DOI:
10.1039/C7SC04768A
(Minireview)
Chem. Sci., 2018,
9, 1424-1432
sp3 C–H activation via exo-type directing groups
Received
4th November 2017
, Accepted 2nd January 2018
First published on 17th January 2018
Abstract
The application of exo-type directing groups (DGs) has led to the discovery of a wide range of novel C(sp3)–H activation methods, which allow efficient and site-selective functionalization of alcohol and amide derivatives. In this mini-review we discuss the challenges of this field and summarize the achievements, including DG designs, reaction discoveries and mechanistic studies.
Introduction
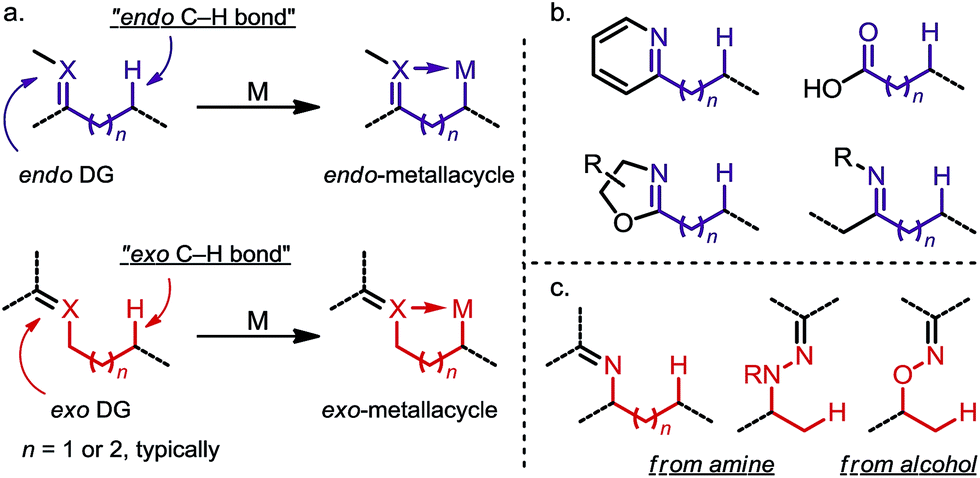
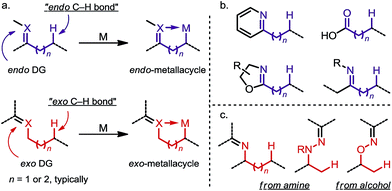
Site-selective functionalization of inert aliphatic C–H bonds has become an increasingly important tool, as it shows promise for streamlining the synthesis of complex organic molecules.1 Among various approaches, cyclometalation aided by an adjacent directing group (DG) is frequently utilized to activate C–H bonds with excellent site-selectivity control. Given that the majority of coordinating groups bear a π-bond moiety,1b,i the DGs can be divided into two categories: endo-DGs that feature an endocyclic π-bond after C–H metalation and exo-DGs that feature an exocyclic π-bond instead (Scheme 1a). The C–H activations assisted by endo-DGs have been widely explored.2 While highly efficient, these C–H activations usually require functional groups (FGs) with double bonds already built in, such as pyridines,2a oxazolines,2b carboxylic acids2c and ketone derivatives2a,d–f (Scheme 1b). In contrast, reactions via an exo-directing mode were historically less developed, but have received considerable attention recently. Compared to their endo counterparts, the exo-DGs are typically derived from more flexible FGs, such as alcohols and aliphatic amines, which opens the door for developing new site-selective C–H functionalizations (Scheme 1c). To better describe the challenges and achievements of this field, this mini-review is divided into two parts, with (i) a brief introduction to the “endo effect” from the organometallic viewpoint and (ii) a discussion of stoichiometric and catalytic transformations in functionalizing unactivated C(sp3)–H bonds via exo-type DGs. Reactions with C(sp2)–H or activated C(sp3)–H bonds will not be included in this article.
 |
| | Scheme 1 (a) Endo-metalation vs. exo-metalation. (b) Representative endo-type directing groups. (c) Representative exo-type directing groups. | |
Endo vs. exo: the “endo effect”
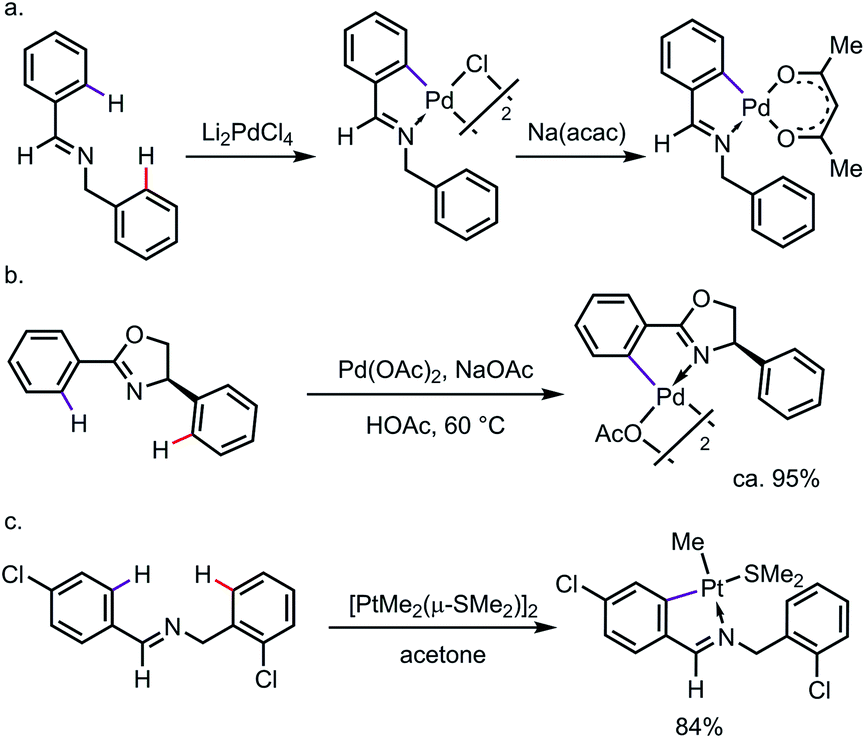
C–H cyclometalation can be highly regioselective when C–H bonds are available for activation through either endo or exo directing modes.3 The selective activation of “endo C–H bonds” over “exo C–H bonds”, which has been widely referred to as the “endo effect”, has been observed with various types of DG, such as imines and oxazolines.4 For example, treating N-benzylbenzaldimine with Li2PdCl4 led to a chloro-bridged endo-palladacycle, which underwent ligand exchange to give the monomer whose structure was unambiguously assigned by X-ray diffraction (Scheme 2a).5 In nearly a quantitative yield, the endo-metallacycle was also formed exclusively when 4-phenyl-2-oxazoline was treated with stoichiometric Pd(OAc)2 (Scheme 2b).6 Such a tendency was also observed for other transition metals, such as platinum (Scheme 2c).7 It is worth pointing out that the endo effect is not restricted to C–H metalation reactions. In the case of the oxidative addition of ortho-brominated N-benzylbenzaldimine to Pd(0), the endo-complex was formed preferentially as well.8 While the detailed reason for this endo effect is yet to be fully elucidated, studies have suggested that a combination of structural factors, e.g. bond angle, distortion of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N π-bond, the planarity of the metallacycle etc., makes the endo-complexes derived from imines or oxazolines more thermodynamically stable than their exo counterparts.9
N π-bond, the planarity of the metallacycle etc., makes the endo-complexes derived from imines or oxazolines more thermodynamically stable than their exo counterparts.9
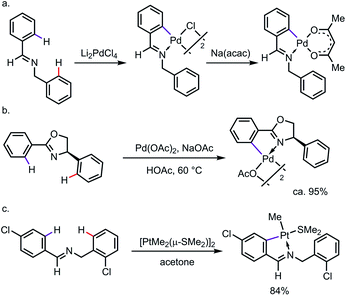 |
| | Scheme 2
Endo preference in regioselective C–H metalation. | |
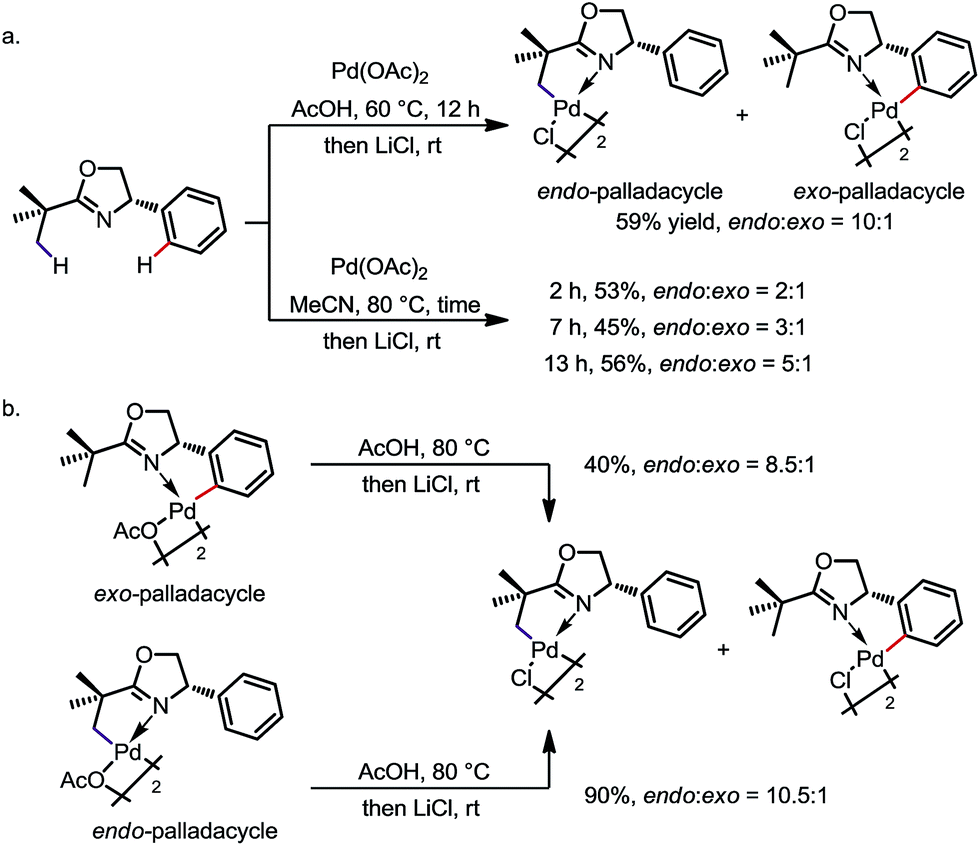
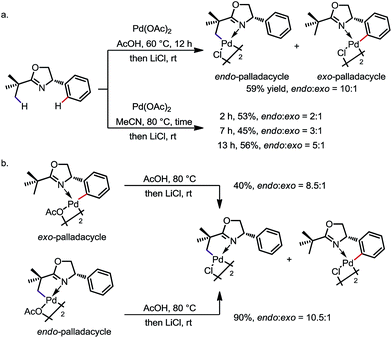
The preference for endo activation can be very strong in certain cases, such that it permits the activation of an endo C(sp3)–H bond in the presence of a more reactive exo C(sp2)–H bond. The reaction between 2-tert-butyl-4-phenyl-2-oxazoline and Pd(OAc)2 is an impressive example (Scheme 3).9a Under various reaction conditions, the endo sp3 palladated complex was obtained as the major product with up to 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 regioselectivity (Scheme 3a). In addition, treatment of the pure exo-complex with AcOH at 80 °C led to an 8.5:1 mixture of the endo- and exo-isomers. Treatment of the pure endo-complex under the same reaction conditions led to a similar 10.5:1 mixture of isomers (Scheme 3b). These experiments suggest that (i) there is an equilibrium between the exo and endo isomers, and (ii) the endo sp3 C–H palladation is thermodynamically favoured, at least in these cases.
:1 regioselectivity (Scheme 3a). In addition, treatment of the pure exo-complex with AcOH at 80 °C led to an 8.5:1 mixture of the endo- and exo-isomers. Treatment of the pure endo-complex under the same reaction conditions led to a similar 10.5:1 mixture of isomers (Scheme 3b). These experiments suggest that (i) there is an equilibrium between the exo and endo isomers, and (ii) the endo sp3 C–H palladation is thermodynamically favoured, at least in these cases.
 |
| | Scheme 3 Selective activation of endo C(sp3)–H bonds over exo C(sp2)–H bonds. | |
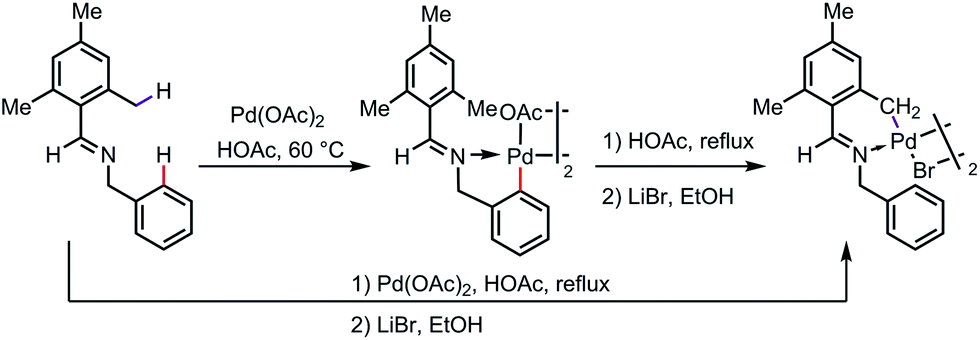
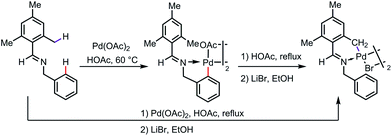
It appears that, in some cases, the exo-complex is the kinetic product, which can be isolated and then isomerized to the more thermodynamically stable endo-complex. As one example, a five-membered exo-palladacycle was obtained by reacting N-mesitylbenzylideneamine with Pd(OAc)2 under mild reaction conditions at 60 °C, which was further transformed to a more stable endo palladacycle in refluxing acetic acid (Scheme 4).10
 |
| | Scheme 4 Isomerization of exo-metallacycles to endo-metallacycles. | |
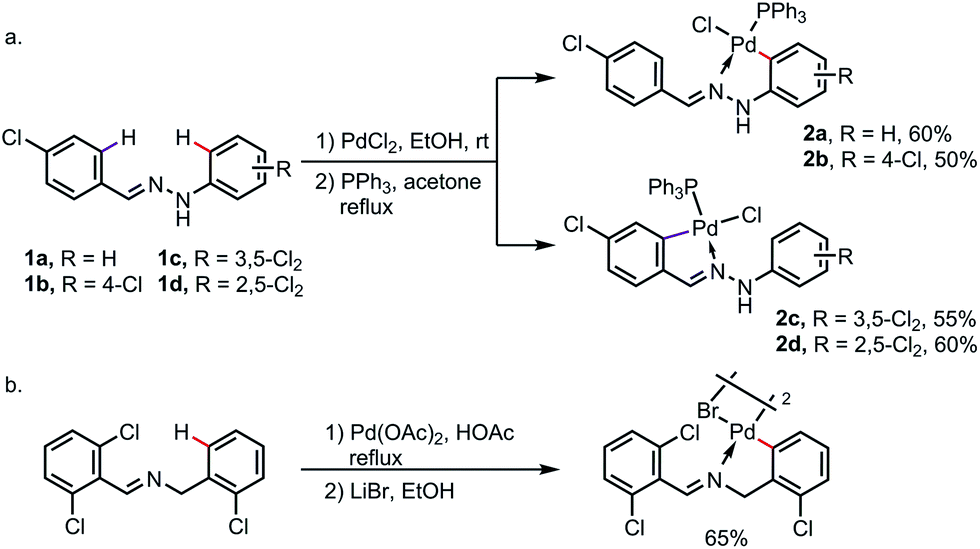
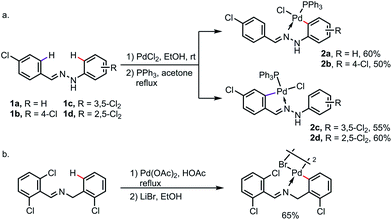
Under a strong electronic or steric bias, the exo-metallacycle can sometimes become the major product. For instance, exo-palladacycles were obtained at room temperature for the reaction of benzaldehyde phenylhydrazones 1a and 1b with PdCl2, likely due to the large difference in electron density between the endo- and exo-rings as a result of the highly electron-donating amine moiety. As expected, when more electron-withdrawing (and also sterically demanding) chlorine substituents were introduced at the ortho- and meta-positions of the phenylhydrazine rings (1c and 1d), only the endo-metallacycles were obtained (Scheme 5a).11 Besides, the exo-metalation can also be accomplished by blocking the endo reaction sites. For example, when both the endo C–H bonds of N-benzylbenzaldimine were replaced by chlorine atoms, the exo-palladacycle was formed exclusively (Scheme 5b).12
 |
| | Scheme 5
Exo-metallacycle formation promoted by (a) electronic bias and (b) blocking of endo reaction sites. | |
sp3 C–H activation via exo-type DGs
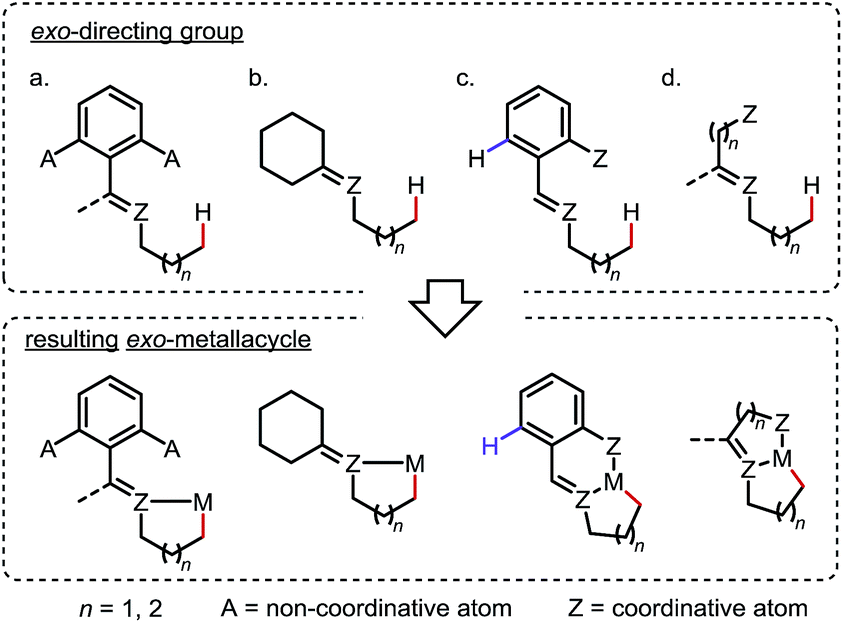
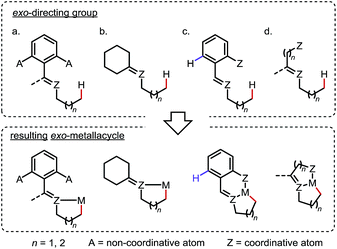
To outcompete the endo preference, two general strategies are commonly applied in the design of exo-DGs for sp3 C–H activation: (i) blocking or obviating potential endo reaction sites (Scheme 6a and b) and/or (ii) adding an extra coordinating entity at a proper position on the endo side to enable chelation (Scheme 6c and d). In the latter case, formation of an additional 5- or 6-membered metallacycle inhibits the activation of the endo C–H bonds (if any) and may further promote the exo metalation due to the increased structural rigidity.
 |
| | Scheme 6 General strategies in the design of exo-directing groups. | |
a. Reactions with stoichiometric metals
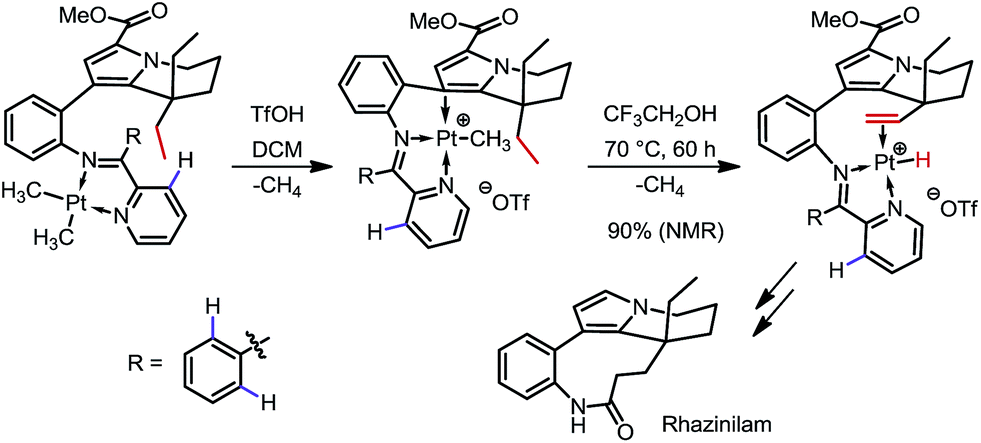
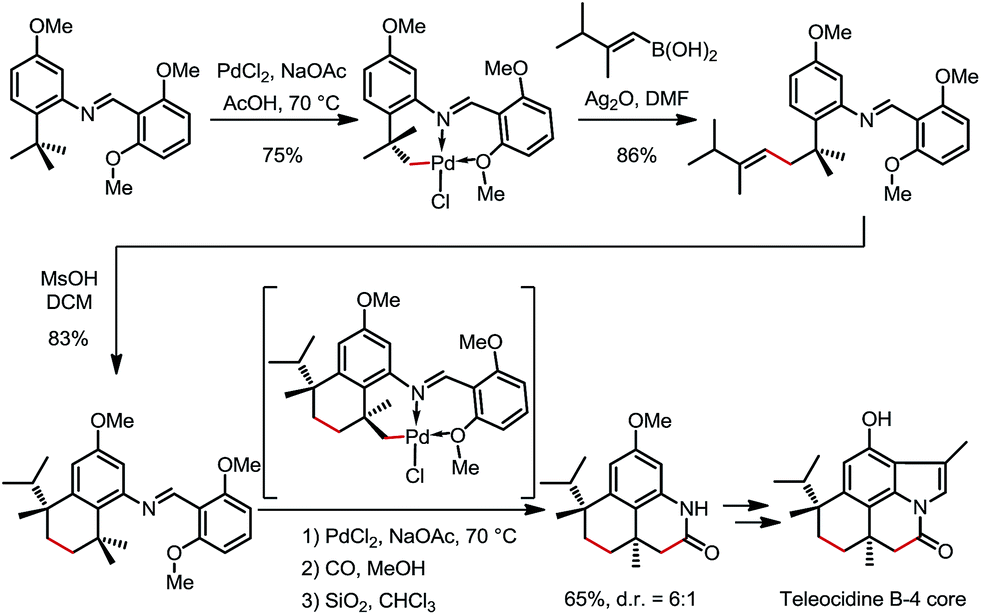
In 2000 and 2002, two seminal stoichiometric examples were reported by Sames and co-workers in the synthesis of rhazinilam13 and the core of teleocidin B4.14 In Scheme 7, the C–H platination of a remote ethyl group was promoted by a phenylpyridyl ketimine-type DG using stoichiometric cationic Pt(II), followed by a β-hydride elimination to afford an alkene–Pt(II)–H complex. The endo C(sp2)–H bonds at the phenyl and pyridyl groups were untouched. In Scheme 8, exo-palladacycles were obtained through the activation of tert-butyl groups when treating 2-tert-butylaniline-derived 2,6-bismethoxybenzaldimines with PdCl2 in acetic acid. The two ortho-OMe substituents not only blocked the endo reaction sites, but also served as additional directing groups. The alkenylation and carbonylation products were further furnished in high yields by treating the resulting Pd complexes with vinyl boronic acid and CO/MeOH, respectively (Scheme 8).
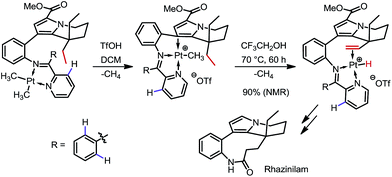 |
| | Scheme 7 Pt-mediated dehydrogenation of a remote ethyl group. | |
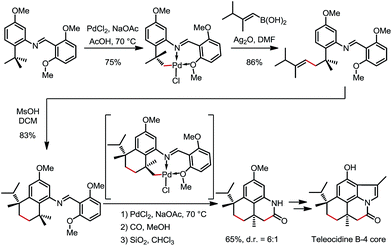 |
| | Scheme 8 Pd-mediated C(sp3)–H alkenylation and carbonylation of 2-tert-butylaniline derivatives. | |
b. Catalytic reactions
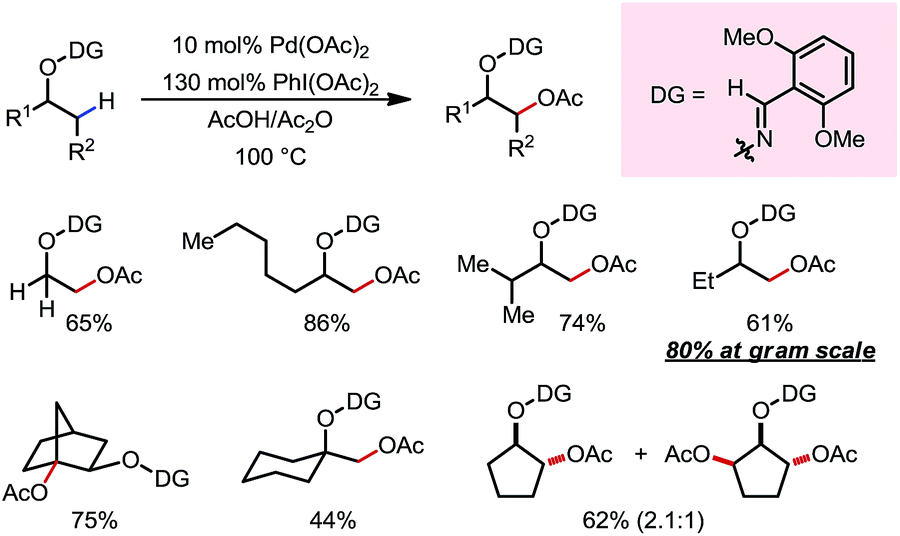
In 2012, Dong and co-workers reported a Pd-catalyzed β-C(sp3)–H acetoxylation reaction of masked alcohols using PIDA as the oxidant (Scheme 9).15 A 2,6-bismethoxybenzaldoxime protecting group was used as an exo-DG, and to mask the hydroxyl group of the alcohol. The ortho-OMe groups once again were found to be indispensable, as they blocked the endo reaction sites and served as potential secondary coordination sites. Methyl, cyclic methylene and even bridgehead methine C–H bonds were readily acetoxylated in a group of primary, secondary and tertiary alcohol derivatives, providing chemically differentiated 1,2-diols after orthogonal deprotection. It is worth mentioning that the DG can be conveniently removed using zinc and acetic acid.
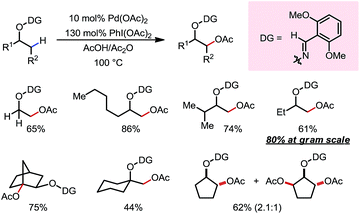 |
| | Scheme 9 Pd-catalyzed β-C–H acetoxylation of masked alcohols. | |
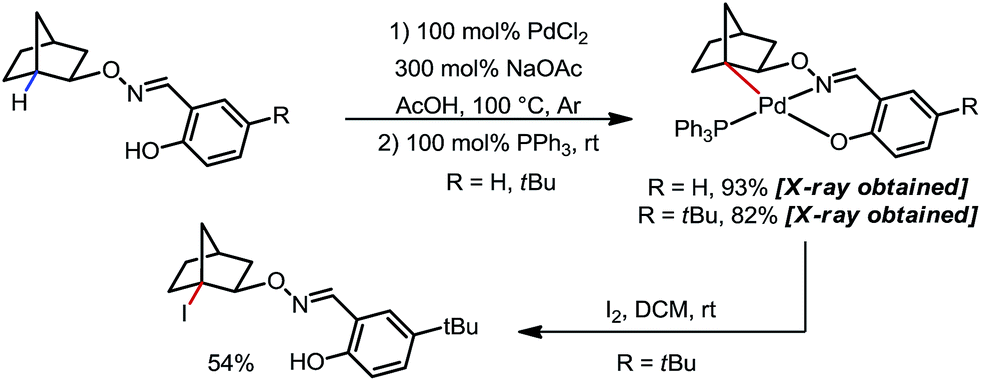
The mechanism was investigated by the same team in 2016 to elucidate the unusual activation of bridgehead C–H bonds.16 With a salicylaldehyde-derived exo-DG, the C–H cyclopalladation occurred smoothly at the bridgehead position, furnishing a [6,5]-fused palladacycle (Scheme 10). Further treatment of the complex with iodine led to iodination at the bridgehead position in a moderate yield. The previously used 2,6-bismethoxybenzaldoxime DG, on the other hand, was found incompatible in this stoichiometric study, due to an elimination to yield 2,6-bismethoxybenzonitrile when no oxidant was applied.
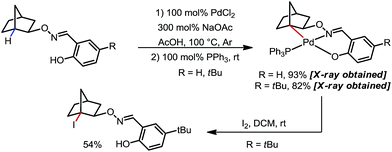 |
| | Scheme 10 C–H cyclopalladation at bridgehead positions enabled by an exo-directing group. | |
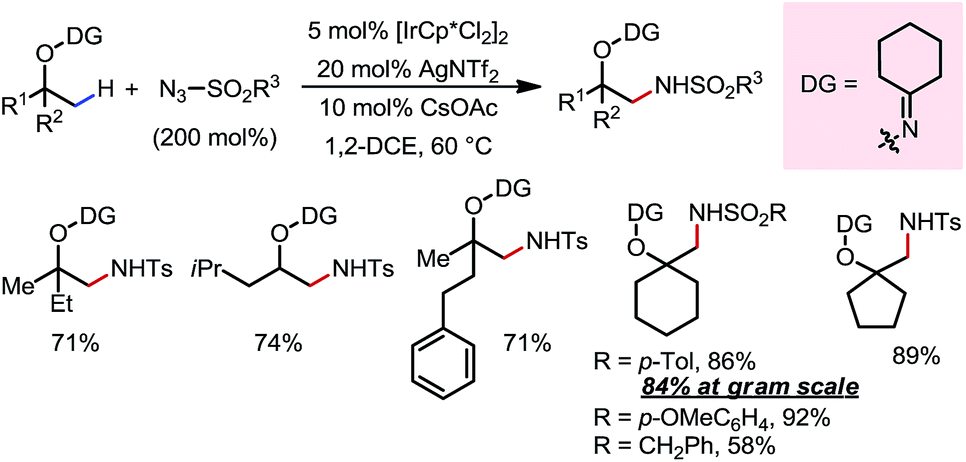
An Ir(III)-catalyzed β-C(sp3)–H amidation reaction of masked alcohols was reported in 2014 by Chang and co-workers using sulfonyl azides as the amine source (Scheme 11).17 A cyclohexyl ketoxime-type DG was introduced on the free alcohol to facilitate the C–H activation through formation of a five-membered exo-iridacycle. A range of primary, secondary and tertiary alcohols can be readily amidated at β-methyl positions using different sulfonyl azides, furnishing β-amino alcohols after removal of the DG through reductive cleavage of the N–O bond using LiAlH4.
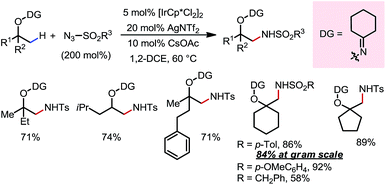 |
| | Scheme 11 Ir-catalyzed β-C–H amidation of masked alcohols. | |
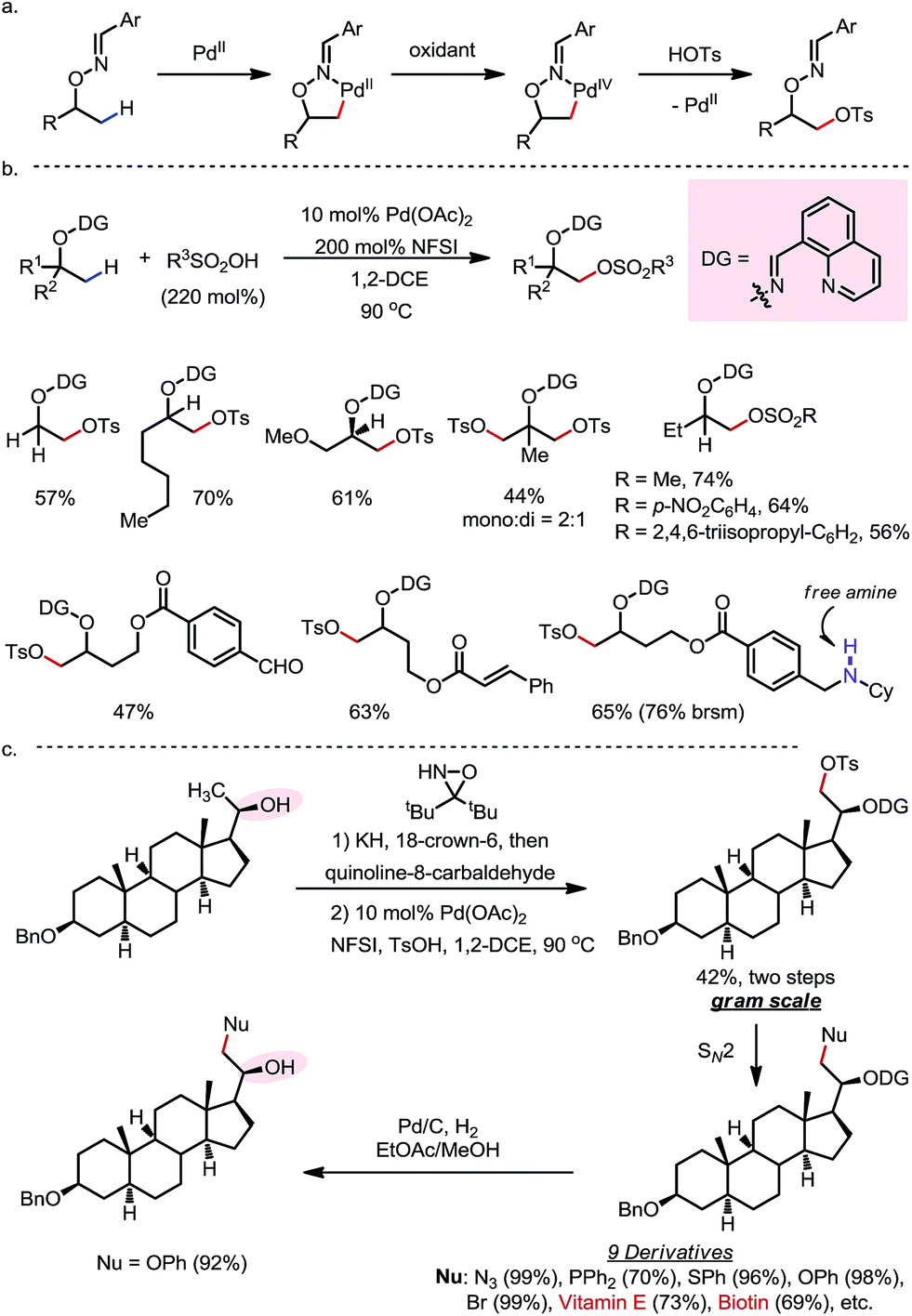
In 2015, a Pd-catalyzed β-C(sp3)–H sulfonyloxylation reaction of masked alcohols was reported by Dong and co-workers, using NFSI as the oxidant and sulfonic acids as the source of the sulfonyloxy groups (Scheme 12).18 An 8-quinolinecarbaldoxime-type exo-DG, which can be installed through a one- or two-pot procedure from the free alcohol, was found to be optimal. The proposed mechanism following a Pd(II)–Pd(IV) cycle is depicted in Scheme 12a, and the reaction proceeds through (i) β-C–H palladation to form a five-membered exo-palladacycle, (ii) oxidation to generate Pd(IV) and (iii) reductive elimination to yield the product and regenerate the Pd(II) catalyst.
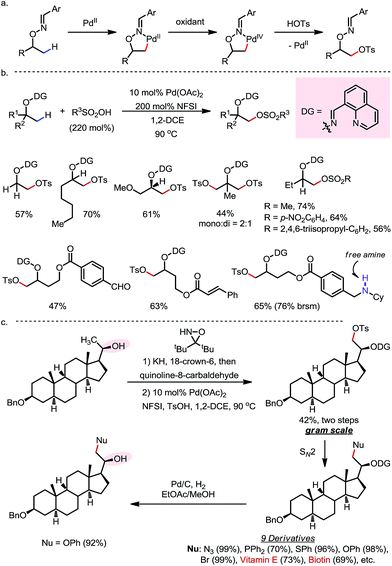 |
| | Scheme 12 Pd-catalyzed β-C–H sulfonyloxylation of masked alcohols. | |
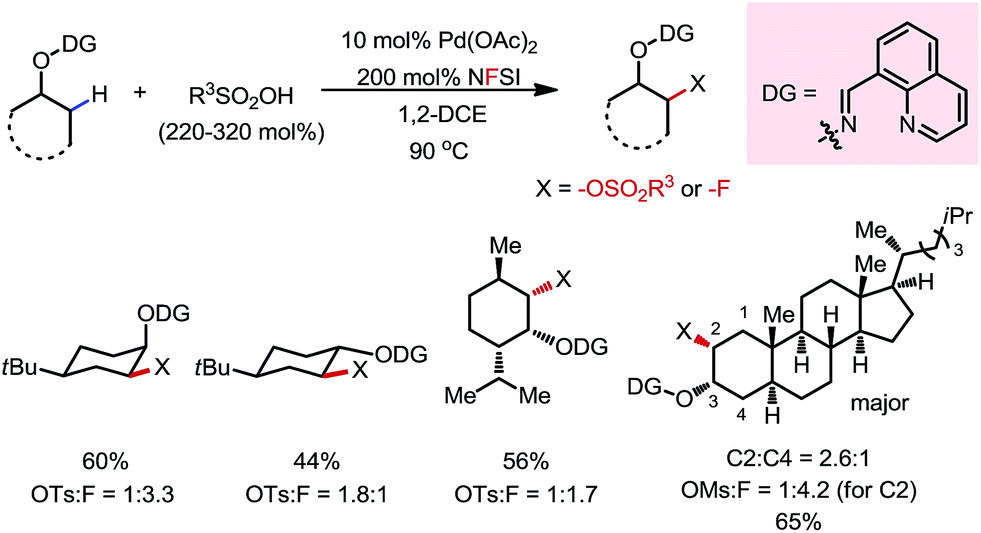
A variety of primary, secondary and tertiary alcohols with different FGs were found to be compatible in this reaction (Scheme 12b). In addition to OTs, other sulfonyloxy groups, such as OMs and ONs, were readily introduced at the β-methyl position and could then serve as a good leaving group for further derivatization. For example, the mono-protected 2H-pregnanediol was rapidly functionalized to give various derivatives at the inert C21 position through sequential β-tosyloxylation and SN2 reactions (Scheme 12c). The DG can be easily removed either under catalytic hydrogenation conditions or using zinc and acetic acid. Moreover, the C–H bonds at the cyclic methylenes could also be functionalized when no β-methyl was available (Scheme 13). Interestingly, competing β-C–H fluorination occurred along with the β-sulfonyloxylation. A preference for functionalizing equatorial C–H bonds was observed in all cases.
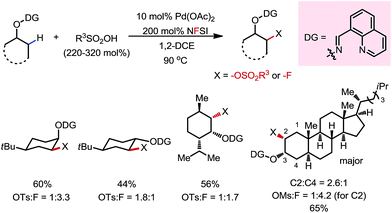 |
| | Scheme 13 Pd-catalyzed β-C–H fluorination/sulfonyloxylation of masked cyclohexanols. | |
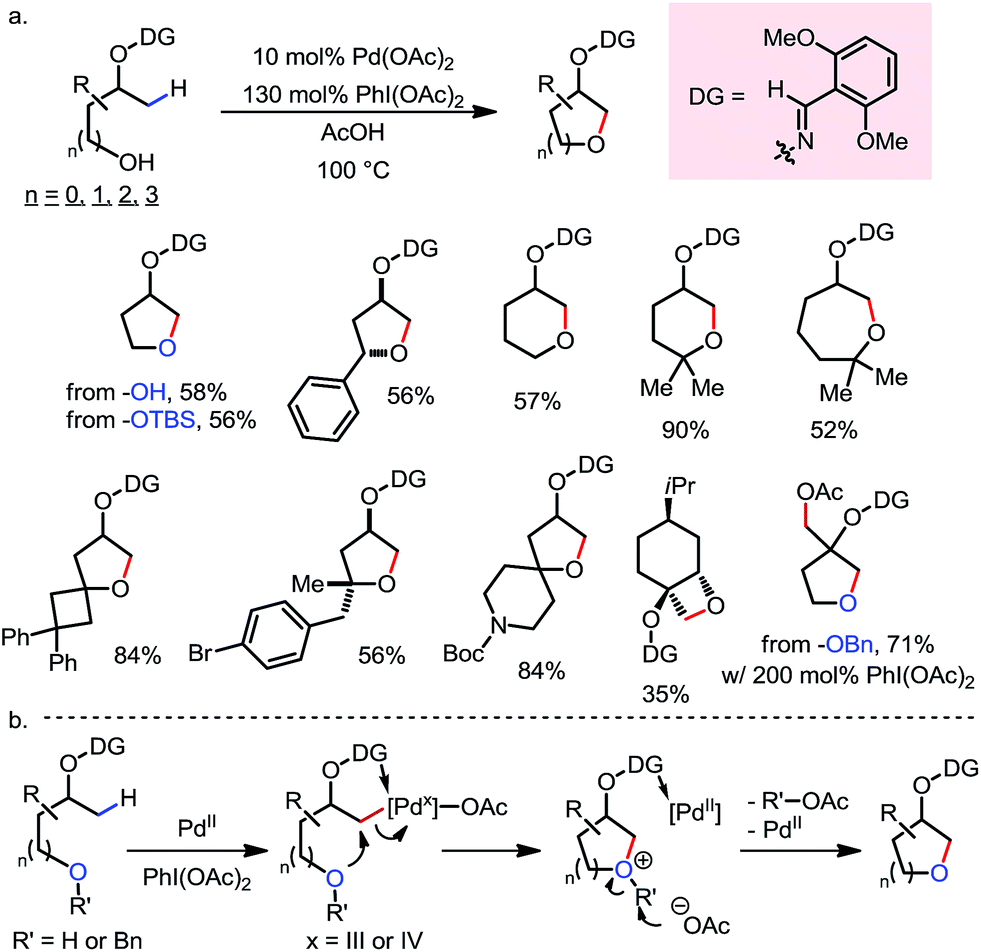
A Pd-catalyzed intramolecular β-C(sp3)–H etherification reaction with internal hydroxyl nucleophiles was reported by Dong and co-workers in 2015 (Scheme 14).19 Directed by an oxime-masked alcohol, various aliphatic cyclic ethers with 4- to 7-membered rings were readily formed through dehydrogenative annulation at β-methyl positions. Tethered primary, secondary and tertiary hydroxyl groups can all be used as nucleophiles, as well as TBS- and Bn-protected alcohols (Scheme 14a). The reaction was proposed to begin with β-C–H palladation, followed by oxidation to form a hypervalent Pd species and then an SN2-type reductive elimination to furnish the ether product (Scheme 14b).
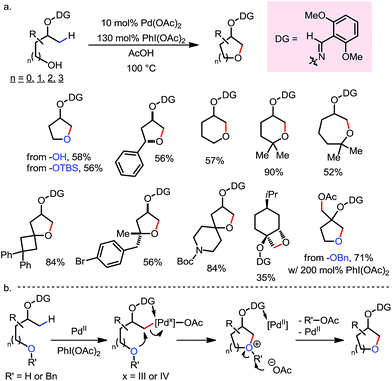 |
| | Scheme 14 Pd-catalyzed intramolecular β-C–H etherification with internal hydroxyl nucleophiles. | |
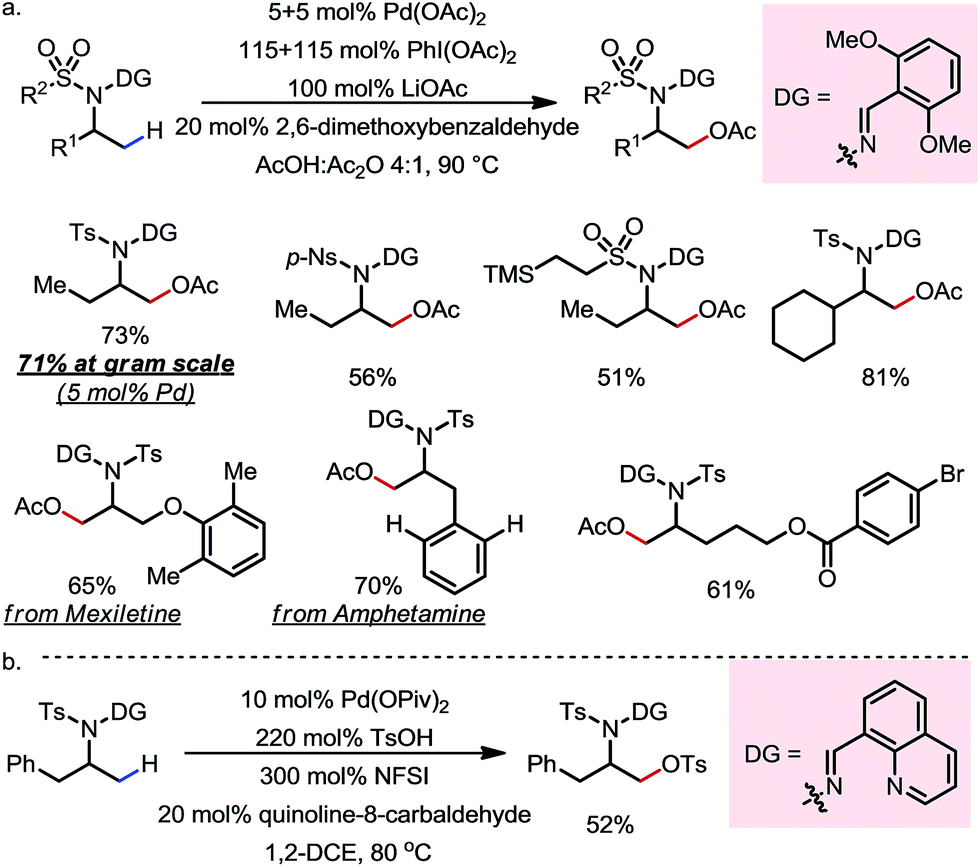
Apart from using alcohol-based substrates, in 2016 Dong and co-workers reported a Pd-catalyzed β-C(sp3)–H oxygenation reaction of sulfonyl-protected primary amines (Scheme 15).20 A removable hydrazone-based exo-DG, which was introduced through a one-pot procedure on the free amine, was capable of promoting the β-C–H activation through formation of a five-membered exo-palladacycle. Similar to the case of oxime-based DGs, 2,6-bismethoxyphenyl- and 8-quinolinyl-derived hydrazones were used for the β-acetoxylation (Scheme 15a) and the β-tosyloxylation (Scheme 15b), respectively. A wide range of primary amines with different skeletons and FGs, including those equipped with removable sulfonyl protecting groups (such as p-nosyl and SES), were readily oxidized at the β-methyl C–H bonds. The β-OTs could be further derivatized through SN2 reactions. The DG can be readily removed through cleavage of the N–N bond using zinc and acetic acid.
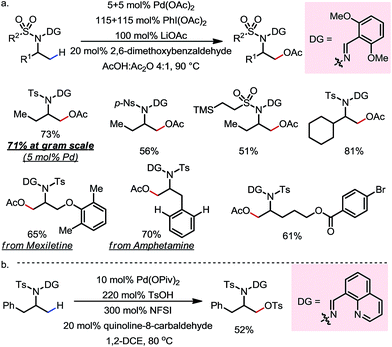 |
| | Scheme 15 Pd-catalyzed β-C–H oxygenation of protected amines. | |
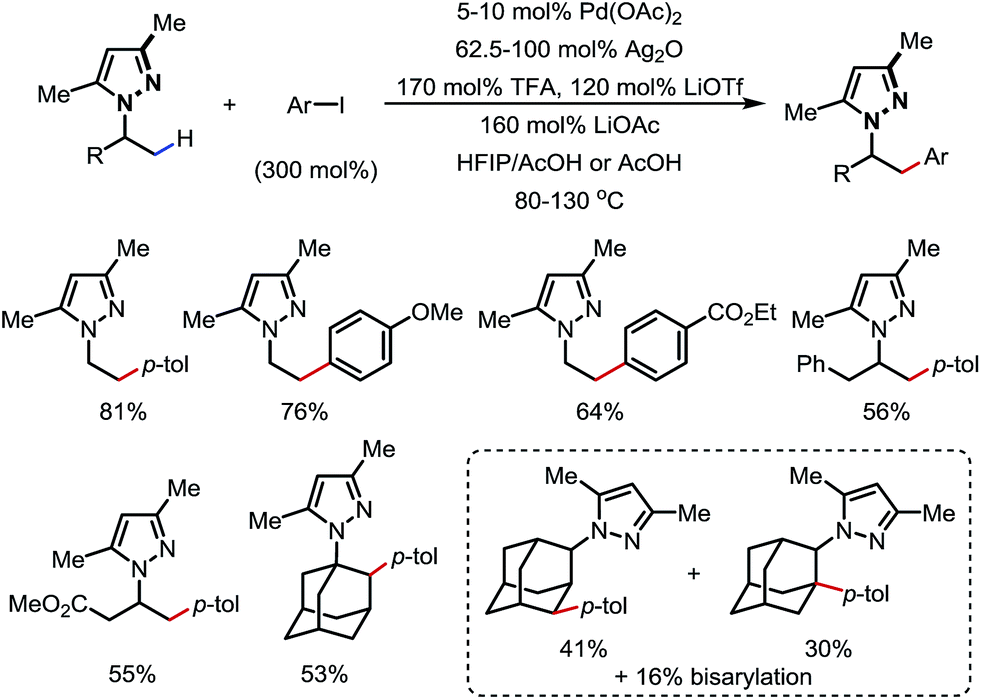
In 2017, Daugulis and co-workers reported a Pd-catalyzed β-C(sp3)–H arylation promoted by a pyrazole DG (Scheme 16).21 Aryl iodides were used as the aryl source, and a high yield was achieved when combining LiOTf/TFA/LiOAc and Ag2O as the additives. Methyl and some methylene/methine C–H bonds were arylated with a good tolerance of FGs. Upon ozonolysis, the pyrazole motif can be converted to an amide moiety.
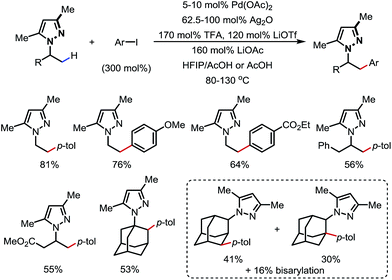 |
| | Scheme 16 Pd-catalyzed β-C–H arylation of 1-alkyl-1H-pyrazoles. | |
In 2017, Liu22a and Liu22b independently reported the Pd-catalyzed β-sulfonimidation of masked alcohols using NFSI as the oxidant and the nitrogen source (Scheme 17). The 8-quinolyl-derived aldoxime was found to be the optimal exo DG in each work. Methyl C(sp3)–H bonds in various skeletons can be activated with tolerance of many FGs, such as free alcohols, olefins, primary alkyl chlorides and tosylates. 1,2-Amino alcohol derivatives were furnished upon removal of the DG and the sulfonyl group.
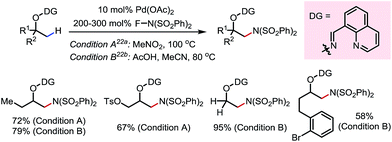 |
| | Scheme 17 Pd-catalyzed β-C–H sulfonimidation of masked alcohols. | |
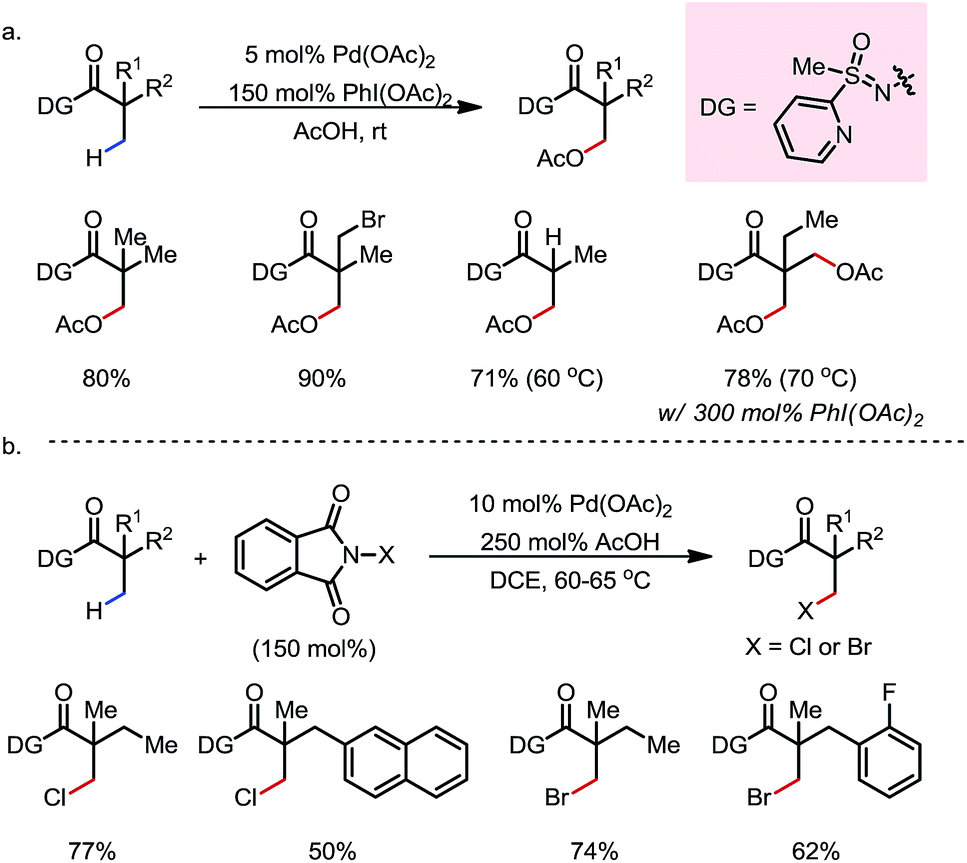
A unique S-methyl-S-2-pyridylsulfoximine-based DG,23 developed by the Sahoo group, was found to be another effective exo-type DG that promotes C(sp3)–H activation (Scheme 18).23b,c Various primary β-C–H bonds of sulfoximine-N-amides, typically derived from the corresponding carboxylic acids or acyl chlorides, underwent facile Pd-catalyzed acetoxylation23b (Scheme 18a) or halogenation23c (Scheme 18b) using PhI(OAc)2 or N-halophthalimide as the oxidant, respectively. A number of FGs were found to be compatible. The DG can be removed and recycled from the products through hydrolysis, furnishing free carboxylic acids in high yields.
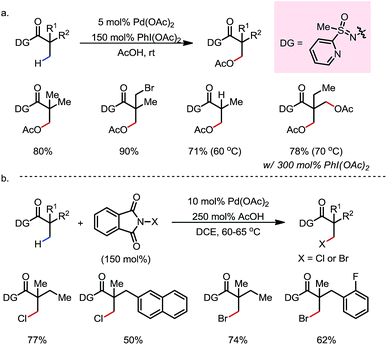 |
| | Scheme 18 Pd-catalyzed β-C–H acetoxylation and halogenation of sulfoximine-N-amides. | |
While significant progress has been made in C–H activation using amide-,24 sulfonamide-,25 hydrazone-20 and urea-type DGs,26 extra synthetic steps are required for DG installation and removal. Hence, direct employment of free amines as the substrates became an ideal approach.27 In 2016, the Dong,28 Ge,29 Yu,30 and Murakami31 groups independently reported the Pd-catalyzed γ-C(sp3)–H arylation of free primary amines using 8-formylquinoline, glyoxylic acid, 2-hydroxynicotinaldehyde and 3,5-di-tert-butylsalicylaldehyde as temporary DGs (TDGs), respectively.
8-formylquinoline was first used by Dong and co-workers (Scheme 19).28 Through in situ condensation with free primary amines, an imine was formed, which promoted the γ-C–H activation as an exo-DG via formation of a [6,5]-fused palladacycle. By coupling with bisaryliodonium salts as the oxidant and the aryl source, methyl and methylene (cyclic and acyclic) C–H bonds at the γ-position of various free primary amines were readily arylated with broad FG tolerance (Scheme 19a). While a one-pot benzoyl protection was generally conducted afterwards for the convenience of isolation, direct isolation of free arylated amines was feasible on a larger scale, along with the recovery of most of the DG component. Catalytic use of the DG component was also demonstrated, albeit with relatively low efficiency (Scheme 19b).
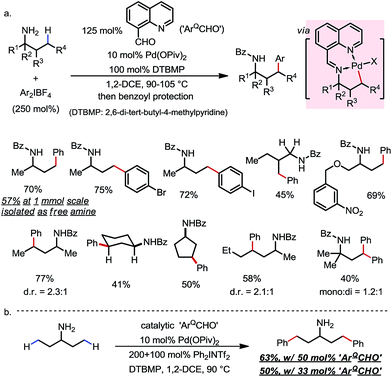 |
| | Scheme 19 Pd-catalyzed γ-C–H arylation of free primary amines using 8-formylquinoline as the transient directing group. | |
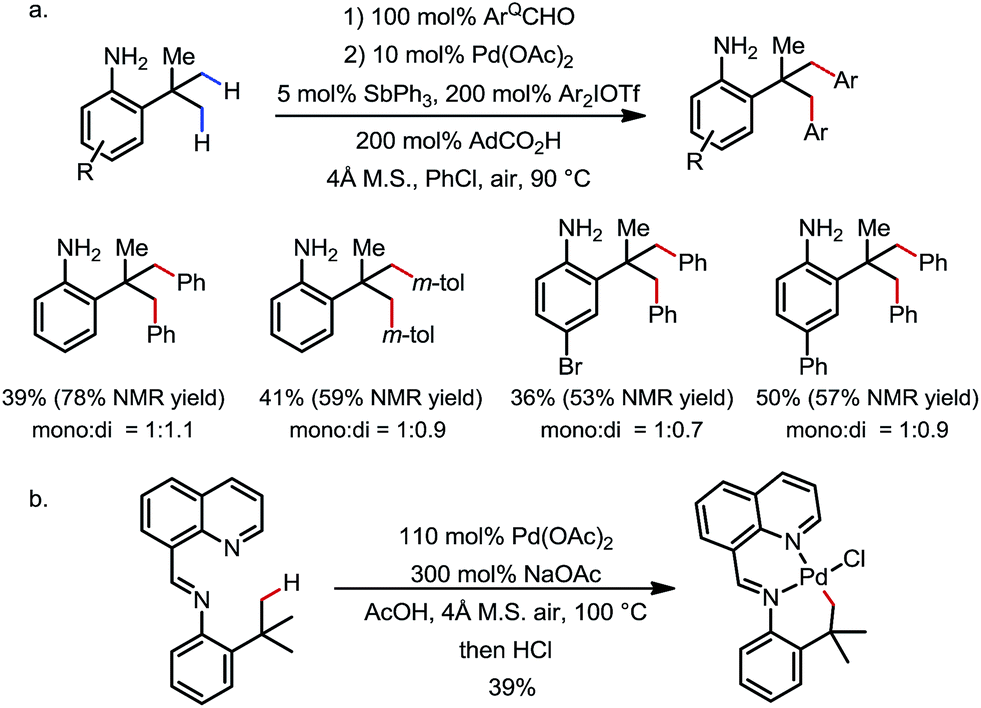
The δ-arylation of 2-tert-butylanilines was also achieved under modified reaction conditions (Scheme 20). To achieve higher yields, the aniline substrates were pre-mixed with 8-formylquinoline for 1 h. A group of FGs were found to be compatible, and the aniline products were directly isolated without derivatization (Scheme 20a). A cyclopalladated complex was isolated by reacting the pre-condensed imine with stoichiometric Pd(OAc)2, providing solid evidence for the exo coordination mode (Scheme 20b).
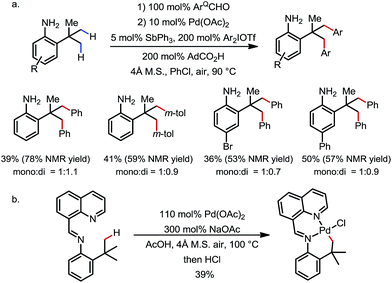 |
| | Scheme 20 Pd-catalyzed δ-C–H arylation of 2-tert-butylanilines. | |
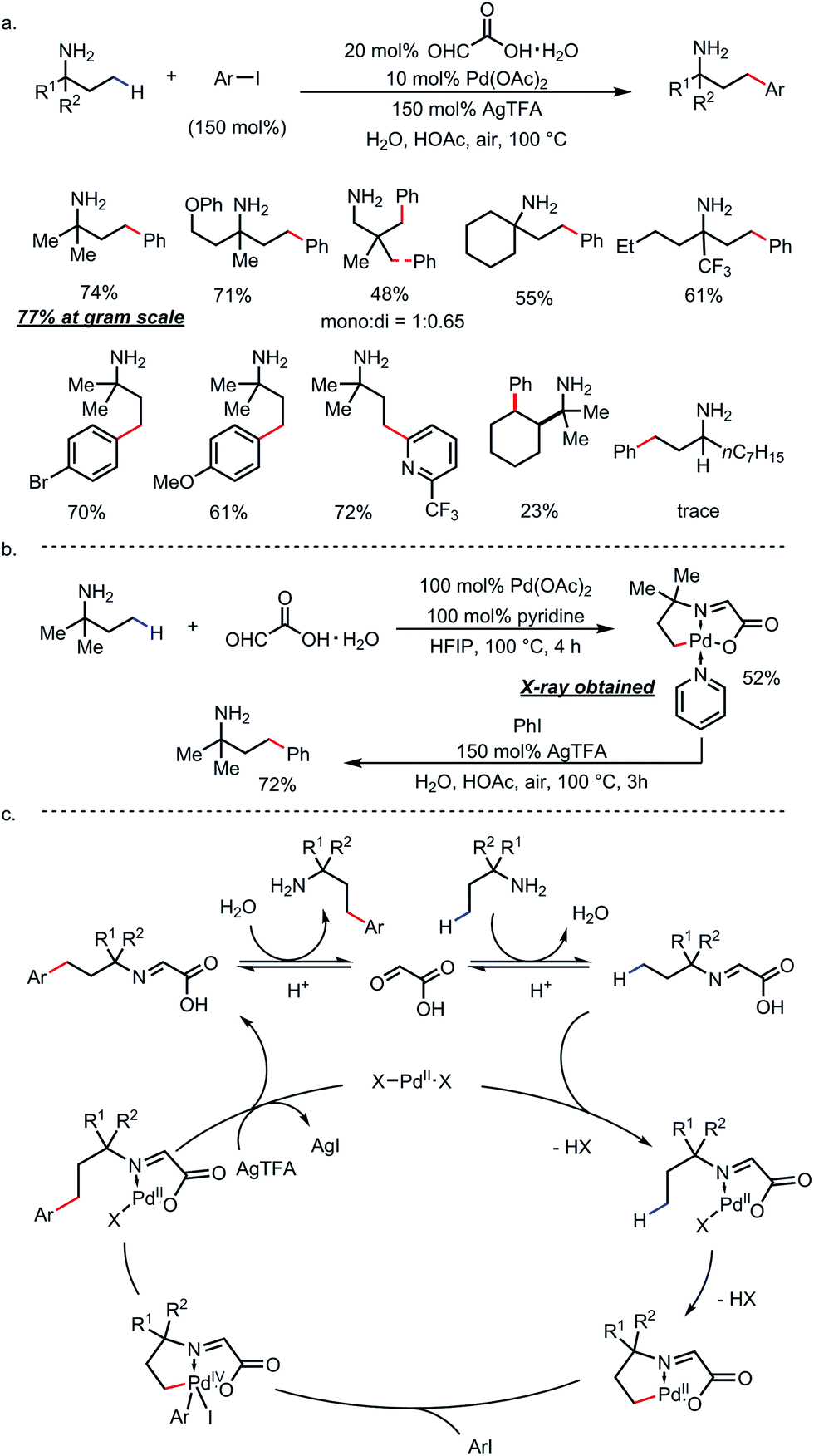
Ge and co-workers identified glyoxylic acid monohydrate as another optimal TDG (Scheme 21).29 Using 20 mol% of the TDG and 10 mol% of Pd(OAc)2 as the catalyst, γ-arylated amines were furnished and directly isolated in high yields from a large number of primary amines and aryl iodides in the presence of stoichiometric silver salt (Scheme 21a). Amines with a secondary α-carbon were found incompatible due to α-oxidation and the lack of the Thorpe–Ingold effect. A C–H palladated complex that featured a [5,5]-fused palladacycle was obtained by reacting tert-amylamine with glyoxylic acid and stoichiometric Pd(OAc)2. Further treatment of this complex under arylation conditions yielded γ-arylated tert-amylamine as the product (Scheme 21b). A detailed reaction mechanism was proposed accordingly (Scheme 21c).
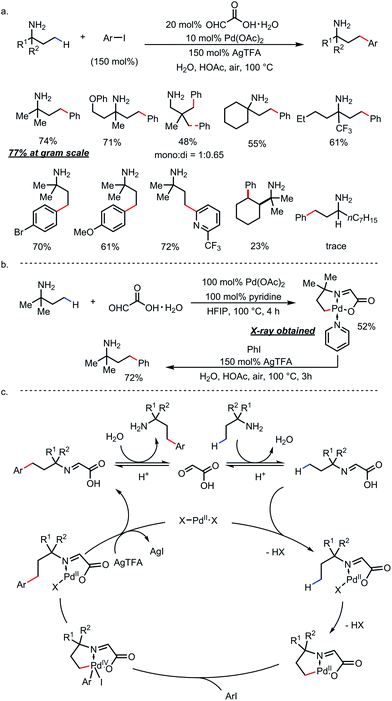 |
| | Scheme 21 Pd-catalyzed γ-C–H arylation of free primary amines using glyoxylic acid as the transient directing group. | |
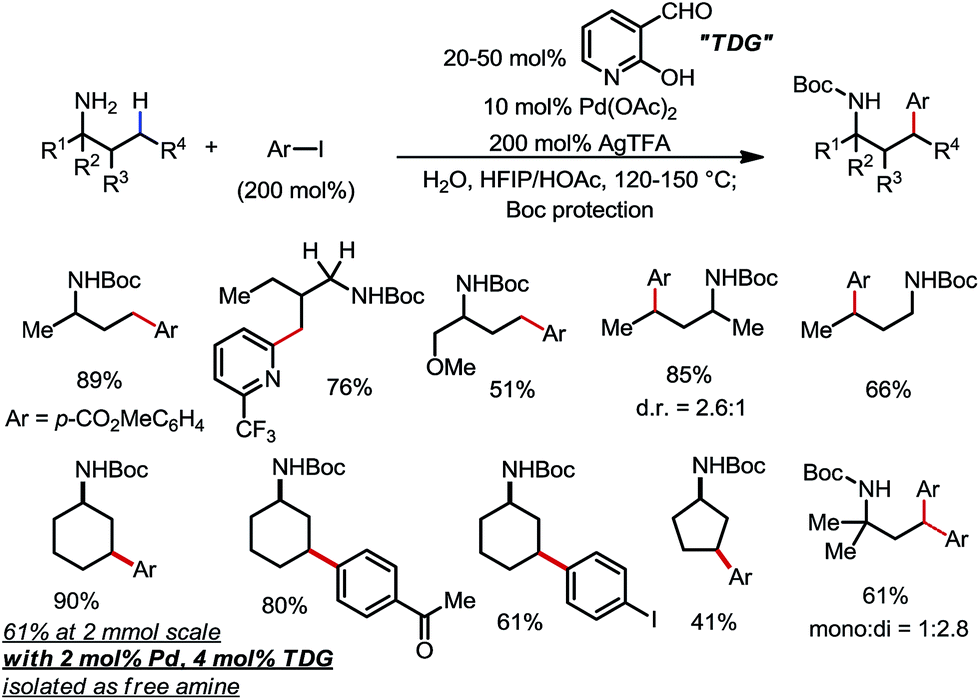
Yu and co-workers discovered that the same type of reaction could also be achieved using 20–50 mol% 2-hydroxynicotinaldehyde as the TDG (Scheme 22).30 A range of different aryl and heteroaryl iodides were readily coupled with primary amines through the activation of the γ-methyl and methylene C–H bonds. Various FGs were well tolerated. In one example, the unprotected γ-arylated cyclohexylamine product was obtained in 61% yield on a 2 mmol scale when only 4 mol% TDG and 2 mol% Pd were employed.
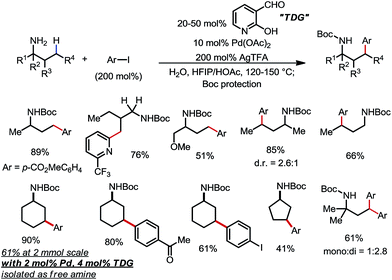 |
| | Scheme 22 Pd-catalyzed γ-C–H arylation of free primary amines using 2-hydroxynicotinaldehyde as the transient directing group. | |
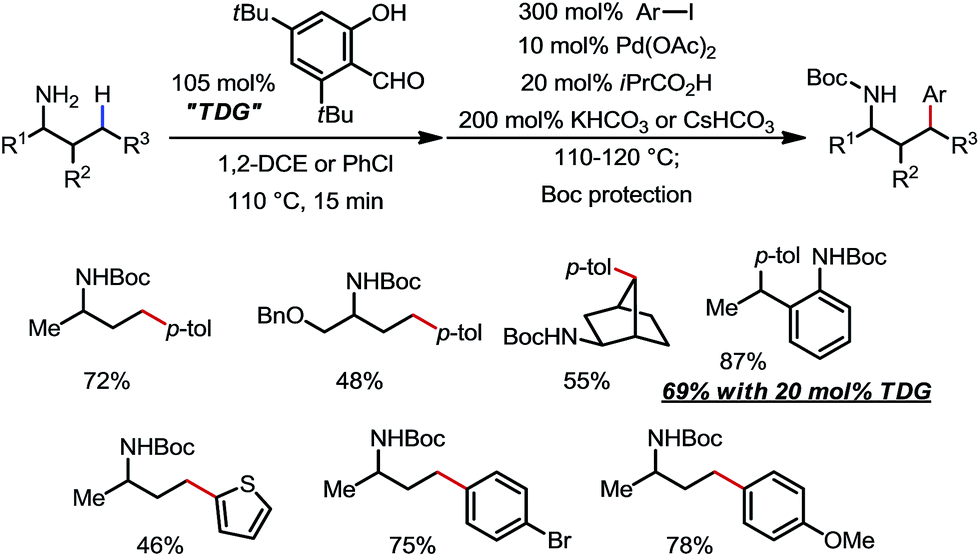
3,5-Di-tert-butylsalicylaldehyde was found to be another effective TDG by Murakami and co-workers (Scheme 23).31 Pre-condensation between the amine substrates and the TDG was carried out at 110 °C for 15 min before the addition of the rest of reagents. Various aryl iodides and unprotected primary amines were compatible. The feasibility of using the DG in a catalytic fashion was also demonstrated in one example with 2-ethylaniline as the substrate.
 |
| | Scheme 23 Pd-catalyzed γ-C–H arylation of free primary amines using 3,5-di-tert-butylsalicylaldehyde as the transient directing group. | |
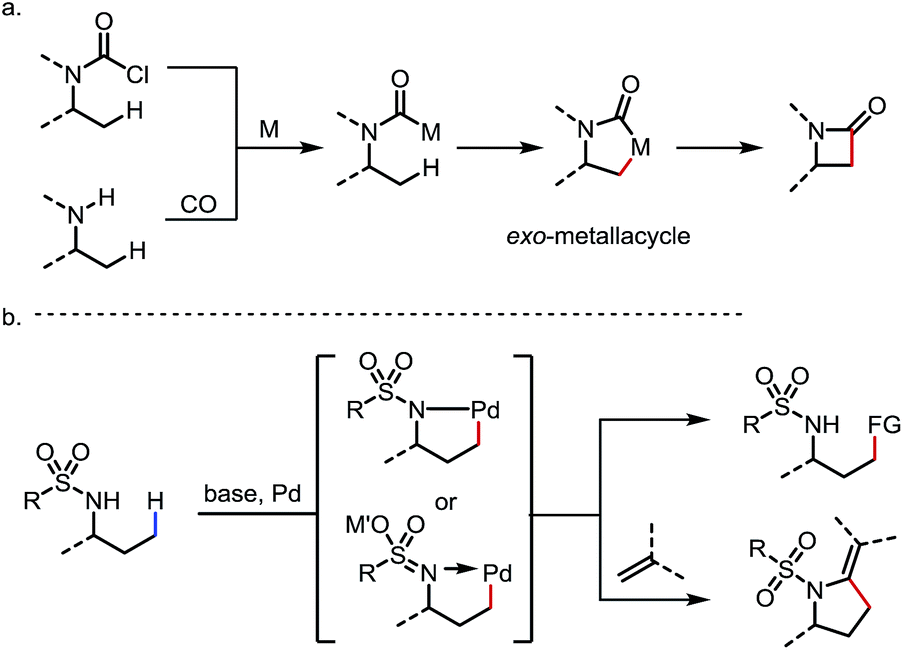
Two other types of sp3 C–H activation via the formation of exo-metallacycles are also worth mentioning, although the DGs or the directing motifs applied in these reactions may not strictly qualify as typical exo-DGs (Scheme 24). In the first case, a carbamoyl-metal species is generated from either (i) oxidative addition of acyl chlorides32 or (ii) carbonylation of free amines.27c,33 Upon cyclometalation at the β-C–H bond, an exo-metallacycle is formed, which further transforms to a β-lactam through reductive elimination32,33 (Scheme 24a). A similar reaction mode was also reported with vinyl bromides through sequential oxidative addition/C–H metalation to induce intramolecular C(sp3)–H activation and the subsequent formation of C–C bonds.1o,34 In the second scenario, it is sometimes proposed that the deprotonation of the sulfonamide under basic conditions generates a sulfonaimine motif, which promotes the cyclopalladation at the γ-position as an exo-DG.25,35 Further transformations of this intermediate lead to various C–C bond forming reactions (Scheme 24b). Given the space limitation, these two types of reaction will not be discussed in detail.
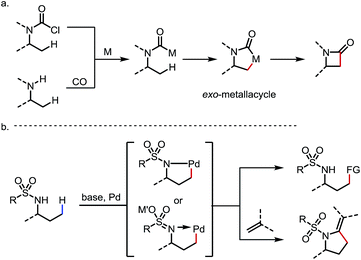 |
| | Scheme 24 Two other types of C(sp3)–H activation via the formation of exo-metallacycles. | |
Conclusions
The exo-type DGs have emerged as a powerful tool for the functionalization of unactivated sp3 C–H bonds, particularly in alcohol or amine-containing compounds. A range of novel directing modes and unconventional site selectivities have therefore been disclosed using exo-type DGs, such as in the β-functionalization of masked alcohols and in the β- and γ-functionalization of amines. Functionalization of other classes of compound, beyond alcohols and amines, using such an exo-directing mode will continue to be discovered. In addition, activation of more remote C–H bonds through the formation of larger metallacycles, as well as new catalytic transformations using other transition metals or new classes of exo-DG, should represent promising directions of research. Finally, it is anticipated that applications of these distinct methods in the syntheses of complex natural products or drug molecules would appear more frequently in future work.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We thank the Frasch Foundation for funding. Y. X. thanks the William Rainey Harper Dissertation Fellowship and the Bristol-Myers Squibb Graduate Fellowship for financial support. Dr Siu Yin (Serena) Lee is acknowledged for proofreading the manuscript.
Notes and references
-
(a) O. Daugulis, H.-Q. Do and D. Shabashov, Acc. Chem. Res., 2009, 42, 1074–1086 CrossRef CAS PubMed;
(b) T. W. Lyons and M. S. Sanford, Chem. Rev., 2010, 110, 1147–1169 CrossRef CAS PubMed;
(c) D. A. Colby, R. G. Bergman and J. A. Ellman, Chem. Rev., 2010, 110, 624–655 CrossRef CAS PubMed;
(d)
J.-Q. Yu and Z. Shi, Topics in Current Chemistry, C–H Activation, Springer, 2010, vol. 292 Search PubMed;
(e) H. M. L. Davies, J. Du Bois and J.-Q. Yu, Chem. Soc. Rev., 2011, 40, 1855–1856 RSC;
(f) J. Yamaguchi, A. D. Yamaguchi and K. Itami, Angew. Chem., Int. Ed., 2012, 51, 8960–9009 CrossRef CAS PubMed;
(g) M. C. White, Science, 2012, 335, 807–810 CrossRef CAS PubMed;
(h) L. Yang and H. Huang, Chem. Rev., 2015, 115, 3468–3517 CrossRef CAS PubMedY. Minami and T. Hiyama, Acc. Chem. Res., 2016, 49, 67–77 CrossRef CAS PubMed;
(i) T. Gensch, M. Hopkinson, F. Glorius and J. Wencel-Delord, Chem. Soc. Rev., 2016, 45, 2900–2936 RSC;
(j) J. He, M. Wasa, K. S. L. Chan, Q. Shao and J.-Q. Yu, Chem. Rev., 2017, 117, 8754–8786 CrossRef CAS PubMed;
(k) Z. Dong, Z. Ren, S. J. Thompson, Y. Xu and G. Dong, Chem. Rev., 2017, 117, 9333–9403 CrossRef CAS PubMed;
(l) J. R. Hummel, J. A. Boerth and J. A. Ellman, Chem. Rev., 2017, 117, 9163–9227 CrossRef CAS PubMed;
(m) D. S. Kim, W. J. Park and C. H. Jun, Chem. Rev., 2017, 117, 8977–9015 CrossRef CAS PubMed;
(n) Y. Park, Y. Kim and S. Chang, Chem. Rev., 2017, 117, 9247–9301 CrossRef CAS PubMed;
(o) O. Baudoin, Acc. Chem. Res., 2017, 50, 1114–1123 CrossRef CAS PubMed.
- For representative works using endo-type DGs, see:
(a) L. V. Desai, K. L. Hull and M. S. Sanford, J. Am. Chem. Soc., 2004, 126, 9542–9543 CrossRef CAS PubMed;
(b) R. Giri, X. Chen and J.-Q. Yu, Angew. Chem., Int. Ed., 2005, 44, 2112–2115 CrossRef CAS PubMed;
(c) R. Giri, N. Maugel, J.-J. Li, D.-H. Wang, S. P. Breazzano, L. B. Saunders and J.-Q. Yu, J. Am. Chem. Soc., 2007, 129, 3510–3511 CrossRef CAS PubMed;
(d) F.-L. Zhang, K. Hong, T.-J. Li, H. Park and J.-Q. Yu, Science, 2016, 351, 252–256 CrossRef CAS PubMed;
(e) K. Yang, Q. Li, Y. Liu, G. Li and H. Ge, J. Am. Chem. Soc., 2016, 138, 12775–12778 CrossRef CAS PubMed;
(f) Y. Xu, M. C. Young and G. Dong, J. Am. Chem. Soc., 2017, 139, 5716–5719 CrossRef CAS PubMed.
- J. Dupont, C. S. Consorti and J. Spencer, Chem. Rev., 2005, 105, 2527–2571 CrossRef CAS PubMed.
-
(a) G. De Munno, M. Ghedini and F. Neve, Inorg. Chim. Acta, 1995, 239, 155–158 CrossRef CAS;
(b) J. Barro, J. Granell, D. Saiz, J. Sales, M. Font-Bardía and X. Solans, J. Organomet. Chem., 1993, 456, 147–154 CrossRef CAS.
- P. W. Clark, S. F. Dyke and G. Smith, J. Organomet. Chem., 1987, 330, 447–460 CrossRef CAS.
- O. N. Gorunova, K. J. Keuseman, B. M. Goebel, N. A. Kataeva, A. V. Churakov, L. G. Kuz’mina, V. V. Dunina and I. P. Smoliakova, J. Organomet. Chem., 2004, 689, 2382–2394 CrossRef CAS.
- M. Crespo, M. Martinez and J. Sales, Organometallics, 1992, 11, 1288–1295 CrossRef CAS.
- J. Albert, J. Barro and J. Granell, J. Organomet. Chem., 1991, 408, 115–123 CrossRef CAS.
-
(a) R. Y. Mawo, S. Mustakim, V. G. Young Jr, M. R. Hoffmann and I. P. Smoliakova, Organometallics, 2007, 26, 1801–1810 CrossRef CAS;
(b) K. J. Keuseman, I. P. Smoliakova and V. V. Dunina, Organometallics, 2005, 24, 4159–4169 CrossRef CAS.
- J. Albert, R. M. Ceder, M. Gomez, J. Granell and J. Sales, Organometallics, 1992, 11, 1536–1541 CrossRef CAS.
- J. Granell, R. Moragas, J. Sales, M. Font-Bardía and X. Solans, J. Chem. Soc., Dalton Trans., 1993, 8, 1237–1244 RSC.
- J. Albert, M. Gomez, J. Granell and J. Sales, Organometallics, 1990, 9, 1405–1413 CrossRef CAS.
- J. A. Johnson and D. Sames, J. Am. Chem. Soc., 2000, 122, 6321–6322 CrossRef CAS.
- B. D. Dangel, K. Godula, S. W. Youn, B. Sezen and D. Sames, J. Am. Chem. Soc., 2002, 124, 11856–11857 CrossRef CAS PubMed.
- Z. Ren, F. Mo and G. Dong, J. Am. Chem. Soc., 2012, 134, 16991–16994 CrossRef CAS PubMed.
- Z. Ren and G. Dong, Organometallics, 2016, 35, 1057–1059 CrossRef CAS.
- T. Kang, H. Kim, J. G. Kim and S. Chang, Chem. Commun., 2014, 50, 12073–12075 RSC.
- Y. Xu, G. Yan, Z. Ren and G. Dong, Nat. Chem., 2015, 7, 829–834 CrossRef CAS PubMed.
- S. J. Thompson, D. Q. Thach and G. Dong, J. Am. Chem. Soc., 2015, 137, 11586–11589 CrossRef CAS PubMed.
- Z. Huang, C. Wang and G. Dong, Angew. Chem., Int. Ed., 2016, 55, 5299–5303 CrossRef CAS PubMed.
- N. Gulia and O. Daugulis, Angew. Chem., Int. Ed., 2017, 56, 3630–3634 CrossRef CAS PubMed.
-
(a) Y. Dong and G. Liu, J. Org. Chem., 2017, 82, 3864–3872 CrossRef CAS PubMed;
(b) L. Jin, X. Zeng, S. Li, X. Hong, G. Qiu and P. Liu, Chem. Commun., 2017, 53, 3986–3989 RSC.
-
(a) M. R. Yadav, R. K. Rit and A. K. Sahoo, Chem.–Eur. J., 2012, 18, 5541–5545 CrossRef CAS PubMed;
(b) R. K. Rit, M. R. Yadav and A. K. Sahoo, Org. Lett., 2012, 14, 3724–3727 CrossRef CAS PubMed;
(c) R. K. Rit, M. R. Yadav, K. Ghosh, M. Shankar and A. K. Sahoo, Org. Lett., 2014, 16, 5258–5261 CrossRef CAS PubMed.
- For a seminal example, see: V. G. Zaitsev, D. Shabashov and O. Daugulis, J. Am. Chem. Soc., 2005, 127, 13154–13155 CrossRef CAS PubMed.
- For selected seminal examples, see:
(a) N. Rodríguez, J. Romero-Revilla, M. A. Fernández-Ibáñez and J. C. Carretero, Chem. Sci., 2013, 4, 175–179 RSC;
(b) K. S. L. Chan, M. Wasa, L. Chu, B. N. Laforteza, M. Miura and J.-Q. Yu, Nat. Chem., 2014, 6, 146–150 CrossRef CAS PubMed;
(c) L. Chu, K.-J. Xiao and J.-Q. Yu, Science, 2014, 346, 451–455 CrossRef CAS PubMed;
(d) M. Yang, B. Su, Y. Wang, K. Chen, X. Jiang, Y.-F. Zhang, X.-S. Zhang, G. Chen, Y. Cheng, Z. Cao, Q.-Y. Guo, L. Wang and Z.-J. Shi, Nat. Commun., 2014, 5, 4707–4712 CrossRef CAS PubMed.
- For a seminal example, see: M. Kim, J. V. Mulcahy, C. G. Espino and J. Du Bois, Org. Lett., 2006, 8, 1073–1076 CrossRef CAS PubMed.
-
(a) C. He and M. J. Gaunt, Angew. Chem., Int. Ed., 2015, 54, 15840–15844 CrossRef CAS PubMed;
(b) A. P. Smalley and M. J. Gaunt, J. Am. Chem. Soc., 2015, 137, 10632–10641 CrossRef CAS PubMed;
(c) A. McNally, B. Haffemayer, B. S. L. Collins and M. J. Gaunt, Nature, 2014, 510, 129–133 CrossRef CAS PubMed. For a non-directed approach, see:
(d) M. Lee and M. S. Sanford, J. Am. Chem. Soc., 2015, 137, 12796–12799 CrossRef CAS PubMed.
- Y. Xu, M. C. Young, C. Wang, D. M. Magness and G. Dong, Angew. Chem., Int. Ed., 2016, 55, 9084–9087 CrossRef CAS PubMed.
- Y. Liu and H. Ge, Nat. Chem., 2017, 9, 26–32 CAS.
- Y. Wu, Y. Q. Chen, T. Liu, M. D. Eastgate and J. Q. Yu, J. Am. Chem. Soc., 2016, 138, 14554–14557 CrossRef CAS PubMed.
- A. Yada, W. Liao, Y. Sato and M. Murakami, Angew. Chem., Int. Ed., 2017, 56, 1073–1076 CrossRef CAS PubMed.
-
(a) C. Tsukano, M. Okuno and Y. Takemoto, Angew. Chem., Int. Ed., 2012, 51, 2763–2766 CrossRef CAS PubMed;
(b) D. Dailler, R. Rocaboy and O. Baudoin, Angew. Chem., Int. Ed., 2017, 129, 7324–7328 CrossRef.
-
(a) D. Willcox, B. G. N. Chappell, K. F. Hogg, J. Calleja, A. P. Smalley and M. J. Gaunt, Science, 2016, 354, 851–857 CrossRef CAS PubMed;
(b) J. R. Cabrera-Pardo, A. Trowbridge, M. Nappi, K. Ozaki and M. J. Gaunt, Angew. Chem., Int. Ed., 2017, 56, 11958–11962 CrossRef CAS PubMed.
- For a seminal example, see: J. Sofack-Kreutzer, N. Martin, A. Renaudat, R. Jazzar and O. Baudoin, Angew. Chem., Int. Ed., 2012, 51, 10399–10402 CrossRef CAS PubMed.
- H. Jiang, J. He, T. Liu and J.-Q. Yu, J. Am. Chem. Soc., 2016, 138, 2055–2059 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence and
Guangbin
Dong
and
Guangbin
Dong