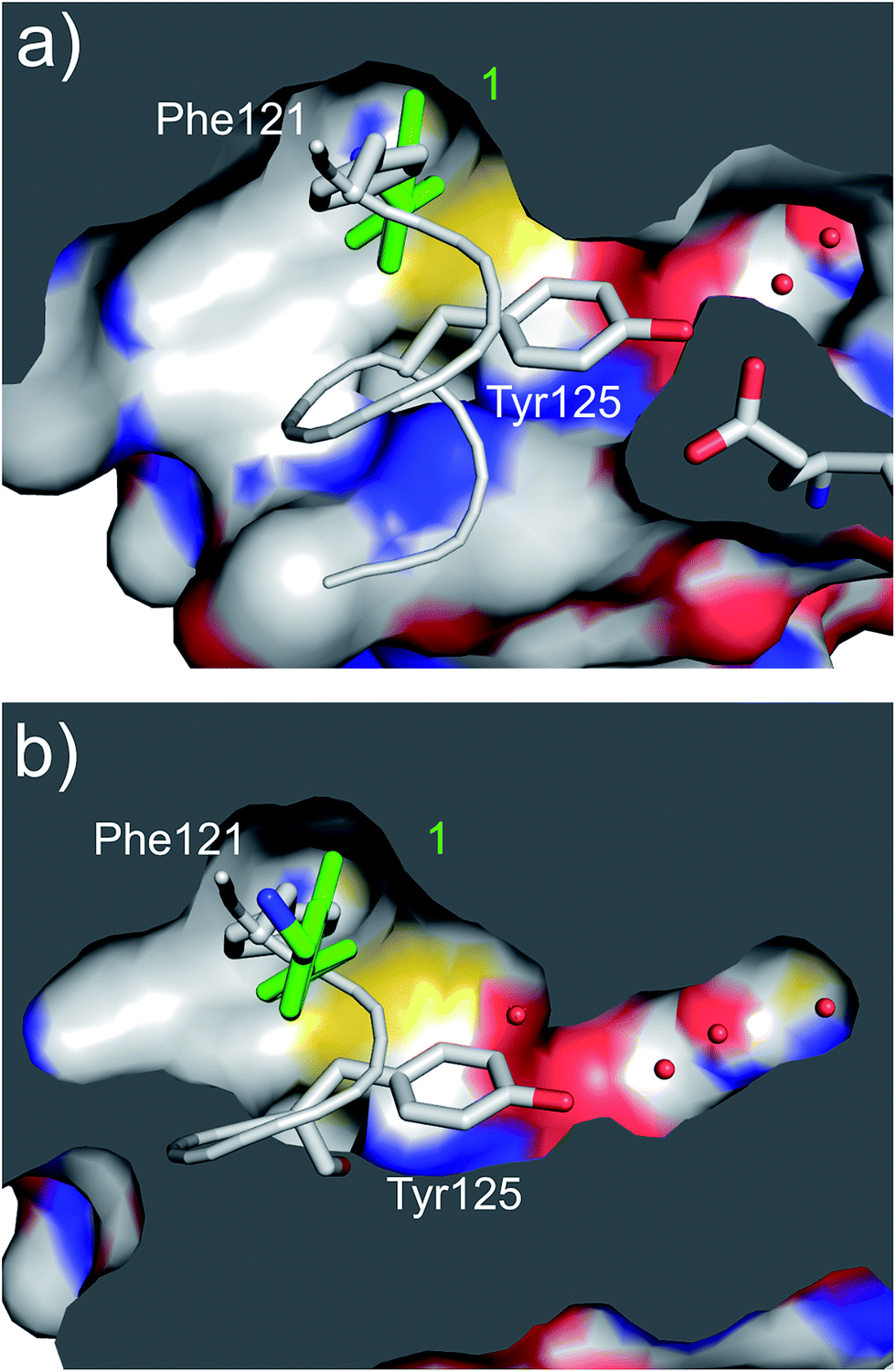 | ||
| Fig. 3 (a) A cross section of the αD pocket of 1 (green) bound to CK2α. The first two water molecules in the water channel are shown (PDB: 5CSH). (b) The structure of the αD loop (in grey) in the closed conformation of the apo protein (PDB: 5CSP).19 Tyr125 fills the mouth of the water channel. The binding mode of 1 (in green) superimposed on the apo structure is shown, with 1 occupying the same position as Phe121. | ||
A robust crystallographic system for CK2α enabled us to use X-ray crystallography as our primary screening technique. Therefore, co-crystal structures of all the compounds were attempted. The majority of the structures showed the ligand bound to CK2 and for all the compounds showing a clear electron density Kd was determined via ITC (overview of the results of the ITC experiments can be found in Table S1†). Several structures that did not show clear electron density for the ligand were also investigated using ITC to provide SAR data and controls. A number of mono- and di-substituted rings and bicyclic systems were investigated (Table 1 and 2, Fig. 4).
|
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| Compound | Ring A | K d (μM) | LEb | PDB | Compound | Ring A | K d (μ M) | LEb | PDB |
| a Measured by ITC. b LE = ligand efficiency*. *Ligand efficiency is defined as the ratio of the Gibbs free energy of binding of a ligand divided by the number of heavy (non-hydrogen) atoms in the molecule (LE = ΔG/[number of heavy atoms]).20 | |||||||||
| 1 |
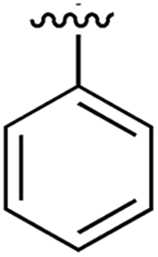
|
267 | 0.33 | 5CSH | 7 |
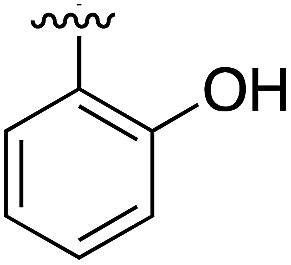
|
375 | 0.30 | 5OSL |
| 2 |
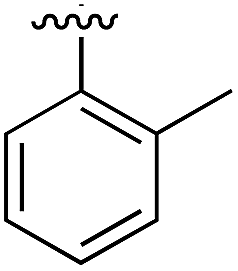
|
41 | 0.38 | 5ORH | 8 |
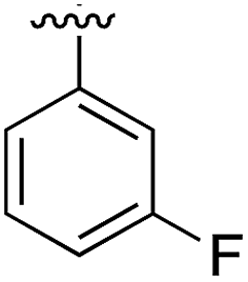
|
>500 | — | n.a |
| 3 |
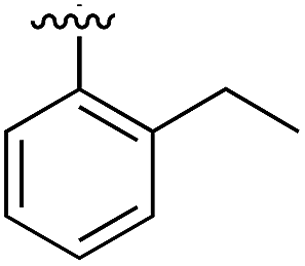
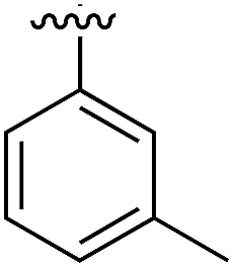
|
17 | 0.39 | 5ORJ | 9 |
|
>500 | — | 5OUL |
| 4 |
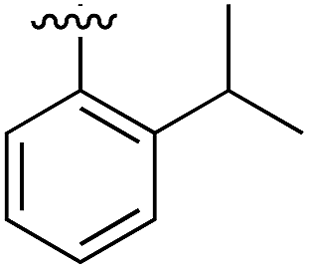
|
205 | 0.29 | 5OS7 | 10 |
|
>500 | — | n.a |
| 5 |
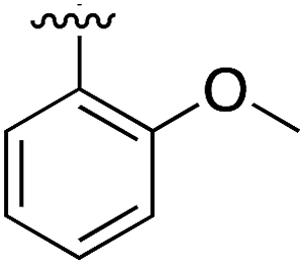
|
244 | 0.30 | 5OQU | 11 |
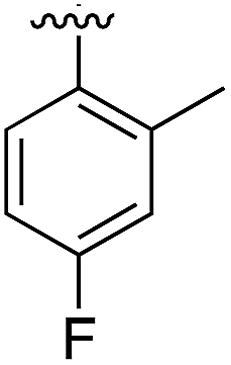
|
105 | 0.33 | 5OS8 |
| 6 |
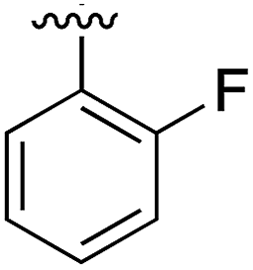
|
234 | 0.32 | 5ORK | 12 |
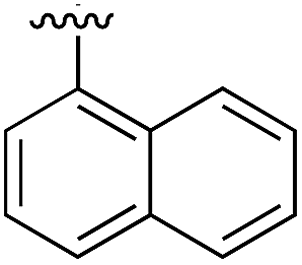
|
250 | 0.27 | n.a |
| 13 |
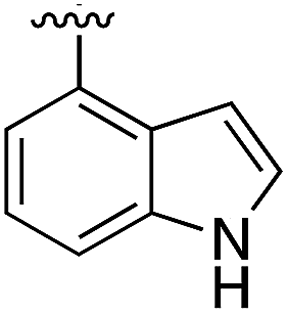
|
>500 | — | n.a | |||||
|
|
||||
|---|---|---|---|---|
| Compound | Ring A | K d (μM) | LEb | PDB |
| a Measured by ITC. b LE = ligand efficiency*. *Ligand efficiency is defined as the ratio of the Gibbs free energy of binding of a ligand divided by the number of heavy (non-hydrogen) atoms in the molecule (LE = ΔG/[number of heavy atoms]).20 | ||||
| 14 |
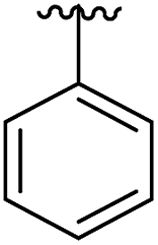
|
12 | 0.43 | 5OTR |
| 15 |
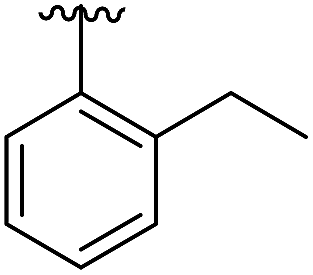
|
6.5 | 0.41 | 5OTZ |
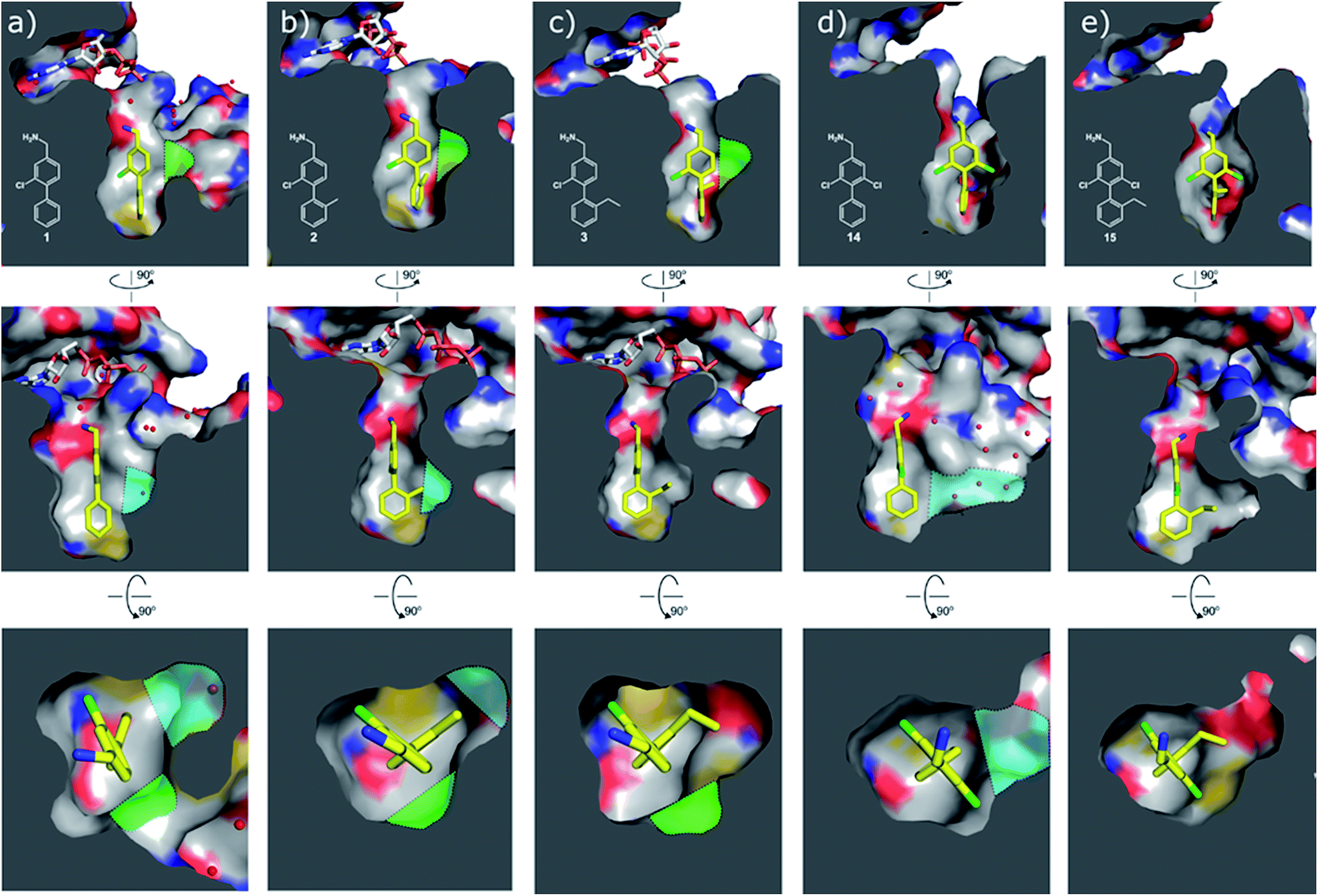 | ||
| Fig. 4 Cross sections of the αD and ATP pockets: first and second rows are lateral views and last row is a view from the top. The water channel is highlighted in blue and the hydrophobic pocket in green for complexes that are not making the most of the space available. (a) Co-crystal structure of 1 with CK2α (PDB: 5CSH); (b) co-crystal structure of 2 with CK2α (PDB: 5ORH); (c) co-crystal structure of 3 with CK2α (PDB: 5ORJ); (d) co-crystal structure of 14 with CK2α (PDB: 5OTR); (e) co-crystal structure of 15 with CK2α (PDB: 5OTZ). | ||
Substitution in the 2 position of ring A was beneficial when compared to the reference compound 1 (Kd = 267 μM); the 2-methyl derivative 2 was promising with a Kd of 41 μM. The introduction of an ethyl group in the same position (3) resulted in the highest affinity (Kd = 17 μM) and highest ligand efficiency (LE = 0.39) of the series and the crystal structure (PDB: 5ORJ) showed how the lateral chain grows towards the water channel, displacing the first water molecule on the left (Fig. 4b and c). The presence of the chlorine on ring B and the ethyl group on ring A enforced an almost orthogonal conformation of the biaryl system (Fig. 4c). Expanding the size of the lateral chain did not improve the binding (Kd = 205 μM) nor the LE of the isopropyl derivative 4. The 2-MeO and 2-F derivatives, 5 and 6 respectively, had a comparable Kd to 1 while the 2-OH group (compound 7) was detrimental to binding even though it formed a hydrogen bond to the conserved water (Fig. S1a,† PDB: 5OSL). Substitution in the 3 position of ring A (compounds 8 to 10) appeared not to be tolerated as only the fluoro-derivative 9 yielded a co-crystal structures. The disubstituted 2-Me-4-F derivative 11 was found to have a Kd of 105 μM, marginally higher than the 2-methyl compound 2, but with a lower ligand efficiency (LE = 0.33). Expanding ring A to a naphthyl group 12 improved the affinity only slightly, while the indolyl derivative 13 did not show any significant binding and we were unable obtain structures of these in complex with CK2α. Therefore, compound 3 represented the fragment with the best LE and it was chosen as the best optimized fragment as far as the ring A is concerned.
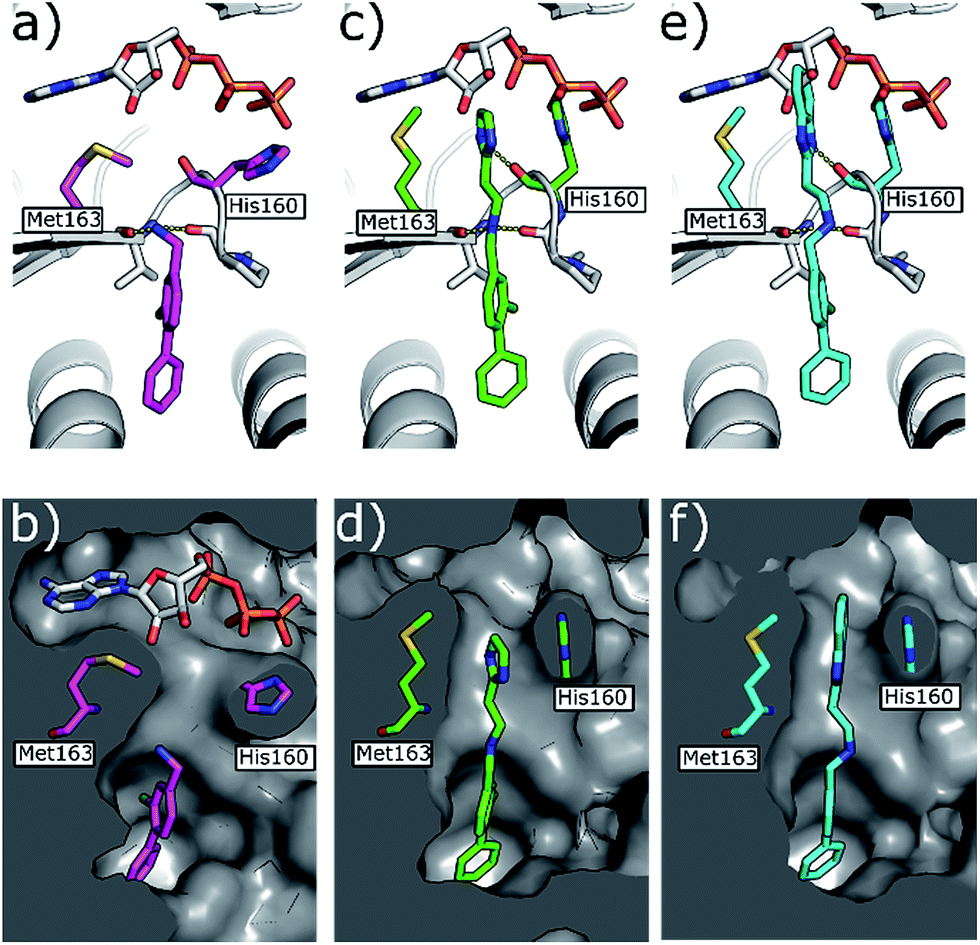 | ||
| Fig. 5 (a) Co-crystal structure of 1 (in pink) and ATP bound to CK2α (PDB: 5CSH)19 and (b) surface representation of CK2α with 1 bound. (c) Co-crystal structure of 21 (in green) bound to CK2α. The binding mode of ATP (grey) in the ATP site when 1 is bound is superimposed upon the structure. (d) Co-crystal structure of 21 (in green) bound to the surface representation of CK2α. (e) Co-crystal structure of 22 (in blue) bound to CK2α. The binding mode of ATP (grey) in the ATP site when 1 is bound is superimposed upon the structure. (f) Co-crystal structure of 22 (in blue) bound to the surface representation of CK2α. | ||
During the development of CAM4066 a series of flexible linkers were designed and tested to join the αD site fragment to an ATP site fragment. These compounds revealed that it was possible to induce the opening of a small channel between the αD and the ATP sites. Our aim was to induce the opening of the channel with shorter, more rigid compounds than CAM4066. The flipping of the side chain of Met163 allows the formation of the channel and results in blocking the ATP site – Met163 is located just underneath the adenine base of ATP. Therefore, these compounds would not need to grow deep into the ATP site to achieve inhibition. The channel from the αD site is lined by Met163 and His160 and we envisioned that compounds with aromatic groups that stacked between these amino acid residues would improve the affinity and cause the conformational change that would lead to inhibition. Toward this end, the effect of several aromatic groups on the amine were investigated using a kinase activity assay (Table 3).
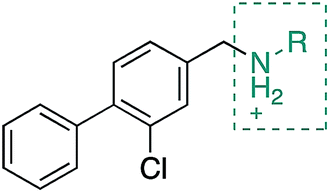
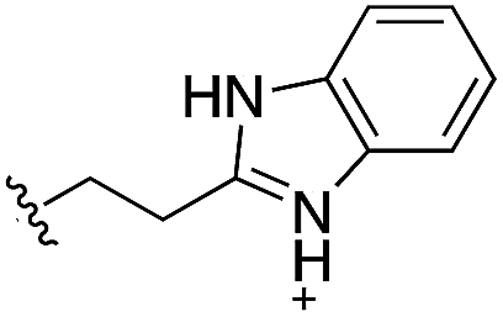
Whilst the reference compound 1, tested at 500 μM, inhibited only 21% of the protein activity, the inhibitory activity of the aromatic derivatives 16–19, were found to be considerably improved with compounds 16 and 19 being the most promising (54 and 52% respectively). In order to pick up additional H-bond interactions the pyrrole derivative 19 was chosen over 16 and we hypothesized that the heterocycle should go further up into the channel. Whilst the imidazole derivative 20 did not show significant improvement, the benzimidazole derivative 22 was found to be the most potent compound with an IC50 of 58 μM. Fig. 5d and f show the co-crystal structure of 21 and 22 with CK2α, respectively. As expected, Met163 flips upon binding of the more extended compounds compared to the co-crystal structure of 1 (Fig. 5b). This explains the displacement of ATP and therefore activity inhibition even without fragments binding directly in the ATP pocket.
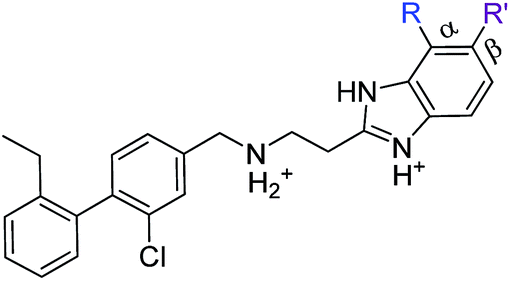
As these compounds showed increased activity, the concentration for the inhibition assay was decreased to 10 μM and cellular activity was investigated (Table 4). Although 15 had the highest affinity of the αD binders (Table 2), 3 was chosen for further studies for synthetic reasons, with the idea of retrieving the substitution pattern of 15 in the final compound. Merging 3 with 22 provided compound 23, which featured higher potency than the original fragments in the inhibition assay and a promising GI50 of 10 μM in HCT116 cells. Therefore, SAR studies around the benzimidazole ring were then performed (compounds 24–30), with both electron withdrawing and donating groups in position α and β of the benzimidazole improving the inhibition activity in respect to the derivative 23. The α and β methoxy derivatives 26 and 29 were found to be the most promising in the respective α and β substituted series with IC50 of 8 and 7 μM, respectively (Table 4, IC50 and GI50 curves are shown in Fig. S4 and S5†).
|
|
||||
|---|---|---|---|---|
| Compound | R | R′ | Inhibition @10 μM (%) | PBD |
| 23 | Cl | H | 33 (IC50 18) | 5OSZ |
| CAM4712 | Cl | Cl | 80 (IC50 7) | 5OTY |
| 31 | Me | H | 30 | 5OYF |
| 32 | CF3 | H | 23 | 6EHU |
| 33 | OMe | H | 41 | 5OTQ |
| 34 | OCF3 | H | 39 | n.a. |
With the optimisation around each aromatic ring in hand, compounds 35 and 36 were designed via a merging strategy so that they contained the most promising substitution patterns. The methoxy derivatives 35 and 36 were synthesized and tested but, disappointingly, gave worse results than CAM4712 (Fig. 6).
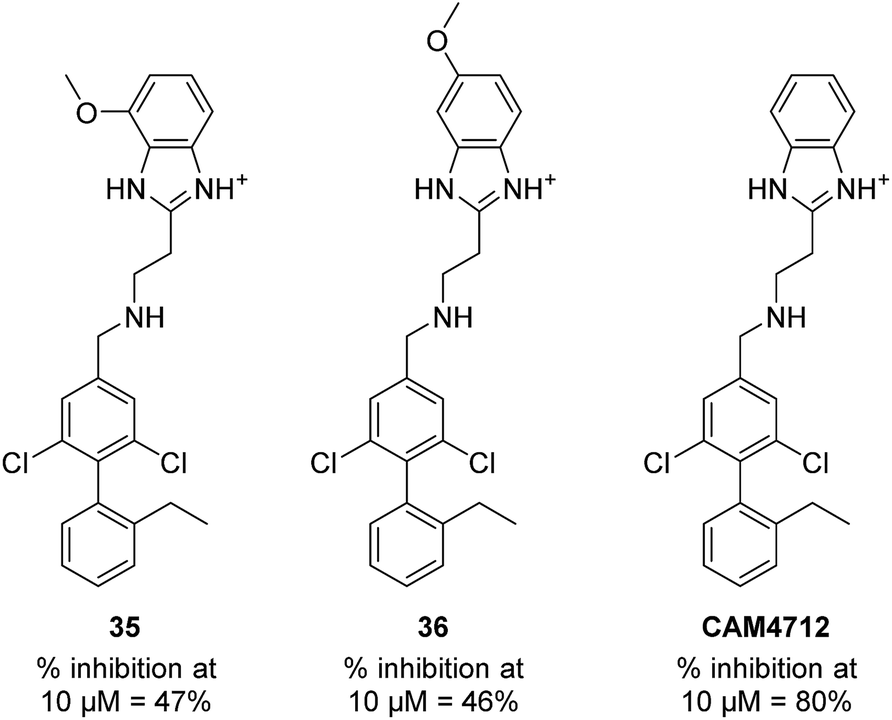 | ||
| Fig. 6 Compounds 35, 36 and CAM4712 with respective data. | ||
Four probes were used for this study in four separate competition experiments, each of them titrated into CK2α in the presence of and absence 20 μM CAM4712 giving the following results:
(1) Inhibition of binding in the αD or ATP site by CAM4712:
The binding of CAM4066 to CK2α was inhibited by 20 μM CAM4712. From this the affinity of CAM4712 was estimated to be 4.0 μM (Fig. S2†). This experiment confirms that CAM4712 binds to CK2α but the binding could partially occur in the ATP site.
(2) Inhibition of binding in the αD site:
15 was titrated into CK2α in the presence of 20 μM CAM4712 (Fig. S3†). This showed that CAM4712 was also able to inhibit the binding of 15 to CK2α and from this the affinity was estimated to be 5.0 μM. As 15 binds in the αD site this experiment confirms that the binding site of CAM4712 is the αD pocket as well as confirming the affinity.
(3) Inhibition of binding to Lys68:
2-Hydroxyl, 5-methyl benzoic acid (37) binds to the conserved Lys68 in the ATP site and occupies the right-hand side of the pocket (PDB: 5CSP). The binding of compound 37 was not inhibited by CAM4712 (Fig. S6†) confirming that the benzimidazole ring does not interact with the right-hand side of the ATP pocket and validates the binding mode derived from the crystal structure (PDB: 5OTZ). This result predicted that it would be possible to generate a crystal structure of CAM4712 and 37 bound simultaneously to CK2α and this was confirmed by a crystal structure showing both compounds binding simultaneously to CK2α (Fig. S6d,† PDB: 6EHK).
(4) Inhibition of binding in the ATP site/hinge region:
CX4945, which from the analyses of crystal structures would clash with CAM4712 in the hinge region, was titrated into CK2α in the presence of CAM4712 (Fig. S7†). CAM4712 was shown to inhibit the binding of CX4945 to CK2α. The affinity of CAM4712 for CK2α was estimated to be 3.0 μM. This confirms that the benzimidazole ring binds in the Met163 channel and blocks access to the ATP site as this would inhibit the binding of CX4945.
In summary, these competition experiments suggest firstly that the Kd of CAM4712 towards CK2α is approximately 4 μM. Secondly, they confirm that the binding mode of CAM4712 in the αD pocket and mouth of the ATP site corresponds to that seen in the crystal structure.
The validation experiments of CAM4712 and the crystal structures allowed us to rationalise the difference in in vitro potency between CAM4066 and CAM4712 (IC50 7 μM and 0.37 μM respectively). Whilst the binding of both compounds to CK2 resulted in the flipping of the Met163, CAM4712 did not H-bond the conserved Lys68 in the ATP binding site. Instead, a low-energy hydrophobic π–π interaction between the His160 and the benzimidazole was introduced (as shown for the related compound 22 in Fig. 5f) resulting in loss of binding affinity and potency compared to CAM4066.
The efficacy of CAM4712 in cellular assays was tested in HCT116 cell line, which is known to overexpress CK2α. A cell growth inhibition assay yielded a GI50 for CAM4712 of 10.0 ± 3.6 μM, which is similar to that of the clinical trials candidate CX4945 (11.3 ± 1.2 μM). It is also similar to pro-CAM4066 (GI50 9.1 ± 1.4 μM, IC50 0.37 μM),18 but, importantly, no drop-off in potency was observed from the functional to the cellular assay. This represents a large step forward compared to CAM4066, which had to be administered as the prodrug pro-CAM4066. The target engagement by CAM4712 was analysed by following the CK2α dependent phosphorylation of Ser129 of Akt1. This showed good inhibition of the phosphorylation of Ser129 by CAM4712 as well as by its close analogues 23 and 26 which confirms that these compounds inhibit CK2α in the cellular environment (Fig. 7).
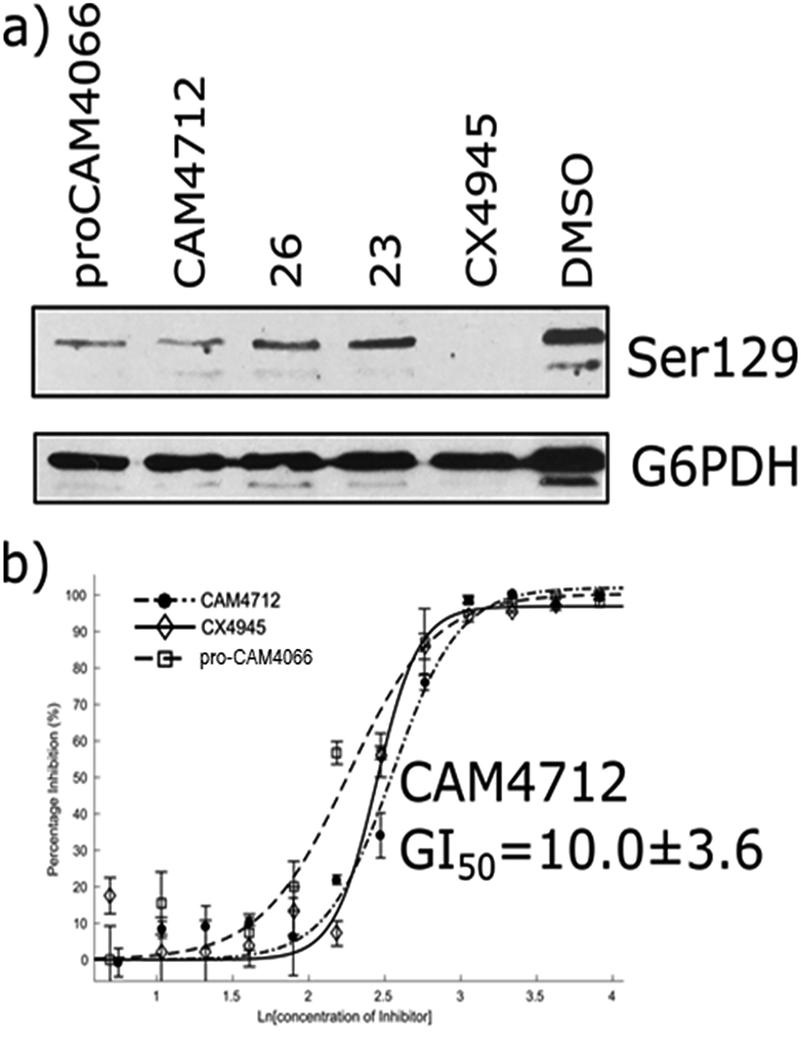 | ||
| Fig. 7 (a) Western blot analysis showing the specific CK2 phosphorylation target: AKT1 serine 129. HCT116 cells were treated with 2 × GI50 of CX4945 (20 μM), CAM4712 (20 μM) or pro-CAM4066 (20 μM) for 72 hours (b) dose response curve for the inhibition of the growth of HCT116 cells by CAM4712, pro-CAM4066 and CX4945. All graphs show the mean ± SEM of not less than three independent experiments with each in triplicate. | ||
CAM4712 showed a 10-fold decrease in potency compared to CAM4066 and therefore the selectivity of CAM4712 was screened against a panel of 140 kinases at a concentration of 30 μM (4 × IC50). CAM4712 showed good selectivity against the 20 closely related CMGC kinases in the panel (Fig. S6a†). However, 4 kinases (CAMK1, SmMLCK, EF2K and SGK1) were inhibited by CAM4712 for more than 50% (Fig. S8b†) and hence, CAM4712 showed a reduced overall selectivity compared to CAM4066 (which was screened at 2 μM).19 This is not surprising considering the high concentration used in the selectivity screen due to CAM4712 being less potent than CAM4066. Nevertheless, CAM4712 showed a more selective profile than other CK2α inhibitors (Fig. S8b†) and represents, therefore, a good starting point for further development of selective CK2α inhibitors. Our aim in this work has not been to gain specificity but rather to demonstrate mechanistically that an inhibitor that is not making significant contacts with the conserved active site is able to inhibit the kinase effectively. To fully exploit the selectivity that αD binding offers, further optimization of CAM4712 is needed to increase its affinity towards CK2α.
One of the aims of this work was to generate an improved inhibitor compared to the previous compound CAM4066. We managed to design a compound with the physico-chemical properties falling into the range for bioavailable compounds according to Lipinski's rules:21 the number of rotatable bonds was reduced, the amide groups were removed and the compound entered the cells and showed activity without the use of a prodrug. Moreover, the number of H-bond donors and acceptors was reduced to 2 and the molecular weight was kept below 500 Da (Table 6). This was all achieved without interacting with the deep and conserved ATP binding site.

|
Conclusions
In conclusion, we have developed a series of second-generation CK2α inhibitors that target the αD site. This was achieved by first optimising the fragments that bound in the αD site, followed by identification of groups that grow towards the mouth of the ATP site to provide potent inhibitors of CK2α. In our previous work we demonstrated that selectivity could be achieved anchoring the inhibitors in the αD pocket and with this work we achieve inhibition with ligands that do not target the active site. CAM4712 showed high cellular activity (10.0 ± 3.6 μM) and target engagement was demonstrated. This second generation of αD pocket inhibitor overcomes the limitations of our first inhibitor, including the fact that it does not need to be administered as a pro-drug to exert anti-proliferative activity. We have also shown that it is not necessary to interact with the ATP site directly, but effective inhibition of the kinase and displacement of ATP can be achieved by blocking the mouth of the ATP site with no need to interact with conserved features of the ATP binding site. These results demonstrate an entirely new approach to CK2α inhibition and will allow the future development of drug-like molecules, lead compounds and chemical tools that utilise the novel properties of the αD site.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to thank the X-ray crystallographic facility and the Biophysics facility at the Department of Biochemistry for support and access to equipment. We thank Diamond Light Source for access to beamlines IO2, IO3, IO4 and I24 (proposals mx9537 and mx9007). We are grateful for academic licence of Instant JChem that was used for structure database management, search and prediction, Instant JChem 16.16.10, 2016, ChemAxon (http://www.chemaxon.com). This work was funded by the European Research Council under the European Union's Seventh Framework Programme (FP7/2007-2013)/ERC grant agreement no. [279337/DOS] (to DRS) and the Wellcome Trust Strategic (090340/Z/09/Z) and Pathfinder (107714/Z/15/Z) Awards (to DRS and MH). In addition, the Spring group research was supported by grants from the Engineering and Physical Sciences Research Council, Biotechnology and Biological Sciences Research Council, Medical Research Council and the Royal Society. CD thanks the Herchel Smith Funds for a postdoctoral fellowship. JI would like to thank Trinity College, University of Cambridge for funding. SLM would like to acknowledge a CASE-EPSRC award in partnership with BASF.Notes and references
- R. Battistutta and G. Lolli, Mol. Cell. Biochem., 2011, 356, 67–73 CrossRef CAS PubMed.
- K. Niefind, B. Guerra, I. Ermakowa and O. G. Issinger, EMBO J., 2001, 20, 5320–5331 CrossRef CAS PubMed.
- F. Meggio and L. A. Pinna, FASEB J., 2016, 17(3), 349–368 CrossRef PubMed.
- J. Zhang, P. L. Yang and N. S. Gray, Nat. Rev. Cancer, 2009, 9, 28–39 CrossRef CAS PubMed.
- R. T. Nitta, S. Gholamin, A. H. Feroze, M. Agarwal, S. H. Cheshier, S. S. Mitra and G. Li, Oncogene, 2015, 34, 3688–3699 CrossRef CAS PubMed.
- M. Ruzzene and L. A. Pinna, Biochim. Biophys. Acta, 2010, 1804, 499–504 CrossRef CAS PubMed.
- B. Zhou, D. A. Ritt, D. K. Morrison, C. J. Der and A. D. Cox, J. Biol. Chem., 2016, 291(34), 17804–17815 CrossRef CAS PubMed.
- G. Di Maira, F. Brustolon, J. Bertacchini, K. Tosoni, S. Marmiroli, L. A. Pinna and M. Ruzzene, Oncogene, 2007, 26, 6915–6926 CrossRef CAS PubMed.
- J. S. Duncan and D. W. Litchfield, Biochim. Biophys. Acta, 2008, 1784, 33–47 CrossRef CAS PubMed.
- S. Sarno and L. A. Pinna, Mol. BioSyst., 2008, 4, 889–894 RSC.
- R. Battistutta, G. Cozza, F. Pierre, E. Papinutto, G. Lolli, S. Sarno, S. E. O'Brien, A. Siddiqui-Jain, M. Haddach, K. Anderes, D. M. Ryckman, F. Meggio and L. A. Pinna, Biochemistry, 2011, 50, 8478–8488 CrossRef CAS PubMed.
- B. Guerra, J. Hochscherf, N. B. Jensen and O.-G. Issinger, Mol. Cell. Biochem., 2015, 406, 151–161 CrossRef CAS PubMed.
- C. Götz, A. Gratz, U. Kucklaender and J. Jose, Biochim. Biophys. Acta, 2012, 1820, 970–977 CrossRef PubMed.
- A. Siddiqui-Jain, D. Drygin, N. Streiner, P. Chua, F. Pierre, S. E. O'Brien, J. Bliesath, M. Omori, N. Huser, C. Ho, C. Proffitt, M. K. Schwaebe, D. M. Ryckman, W. G. Rice and K. Anderes, Cancer Res., 2010, 70, 10288–10298 CrossRef CAS PubMed.
- H. J. Chon, K. J. Bae, Y. Lee and J. Kim, Front. Pharmacol., 2015, 6(70), 1–5 CAS.
- F. Pierre, P. C. Chua, S. E. O'Brien, A. Siddiqui-Jain, P. Bourbon, M. Haddach, J. Michaux, J. Nagasawa, M. K. Schwaebe, E. Stefan, A. Vialettes, J. P. Whitten, T. K. Chen, L. Darjania, R. Stansfield, K. Anderes, J. Bliesath, D. Drygin, C. Ho, M. Omori, C. Proffitt, N. Streiner, K. Trent, W. G. Rice and D. M. Ryckman, J. Med. Chem., 2011, 54, 635–654 CrossRef CAS PubMed.
- H. Kim, K. Choi, H. Kang, S. Y. Lee, S. W. Chi, M. S. Lee, J. Song, D. Im, Y. Choi and S. Cho, PLoS One, 2014, 9, 1–8 Search PubMed.
- P. Brear, C. De Fusco, K. H. Georgiou, N. J. Francis-Newton, C. J. Stubbs, H. F. Sore, A. R. Venkitaraman, C. Abell, D. R. Spring and M. Hyvönen, Chem. Sci., 2016, 7, 6839–6845 RSC.
- C. De Fusco, P. Brear, J. Iegre, K. H. Georgiou, H. F. Sore, M. Hyvönen and D. R. Spring, Bioorg. Med. Chem., 2017, 25, 3471–3482 CrossRef CAS PubMed.
- A. L. Hopkins, G. M. Keserü, P. D. Leeson, D. C. Rees and C. H. Reynolds, Nat. Rev. Drug Discovery, 2014, 13, 105–121 CrossRef CAS PubMed.
- C. A. Lipinski, F. Lombardo, B. W. Dominy and P. J. Feeney, Adv. Drug Delivery Rev., 1997, 23, 3–25 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: All experimental details, crystallographic data collection and refinement statistics, details of chemical synthesis, additional figures and tables. See DOI: 10.1039/c7sc05122k |
| ‡ These authors contributed equally to the work presented here. |
| This journal is © The Royal Society of Chemistry 2018 |