
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTuning protein folding in lysosomal storage diseases: the chemistry behind pharmacological chaperones
David M.
Pereira
 *,
Patrícia
Valentão
and
Paula B.
Andrade
*,
Patrícia
Valentão
and
Paula B.
Andrade
REQUIMTE/LAQV, Laboratório de Farmacognosia, Departamento de Química, Faculdade de Farmácia, Universidade do Porto, Rua de Jorge Viterbo Ferreira 228, 4050-313 Porto, Portugal. E-mail: dpereira@ff.up.pt
First published on 10th January 2018
Abstract
Misfolding of proteins is the basis of several proteinopathies. Chemical and pharmacological chaperones are small molecules capable of inducing the correct conformation of proteins, thus being of interest for human therapeutics. The most recent developments in medicinal chemistry and in the drug development of pharmacological chaperones are discussed, with focus on lysosomal storage diseases.
 David M. Pereira | David M. Pereira obtained his PhD in Pharmaceutical Sciences at the University of Porto for his work on Phytochemistry and Pharmacognosy. In 2014, he was appointed Assistant Professor at the Faculty of Pharmacy at the same university. His research interests focus on natural products chemistry and biological assessment of small molecules. In particular, he is interested in small molecular modulators of endoplasmic reticulum stress and the proteostasis network, with connections to cancer and inflammation. |
 Patrícia Valentão | Patrícia Valentão obtained her PhD in Pharmaceutical Sciences at the University of Porto for her work on pharmacognosy. She was appointed Assistant Professor at the same university in 2010. Her research focuses on qualitative and quantitative metabolite profiling of terrestrial and marine natural matrices with the goal to reveal the composition/activity relationships of these compounds in various clinically relevant areas. |
 Paula B. Andrade | Paula B. Andrade is an Associate Professor at the Faculty of Pharmacy of the University of Porto. She coordinates the Natural Products group of the REQUIMENTE/LAQV, an institute active in Green Chemistry, and evaluates research projects for various international entities. Her research on bioactive substances originating from matrices of marine and terrestrial origin was published in more than 30 book chapters and over 300 articles in international journals. |
1 Introduction
Proteins are essential for life, with protein function being largely affected by their tridimensional structure. Folding of proteins may take place in several cellular locations, including the cytoplasm mitochondria, nucleus and also the endoplasmic reticulum (ER), the latter retaining polypeptides in the event that they are unable to correctly fold and being unable to be further trafficked to the Golgi apparatus and other organelles.When a misfolding event takes place, which may be a consequence of either environmental factors or mutations that impact protein conformation, proteins may expose hydrophobic segments that would not normally be available, which may result in intermolecular binding with other proteins and subsequent aggregation (Fig. 1, left). This aggregation has a wide range of deleterious effects and has been associated with the pathophysiology of several diseases, notably Alzheimer's disease (AD), Parkinson's disease (PD) and Huntington's.1,2 In other cases, the misfolding of proteins results in a lack of catalytic activity towards their corresponding substrates. To cope with these misfolding events, cells have developed a protein quality control (PQC) system that oversees protein folding and is ultimately aimed to induce or restore proper protein folding.3 This complex system relies on the unfolded protein response (UPR) in the ER, which attenuates protein synthesis and, in advanced stages, the proteolytic activity of the 26S proteasome via endoplasmic reticulum-associated protein degradation (ERAD, Fig. 2).4
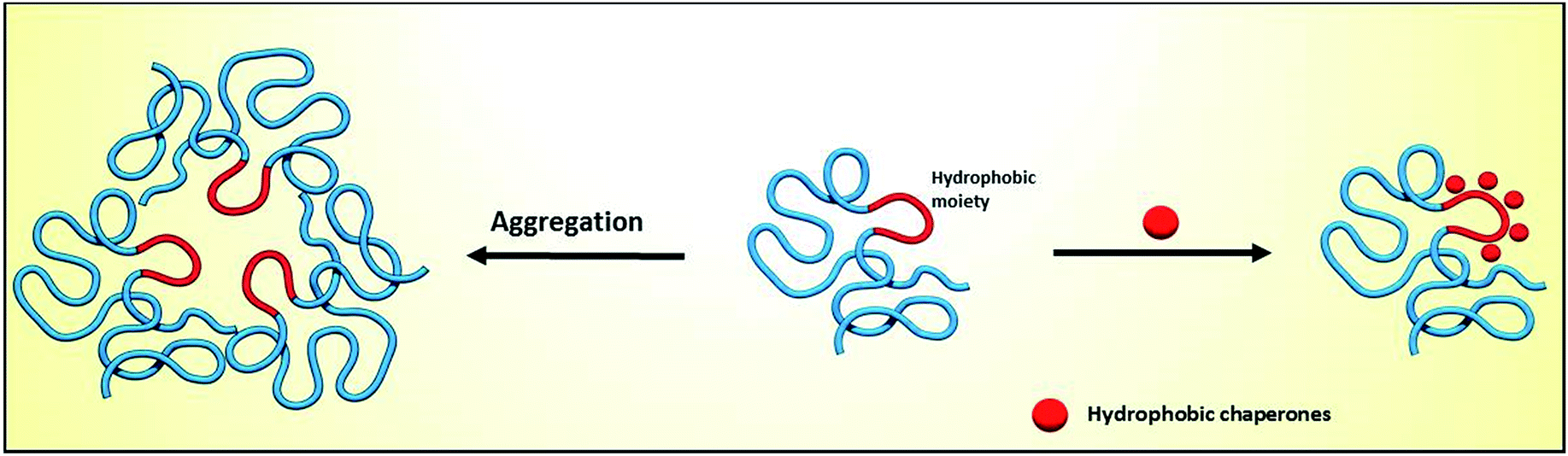 | ||
| Fig. 1 Impact of protein misfolding on protein aggregation (left) and rescue by hydrophobic chaperones (right). | ||
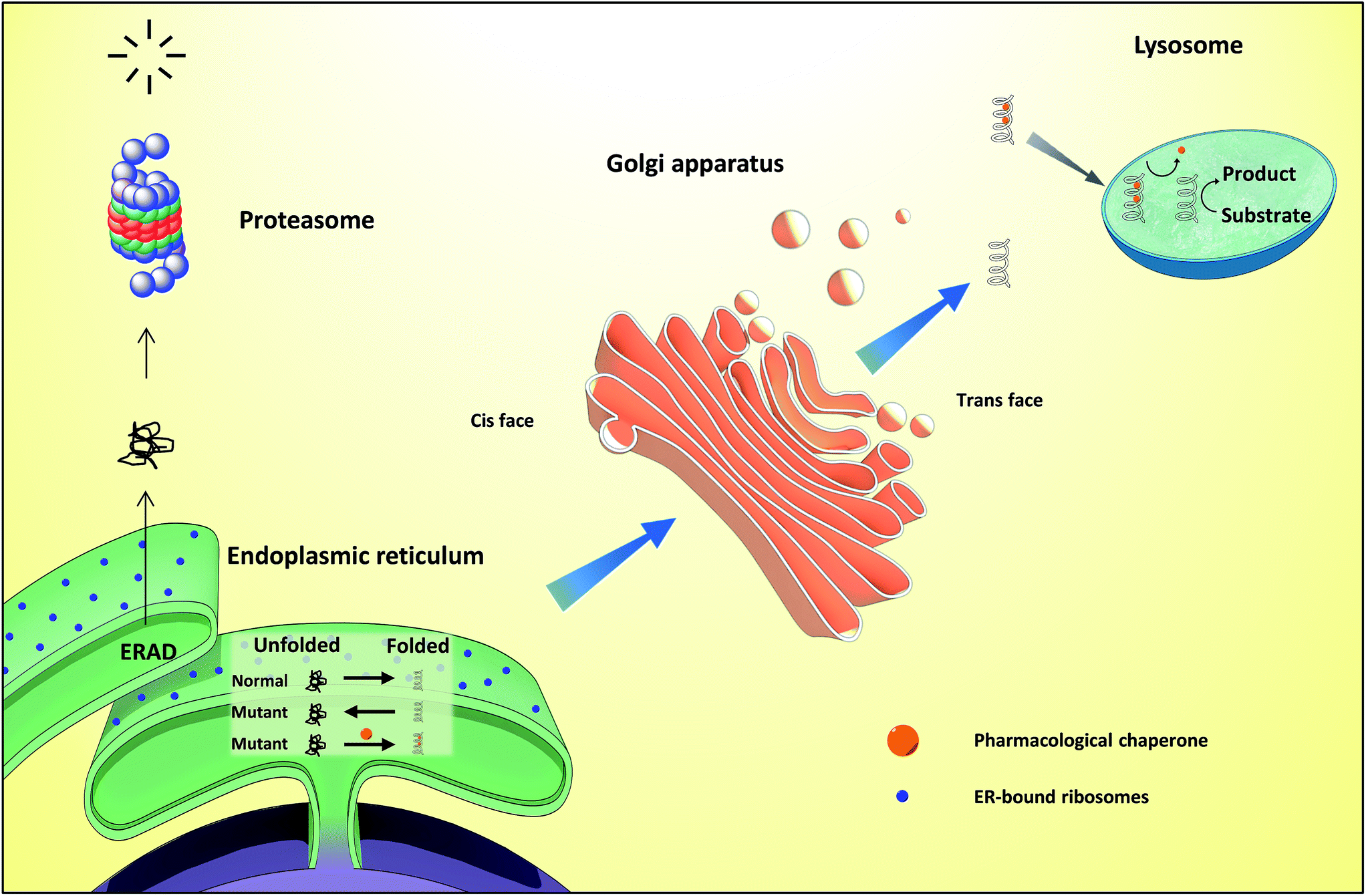 | ||
| Fig. 2 Overall view of the folding and quality control check of proteins in the ER, followed by subsequent trafficking to the Golgi apparatus. In the case of lysosomal storage diseases, pharmacological chaperones (PC) promote the correct folding of proteins in the ER, allowing the protein to reach the lysosome where, because of acidic pH and high substrate concentration, PC dissociates from the enzyme, thus delivering a functional protein. ERAD – endoplasmic reticulum-associated protein degradation. | ||
Chaperones are molecules capable of assisting proteins into their correct conformation. Considering that in several proteinopathies, the restoration of just 10–20% of protein activity is enough to prevent clinical manifestations of the disease,5–7 chaperone-based therapy emerges as promising targets for countering diseases that result from protein misfolding.
Chaperones can be sorted in three groups according to their chemical structure and mechanism of action: molecular, chemical and pharmacological. Molecular chaperones that are themselves proteins will not be discussed here.
2 The chemistry of chaperones
2.1 Chemical chaperones
Chemical chaperones are low molecular weight molecules that display a nonspecific mode of action and are frequently unable to bind directly to proteins. From a functional point of view, these molecules can be sorted either as osmolytes or hydrophobic compounds.3Osmolytes can alter solvent properties, thus forcing thermodynamically unstable proteins to fold and stabilize.10 The physical chemistry behind the ability of osmolytes to affect protein stability is elegantly simple: due to their ability to sequester water molecules, osmolytes create a hydrophobic environment around proteins, hence increasing the free energy of the unfolded state and favoring the folded state because the hydrophilic protein backbone minimizes its exposure to the hydrophobic surroundings.10 In this light, the osmolyte mechanism of action is generally universal, being mostly independent of the primary sequence and affecting all proteins. From a chemical point of view, several classes of compounds may act as osmolytes, including free amino acids and their derivatives (taurine, β-alanin, glycine, etc.), polyols (glycerol, trehalose, sucrose, etc.) or methylamines (trimethylamine N-oxide [TMAO]).
Physiologically, the contribution of each osmolyte to the overall stability of proteins seems to be dependent on the specific stress environment. For instance, polyols protect cells against dehydration and extreme temperatures, amino acids are useful in situations involving high salinity and methylamines are found predominantly in urea-rich cells because of urea's deleterious effects on protein structure.9
4-Phenylbutyrate (4-PBA, 1) is one of the most well-known chemical chaperones. This short-chain fatty acid is orally bioavailable and blood–brain barrier (BBB) permeable, the latter property being particularly interesting in light of its significant neuroprotective effect in animal models of neurodegenerative diseases, such as AD11,12 and PD.13,14 Several derivatives have already been obtained (2–4), and their structure–activity relationship (SAR) investigated.4
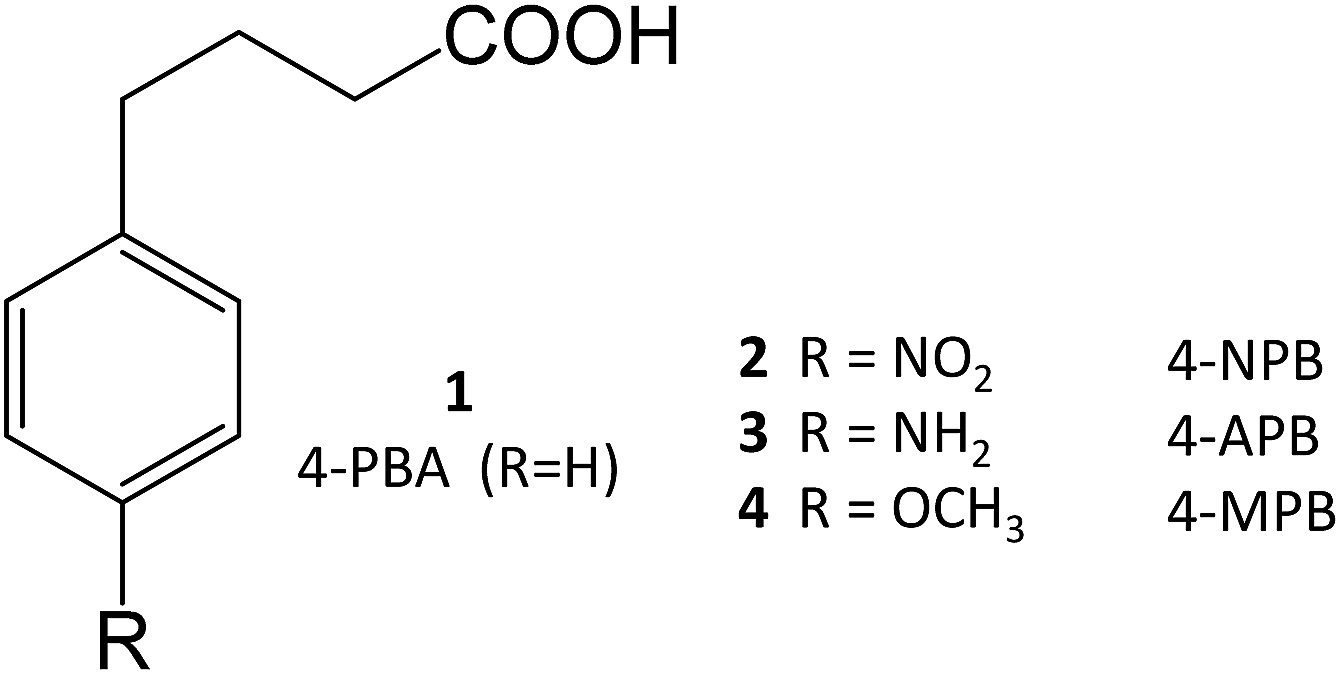
4-PBA was approved by the Food and Drug Administration as an ammonium scavenger for the treatment of urea cycle disorders in children.5 Clinical trials have also shown its potential in the treatment of sickle cell disease and thalassemia, which is associated to its ability to activate β-globin transcription.6
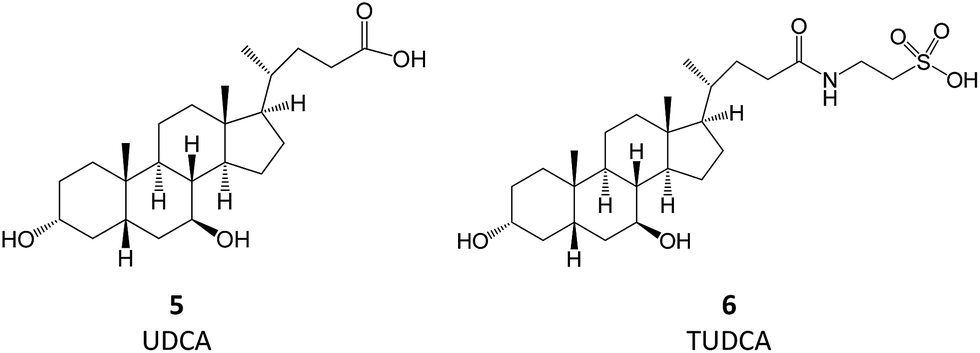
Bile acids (BAs) are (acidic) steroids biosynthesized from cholesterol in the liver. Upon secretion in the intestine, they can be metabolized by bacteria that perform dehydroxylation reactions, thus forming secondary BAs, of which deoxycholic acid and ursodeoxycholic acid (UDCA, 5) are examples. Upon their return to the liver, secondary BAs are conjugated with amino acids, the most relevant resulting molecule being tauroursodeoxycholic acid (TUDCA, 6).7 TUDCA and UDCA have been extensively investigated for their potential use in aggregation-associated events.
2.2 Pharmacological chaperones
Pharmacological chaperones (PCs) are molecules of low molecular weight that bind to proteins and can induce thermodynamic stabilization, template-based induction of correct folding or changes in the folding/unfolding kinetics,8 thus contributing to correct protein function.9In a rather contra-intuitive discovery in 1990, it was found that the residual activity of galactosidase A (α-Gal) could be markedly enhanced by using sub-inhibitory concentrations of iminosugar-based inhibitors of this enzyme. This mechanism of action relies on the fact that reversible competitive inhibitors promote the folding state of the enzyme (which is still catalytically active despite its misfolded conformation) by lowering the free energy of the protein–PC complex (Fig. 3).10 In addition to inhibitors, protein ligands are also capable of acting as PCs.11
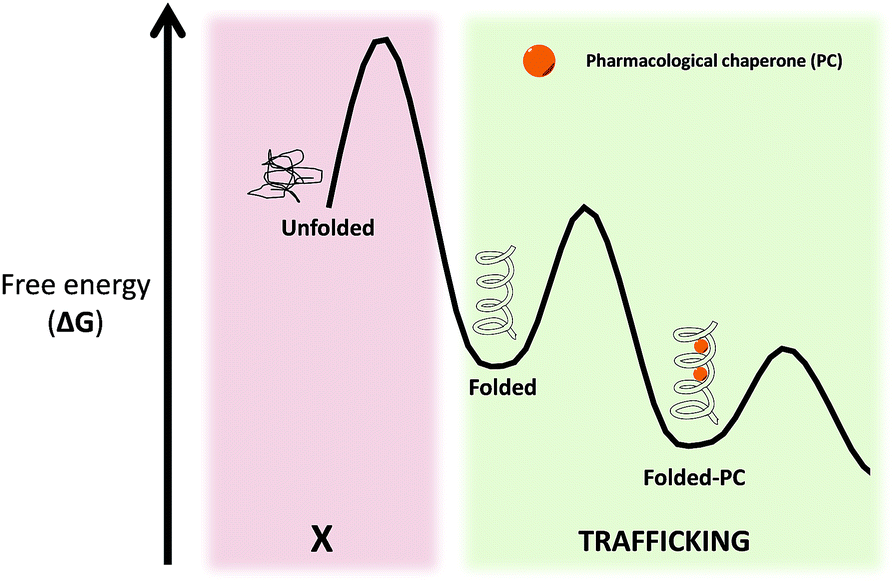 | ||
| Fig. 3 Free energy (ΔG) diagram of unfolded/folded proteins, as well as complexes of folded proteins with PC. | ||
By inducing or stabilizing the correct conformation of misfolded proteins, PCs prevent retention and/or degradation by the ER quality-control system and facilitate protein trafficking into other organelles, thereby frequently restoring protein activity.
Although conceptually similar to chemical chaperoning in the way they promote protein folding, PCs have the advantage of introducing selectivity in the form of a small molecule binding specifically to a given target.
3 Chaperones as a clinical strategy for LSDs: contributions from medicinal chemistry
Lysosomes are organelles involved in the degradation of several cellular molecules, for which they have developed a number of glycosidic and proteolytic enzymes that function in the acidic environment of these cellular compartments.12 These enzymes are synthesized in the neutral pH of the ER and subsequently translocated to the lysosome. Disruption of these enzymes results in the accumulation of substrates or metabolic intermediates in lysosomes and, frequently, in pathology. The group of resulting diseases, comprising over fifty conditions, are generically known as lysosome storage diseases (LSDs), which have been thoroughly reviewed before.13,14Current therapeutic options for LSDs include enzyme replacement therapy (ERT), which involves the infusion of a recombinant enzyme on a weekly or biweekly basis to catalyse the reactions that the residual activity of the patients' misfolded enzymes are unable to mediate. This approach suffers from the generation of anti-protein antibodies that can result in hypersensitivity reactions, as well as in the inactivation/clearance of the infused enzyme.15 Furthermore, the exogenous enzyme is unable to cross the BBB, thus being unsuitable to counter the neurological symptoms of the disease. In a distinct approach, substrate reduction therapy (SRT) is based on the inhibition of the enzymes required for the production of the corresponding storage product.15
Differently from ERT and SRT, the use of chaperones in LSDs, notably PCs, allows mutant enzymes to be correctly folded, thus avoiding ER retention and paving the way to transportation to the Golgi apparatus for maturation and subsequently to the lysosomes (Fig. 2), where, because of low pH and high substrate concentrations, the enzyme–chaperone complex dissociates, and the mutant enzyme hydrolyses its natural substrate according to its residual activity. It should be highlighted that most drugs target defective enzymes in the ER given the pivotal play of this organelle in the folding and trafficking of lysosome-bound glycosidases.
A major strategy for the development of PCs for the treatment of LSDs relies on PC glycomimetics, in which small molecules emulate the structure of the monosaccharide moiety that is cleaved by the target lysosomal glycosidase.16 When surveying the bibliography for PC chaperones for LSDs, it becomes apparent that it is dominated by three different pyranose sugar mimics, which can be sorted as follows: (a) 1-deoxynojirimycin-based, (b) isofagomine-based and (c) 1,5-dideoxy-1,5-imino-D-xylitol (DIX)-based. Selectivity is a crucial factor for the PCs used in LSDs, as a given molecule should be able to interfere with a particular glycosidase without affecting others. For this reason, PCs must be able to exert conformational (gluco vs. galacto substrates) and anomeric (α- or β-galactopyranosil substrates) selectivity.16
3.1 Gaucher disease
Gaucher disease is the most common autosomal recessive LSD, being caused by mutations in gene coding lysosomal β-glucocerebrosidase (GCase) and ultimately resulting in the accumulation of glucosylceramide in macrophages and the development of hepatosplenomegaly, anemia, skeletal lesions and central nervous system dysfunctions.17,18GCase is synthesized in ER-bound polyribosomes and, after being subjected to ER quality control, is trafficked to lysosomes. Mutant GCase, by failing this quality control check, is retained in the ER lumen for refolding attempts that, if failed, will result in retrotranslocation to the cytoplasm for subsequent degradation by the ubiquitin-proteasome system.19 N370S and L444P are the most important mutant forms of the enzyme (prevalence of 53% and 18%, respectively)20 and are the reason for which most research is conducted using cells from patients bearing these mutations.
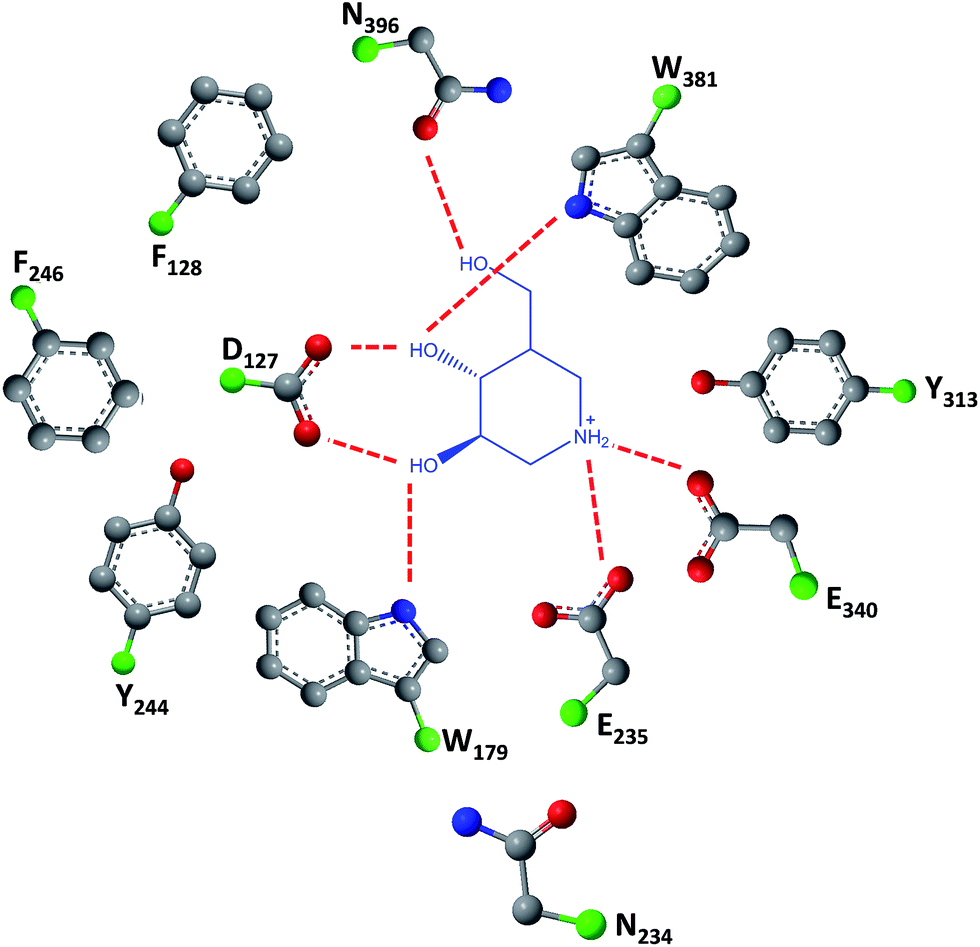 | ||
| Fig. 4 Schematic representation of hydrogen bond interactions (red dashed lines) between GCase and isofagomine (7). Data from Protein Data bank (https://www.rcsb.org/pdb) and ref. 2. | ||
Taking 1-deoxynojirimycin (10) as lead molecule, several N-alkyl derivatives have been synthesized, and their impact upon human GCase activity assessed.26 In a seminal work, Complain and colleagues compared several iminosugars resulting from different substitution strategies. The IC50 found for the parent molecule, 1-deoxynojirimycin, was 240 μM and was markedly reduced to 0.66 μM in the case of the N-nonyl derivative (12).26 Building on previous results suggesting that a simple 1,2-shift of the alkyl chain from the endocyclic nitrogen to the “anomeric” carbon in an α-configuration could lead to increased potency towards glucosidase inhibition,27 they evaluated several α-1-C-alkyl-1-deoxynojirimycin derivatives that exhibit these molecular traits.26 The results showed that the inhibitory potency of the α-1-C-alkyl-1-deoxynojirimycin derivatives increased with the length of the alkyl chain, reaching an IC50 of 0.27 μM for α-1-C-nonyl-1-deoxynojirimycin (13),26 thus being more potent than the corresponding N-nonyl analogue. In a parallel strategy, the same group also evaluated the inhibitory activity of DIX (14), which was more than 100 times more potent than 1-deoxynojirimycin (2.3 vs. 240 μM). When assessing the effect of N- and C-alkylation on the inhibitory activity, a similar trend was found, with N-nonyl-DIX (15) exhibiting an IC50 of 1.5 and α-1-C-nonyl-DIX (16) a remarkable IC50 of 6.8 nM, thus being over 35![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 times more potent than the reference molecule 1-deoxynojirimycin.26 In addition to this remarkable potency, these molecules also present selectivity towards GCase, as shown by the lack of inhibitory activity towards other glucosidases, such as maltase, sucrose and isomaltase.26
000 times more potent than the reference molecule 1-deoxynojirimycin.26 In addition to this remarkable potency, these molecules also present selectivity towards GCase, as shown by the lack of inhibitory activity towards other glucosidases, such as maltase, sucrose and isomaltase.26
Other groups have continued the investigation of the inhibitory properties of these molecules. Brumshtein and collaborators have studied the crystal structures of complexes of N-butyl- and N-nonyl-deoxynojirimycin bound to acid GCase as a strategy to understand the chemical basis of the inhibitory activity of these iminosugars.28 The results showed that both inhibitors bind at the active site of GCase, the iminosugar moiety establishing hydrogen bonds with the side chains of the active site residues, while the alkyl chains are oriented towards the entrance of the active site where they undergo hydrophobic interactions, ultimately stabilizing the complex and promoting the correct folding and transport of mutant GCase to lysosomes.28
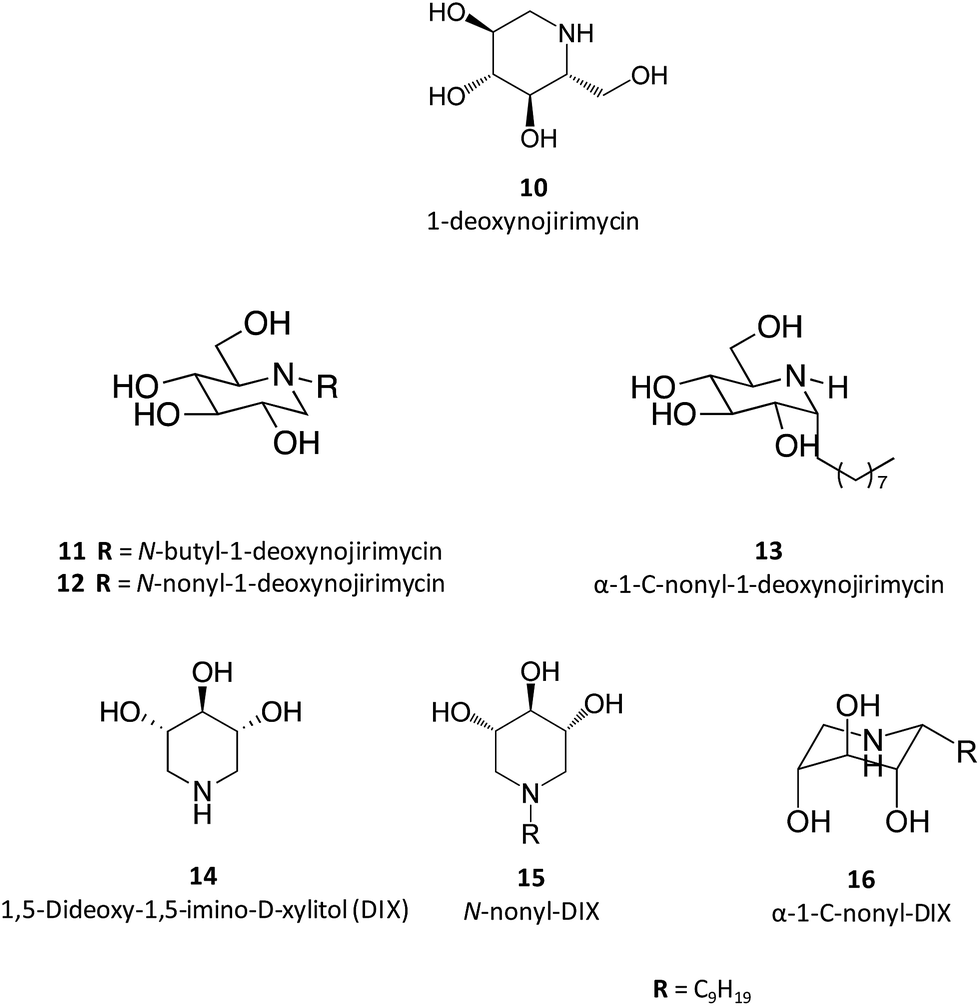
The interest in α-1-C-nonyl-DIX analogues has driven several groups to develop a plethora of synthetic strategies. For example, Goodard-Borger and co-workers described a highly divergent route to generate a collection of 16 analogues by using the thiol–ene reaction.29
As easily depicted by the molecules discussed above, pharmacological chaperones currently investigated for GCase are largely dominated by piperidine structures as pyranose analogues. In a rather innovative approach, Kato and colleagues shifted the attention towards pyrrolidine-based PCs, which allowed the selection of 1,4-dideoxy-1,4-imino-D-arabinitol (DAB) with different alkyl chain lengths.2 The impact of the configuration of the hydroxyl groups on the pyrrolidine ring and of the alkyl chain length on the stabilization of GCase under heat-shock conditions and their effects as pharmacological chaperone in N370S Gaucher fibroblasts was investigated. Moderate inhibitory activity was found up to the – heptyl derivative (IC50 higher than 30 μM); however, an extension of the α-1-C-alkyl chain length gave a series of highly potent and selective inhibitors, with α-1-C-tridecyl-DAB displaying sub-micromolar IC50 (0.77 μM). This same molecule was also shown to be the most effective in stabilizing GCase in a heat-shock in vitro assay.2 Subsequent molecular docking studies showed that the α-1-C-tridecyl group has a favourable interaction with the hydrophobic pocket, while the sugar analogue interacts via hydrogen bonds with Asp127, Glu235 and Glu340 (Fig. 5).2 This pyrrolidine-type amphiphilic iminosugar can probably accommodate either one of two orientations, namely the nitrogen atom aligned with C-1 (lipophilic substituent attached to the nitrogen atom) or O-5 (lipophilic substituent attached to an adjacent carbon atom) in glucopyranosides.
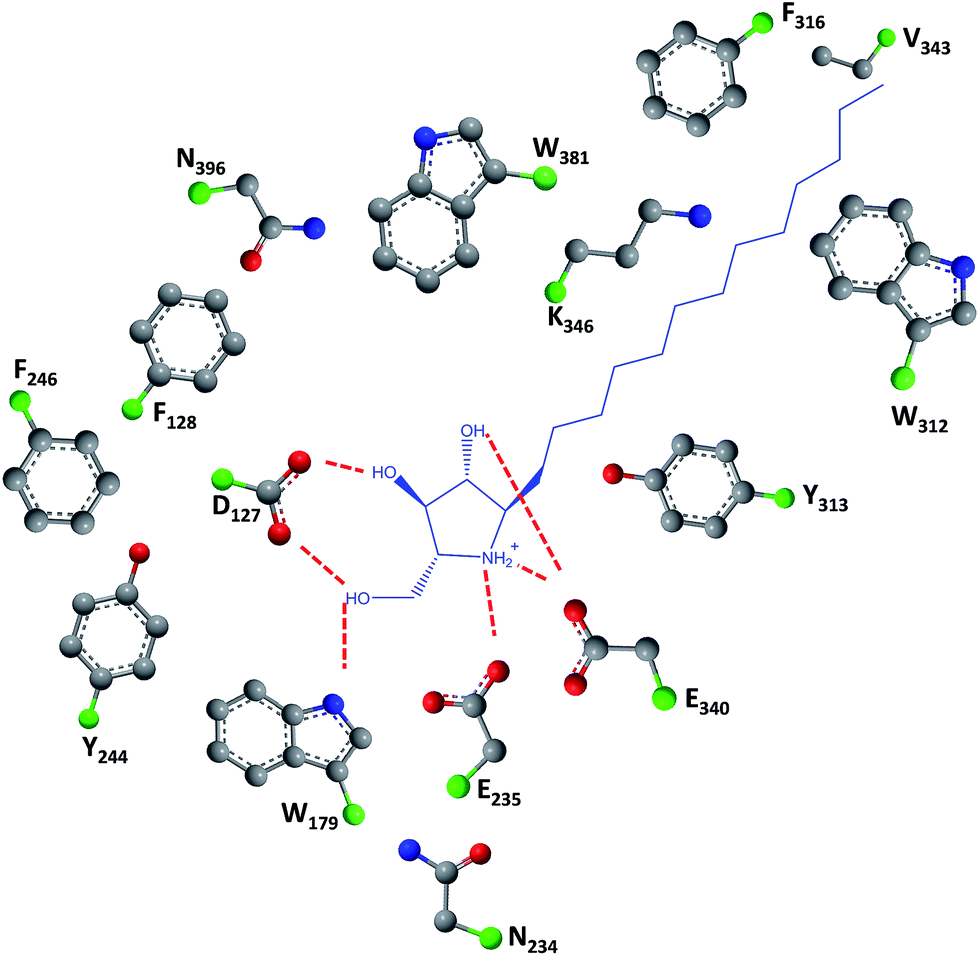 | ||
| Fig. 5 Schematic representation of hydrogen bond interactions (red dashed lines) between GCase and α-1-C-tridecyl-DAB. Data from Protein Data bank (https://www.rcsb.org/pdb) and ref. 2. | ||
A series of N-substituted δ-lactams was designed with a carbonyl group instead of a hydroxyl methyl group in the 1-deoxynojirimycin scaffold. The molecules were synthesized via a concise route with a one-pot tandem reaction comprising amination, cyclization and introduction of N-substituents as key step, as described by Wang and colleagues.30 Interestingly, these non-toxic molecules displayed weak inhibition against human native GCase. However, one third of the molecules was capable of activating the mutant N370S GCase, one of the molecules eliciting more than 6-fold increase in activity, a trait that was lost upon reduction of the carbonyl group, hence showing the importance of this function.30
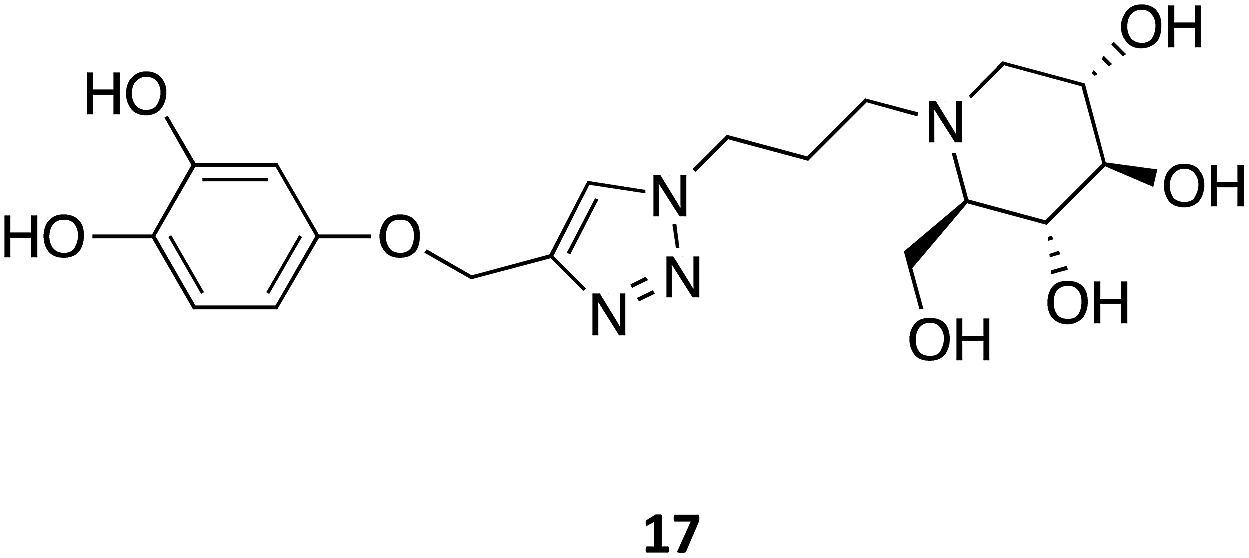
Other N-substitutions have been tried by other groups. For example, Diot et al. have employed click strategies to obtain amphiphilic 1-deoxynojirimycin derivatives, specifically triazoles. Among the numerous molecules tested, compound 17 exhibited the highest capacity to chaperone GCase, reaching almost a 2-fold increase in activity in Gaucher lymphoblasts with the most common N370S mutation.31
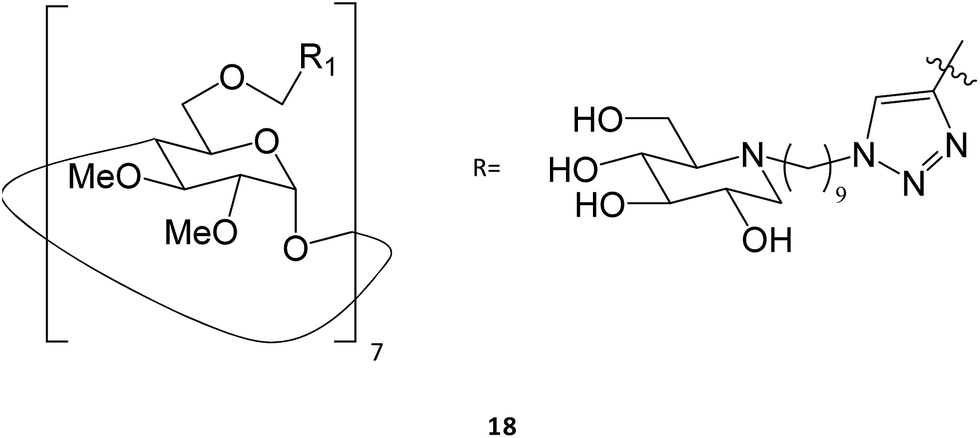
Multivalence has also been a valid strategy to obtain new iminosugars that have applications as PC. Joosten and colleagues described a systematic investigation of structure–activity relationships by synthesizing a panel of tri- to 14-valent systems with different alkyl spacer lengths and evaluating the effects of size, valence, ligand topology, and scaffold structure on the GCase binding affinity and chaperoning activity.32 Among the eighteen molecules tested, the heptavalent iminosugar 18 increased the activity of recombinant GCase by one order of magnitude when compared with the corresponding monovalent analogue, causing a change in potency from μM to nM.
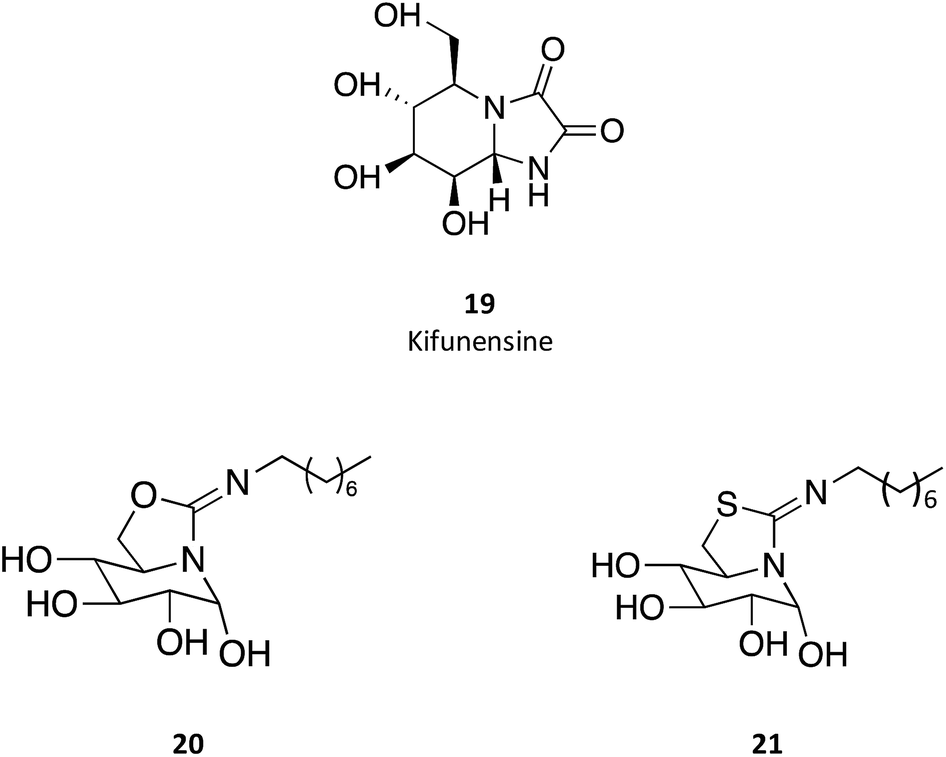
sp2-Iminosugar analogues of nojirimycin that bear lipophilic substituents, such as 5-N,6-O-(N′-octyliminomethylidene)nojirimycin (20) or its 6-thio derivative (21), are anomeric-specific inhibitors of β-glucosidases33—including GCAse. X-ray structural studies suggest that the rigid bicyclic framework imposes a restricted orientation of the hydrophobic substituent, which could be responsible for the anomeric selectivity. For the synthesis of these iminosugars, the authors exploited the capacity of nitrogen atoms in pseudoamide functionalities, such as cyclic isourea, isothiourea and guanidine, to be involved in intramolecular nucleophilic addition reactions to the masked aldehyde group of monosaccharides through the open-chain form.33 Another interesting trait of these molecules is that they are stronger inhibitors of GCase at pH 7.0 than at pH 5.2 by about one order of magnitude, which is a desirable characteristic for chaperones, as they should bind GCase in the ER (pH 7) to promote folding and trafficking and dissociate upon arrival to the lysosome (pH 5). A rather important information provided by this study is that the in vitro inhibition of GCase measured for the set of molecules synthesized did not correlate with the cellular chaperone activity.
Calystegines (example: 22) are polyhydroxy nortropane alkaloids first isolated from the extracts of Calystegia sepium; they had already been identified in several species of the Convolvulaceae, Solanaceae, and Moraceae families, notably in species relevant for human nutrition, such as potatoes (Solanum tuberosum) and eggplant (Solanum melongena).34 These alkaloids were new additions to the azosugar and iminosugar families of glycomimetis owing to their ability to inhibit glycosidases.35
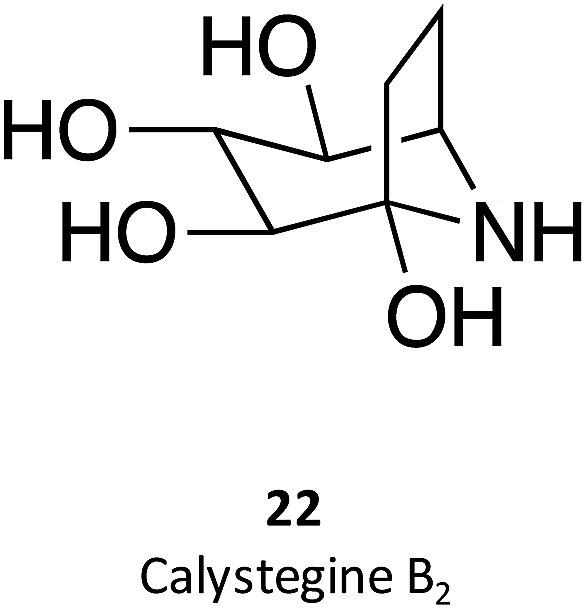
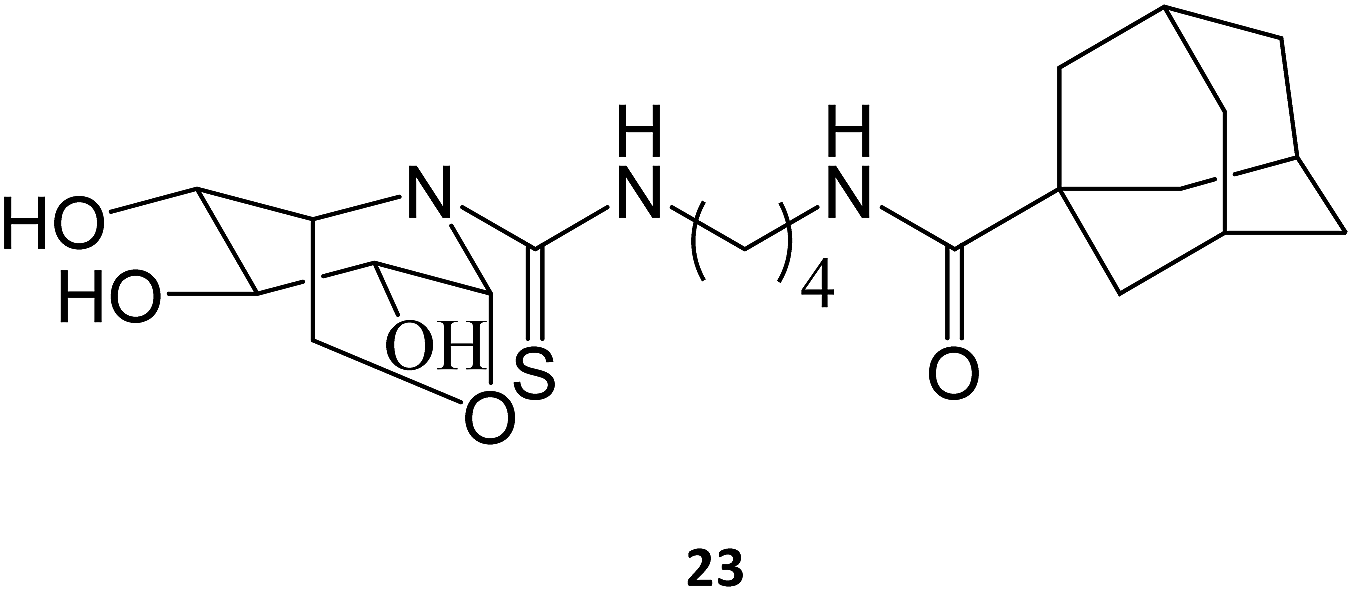
The synthesis and assessment of new bicyclic L-idonojirimycin derivatives related to calystegine yielded the identification of some chemical entities with potential to be used in Gaucher disease. The sp2 sugar glycomimetic, N-[N′-(4-adamantan-1-ylcarboxamidobutyl)thiocarbamoyl]-1,6-anhydro-L-idonojirimycin (23), was shown to be a promising PC, with 25 μM eliciting a 2.4-fold increase in GCase activity in fibroblasts from patients harboring the L444P/L444P genotype.36
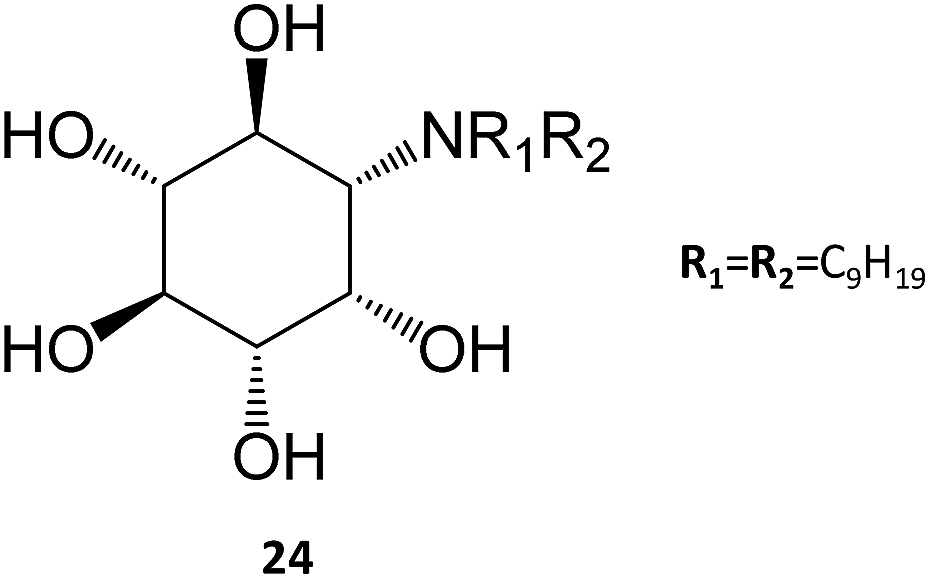
In a remarkable result, the N,N-dinonyl substituted amino-myo-inositol (24) displayed a Ki of 1 nM with the isolated enzyme and an IC50 of 4.3 nM when assayed in human fibroblasts. Assessment of GCase activity in N370S (1 nM) and L444P (0.01 nM) lymphoblasts resulted in an increase of 90% and 40%, respectively. These picomolar-range PCs are certainly among the most potent molecules described so far in literature and, in light of their adequate permeability, subcellular distribution, and cell metabolism characteristics, they are likely to be further developed in the near future.
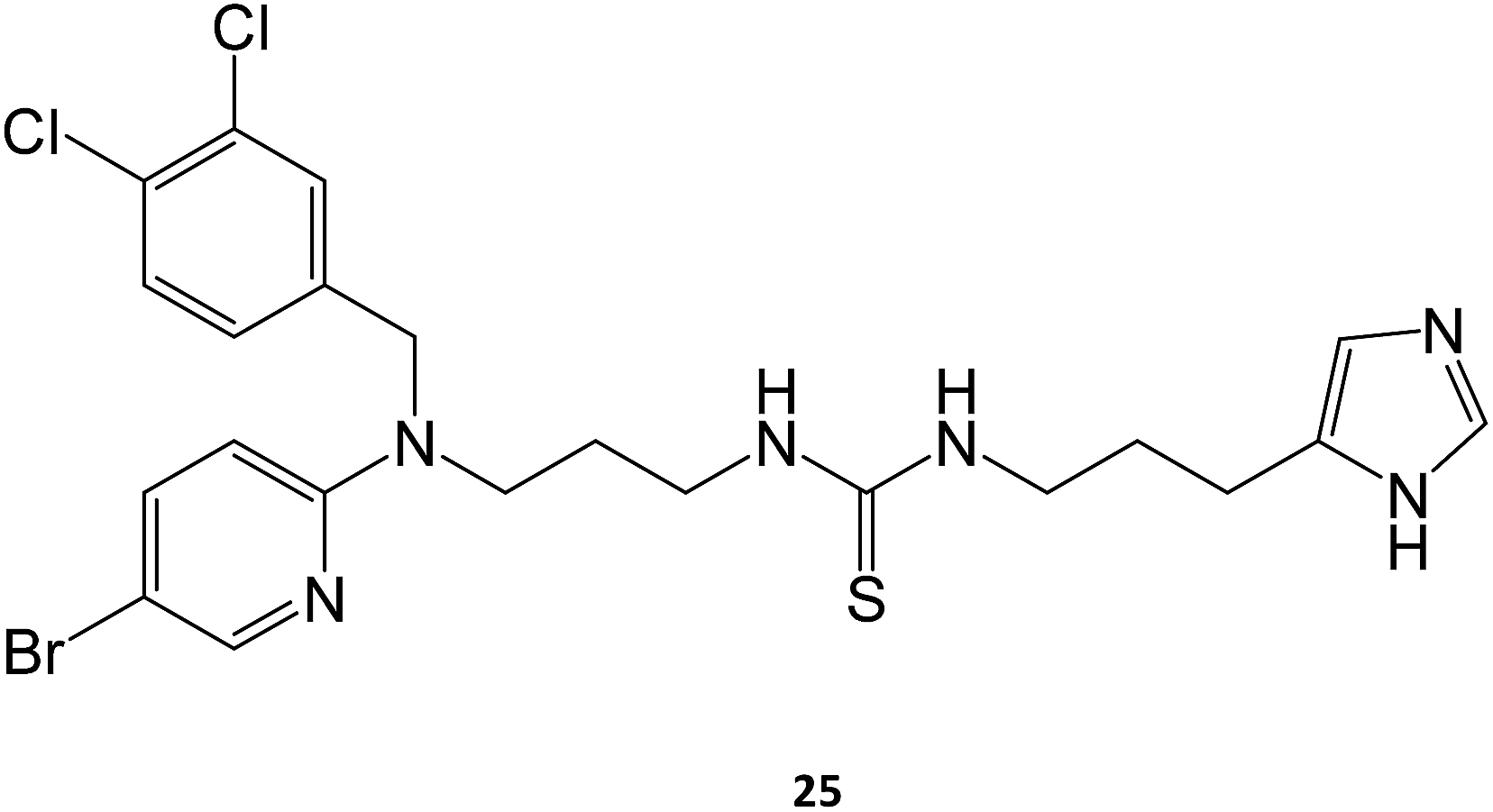
Marugan and collaborators synthesized a series of over twenty molecules and evaluated their inhibitory potential towards GCase. Among them, compound 25 was the most potent, displaying an IC50 of 0.6 μM.40 The chaperone capacity of the molecule was tested and demonstrated in a thermostabilization assay, where the molecule displayed higher potency than isofagomine.
Zheng and colleagues have screened a library of several non-iminosugar inhibitors of GCase.41 Molecules belonging to the class of aminoquinolines, sulfonamides and triazines were selected for further studies owing to their potency and efficacy and, importantly, high selectivity against closely-related hydrolases. Among these molecules, two were selected, namely compounds 26 and 27, which presented a Ki of 0.021 μM and 0.052 μM, respectively, in an assay using the pure enzyme. Also important, the incubation of Gaucher fibroblasts with the molecules (40 μM) resulted in a 40–90% increase in GCase activity in N370S mutant cells.41
3.2 Krabbe disease
Krabbe disease is an inherited disorder that destroys the myelin of nerve cells in the brain and throughout the nervous system. It is caused by mutations in the GALC gene (14q31), ultimately causing a deficiency in galactosylceramidase (GALC). Over one hundred mutations that affect GALC mRNA processing have been identified, involving deletions, frameshifts and missense mutations.42–44The absence of a GALC function results in the accumulation of the cytotoxic metabolite psychosine, which triggers cell death and overall demyelination throughout the central and peripheral nervous systems. Currently, there is no cure, leukoencephalopathy taking place early in life and infants dying before the age of two. As in the case of the Gaucher and Fabry diseases, ERT can relieve the peripheral symptoms of this LSD; however, the inability of this therapy to cross the BBB means that it is unsuitable for treating the pathological events in the central nervous system. Hematopoietic stem-cell transplantation is currently the best treatment in pre-symptomatic individuals, although it carries a significant mortality risk.45
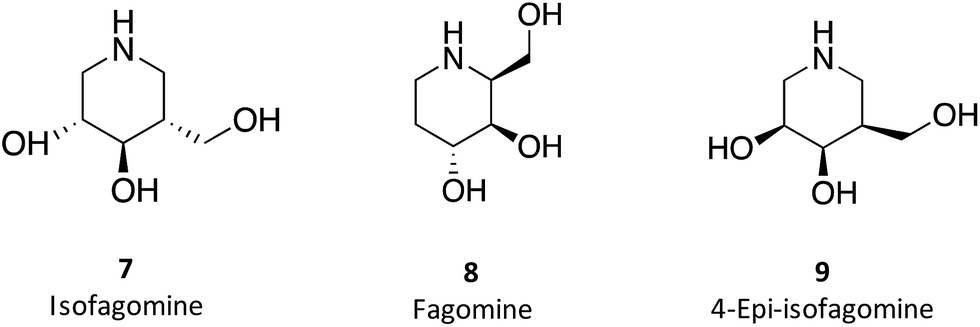
1-N-Iminosugars were proved good GALC inhibitors, in particular 4-epi-isofagomine (9, Fig. 6), which was able to attain 98% inhibition and displayed an IC50 of 1 μM. A pressing problem with this molecule is the lack of selectivity, as it was also able to inhibit lysosomal β-Gal; however, the available knowledge of the X-ray structures of the two enzymes46,47 should be able to guide the further design of molecules with enhanced selectivity.
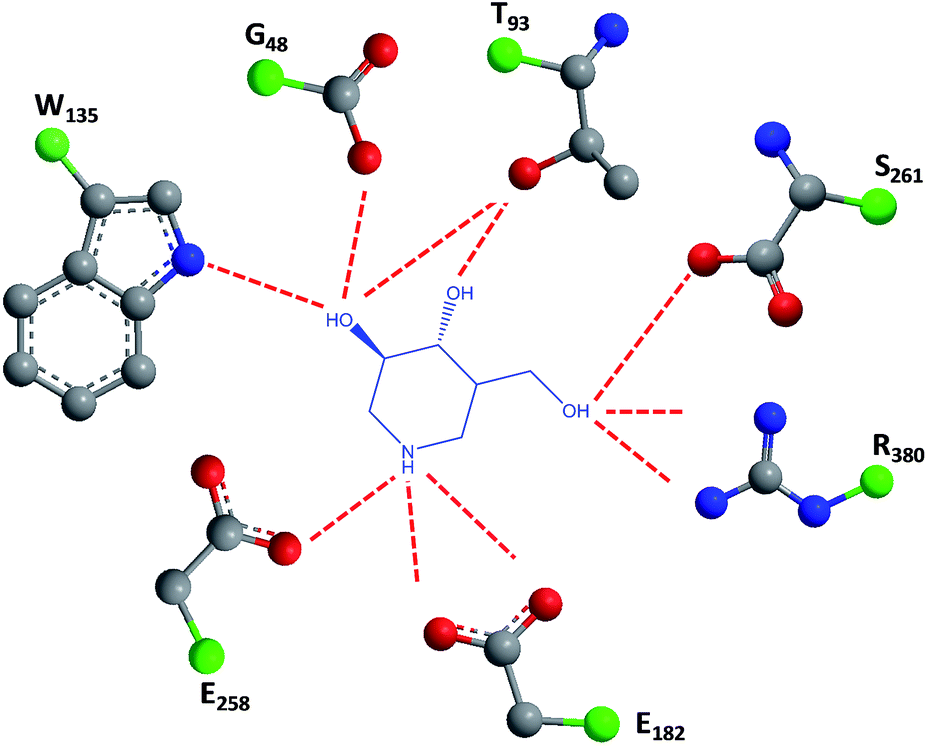 | ||
| Fig. 6 Schematic representation of hydrogen bond interactions (red dashed lines) between GALC and isofagomine (7). Data from Protein Data bank (https://www.rcsb.org/pdb) and ref. 1. | ||
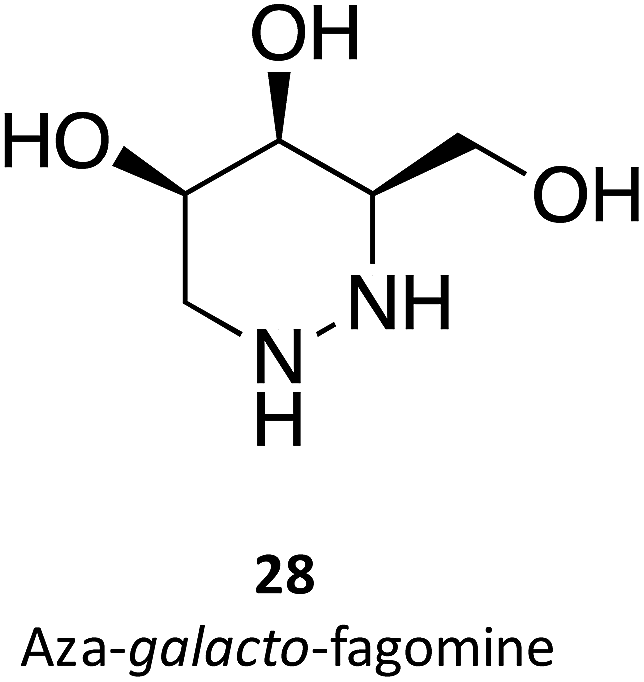
Hill and co-workers have synthesized a number of galacto-configured azosugars, whose interaction with GALC was studied, thus paving the way to understand the impact of the chemical structure and physical properties of azosugars as pharmacological chaperones for Krabbe disease.1 In addition to their inhibitory activity towards GALC, the molecules synthesized were also capable of increasing the stability of the enzyme, as assessed in a thermal denaturation assay by differential scanning fluorimetry, the most promising molecules being aza-galacto-fagomine (28) and iso-galacto-fagomine (also known as 4-epi-isofagomine, 9). In the near future, it would be interesting to know if these same molecules are also capable of increasing GALC activity in cells or animal models of the disease.
As in the case of Gaucher disease, glycomimetics is the most predominant strategy for obtaining new molecules to be used as PCs, which are in this case competitive inhibitors of their target enzyme.
Allosteric chaperones have been explored as an alternative chaperone strategy. By binding to an allosteric site, the functional state of the enzyme may be rescued without competitive inhibition. Only a few cases of this mechanism of action can be found in literature, including the piperidine alkaloid α-lobeline (29) found in several plants of the genus Lobelia. This molecule has been described as a putative PC towards the hyperglycosylation mutant D528N, as it significantly increased GALC activity (over 60% at 240 μM) but is ineffective with other tested Krabbe disease mutations.48
Another study also reported the PC activity of α-lobeline in fibroblasts from a Krabbe patient bearing the missense mutations E130K + N295T, which resulted in a 3-fold increase in activity at 50 μM, an effect also found for 3′,4′,7-trihydroxyisoflavone (30).49 The two molecules were shown to bind to multiple sites, which, in tandem with the high concentrations required, could hinder their subsequent clinical development.
3.3 Fabry disease
Fabry disease is a lysosomal storage disorder linked to the X chromosome that results in α-Gal deficiency, which, in turn leads to the accumulation of globotriaosylceramide (GL-3) and, eventually, to renal failure, myocardial infarction and stroke.50,51 Contrarily to Gaucher and Krabbe disease, very few examples of PCs for Fabry disease can be found in literature.The roots of Adenophora triphylla have been the origin of a series of iminosugars whose activity was assessed against several glycosidases, such as 2,5-dideoxy-2,5-imino-D-altritol bonded to α-Gal with an Ki of 0.5 μM.52 In addition to improving the in vitro thermostability of α-Gal, the molecule was able to increase the α-Gal A activity by 9.6-fold in Fabry R301Q lymphoblasts after incubation for three days.52
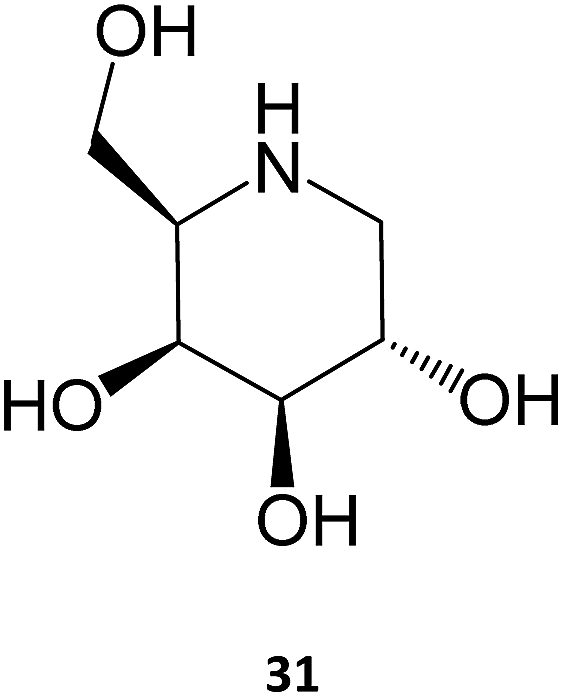
The iminosugar 1-deoxygalactonojirimycin (DGJ, migalastat, 31), initially isolated from Streptomyces lydicus (strain PA-5726) in 1988, is a reversible, competitive inhibitor of α-Gal A that has been shown to bind to the active site of the enzyme and improve folding, stability, and lysosomal trafficking in cultured cells from males with Fabry disease.10,53 This PC has also been shown to be effective in vivo, with oral administration of DGJ for 7 days in transgenic mice expressing a mutated human α-Gal A leading to increased α-Gal A levels and total enzymatic activity in tissues, such as heart, kidney, spleen, and liver.54 Even more important for the clinical outcome, a 4-week treatment resulted in reduced levels of GL-3 in the kidneys of the animals.55 As it will be further discussed in Section 4, this molecule is currently being marketed for treating Fabry disease.
3.4 Other LSDs
β-Gal is an enzyme that hydrolyzes terminal galactose residues from cell constituents, including GM1-gangliosides, glycoproteins, oligosaccharides, and the glycosaminoglycan keratan sulfate. Deficiency in this enzyme activity is the basis of two clinically distinct LSDs, namely GM1-gangliosidosis and Morquio disease type B. Depending on the mutation, distinct substrates accumulate in lysosomes, cells, organs and body fluids. When degradation of GM1-gangliosides is compromised, patients develop GM1-gangliosidosis, while in the case of Morquio disease type B accumulation of keratin sulfate takes place.Although the biochemical basis of both diseases is similar, their clinical manifestations are markedly different. While GM1-gangliosidosis is a neurodegenerative disease with several clinical manifestations that depend on the time of onset of the symptoms and residual β-Gal activity,56,57 Morquio disease type B is a mucopolysaccharidose that manifests itself as skeletal dysplasia, resulting in skeletal abnormalities and short stature.58,59 Unfortunately, there is no cure for either disease, with few therapeutic options being available besides palliative treatments.
Several molecules have been studied for their potential use as PC for this disease, notably sugar-based compounds. DGJ (31) has been shown to increase the activity of β-Gal up to 6-fold in fibroblasts from patients expressing several distinct mutations, being particularly effective against the R201C and R457Q mutations. The DGJ analog N-butyl-deoxy-galactonojirimycin (NB-DGJ) was equally potent, however it was also effective against the I51T mutation.60
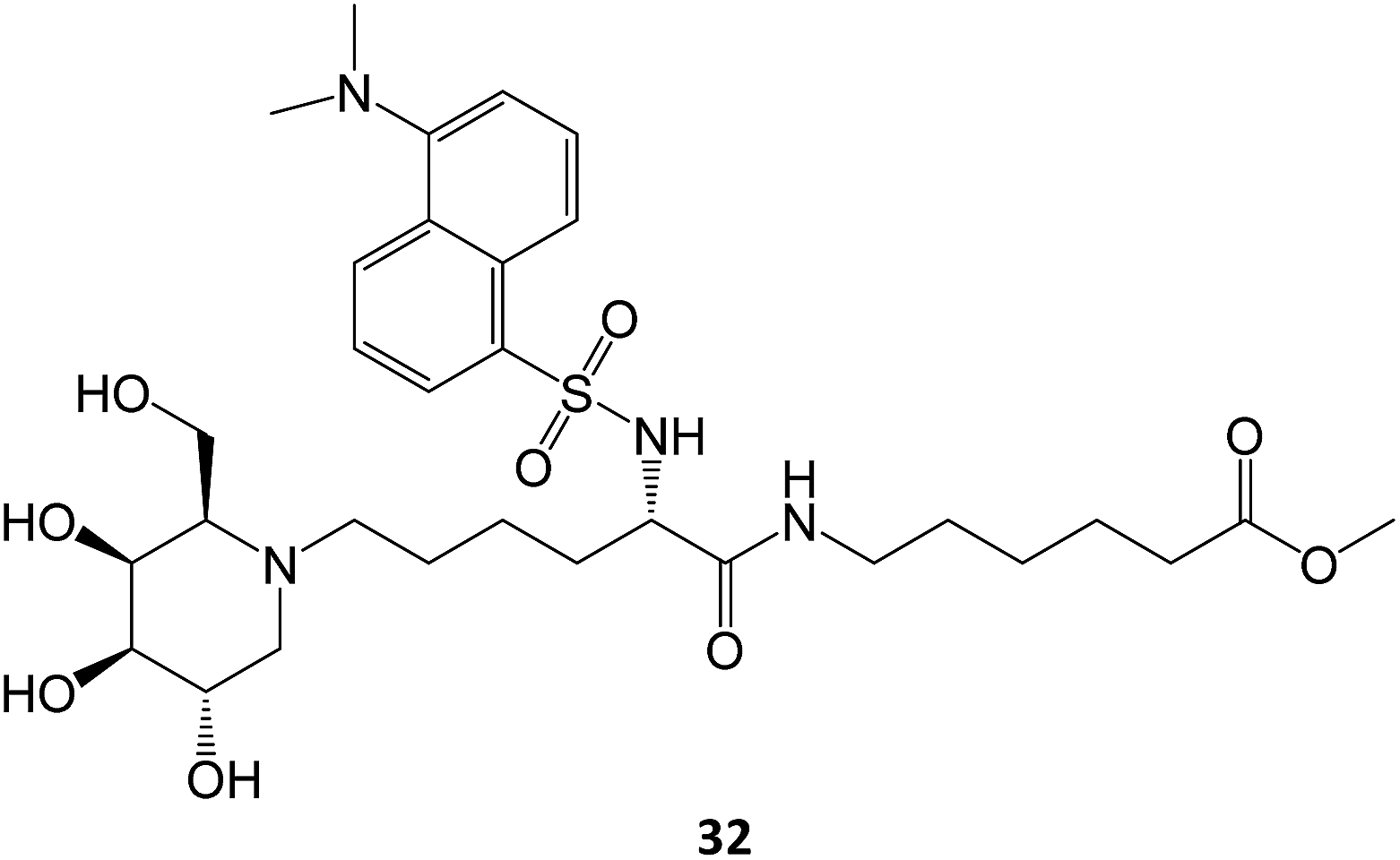
A N-alkylated derivative of 1-deoxygalactonojirimycin, DLHex-DGJ (methyl 6-{[N2-(dansyl)-N6-(1,5-dideoxy-D-galactitol-1,5-diyl)-L-lysyl]amino} hexanoate, 32), has also been described. The presence of 20 μM DLHex-DGJ was capable of yielding a 10% increase in enzyme activity in patient cells harboring the R201C mutation. Up to an 18-fold increase in β-Gal activity was achieved at the concentration previously described for DGJ (500 μM).61
N-Octyl-4-epi-beta-valienamine (NOEV) has been shown to effectively increase the activity of β-gal in the 0.2–2 μM range after an incubation period of 4 days. Interestingly, the molecule exhibited mutation-dependent potency, being more effective towards R201C and R201H than R457Q, W273L, and Y83H mutations.62
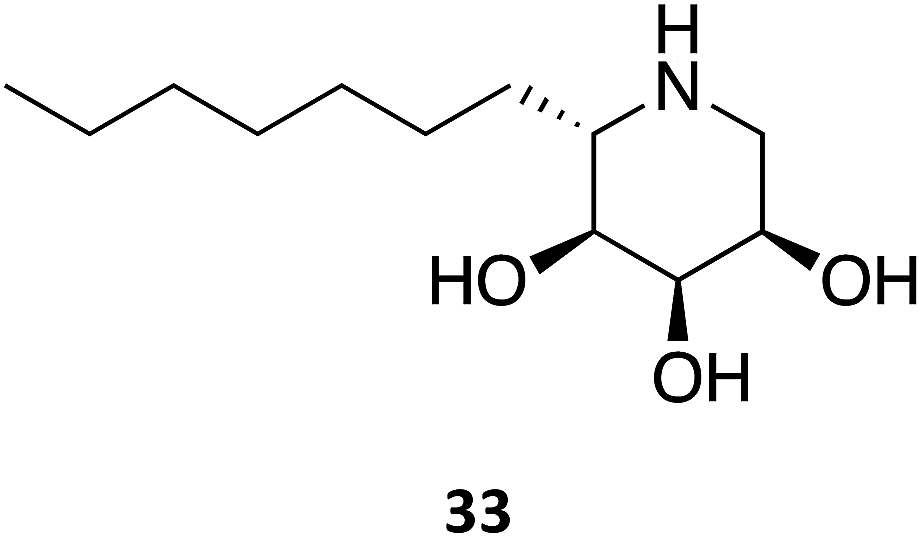
A series of 1,5-dideoxy-1,5-imino-(L)-ribitol (DIR) derivatives carrying alkyl or functionalized alkyl groups were synthesized and evaluated for their glycosidase inhibitory capacity.63 These molecules, which were designed as 4-epi-isofagomine (9) mimics, were found to be highly selective β-Gal inhibitors when compared to α-glycosidases; however, when considering the origin of the enzyme, the activity diminished in the coffee > almond > bovine > human series. The activity was markedly increased upon alkylation of the C5, with data suggesting that the “pseudo-anomeric” configuration of the series does not play a significant role. The molecules were evaluated for their PC activity in fibroblasts bearing the R201C mutation and, surprisingly, one of the molecules (33) was capable of enhancing human β-Gal activity despite not being a good inhibitor, with 10 μM eliciting an increase in enzymatic activity by over 2-fold.
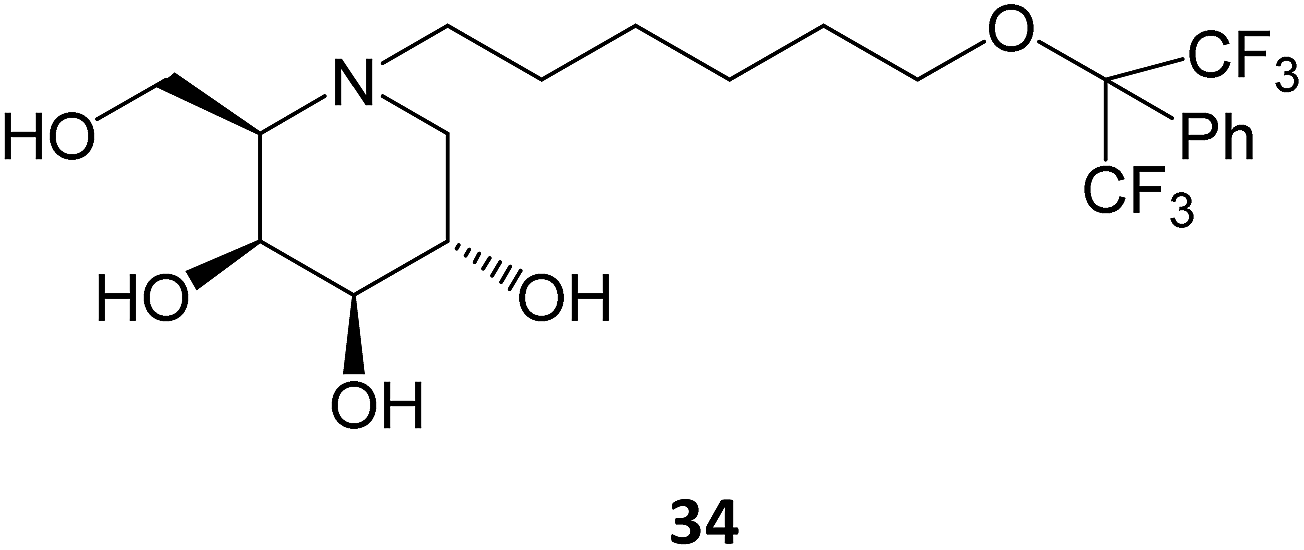
Novel medicinal chemistry strategies have been tested, such as iminoalditol-type molecules featuring N-substituents containing perfluorinated regions.64 In particular, the D-galacto series were shown to be pharmacological chaperones towards the lysosomal β-Gal mutants associated with GM1 gangliosidosis and Morquio B disease, with 34 causing an increase in enzyme activity up to 5-fold in concentrations in the 5–20 μM range.64
4 Current chaperone therapeutic arsenal & ongoing clinical trials
ERT and SRT remain in most cases the only therapeutic options for LSDs, particularly in the case of Gaucher and Fabry diseases.DGJ (31) was designated an orphan drug for Fabry disease in 2004 in the US and in 2006 in the European Union. Two phase III clinical trials with a total of about 110 patients were conducted between 2009 and 2015, with DGJ showing efficacy in stabilizing kidney and heart function over the 30-month period.65,66 The drug was approved in the European Union in May 2016 as an increasing number of clinical trials was concluded.67,68 The company responsible for the drug has published a list of over 265 amenable mutations that can be treated with the drug,69 a very promising information considering that 35 to 50% of Fabry patients have an amenable mutation.
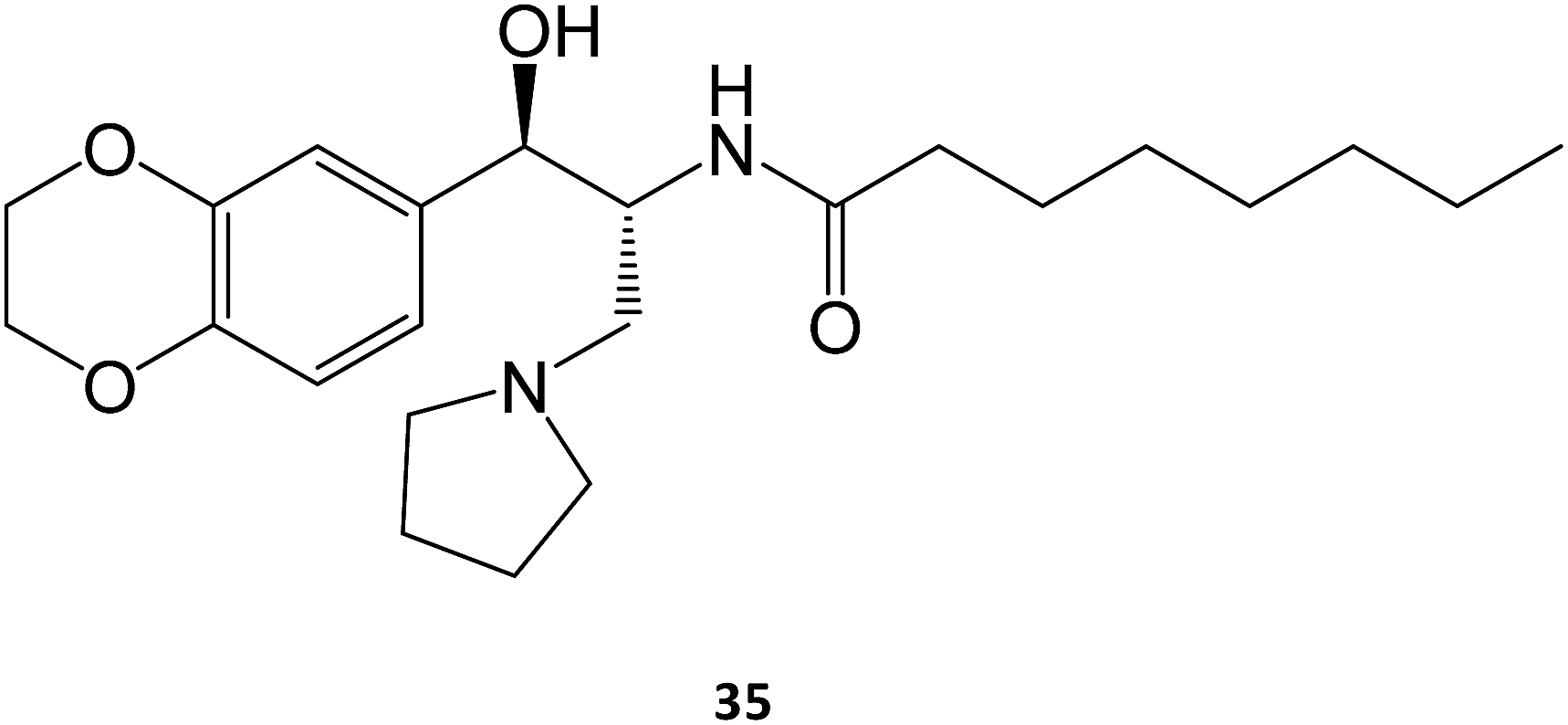
In the case of Gaucher disease, the small molecules N-butyl-1-deoxynojirimycin (miglustat)21–25 and eliglustat70–72 (35) are being marketed and well received owing to their high efficacy and lower side-effects. Interestingly, there have been recent reports (2017) that miglustat may reverse disease progression in juvenile/adult GM1-gangliosidosis, with juvenile patients regaining the ability to walk without assistance for few meters.73 Although isofagomine generated great interest in clinical trials, it unfortunately did not advance from phase II in 2009, owing to its low in vivo efficacy, which can arise from its high hydrophilicity that compromises pharmacokinetics, namely membrane crossing. The first patents on this molecule expired in 2015.
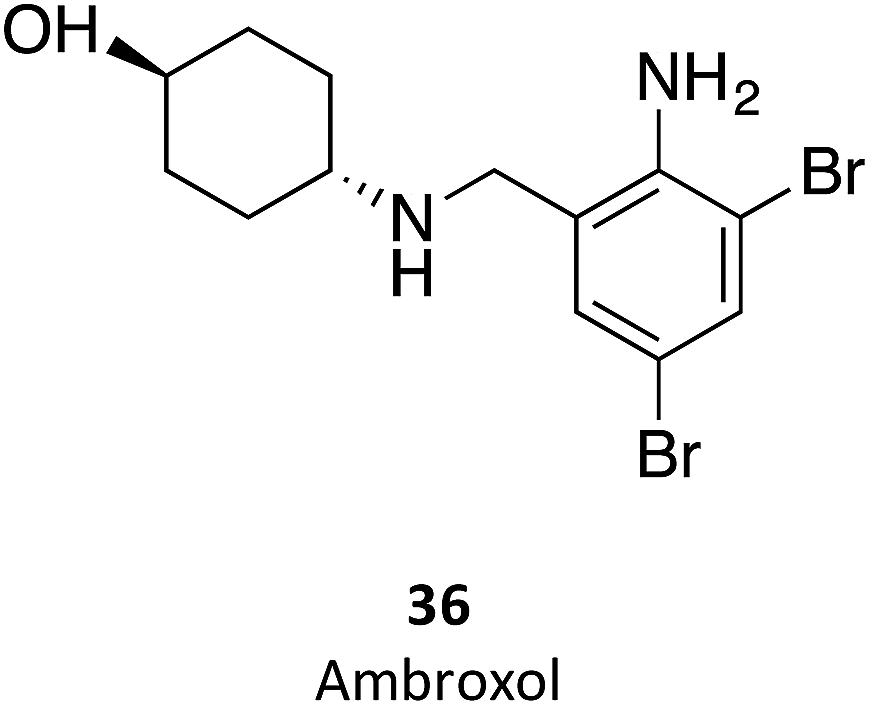
Ambroxol (36) is a secretolytic agent used in the treatment of respiratory diseases associated with viscid or excessive mucus.16 In a screening involving over a thousand FDA-approved drugs, ambroxol was reported to be an excellent chaperone candidate for Gaucher disease.74 Pilot studies have been conducted in 2013 (ref. 75) and 2016 (ref. 76) to assess the tolerability and efficacy of ambroxol in patients with type 1 Gaucher disease; some studies with non-human primates are also available.77 All results point to the efficacy of ambroxol in increasing GCase activity while displaying a safe toxicological profile. A rather interesting property of this molecule is that its activity is pH dependent, being maximum at the neutral pH of the ER and negligible in the acidic pH of the lysosome.74
5 Future perspectives and novel strategies
The use of small molecules to treat pathologies that are caused by misfolded proteins is a game changer for several diseases for which no cure is yet available. The process of discovering such chemical entities has been largely driven by screening large libraries of compounds or, alternatively, through the rational design of molecules based on the structure and thermodynamics of the target proteins.1,78,79When anticipating future directions for the field, drug repurposing is likely to be an effective strategy, as seen in the case of ambroxol. Drugs that are already on the market have generated enough safety information to fast-forward their future application in other diseases.
Deficient pharmacokinetics is another problem that must be properly addressed to position pharmacological chaperoning as an ever-efficient clinical strategy. In addition to the issues of absorption, distribution, metabolism and excretion that affect most drugs, PCs for treating LSDs have to face yet another challenge, namely the markedly distinct pH of their target sites at the cellular level: neutral in the ER and acidic in lysosomes. In this way, novel delivery strategies will also play a major role in tuning the selectivity of new drug candidates. For example, in a rather elegant work, Mena-Barragán reports the use of pH-responsive molecules capable of targeting human GCase or α-Gal, hence showing potential as pharmacological chaperones for Gaucher or Fabry diseases.80 By incorporating an orthoester segment into the iminosugar conjugates, the nature of the aglycone moiety is modulated from hydrophobic to hydrophilic in the pH range 5–7, thus being adequate for lysosome uptake. Once in the organelle, the ester is hydrolysed, and the free inhibitor is delivered. The concept of chaperone prodrug has also been increasingly present in the literature.79
The selectivity of the molecules towards different glycosidases is another aspect of pivotal importance for new PCs, as novel molecules must be able to target a group of enzymes that sometimes overlap in their substrates. This is however a two-edged sword. For one, lack of selectivity can result in undesired side-effects that arise from the inhibition of closely-related, unaffected enzymes. However, in some situations, this lack of selectivity has been shown to exhibit clinical advantages. For example, the above-mentioned molecule miglustat, marketed for the treatment of Gaucher disease, has been described as effective against other LSDs, such as GM1 gangliosidosis and Morquio B disease. For this reason, in the near future, we may witness the repurposing of drugs marketed for LSDs into other diseases of the same group on the grounds of the pan-inhibitory spectrum that some of these molecules exhibit.
In addition to their increasing use in LSDs, PCs are likely to continue to be used in a myriad of other diseases, with a survey in literature showing their efficacy in in vitro models of diseases such as homocystinuria,81 colitis,82 asthma83 and type 2 diabetes,84 to name a few.
Regardless of the target, we can expect that, in the next few years, the exciting advances in organic and medicinal chemistry we are experiencing today, together with the growing body of X-ray crystal structure information of target proteins, will likely result in the advent of new chemical entities capable of treating several still-incurable diseases.
Conflicts of interest
The authors declare no conflicts of interest.References
- C. H. Hill, A. H. Viuff, S. J. Spratley, S. Salamone, S. H. Christensen, R. J. Read, N. W. Moriarty, H. H. Jensen and J. E. Deane, Chem. Sci., 2015, 6, 3075–3086 RSC.
- A. Kato, I. Nakagome, K. Sato, A. Yamamoto, I. Adachi, R. J. Nash, G. W. Fleet, Y. Natori, Y. Watanabe and T. Imahori, Org. Biomol. Chem., 2016, 14, 1039–1048 CAS.
- L. Cortez and V. Sim, Prion, 2014, 8, 197–202 CrossRef CAS.
- S. Mimori, H. Ohtaka, Y. Koshikawa, K. Kawada, M. Kaneko, Y. Okuma, Y. Nomura, Y. Murakami and H. Hamana, Bioorg. Med. Chem. Lett., 2013, 23, 6015–6018 CrossRef CAS PubMed.
- S. W. Brusilow, Adv. Pediatr., 1996, 43, 127–170 CAS.
- A. F. Collins, H. Pearson, P. Giardina, K. McDonagh, S. Brusilow and G. Dover, Blood, 1995, 85, 43–49 CAS.
- E. M. Sánchez-Fernández, J. M. G. Fernández and C. O. Mellet, Chem. Commun., 2016, 52, 5497–5515 RSC.
- N. J. Leidenheimer and K. G. Ryder, Pharmacol. Res., 2014, 83, 10–19 CrossRef CAS PubMed.
- V. Bernier, M. Lagace, D. G. Bichet and M. Bouvier, Trends Endocrinol. Metab., 2004, 15, 222–228 CrossRef CAS PubMed.
- J.-Q. Fan, Trends Pharmacol. Sci., 2003, 24, 355–360 CrossRef CAS PubMed.
- T. Arakawa, D. Ejima, Y. Kita and K. Tsumoto, BBA, Biochimica et Biophysica Acta, Proteins and Proteomics, 2006, 1764, 1677–1687 CrossRef CAS PubMed.
- J. P. Luzio, Y. Hackmann, N. M. Dieckmann and G. M. Griffiths, Cold Spring Harbor Perspect. Biol., 2014, 6, a016840 CrossRef PubMed.
- R. E. Boyd, G. Lee, P. Rybczynski, E. R. Benjamin, R. Khanna, B. A. Wustman and K. J. Valenzano, J. Med. Chem., 2013, 56, 2705–2725 CrossRef CAS PubMed.
- K. J. Valenzano, R. Khanna, A. C. Powe Jr, R. Boyd, G. Lee, J. J. Flanagan and E. R. Benjamin, Assay Drug Dev. Technol., 2011, 9, 213–235 CrossRef CAS PubMed.
- G. Parenti, G. Andria and A. Ballabio, Annu. Rev. Med., 2015, 66, 471–486 CrossRef CAS PubMed.
- M. Malerba and B. Ragnoli, Expert Opin. Drug Metab. Toxicol., 2008, 4, 1119–1129 CrossRef CAS PubMed.
- R. S. Kamath, E. Lukina, N. Watman, M. Dragosky, G. M. Pastores, E. A. Arreguin, H. Rosenbaum, A. Zimran, R. Aguzzi and A. C. Puga, Skeletal Radiol., 2014, 43, 1353–1360 CrossRef PubMed.
- A. S. Thomas, A. Mehta and D. A. Hughes, Br. J. Haematol., 2014, 165, 427–440 CrossRef CAS PubMed.
- I. Ron and M. Horowitz, Hum. Mol. Genet., 2005, 14, 2387–2398 CrossRef CAS PubMed.
- J. Charrow, H. C. Andersson, P. Kaplan, E. H. Kolodny, P. Mistry, G. Pastores, B. E. Rosenbloom, C. R. Scott, R. S. Wappner and N. J. Weinreb, Arch. Intern. Med., 2000, 160, 2835–2843 CrossRef CAS PubMed.
- E. Canda, M. Kose, M. Kagnici, S. K. Ucar, E. Y. Sozmen and M. Coker, Blood Cells, Mol., Dis., 2018, 68, 180–184 CrossRef PubMed.
- P. Giraldo, M. Andrade-Campos, P. Alfonso, P. Irun, K. Atutxa, A. Acedo, A. Barez, M. Blanes, V. Diaz-Morant and M. A. Fernández-Galán, Blood Cells, Mol., Dis., 2018, 68, 173–179 CrossRef CAS PubMed.
- C. E. Hollak, D. Hughes, I. N. van Schaik, B. Schwierin and B. Bembi, Pharmacoepidemiol. Drug Saf., 2009, 18, 770–777 CrossRef CAS PubMed.
- M. J. Peterschmitt, G. F. Cox, J. Ibrahim, J. MacDougall, L. H. Underhill, P. Patel and S. J. Gaemers, Blood Cells, Mol., Dis., 2018, 68, 185–191 CrossRef PubMed.
- Y. Wang, M. C. Bartlett, T. W. Loo and D. M. Clarke, Mol. Pharmacol., 2006, 70, 297–302 CrossRef CAS PubMed.
- P. Compain, O. R. Martin, C. Boucheron, G. Godin, L. Yu, K. Ikeda and N. Asano, ChemBioChem, 2006, 7, 1356–1359 CrossRef CAS PubMed.
- G. Godin, P. Compain, O. R. Martin, K. Ikeda, L. Yu and N. Asano, Bioorg. Med. Chem. Lett., 2004, 14, 5991–5995 CrossRef CAS PubMed.
- B. Brumshtein, H. M. Greenblatt, T. D. Butters, Y. Shaaltiel, D. Aviezer, I. Silman, A. H. Futerman and J. L. Sussman, J. Biol. Chem., 2007, 282, 29052–29058 CrossRef CAS PubMed.
- E. D. Goddard-Borger, M. B. Tropak, S. Yonekawa, C. Tysoe, D. J. Mahuran and S. G. Withers, J. Med. Chem., 2012, 55, 2737 CrossRef CAS PubMed.
- G.-N. Wang, G. Reinkensmeier, S.-W. Zhang, J. Zhou, L.-R. Zhang, L.-H. Zhang, T. D. Butters and X.-S. Ye, J. Med. Chem., 2009, 52, 3146–3149 CrossRef CAS PubMed.
- J. D. Diot, I. G. Moreno, G. Twigg, C. O. Mellet, K. Haupt, T. D. Butters, J. Kovensky and S. b. G. Gouin, J. Org. Chem., 2011, 76, 7757–7768 CrossRef CAS PubMed.
- A. Joosten, C. Decroocq, J. de Sousa, J. P. Schneider, E. Etamé, A. Bodlenner, T. D. Butters and P. Compain, ChemBioChem, 2014, 15, 309–319 CrossRef CAS PubMed.
- Z. Luan, K. Higaki, M. Aguilar-Moncayo, H. Ninomiya, K. Ohno, M. I. García-Moreno, C. Ortiz Mellet, J. M. García Fernández and Y. Suzuki, ChemBioChem, 2009, 10, 2780–2792 CrossRef CAS PubMed.
- B. Dräger, Nat. Prod. Rep., 2004, 21, 211–223 RSC.
- M. Aguilar, T. M. Gloster, M. I. García-Moreno, C. Ortiz Mellet, G. J. Davies, A. Llebaria, J. Casas, M. Egido-Gabás and J. M. García Fernandez, ChemBioChem, 2008, 9, 2612–2618 CrossRef CAS PubMed.
- M. de La Mata, D. Cotán, M. Oropesa-Ávila, J. Garrido-Maraver, M. D. Cordero, M. V. Paz, A. D. Pavón, E. Alcocer-Gómez, I. De Lavera and P. Ybot-González, Sci. Rep., 2015, 5, 10903 CrossRef CAS PubMed.
- A. Trapero, P. González-Bulnes, T. D. Butters and A. Llebaria, J. Med. Chem., 2012, 55, 4479–4488 CrossRef CAS PubMed.
- H. Dvir, M. Harel, A. A. McCarthy, L. Toker, I. Silman, A. H. Futerman and J. L. Sussman, EMBO Rep., 2003, 4, 704–709 CrossRef CAS PubMed.
- R. L. Lieberman, Enzyme Res., 2011, 2011, 973231 Search PubMed.
- J. J. Marugan, W. Huang, O. Motabar, W. Zheng, J. Xiao, S. Patnaik, N. Southall, W. Westbroek, W. A. Lea and A. Simeonov, MedChemComm, 2012, 3, 56–60 RSC.
- W. Zheng, J. Padia, D. J. Urban, A. Jadhav, O. Goker-Alpan, A. Simeonov, E. Goldin, D. Auld, M. E. LaMarca and J. Inglese, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 13192–13197 CrossRef CAS PubMed.
- L. Fu, K. Inui, T. Nishigaki, N. Tatsumi, H. Tsukamoto, C. Kokubu, T. Muramatsu and S. Okada, J. Inherited Metab. Dis., 1999, 22, 155–162 CrossRef CAS PubMed.
- B. Tappino, R. Biancheri, M. Mort, S. Regis, F. Corsolini, A. Rossi, M. Stroppiano, S. Lualdi, A. Fiumara and B. Bembi, Hum. Mutat., 2010, 31(12), E1894–E1915 CrossRef PubMed.
- C. Xu, N. Sakai, M. Taniike, K. Inui and K. Ozono, J. Hum. Genet., 2006, 51, 548–554 CrossRef CAS PubMed.
- W. Krivit, E. G. Shapiro, C. Peters, J. E. Wagner, G. Cornu, J. Kurtzberg, D. A. Wenger, E. H. Kolodny, M. T. Vanier and D. J. Loes, N. Engl. J. Med., 1998, 338, 1119–1127 CrossRef CAS PubMed.
- J. E. Deane, S. C. Graham, N. N. Kim, P. E. Stein, R. McNair, M. B. Cachón-González, T. M. Cox and R. J. Read, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 15169–15173 CrossRef CAS PubMed.
- U. Ohto, K. Usui, T. Ochi, K. Yuki, Y. Satow and T. Shimizu, J. Biol. Chem., 2012, 287, 1801–1812 CrossRef CAS PubMed.
- W. C. Lee, D. Kang, E. Causevic, A. R. Herdt, E. A. Eckman and C. B. Eckman, J. Neurosci., 2010, 30, 5489–5497 CrossRef CAS PubMed.
- A. S. Berardi, G. Pannuzzo, A. Graziano, E. Costantino-Ceccarini, P. Piomboni and A. Luddi, Mol. Genet. Metab., 2014, 112, 294–301 CrossRef CAS PubMed.
- S. M. Rombach, B. E. Smid, G. E. Linthorst, M. G. Dijkgraaf and C. E. Hollak, J. Inherited Metab. Dis., 2014, 37, 341–352 CrossRef CAS PubMed.
- S. Ishii, R. Kase, T. Okumiya, H. Sakuraba and Y. Suzuki, Biochem. Biophys. Res. Commun., 1996, 220, 812–815 CrossRef CAS PubMed.
- A. Kato, Y. Yamashita, S. Nakagawa, Y. Koike, I. Adachi, J. Hollinshead, R. J. Nash, K. Ikeda and N. Asano, Bioorg. Med. Chem., 2010, 18, 3790–3794 CrossRef CAS PubMed.
- E. Benjamin, J. Flanagan, A. Schilling, H. Chang, L. Agarwal, E. Katz, X. Wu, C. Pine, B. Wustman and R. Desnick, J. Inherited Metab. Dis., 2009, 32, 424–440 CrossRef CAS PubMed.
- J.-Q. Fan, S. Ishii, N. Asano and Y. Suzuki, Nat. Med., 1999, 5, 112–115 CrossRef CAS PubMed.
- S. Ishii, H.-H. Chang, H. Yoshioka, T. Shimada, K. Mannen, Y. Higuchi, A. Taguchi and J.-Q. Fan, J. Pharmacol. Exp. Ther., 2009, 328, 723–731 CrossRef CAS PubMed.
- J. S. Kannebley, L. Silveira-Moriyama, L. O. D. Bastos and C. E. Steiner, in JIMD Rep., Springer, 2015, vol. 24, pp. 115–122 Search PubMed.
- K. Sandhoff and K. Harzer, J. Neurosci., 2013, 33, 10195–10208 CrossRef CAS PubMed.
- J. W. Callahan, Biochim. Biophys. Acta, Mol. Basis Dis., 1999, 1455, 85–103 CrossRef CAS.
- F. Kubaski, H. H. Kecskemethy, T. Harcke and S. Tomatsu, Mol. Genet. Metab. Rep., 2016, 117, S68–S69 CrossRef.
- L. Tominaga, Y. Ogawa, M. Taniguchi, K. Ohno, J. Matsuda, A. Oshima, Y. Suzuki and E. Nanba, Brain Dev., 2001, 23, 284–287 CrossRef CAS PubMed.
- K. Fantur, D. Hofer, G. Schitter, A. J. Steiner, B. M. Pabst, T. M. Wrodnigg, A. E. Stütz and E. Paschke, Mol. Genet. Metab. Rep., 2010, 100, 262–268 CrossRef CAS PubMed.
- J. Matsuda, O. Suzuki, A. Oshima, Y. Yamamoto, A. Noguchi, K. Takimoto, M. Itoh, Y. Matsuzaki, Y. Yasuda and S. Ogawa, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 15912–15917 CrossRef CAS PubMed.
- S. Front, E. Gallienne, J. Charollais-Thoenig, S. Demotz and O. R. Martin, ChemMedChem, 2016, 11, 133–141 CrossRef CAS PubMed.
- G. Schitter, A. J. Steiner, G. Pototschnig, E. Scheucher, M. Thonhofer, C. A. Tarling, S. G. Withers, K. Fantur, E. Paschke and D. J. Mahuran, ChemBioChem, 2010, 11, 2026–2033 CrossRef CAS PubMed.
- M. Adera, A. Gladden and P. Boudes, Mol. Genet. Metab. Rep., 2011, 102, S4 Search PubMed.
- M. Adera, C. Overton and P. Boudes, Mol. Genet. Metab. Rep., 2011, 102, S4–S5 Search PubMed.
- U. Feldt-Rasmussen, R. Giugliani, D. P. Germain, D. A. Hughes, W. R. Wilcox, R. Schiffmann, D. G. Bichet, A. Jovanovic, D. Bratkovic and J. P. Castelli, Mol. Genet. Metab. Rep., 2017, 120, S45–S46 CrossRef.
- D. A. Hughes, K. Nicholls, S. P. Shankar, G. Sunder-Plassmann, D. Koeller, K. Nedd, G. Vockley, T. Hamazaki, R. Lachmann and T. Ohashi, J. Med. Genet., 2017, 54, 288–296 CrossRef PubMed.
- A. Therapeutics, Migalastat Amenability Table, http://www.galafoldamenabilitytable.com/hcp/, accessed 25-12-2017 Search PubMed.
- M. Balwani, T. A. Burrow, J. Charrow, O. Goker-Alpan, P. Kaplan, P. S. Kishnani, P. Mistry, J. Ruskin and N. Weinreb, Mol. Genet. Metab. Rep., 2016, 117, 95–103 CrossRef CAS PubMed.
- A. Broomfield, J. Mercer, H. J. Church, E. Chronopoulou, G. Pierre, A. Wiskin, K. Booth, S. A. Jones and A. Sutherland, Mol. Genet. Metab. Rep., 2017, 120, S32 Search PubMed.
- R. Pleat, T. M. Cox, T. A. Burrow, P. Giraldo, O. Goker-Alpan, B. E. Rosenbloom, L. R. Croal, L. H. Underhill, S. J. Gaemers and M. J. Peterschmitt, Mol. Genet. Metab. Rep., 2016, 9, 25–28 CrossRef CAS PubMed.
- F. Deodato, E. Procopio, A. Rampazzo, R. Taurisano, M. A. Donati, C. Dionisi-Vici, A. Caciotti, A. Morrone and M. Scarpa, Metab. Brain Dis., 2017, 1–8 Search PubMed.
- G. H. Maegawa, M. B. Tropak, J. D. Buttner, B. A. Rigat, M. Fuller, D. Pandit, L. Tang, G. J. Kornhaber, Y. Hamuro and J. T. Clarke, J. Biol. Chem., 2009, 284, 23502–23516 CrossRef CAS PubMed.
- A. Zimran, G. Altarescu and D. Elstein, Blood Cells, Mol., Dis., 2013, 50, 134–137 CrossRef CAS PubMed.
- A. Narita, K. Shirai, S. Itamura, A. Matsuda, A. Ishihara, K. Matsushita, C. Fukuda, N. Kubota, R. Takayama and H. Shigematsu, Ann. Clin. Transl. Neurol., 2016, 3, 200–215 CrossRef CAS PubMed.
- A. Migdalska-Richards, W. K. D. Ko, Q. Li, E. Bezard and A. H. Schapira, Synapses, 2017, 71(7), e21967 CrossRef PubMed.
- N. C. Ferreira, I. A. Marques, W. A. Conceição, B. Macedo, C. S. Machado, A. Mascarello, L. D. Chiaradia-Delatorre, R. A. Yunes, R. J. Nunes and A. G. Hughson, PLoS One, 2014, 9, e84531 Search PubMed.
- E. Laigre, D. Hazelard, J. Casas, J. Serra-Vinardell, H. Michelakakis, I. Mavridou, J. M. Aerts, A. Delgado and P. Compain, Carbohydr. Res., 2016, 429, 98–104 CrossRef CAS PubMed.
- T. Mena-Barragán, A. Narita, D. Matias, G. Tiscornia, E. Nanba, K. Ohno, Y. Suzuki, K. Higaki, J. M. Garcia Fernández and C. Ortiz Mellet, Angew. Chem., Int. Ed., 2015, 54, 11696–11700 CrossRef PubMed.
- P. Melenovská, J. Kopecká, J. Krijt, A. Hnízda, K. Raková, M. Janošík, B. Wilcken and V. Kožich, J. Inherited Metab. Dis., 2015, 38, 287–294 CrossRef PubMed.
- S. S. Cao, E. M. Zimmermann, B. M. Chuang, B. Song, A. Nwokoye, J. E. Wilkinson, K. A. Eaton and R. J. Kaufman, Gastroenterology, 2013, 144, 989–1000 CrossRef CAS PubMed.
- L. Makhija, V. Krishnan, R. Rehman, S. Chakraborty, S. Maity, U. Mabalirajan, K. Chakraborty, B. Ghosh and A. Agrawal, Am. J. Respir. Cell Mol. Biol., 2014, 50, 923–931 CrossRef PubMed.
- L. Cadavez, J. Montane, G. Alcarraz-Vizán, M. Visa, L. Vidal-Fàbrega, J.-M. Servitja and A. Novials, PLoS One, 2014, 9, e101797 Search PubMed.
| This journal is © The Royal Society of Chemistry 2018 |