
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInedible saccharides: a platform for CO2 capturing
Abdussalam K.
Qaroush
 *a,
Hiba S.
Alshamaly
b,
Shrouq S.
Alazzeh
a,
Ream H.
Abeskhron
a,
Khaleel I.
Assaf
*c and
Ala’a F.
Eftaiha
*b
*a,
Hiba S.
Alshamaly
b,
Shrouq S.
Alazzeh
a,
Ream H.
Abeskhron
a,
Khaleel I.
Assaf
*c and
Ala’a F.
Eftaiha
*b
aDepartment of Chemistry, Faculty of Science, The University of Jordan, Amman 11942, Jordan. E-mail: a.qaroush@ju.edu.jo
bDepartment of Chemistry, The Hashemite University, P. O. Box 150459, Zarqa 13115, Jordan. E-mail: alaa.eftaiha@hu.edu.jo
cDepartment of Life Sciences and Chemistry, Jacobs University Bremen, Campus Ring 1, 28759 Bremen, Germany. E-mail: k.assaf@jacobs-university.de
First published on 5th January 2018
Abstract
The economic viability of eco-friendly and renewable materials promotes the development of an alternative technology for climate change mitigation. Investigations reported over the past few years have allowed understanding the mechanism of action for a wide spectrum of saccharides toward carbon dioxide (CO2), in terms of reactivity, reversibility, stability and uptake. Exploiting bio-renewables, viz., inedible saccharides, to reduce the anthropogenic carbon footprint upon providing a sustainable and promising technology that is of interest to different groups of scientists, to overcome demerits associated with the current state-of-the-art aqueous amine scrubbing agents, following a “green chemistry guideline”, by employing materials with properties relevant to the environment toward sustainable development. The interdisciplinary nature of research in this area provides a large body of literature that would meet the interest of the broad readership of different multidisciplinary fields. Although many reports emphasize the use of biomass in various industrial products ranging from pharmaceutics, medical preparations, soaps, textiles, cosmetics, household cleaners, and so on, to our knowledge there is no focused article that addresses the application of saccharides for CO2 sequestration. In this review, we highlight the recent advances on the use of oligo-, poly- and cyclic saccharides to achieve a reversible binding of CO2. The future research directions are discussed to provide insight toward achieving sustainable development through implementing bio-renewables.
 Abdussalam K. Qaroush | Abdussalam K. Qaroush received both BSc and MSc degrees from the Hashemite University, Jordan. While performing his PhD, he joined the group of Professor Bernhard Rieger (TUM) developing the concept “Green Sorbents for CO2 Activation”. In 2015, he was called by the Higher Council for Science and Technology in Jordan as a Jordanian Scientist and Technologist Abroad (JoSTA) for technology transfer regarding CO2 activation where he established the Jordanian CO2 team (JCO2T). In 2017, Dr Qaroush started his tenure track at the University of Jordan, working on CO2 mitigation (storage, utilisation, recycling) upon applying green technology, polymer chemistry, and organic synthesis. |
 Hiba S. Alshamaly | Hiba S. Alshamaly received her BSc in chemistry from the University of Jordan in 2009. Currently, Hiba is pursuing her MSc degree at the Hashemite University upon a joint project under the supervision of both Dr Eftaiha and Dr Qaroush. Her MSc thesis involves CO2 activation utilising commercially available materials as green sorbents for CO2 capturing. |
 Shrouq S. Alazzeh | Shrouq S. Alazzeh is a bachelor student at the University of Jordan who is interested in green and analytical chemistry. She is keen to participate in scientific events, conferences and workshops to improve her skills in order to help her continue in the advancement of her studies after graduating from her BSc degree. She always has the curiosity to discover unknown chemical reactions which motivated her to start improving her scientific skillfulness at a young age. Shrouq is looking forward to being a distinctive researcher. |
 Ream H. Abeskhron | Ream H. Abeskhron was born and raised in Amman, Jordan. Currently, she is a third-year bachelor student at the University of Jordan. She has developed a passion for chemistry from a very young age, especially in exploring the mechanisms of chemical reactions. Now, she is a research fellow who is passionate to work with green chemistry. Ream is looking forward to getting her MSc and PhD degrees in organic chemistry. |
 Khaleel I. Assaf | Khaleel I. Assaf was born in Amman, Jordan, in 1985. He received his BSc and MSc degrees in chemistry at the Hashemite University, Jordan. In 2015, he graduated with a PhD degree from Jacobs University Bremen, Germany, under the supervision of Prof. Dr Werner Nau, and has subsequently proceeded as a postdoctoral fellow. His research interests lie in the areas of computational and supramolecular chemistry. |
 Ala’a F. Eftaiha | Ala’a Eftaiha has been an assistant professor of physical chemistry at the Hashemite University, Jordan since 2014. He obtained his BSc and MSc in chemistry from the Hashemite University. Prior to doctoral studies, he was a researcher at the Jordanian Pharmaceutical Manufacturing Company. In 2013, Ala’a earned his PhD at the University of Saskatchewan under the supervision of Prof. Dr Matthew F. Paige, and subsequently worked at Dalhousie University as an NSERC-DREAMS postdoctoral fellow. In 2016, he was a Fulbright visiting scholar at the University of California Santa Barbara. His research interests include CO2 capturing, emerging photovoltaic technologies and surfactant monolayers. |
Introduction
The on-going rise of the anthropogenic greenhouse gas (GHG) footprint has a substantial impact on ecological systems,1–3 particularly in the absence of stringent international policies for climate change mitigation. Although the ultimate goal of the intended nationally determined contributions, which are adopted in the Paris agreement, is to maintain the average temperature of the globe at two degrees Celsius above that of the pre-industrial revolution era,4,5 preliminary indicators demonstrate that advanced economies have failed to meet their commitments to depress GHG emissions,6 which keeps the high concentration of CO2 (the most dominant GHG) as one of the main environmental concerns that should be addressed worldwide through boosting carbon offset strategies7,8 toward a concrete international climate policy for the benefit of future generations.9One of the most efficient approaches for climate change mitigation is carbon capture and storage (CCS), which allows the reduction of CO2 emissions while facing the sustained increase in the global energy demand.10–12 The limitations associated with the state-of-the-art monoethanolamine (MEA, Scheme 1A) solvents such as corrosiveness, volatility and the energy penalty required for solvent regeneration, together with other related CCS based technologies, make implementing the principles of green chemistry,13–15 which were introduced by Paul Anastas and John Warner, of particular importance to design environmentally benign materials and innovative routes to ultimately achieve sustainable development by eliminating hazardous chemicals and energy consuming processes.
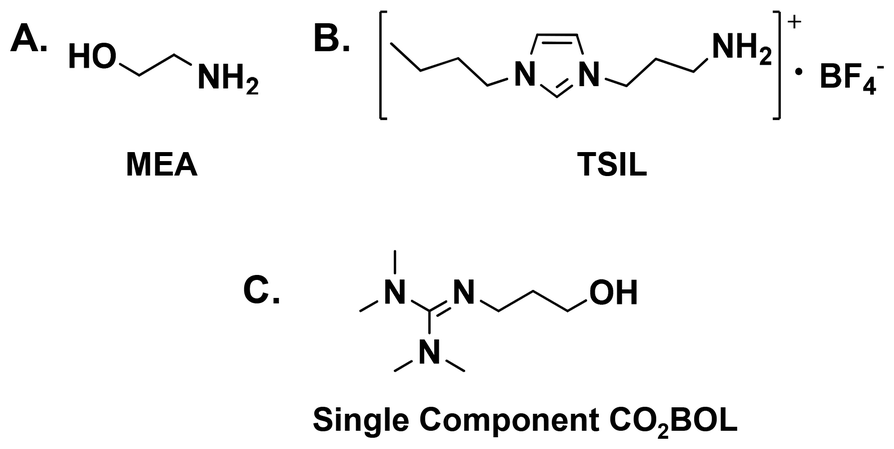 | ||
| Scheme 1 The chemical structures of (A) monoethanolamine (MEA); (B) 1-n-propylamine-3-butylimidazolium tetrafluoroborate, an example of TSIL prepared by the Jr Davis group;16 and (C) 2-(3-hydroxypropyl)-1,1,3,3-tetramethylguanidine (a single component CO2BOL).17 | ||
Following the seventh principle of green chemistry, our group established “renewables for renewables approach” which offers an eco-friendly platform to resolve environmental problems using renewable, inedible polymeric feedstocks for CO2 capturing to promote future development benefiting from their environmental and economic impacts over petroleum-based feedstocks. Examples of renewable feedstocks include, but are not limited to, cellulose, cyclodextrin, chitin and chitosan (chemical structures are shown in Scheme 2).18–20 A major inherent obstacle that hinders the use and processing of cellulose and chitin, despite their abundance in nature, is the limited solubility in common solvents,21,22 which represents a significant technical challenge for both academic and sectors that demand the implementation of alternative greener strategies23,24 other than those reported in the affiliated literature associated with dissolution and activation.25
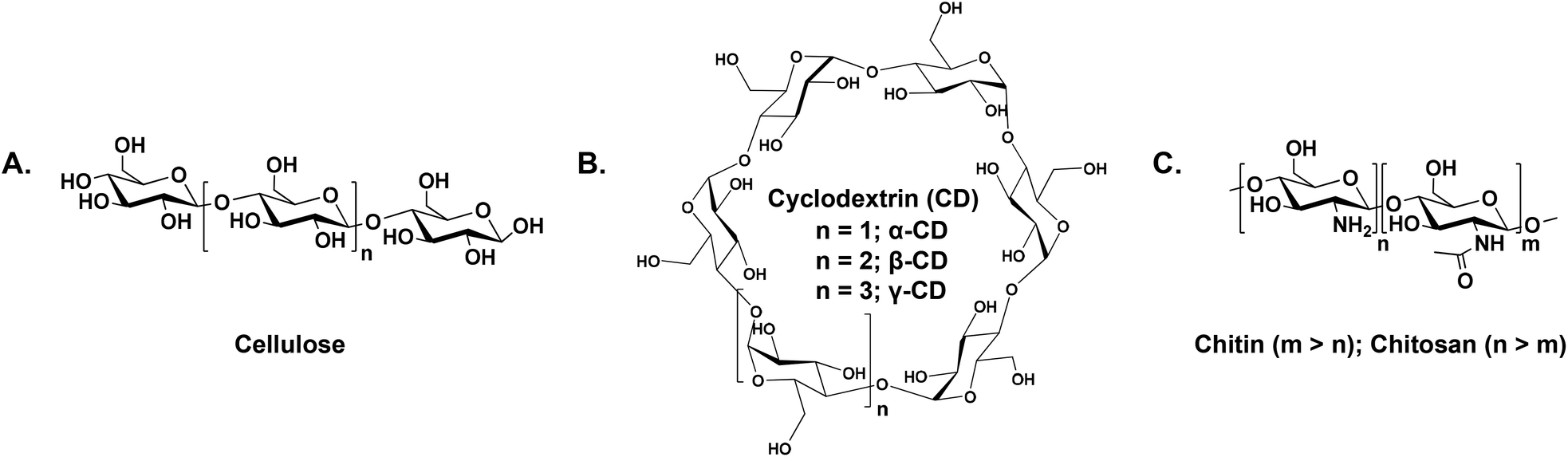 | ||
| Scheme 2 The chemical structures of (A) cellulose, (B) cyclodextrins, and (C) chitin (m > n) and chitosan (m < n). | ||
The fundamental aspects of liquid or solution based sorbents (typically utilised in the most mature capture technology) control the overall process in terms of sorption efficiency and kinetics together with regeneration.26 It is expected that CO2 is absorbed into the condensed phase in the low viscosity regimes, while it adsorbs at the surface of the sorbing material when the mass transfer is restricted to the bulk for high viscosity sorbents. Regarding the mechanism of action, when physical dissolution or physisorption is the prevailing mechanism, the uptake kinetics is expected to be slow and the desorption process is less energy demanding due to the weak, non-bonding interactive forces. If chemisorption is the dominant mechanism, the carbonation reaction is anticipated to be rapid and accompanied by a large energy requirement to regenerate the sequestered species. Chemisorption is accomplished via two different pathways: the first one is CO2 capturing via “amine scrubbing” through the formation of carbamic acid/carbamate (RR′NH–CO2− +NH2−RR′) following a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1/1:2 reaction mechanism, respectively,27,28 in which the nucleophilic attack over CO2 by the amine nitrogen results in the formation of carbamic acid, which reacts with a second mole of amine (sacrificial base) to produce the carbamate salt. In the presence of water, a 1:1 reaction pathway takes place resulting in the formation of a bicarbonate (RR′NH2+ −O2COH) zwitterionic adduct.29–31 It is noteworthy that this pathway is followed when a benzylic amine (o, m, p-xylylenediamines) is used.32 The proposed chemical reaction between amines and CO2 is summarized in Scheme 3A. Similarly to conventional amine scrubbing agents, task-specific ionic liquids (TSILs and amine-functionalized ILs, Scheme 1B) form carbamate salts when exposed to CO2. Although TSILs have several merits over MEA aqueous solution in terms of lower CO2 binding energies, milder regeneration conditions and higher capture capacities, they are not commercially available as MEA33 (commercial sources of MEA (≥98%) list the retail price at $25–$40 per kilogram34,35). TSILs suffer from a huge increase in viscosity when loaded with CO2 due to the hydrogen bonding (H-bonding) network between the carbamate and ammonium counterparts. This could be overcome by following different strategies such as incorporating an aprotic heterocyclic anion36 or mixing TSILs with low-viscosity solvents37 (more reviews regarding using TSILs for capturing CO2 can be found elsewhere33,38).
:1/1:2 reaction mechanism, respectively,27,28 in which the nucleophilic attack over CO2 by the amine nitrogen results in the formation of carbamic acid, which reacts with a second mole of amine (sacrificial base) to produce the carbamate salt. In the presence of water, a 1:1 reaction pathway takes place resulting in the formation of a bicarbonate (RR′NH2+ −O2COH) zwitterionic adduct.29–31 It is noteworthy that this pathway is followed when a benzylic amine (o, m, p-xylylenediamines) is used.32 The proposed chemical reaction between amines and CO2 is summarized in Scheme 3A. Similarly to conventional amine scrubbing agents, task-specific ionic liquids (TSILs and amine-functionalized ILs, Scheme 1B) form carbamate salts when exposed to CO2. Although TSILs have several merits over MEA aqueous solution in terms of lower CO2 binding energies, milder regeneration conditions and higher capture capacities, they are not commercially available as MEA33 (commercial sources of MEA (≥98%) list the retail price at $25–$40 per kilogram34,35). TSILs suffer from a huge increase in viscosity when loaded with CO2 due to the hydrogen bonding (H-bonding) network between the carbamate and ammonium counterparts. This could be overcome by following different strategies such as incorporating an aprotic heterocyclic anion36 or mixing TSILs with low-viscosity solvents37 (more reviews regarding using TSILs for capturing CO2 can be found elsewhere33,38).
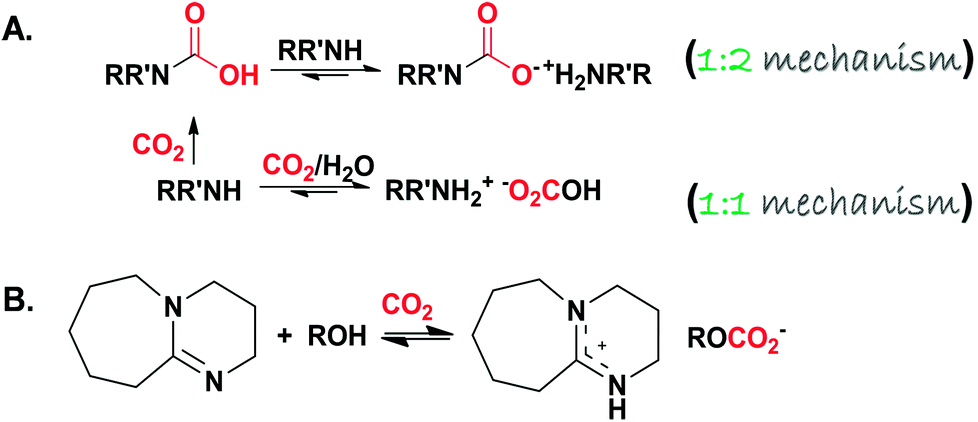 | ||
| Scheme 3 (A) Proposed chemisorption of CO2 by amine based substrates (primary and secondary, the general formula is given for the secondary for clarification) through the formation of RR′N–CO2− +NH2−R′R and RR′NH2+ −O2COH in the absence and presence of water, respectively. (B) The reversible reaction between an alcohol (ROH) and 1,8-diazabicyclo-[5.4.0]-undec-7-ene (DBU) in the presence of CO2. | ||
The second pathway results in the formation of an inorganic or organic carbonate (M2(CO3)X, M+x: a metal cation, and ROCO2−, respectively) following a 1:1 reaction mechanism. Aside from M2(CO3)X, the formation of ROCO2− by activating alcohols to react with CO2 was reported in the literature by two research groups. Firstly, Jessop and co-workers39,40 devised CO2 binding organic liquids (CO2BOLs) which comprised of anhydrous alcohol and a superbase (SB). An example reaction is shown in Scheme 3B. The high volatility associated with CO2BOLs and the hardship of drying/handling, together with the high corrosive character, paved the way toward single component CO2BOLs17 (Scheme 1C) that unfortunately exhibited high viscosity upon chemisorption. Afterwards, Sir J. F. Stoddart’s group41–44 reported that the hydroxyl groups located at the rim of γ-cyclodextrin-based metal–organic frameworks (CD-MOFs) are capable of reacting with CO2 reversibly to form a metal-stabilized organic alkyl-carbonate (a detailed discussion is provided, vide infra).
Most of the literature reports focus on the capturing process highlighting the high-energy penalty associated with the regeneration of conventional aqueous amine solvents in comparison with alcohol/SB binary mixtures that suffer from high volatility and corrosiveness, which makes the use of aprotic solvents a reasonable alternative to increase the efficiency of carbon dioxide desorption as they have a lower heat capacity than aqueous based sorbents and a higher vapor pressure as introduced by Park and colleagues,45 and followed-up by our research group.28,46,47 As a matter of fact, the solvation of ions is of great importance when forming CO2 adducts from neutral solutions. Accordingly, cations are well solvated in polar aprotic solvents, while anions are poorly solvated, therefore, this might favour a shift in the equilibrium towards the neutral species and, consequently, pKa values greater than that formed while working in aqueous solutions.45
In this minireview, recent advances on the sorption of CO2 by inedible saccharides is discussed as follows: (1) a review of the reversible reaction between cellulose and CO2, and the subsequent dissolution and regeneration; (2) the capture and storage of CO2 by cyclodextrins; (3) the chemisorption of CO2 by chitin and chitosan using ILs, oligomers and nanoparticles; and (4) CO2 capturing using functionalised cellulosic materials. We conclude with the challenges and future outlooks of exploiting bio-renewables to be implemented in the area of CCS research.
Cellulose
Cellulose, a low-value agricultural by-product, is the most naturally occurring biopolymer, with a production capacity of ca. 180 billion tons per annum by plants.48,49 It is a linear polymer consisting of β-(1,4)-linked glucopyranose units (Scheme 2A). It is characterized by strong inter- and intramolecular H-bonding which play a major role in its structural stability, chemical properties and resistance to enzymatic hydrolysis. One major obstacle that hinders the effective utilisation of cellulose is its insolubility in conventional solvents due to the semicrystalline structure.21 Over the past few years, several solvent systems for the dissolution of cellulose have been reported50–54 that suffered from serious drawbacks such as corrosiveness, high cost, and reduced dissolution capacity in the presence of polar solvents as well as recycling and purification difficulties which required alternative eco-friendly approaches for cellulose processing such as the use of pressurised CO2 to precipitate cellulose from ILs.55–59Following the concept of switchable solvents, Jérôme and co-workers60 reported on the dissolution of microcrystalline cellulose (MCC) suspended in a mixture of dimethyl sulfoxide (DMSO) and 1,5-diazabicyclo[4.3.0]non-5-ene (DBN) upon bubbling CO2. In the presence of a SB, the primary hydroxyl groups along the cellulose backbone reacted with CO2 resulting in the formation of carbonate species. The process could be reversed by heating the solution at 30 °C under vacuum, by which the cellulose was precipitated (Fig. 1). After exposing this precipitate to CO2, the cellulose was dissolved again which highlighted the reversibility of the system. It is noteworthy that the use of weak bases was not efficient to dissolve cellulose under the investigated conditions, as they were incapable of activating the hydroxyl groups.
 | ||
| Fig. 1 Photographs of (A) a suspension of MCC in DMSO containing 9 wt% of DBN and (B) a transparent, viscous solution with a maximum concentration of 6 wt% cellulose, obtained after exposing the suspension at 40 °C to 2 bar of CO2 for 1 hour. Reprinted with permission from ref. 60. | ||
Another example of a reaction between cellulose and CO2 in the presence of a SB (namely 1,8-diazabicyclo-[5.4.0]-undec-7-ene, DBU), yielding [DBUH]+[cellulose-OCO2]−, was reported by Yang et al.61 The reaction allowed a successful dissolution of the cellulose pulp, and subsequently allowed its modification into cellulose acetate using acetic anhydride (Scheme 4A). Mu and co-workers62 monitored the formation of the aforementioned adduct via in situ infrared spectroscopy. The formed carbonated cellulose underwent a chemical reaction with a silane-based reagent (TEOS: tetraethyl orthosilicate) which provided a green method for the preparation of a super-hydrophobic nanocoated material (Scheme 4B). Moreover, a cellulose-graft-poly(L-lactide) copolymer was synthesized, exploiting the reversible character of the aforementioned reaction. DBU had a dual action in cellulose dissolution with its role as an organocatalyst for the subsequent ring-opening polymerization process (Scheme 4C).63
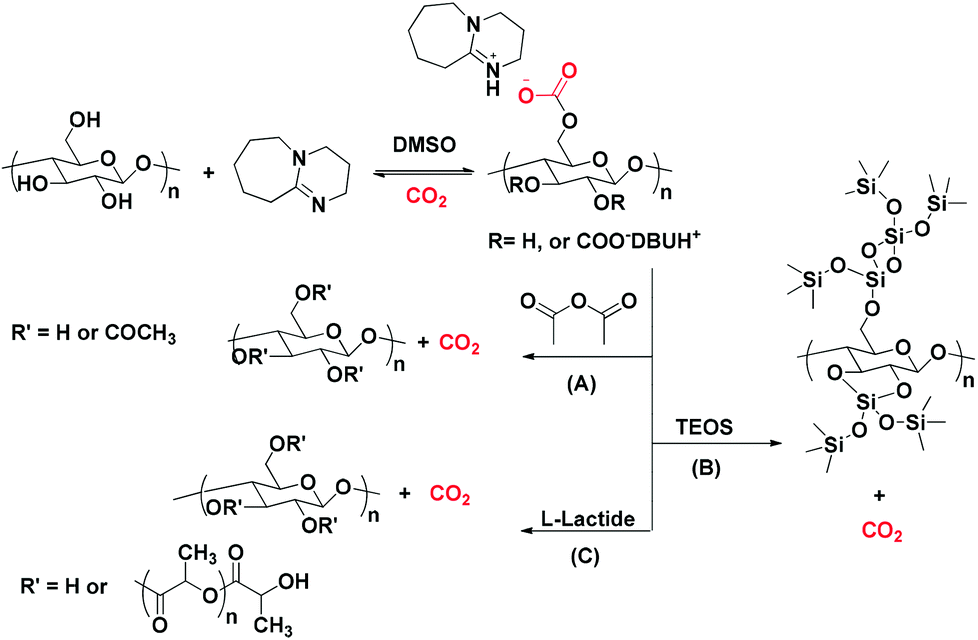 | ||
| Scheme 4 The versatility of cellulose in several applications upon activation through its reaction with CO2 in the presence of DBU. | ||
Once again, Mu’s research group64 reported a detailed study of the dissolution process to understand the mechanism for the formation of a MCC carbonated/SB adduct in DMSO upon bubbling CO2. It was found that DMSO helped in the dissociation of the adduct into free ions by balancing the concentration of the ions and the number of hydrogen bonds. In this context, density functional theory (DFT) calculations revealed multiple interactions between the protonated base and the carbonated cellulose. Very recently, the chemisorption of CO2 onto cellulose in aqueous NaOH solution in the absence of a SB has been reported by Gunnarsson et al.65 and confirmed using both nuclear magnetic resonance (NMR) and attenuated total reflectance Fourier transform infrared spectroscopy (ATR-FTIR). The reversibility of the reaction was achieved by the addition of water. In addition, treating cellulose with NaOH and CO2 in dimethylacetamide (DMAc) resulted in the formation of carbonated cellulose.66 FTIR spectroscopy supported the introduction of the new functionality of cellulose via the existence of a band at 1593 cm−1, as well as a decrease in intensity of the peak associated with hydrogen bonded hydroxyl groups.66
The use of 1,1,3,3-tetramethylguanidine (TMG), in combination with an alcohol dissolved in DMSO revealed a new switchable solvent system that was capable of dissolving cellulose after exposure to CO2.67 The authors attributed the dissolution phenomenon to the formation of a guanidinium alkylcarbonate salt, as identified by both 13C NMR (the emergence of a new peak at 159.2 ppm) and in situ FTIR (newly formed asymmetric and symmetric carbonyl stretching bands at 1663 and 1410 cm−1, respectively). The dissolution of cellulose was alcohol dependent. In the case of ethylene glycol, a fully transparent cellulose solution was observed, allowing for a maximum de-crystallization up to 10 wt% concentration with high stability. In accordance with a previous report,60 no dissolution of cellulose was observed when weak bases (pKa < 12) were utilised.67 In the same context, Nanta et al.68 applied a similar switchable solution consisting of DBU and ethylene glycol in DMSO under high-pressure carbon dioxide to facilitate the dissolution and modification of cellulose derived from cassava pulp waste.
Cyclodextrin
Cyclodextrins (CDs, Scheme 2B) are a class of naturally occurring cyclic oligosaccharide that are obtained via the enzymatic degradation of starch. They consist of D-glucopyranose units linked via α-1,4 linkages in a truncated cone shape, which has a hydrophobic cavity and a hydrophilic outer surface. While the secondary hydroxyl groups are located on the wider rim of the cone, the primary counterparts are on the narrower one.69 The supramolecular chemistry of CDs makes them useful for a wide range of applications in different fields such as pharmacy, food, medicine, the environment and engineering.70Sir J. F Stoddart and his research group exploited the symmetry of γ-CD to synthesize nanoporous materials, which is a prerequisite to induce crystallization upon coordinating the macrocyclic motif to a metal centre, with surface areas of ∼1200 m2 g−1 under mild conditions. It has been reported that γ-CD tends to assemble into crystalline MOFs in the presence of alkali metal salts, and are grown from the vapour diffusion of methanol (MeOH).71,72 Among all investigated MOFs, CD-MOF-2, comprised of γ-CD and rubidium cations (Rb+), showed an outstanding stability after the removal of the solvent compared with other cubic CD-MOFs prepared from potassium (K, CD-MOF-1) and caesium (Cs, CD-MOF-3) salts,73 which makes it useful for subsequent investigations as a CO2 sorbent.41–43Fig. 2A and B show ball-and-stick and space filling representations of CD-MOF-2, respectively.
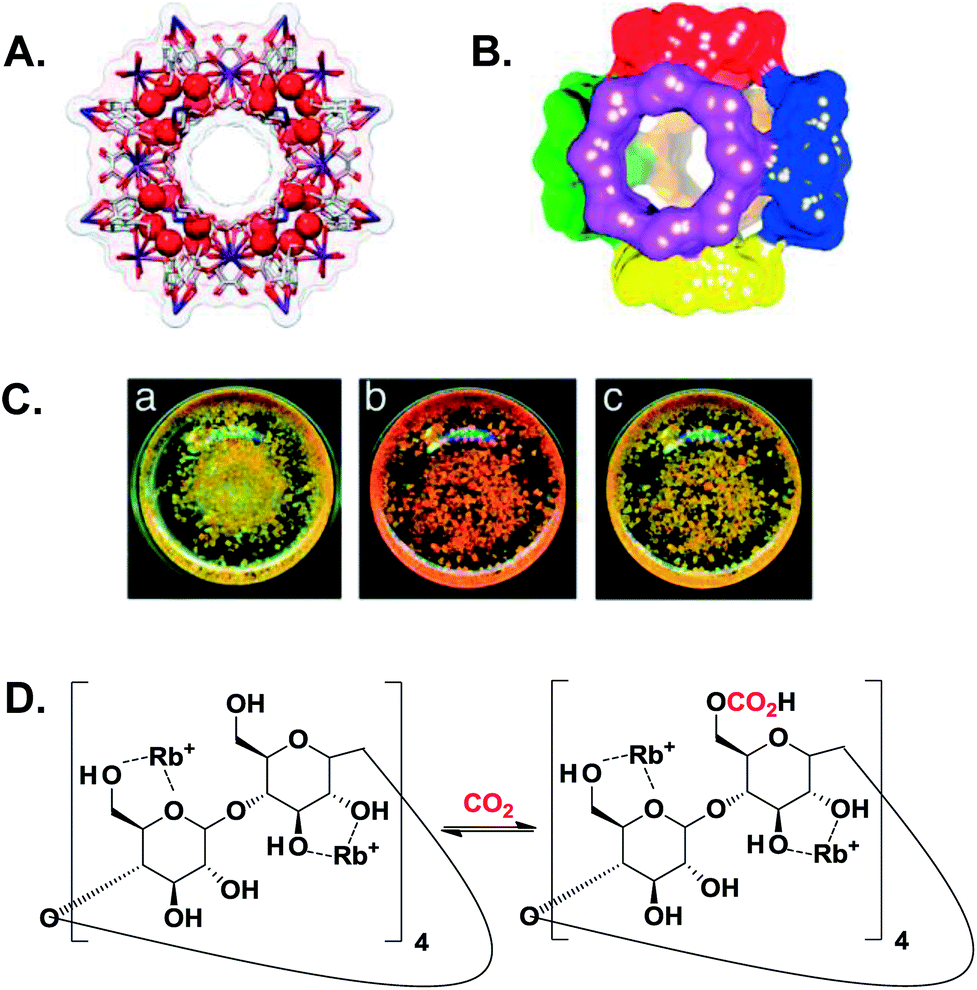 | ||
| Fig. 2 (A) A ball-and-stick view of a cubic array made of (γ-CD)6 units coordinated to 24 Rb+ (red spheres) through the ring oxygen atoms of the secondary and primary hydroxyl groups. (B) A space-filling depiction of six (γ-CD)6 units that link together in a body-centered cubic extended framework. (C) Photographs of CD-MOF-2 incorporating methyl red before (a) and after (b) exposure to CO2. Removal of the CO2 atmosphere restored the colour back to yellow (c). A–C are adapted with permission from ref. 41. (D) A proposed carbonation reaction of activated CD-MOF-2 at the less reactive primary hydroxyl groups, where the free hydroxyls act as nucleophiles to attack CO2.41 | ||
The chemisorption of CO2 by CD-MOF-2 was proved using cross-polarization magic-angle-spinning (CP/MAS) 13C NMR spectroscopy through the emergence of a new peak at 158 ppm that was attributed to the formation of the alkyl carbonic acid based CD-MOF, which was further supported calorimetrically. As shown in Fig. 2C, the addition of methyl red to CD-MOF-2 crystals resulted in a yellow colour due to the basic environment within the framework. Upon exposure to CO2, the yellow colour turned red which was attributed to the formation of carbonic acid throughout the framework, and returned back to yellow after the removal of CO2. It was noteworthy that the CP/MAS spectra of iso-structural CD-MOFs prepared from rubidium fluoride or potassium benzoate (rather than rubidium hydroxide as in the case of CD-MOF-2) revealed identical resonances at 158 ppm, which discard the importance of the hydroxide anion in facilitating chemisorption in comparison with weaker nucleophiles such as fluoride and benzoate anions.41 The same conclusion was obtained from the substantial difference between the adsorption enthalpy on CD-MOF-2 at near-zero coverage and an analogous acid–base reaction that involved a significant concentration of free hydroxide anions (OH−), which suggested that the adsorption took place through the alcoholic functional groups, rather than the interstitial OH− (vide infra).42 Adsorption isotherms indicated that chemisorption was the dominant mechanism between 0 and 25 °C with a sorption capacity of ∼23 cm3 CO2 per g sorbent.41
Mechanistic insight into the adsorption of CO2 on CD-MOF-2 was obtained using calorimetric measurements. The differential enthalpy of the carbonation reaction confirmed that the chemisorption followed two distinct patterns. Firstly, at near-zero coverage with a highly exothermic reaction (−113.5 kJ mol−1 CO2) via the most reactive primary hydroxyl groups, followed by a milder reaction (−65.4 kJ mol−1 CO2) through the less reactive primary and secondary hydroxyl groups at a surface coverage between 0.1 and 0.3. At a higher surface coverage (>0.4), physisorption is the dominant adsorption mechanism with a differential enthalpy of −40.1 kJ mol−1 CO2.42 In our consensus regarding the reaction reversibility, there was unsurprising inconsistency between the results obtained from the isothermal gas uptake and the calorimetric data. The former designated a reversible chemical fixation of CO2,41 the difference between the enthalpy of adsorption at near-zero coverage and the subsequent adsorption cycles indicated an irreversible binding character between the most reactive primary hydroxyl groups and CO2, followed by a reversible binding on the less reactive hydroxyls together with the physisorbed CO2.42 The carbonation reaction of CD-MOF-2 is shown in Fig. 2D.74
The challenge in synthesizing CD-MOFs with cations other than K+, Rb+ and Cs+, is the inability to obtain porous, crystalline frameworks. Very recently, Patel et al.44 demonstrated the partial substitution of K+ with Li+ through co-crystallizing KOH and LiOH with γ-CD. As shown in Table 1, the most stable Li/K–CD-MOF with the highest Li+:K+ ratio (0.61:1.18) had a comparable Brunauer–Emmett–Teller (BET) surface area and a characteristic better sorption in comparison with those obtained for other iso-structural CD-MOFs.
| Sorbent | BET surface area (m2 g−1 sorbent) | Sorption mechanism | Sorption capacity (mmol CO2 per g sorbent) | ||
|---|---|---|---|---|---|
| 0 °C | 25 °C | ||||
| a The uptake was measured at 1 bar. b The uptake was measured at 3 bar. c The CO2 adsorption experiment was carried out at 30 °C. | |||||
| CD-MOF-1 | 1145 (ref. 44) (1320 (ref. 71)) | Chemi & physisorption | 4.2 (ref. 44) | 3.0 (ref. 44) | |
| CD-MOF-2 | 1110 (ref. 71) | 1.7 (ref. 44) | 1.4 (ref. 44) | ||
| Li/K–CD-MOF | 1205 (ref. 44) | 4.5 (ref. 44) | 3.1 (ref. 44) | ||
| CD–aniline | NA | Chemisorption | 0.69c (ref. 75) | ||
| α-CDPC | 703 (ref. 76) | Physisorption | 3.3 (ref. 76) | NA | |
| β-CDPC | 699 (ref. 76) | 2.9 (ref. 76) | NA | ||
| γ-CDPC | 678 (ref. 76) | 2.8 (ref. 76) | NA | ||
| CD–SO3H | 211 (ref. 77) | NA | 1.2a(ref. 77) | 1.7b(ref. 77) | |
The reversible chemisorption of CO2 by CD-MOF-2 was utilised for the electrochemical sensing of CO2. Unexpectedly, it has been reported that CD-MOF-2 (either as a pristine powder or pellet76) displayed a ∼550-fold decrease in ionic conductivity upon exposure to CO2 for 5 minutes in the presence of MeOH or n-hexane, although carbonic acid is relatively more acidic than primary alcohols. This was attributed to the sealing of the β-windows (small triangular-shape voids of diameter 4.2 Å, Fig. 3) of the [(Rb+)4(γ-CD)]6 cubic cages after the carbonation reaction, where the reactive primary hydroxyl groups are located, thus reducing the mass transfer of intermediate molecules. The sensor reusability was tested by measuring the change in conductivity of a CD-MOF-2 pellet following sequential CO2-sorption and desorption (by heating at ∼80 °C for several cycles). It was noteworthy that the conductivity change was higher at low CO2 concentration (less than 20% in the presence of N2 gas).40
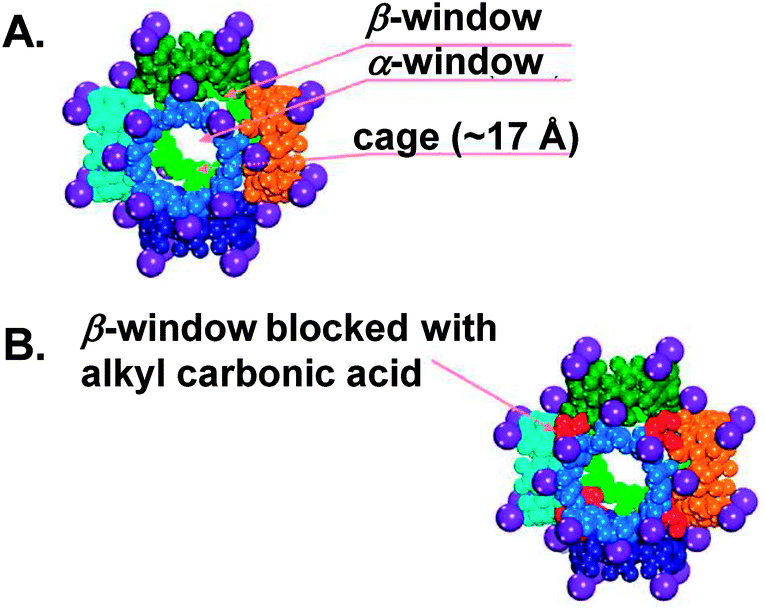 | ||
| Fig. 3 A space-filling representation of [(Rb+)4(γ-CD)]6 cubic cages. Purple spheres indicate Rb+; the sides of the cube (six-CD rings) are depicted in different colours. Adapted with permission from ref. 43. | ||
Aside from CD-MOFs, it has been reported that the inclusion complex of 1:1 β-CD and aniline (CD–aniline) in association with residual water molecules inside the CD cavity, chemisorbed CO2 with a sorption efficiency of 0.69 mmol of CO2 per g sorbent through the formation of bicarbonate according to the following reaction: C6H5NH2 + CO2 + H2O ⇌ C6H5NH3+ HCO3−. The sorption capacity was decreased by 37% when the sorbent was heated to 100 °C under vacuum, which emphasized the importance of the water molecules to the sequestration process.75 Although the performance characteristics of the CD–aniline complex were much lower than the corresponding CD-MOFs, it sorbed 0.85 mol CO2 per mol nitrogen (N) in comparison with the amine efficiency of 0.5 mol CO2 per mol N following a 2:1 sorption mechanism under dry conditions, which was unlikely to take place in the case of the explored inclusion complex. Unlike CD-MOFs (vide supra), the sorption capacity of the amorphous CD–aniline complex (obtained upon grinding) was almost similar to the as-synthesised crystalline material.
Han and co-workers76 reported the facile and elegant synthesis of CD-based microporous carbon (CDPC) using acid catalysed solvothermal carbonization at 180 °C. The obtained materials were regular spheres with diameters between 300 and 800 nm, BET surface areas ranging from 600 to 700 m2 g−1 and high content of oxygen-containing functional groups, such as carboxyl, carbonyl, and hydroxyl as inferred from FTIR and 13C CP/MAS spectra. As shown in Table 1, the CO2 gravimetric uptake of α-, β- and γ-CDPC measured at 0 °C and 1.0 bar were between 2.8 and 3.5 mmol CO2 per g sorbent. Similarly, it has been reported that grafting β-CD with sulfonic acid functional groups (–SO3H) led to the formation of a porous structure (CD–SO3H) with a low oxygen content due to dehydration, which was able to physisorb CO2 30 times more than β-CD at the same investigated conditions (1.2 of the former against 0.04 mmol CO2 per g sorbent of β-CD) with high selectivity over O2, N2 and CH4, which makes it a potential candidate for CO2 separation from flue gas.77
Turning to wet scrubbing applications, the hydroxyl groups of the CD macrocycle should be activated to become susceptible to nucleophilic attack prior the exposure to CO2. It has been reported that a mixture of β-CD and an organic SB, namely DBU, was able to fix CO2 chemically as an amidinium alkylcarbonate. Due to viscosity issues, the ratio (in equivalents) of β-CD:DBU was kept at 0.5:1 throughout the work. The carbonation reaction was followed by 1H and 13C NMR together with FTIR spectroscopy, following the peaks associated with the quaternary carbon of DBU. The relatively low uptake of CO2 (1.8 wt%) was explained by the excessive use of the SB together with the inefficient stirring during the synthesis of the sequestered species. Notably, the sorption capacity was 7.9 wt% in the case of glucose:DBU mixture.78
Chitin and chitosan
Among the various, spread polysaccharides, amine containing sugars, in which the –OH group at C-2 of the hexose monomeric unit is replaced by –NH2, are the main component of chitin (Scheme 2C, m > 50%), which is mainly found in crustaceans, insects, molluscs, bacteria and fungi. The insolubility of chitin in common solvents represents a considerable barrier that confronts its employment in various fields. In the early eighties of the last century, Austin79 explored the solubility of a chitin–lithium chloride (LiCl) complex in several amide solvents such as DMAc and N-methyl-2-pyrrolidone (NMP), followed by several attempts to dissolve chitin using salts and acids (for more details, see the review by Rinaudo22). Deacetylation of chitin either enzymatically or under alkaline conditions80 results in chitosan (Scheme 2C, n > 50%). The solubility of chitosan in aqueous acidic media is dependent on several structural parameters such as the degree of deacetylation, the distribution of the acetyl groups along the polymer backbone and the molar mass.In 2006, Zhang and co-workers81 reported on the use of ILs, namely 1-butyl-3-methyl-imidazolium chloride (Bmim)Cl, as an alternative solvent for chitin and chitosan due to its ability to break down the polymer crystallinity under mild conditions as inferred from wide-angle X-ray diffraction (WAXD) measurements. Interestingly, both chitin and chitosan were recovered from the IL solution using methanol or water. The sorption efficiency of 10 wt% solutions of chitosan/IL and chitin/IL were measured gravimetrically using a microbalance at 30 °C and 1 atm CO2 pressure. Results indicated that the efficiency of the former was 8.1% compared to 3.8% for the latter. Although no spectroscopic evidence for chemisorption was provided, the authors justified the higher sorption capacity of the chitosan-based solution by the chemical fixation of CO2 due to the presence of free amine groups which are either blocked or presented in a lower fraction in the case of chitin. Regarding regeneration, it was reported that heating the chitosan/IL solution at 100 °C for 30 minutes reduced the sorption efficiency to 1.4%, afterwards the IL solution can be used for subsequent capture/release cycles.
The sorption capacity measurements performed by Mu’s research group82 using 6 wt% of chitosan or chitin dissolved in 1-butyl-3-methylimidazolium acetate (Bmim)OAc were consistent with those reported previously by Zhang’s group.81 The values obtained using thermogravimetric analysis (TGA) were 6.7 and 3.5% for chitosan and chitin, respectively, whereas it was 1.3% for neat (Bmim)OAc. The almost doubled capacity of the former suggested that chemisorption was the main mechanism for CO2 capturing through carbamate formation in the case of chitosan, while physical dissolution was responsible for the capturing in the case of chitin. The chemisorption was proved using ATR-FTIR spectroscopy through the disappearance of the peak at 1680 cm−1 which belongs to the bending mode of N–H, the appearance of a new peak centred at 1652 cm−1 ascribed to –N–C(O)– amide moiety of the carbamate ion. The sequestered species was regenerated under 1 atm N2 at 80 °C. The ability of the compressed CO2 to act as an anti-solvent56,83 was exploited to precipitate chitosan through the reaction between CO2 and the acetate-based ILs to end up with the formation of a carboxylate zwitterionic dimer that changes the hydrogen-bonding properties of the IL as inferred from the measurements of the solvatochromic parameters.
Recently, Wasserscheid and co-workers introduced a new type of immobilized IL by encapsulating IL84 or ionogel droplets85 (chitosan dissolved in IL and diluted with acetonitrile (ACN)) in fumed silica forming a free-flowing IL (ionogel)-in-air powder or inverse supported ionic liquid phase (SILP) material (Fig. 4) after evaporating the ACN. The key advantages of the inverse SILP is the high ratio of IL (ionogel) to the nanoporous material together with relatively short gas diffusion distances.
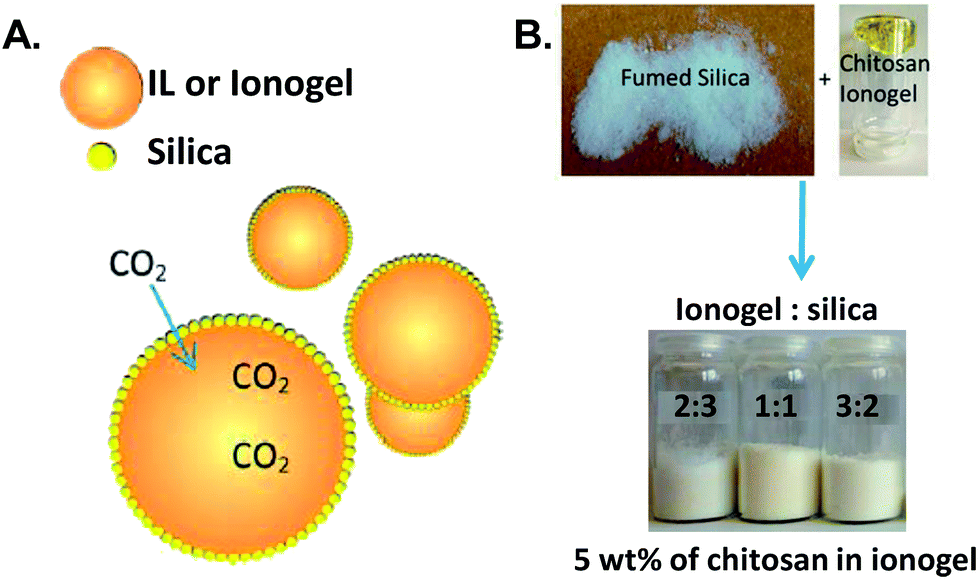 | ||
| Fig. 4 (A) Depiction of an inverse SILP. (B) A photograph of inverse SILPs prepared from an ionogel comprised of 5 wt% chitosan dissolved in 1-ethyl-3-methylimidazolium acetate ([EMIM][OAc]) with different ionogel to silica ratios. Reprinted with permission from ref. 84 and 85. | ||
The gravimetric absorption capacity of the inverse SILP made of 40 wt% IL or ionogel (with 5 wt% of chitosan) and 60 wt% silica measured at 1 bar and 40 °C ranged between 0.1 and 0.8 mmol CO2 per g sorbent. The results indicated that the CO2 uptake was increased when an IL with a weakly basic anion such as chloride or 2-methoxyethyl methylphosphonate were used due to the extra carbamation reaction provided by chitosan together with the enhanced solubility of CO2 in an ionogel compared with the corresponding IL. However, employing a more basic anion such as acetate obstructed the chemistry of sorption by chitosan due to an acidic side product obtained from the carbonation reaction of the IL itself. In comparison with neat chitosan powder, the absorption capacity calculated based on the number of amino groups present in chitosan was improved by 10 times in the inverse SILP. The low absorption efficiency of the neat chitosan was attributed to the restriction of the carbamation reaction at the interface of the powder.
Amine-functionalized porous materials represent an alternative technology for CO2 capturing.12 Chitosan represents a promising alternative for mesoporous silica in the quest to find eco-friendly and low-cost materials to be used as an amine support. Fujiki and Yogo86,87 reported on porous chitosan beads impregnated with polyethyleneimine (PEI) working under both dry and humid conditions with CO2 adsorption capacity of 2.38 and 3.61 mmol per g sorbent, respectively, at 15 kPa of CO2 and 66 °C through the formation of ammonium carbamate as confirmed by in situ diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS). The enhanced CO2 adsorption under wet conditions was explained by the swelling of the chitosan framework, thus exposing buried amino groups to CO2, which resulted in the formation of two adsorption states of CO2 (whereas one state was recognised under dry conditions) as indicated by TGA measurements.
Although Zhang81 opened new horizons for exploiting chitin for CO2 capturing through physical dissolution, our research group published the first report on the chemisorption of CO2 by an oligomeric chitin acetate (CA)/DMSO binary mixture.88,89 The only functional groups along this oligomeric material available for the capturing process were the hydroxyl groups whereas the amino groups at C-2 of the amine pyranose rings were blocked either by protonation or acetylation. Herein, the use of DMSO was a prerequisite for the carbonation reaction to take place through the intermolecular hydrogen bonding between the polar aprotic solvent and primary hydroxyl group at C-6 of the pyranose ring which prompted the nucleophilic attack at CO2 to form the organic alkyl-carbonate as follows: RCH2OH + CO2 ⇌ RCH2OCO2− + H+. The formation of the CO2 sequestered species was confirmed using 13C NMR and in situ ATR-FTIR spectroscopy, through the appearance of the benchmark chemical shift at ca. 157 ppm together with a peak centred at 1555 cm−1 which was ascribed to the vibrational mode of the carbonate. Notably, glucosamine hydrochloride (the monomeric unit of the oligomer) showed no chemisorption upon bubbling CO2. The stability aspects of the organic carbonate adduct were understood using 1H NMR spectroscopy and DFT calculations that indicated that the product was stabilized through non-bonding interactions, namely H-bonding and ionic interaction along the oligomer backbone. This indicated that the supramolecular stabilization was the driving force for the chemisorption, which was absent in the case of glucosamine hydrochloride. The CO2 sorption uptake was measured volumetrically using in situ ATR-FTIR autoclave with a maximum capacity of 3.63 mmol CO2 per g sorbent for a 10.0% (w/v) CA/DMSO solution. The decrease in the sorption capacity with increased concentration was attributed to the huge increase in viscosity at higher concentration of the oligosaccharide solution, which may have hindered the diffusion of CO2 to the bulk of the solution.
To ultimately maximize the sorption capacity of CO2, while using bio-renewable and inedible materials, we moved our attention over oligomeric chitosan due to the presence of two potential active sites to interact with CO2, through the formation of carbonate and carbamate. In this context, our group reported on the use of a low molecular weight chitosan hydrochloride (CS·HCl) dissolved in DMSO for CO2 sequestration.90 Upon bubbling CO2, the formation of the organic carbonate adduct was consistent with the previously affiliated system and ascribed to the supramolecular chemisorption. Ex situ ATR-FTIR spectroscopy of the CS·HCl/NaOH/DMSO solution confirmed the formation of the other sequestered functionality, viz. ammonium carbamate, after bubbling CO2, through the appearance of the fingerprint frequency at 1709 cm−1 which was further elucidated using 1H–15N heteronuclear single quantum coherence (HSQC) NMR spectroscopy, with coupled cross-peaks at 6.8 (correlated to the ammonium counterpart chemical shift) and 84.7 ppm. The CO2 uptake of the CS·HCl/NaOH/DMSO solution was quantified volumetrically using an in situ ATR-FTIR autoclave. A 10% (w/v) solution chemisorbed 1.60 mmol CO2 per g sorbent. Remarkably, the formation of the carbonate was four times that of the carbamate, with faster kinetics for the latter as inferred from the sorption profiles. To our knowledge, this was the first organic carbamato–carbonato bond formation.
One of the major concerns regarding the greenness of DMSO was answered by measuring the biodegradability of the explored system. Following the guidelines of the International Organization for Standardization (ISO), the relatively fast degradation of chitosan dissolved in DMSO (80% in 33 days) provided clear-cut evidence for the absence of any tangible negative impact of using DMSO on the biodegradation of chitosan.
Cellulose-based functionalized sorbent materials
The main uses of cellulose as an abundant biopolymer that functions as a mechanical support and in sustainable porous carbon-based materials, as well as for the preparation of hollow fibres as in membranes for the capturing of CO2, are reported in this section.In 2011, Steinfeld and co-workers91 reported a novel, eco-friendly, benign synthesis for an amine-based nanofibrillated cellulose (NFC) sorbent applicable for CO2 capture from air using N-(2-aminoethyl)-3-aminopropylmethyldimethoxysilane (AEAPDMS). At 25 °C and after 12 h, 1.39 mmol CO2 per g sorbent was sorbed with a relative humidity of 40% and 506 ppm of CO2 concentration in air. The studied material showed persistent stability over 20 consecutive 2-hour adsorption/1-hour desorption cycles. The cyclic capacity of the resultant cycles gave 0.695 mmol CO2 per g sorbent. Once again, as an augmentation to Steinfeld’s92 early work on NFC, the stability of amine-tethered-NFC for CO2 capturing from air (400–530 ppm) under temperature–vacuum swing (TVS) conditions was tested 100 times at 30 °C and 60% humidity for adsorption, followed by desorption at 90 °C and 30 mbar. The average adsorption capacity under the applied conditions was 0.90 mmol CO2 per g sorbent. Due to the presence of O2, the sorbent degraded at 90 °C, reducing its adsorption capacity by 30% (after 15 h and dew point of 22 °C). On the other hand, if moist CO2 was introduced to the sample at the same applied conditions, no tangible loss in CO2 adsorption capacity was recorded. Sevilla and Fuertes93 reported on the utilisation of sustainable porous carbon materials that were prepared using the chemical activation of hydrothermally carbonized starch, cellulose and sawdust (one type of biomass) and further tested for capturing purposes. The activation process was carried out under both severe and mild conditions (KOH/precursor = 4 and 2, respectively, within a set of temperatures in the range of 600–800 °C). Samples obtained under mild activating conditions showed smaller pore sizes and surface areas than those materials obtained under harsher conditions. However, they exhibited the best capacity to store CO2 which is equivalent to 4.8 mmol g−1 (at 25 °C with KOH/precursor = 2 and activated at 600 °C). According to the authors, this was the highest value for any activated carbon. The prepared materials showed fast adsorption kinetics and easy regeneration with good CO2/N2 selectivity measured under equilibrium conditions ∼5.4. Pacheco et al.94 reported on the modification of an aminosilane-functionalized cellulosic polymer sorbent by the anhydrous grafting of N-(2-minoethyl)-3-aminoisobutyldimethylmethoxysilane. The sorption capacity under dry conditions (5 atm, 308 K) was 1.46 mmol per g sorbent with an amine loading of 5.18 mmol amine(nitrogen) per g sorbent. Working under humid conditions facilitated the uptake up to 12% at 1 atm. Moreover, Koros and co-workers95 reported on the use of another type of cellulose acetate fibres that were aminated with (N-(2-aminoethyl)-3-aminoisobutyldimethylmethoxysilane). Using a pressure decay apparatus, the CO2 sorption capacity was a bit low (0.73 mmol CO2 per g sorbent at 1 atm).
Heo and Park96 reported on the preparation of ultra-microporous carbons from cellulose fibres by means of physical activation at different temperatures. Using steam, the physical activation influenced the development of new micropores for CO2 storage. The adsorption capacity for the synthesized materials was 3.776 mmol CO2 per g sorbent at 298 K and 1 atm with a good CO2/N2 selectivity of 47.1. The studied material was regenerated 10 times after repeated sorption/desorption cycles. Their work showed the importance of carbon materials with good adsorption abilities without the need for chemical activation. In 2016, Einloft and co-workers97 reported on the use of rice-husk cellulose-supported ionic liquids for the capture of CO2. The uptake experiments were gravimetric with optimal values for cellulose-modified tetrabutylammonium (CL-TBA) of 44 and 71 mg CO2 per g sorbent at 25 °C and 0.1 and 3 MPa, respectively.
Organoclays, once hybridized with cellulose nanofibers, might be a plausible solution for the incorporation and storage of gases. Such an idea was enhanced by the work of Shah and Imae98 who reported on the utilisation of 2,2,6,6-tetramethylpiperidine-1-oxyl radical cellulose nanofibers (TOCNF) modified with anion and cation-exchange organoclays, viz. hydrotalcite, laponite and sericite. The order of the CO2 storage ranked among the clays applied is as follows: laponite > sericite > hydrotalcite, with a maximum CO2 uptake for the laponite-based hybrid material of 24 mg CO2 per g TOCNF. Hu et al.99 reported on another kind of hierarchical porous N-doped carbon from cellulose in an effective strategy to be utilised as a supercapacitor and in CO2 capture applications (Fig. 5). The application of such a method emerged from the use of a dissolving-gelling process followed by carbonization in an NH3 atmosphere, which enhanced the production of both macro- and micropores. The synthesized aerogels had a high adsorption capacity of 4.99 mmol CO2 per g sorbent which was much higher than those in other porous carbon materials studies as reported accordingly.
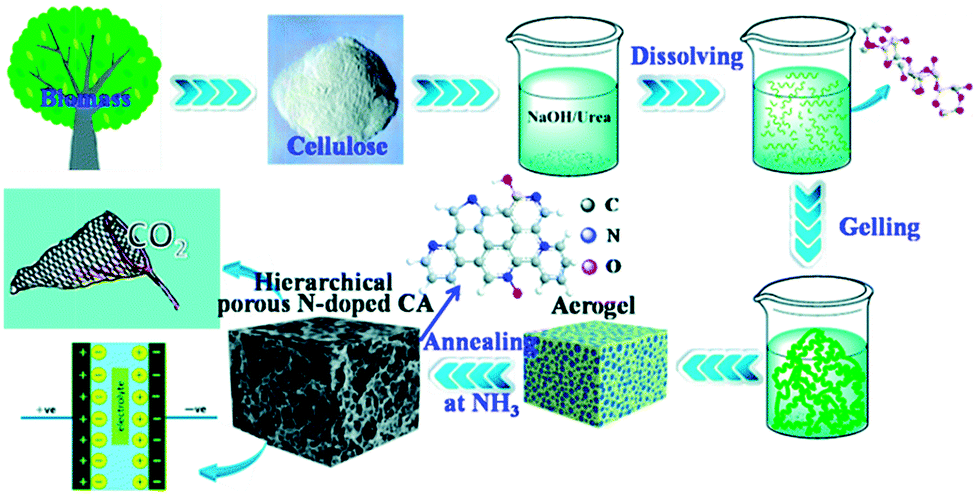 | ||
| Fig. 5 Preparation and applications of a hierarchical porous N-doped carbon aerogel. Adapted with permission from ref. 99. | ||
ILs and cellulose acetate (CAct),100 another type of biodegradable polymer as the parent macromolecule, were also exploited for the purposes of CO2 capturing and separation. Zhang and co-workers101 reported very recently on the introduction of an ether-functionalized pyridinium-based ionic liquid ([EnPy][NTf2]/CAct) which was fabricated into membranes via a casting methodology. The mechanical properties and thermal stability of the composites were a function of the plasticizing effect together with the gas affinity of the IL used. Its addition improved the permselectivities of the membranes but lowered the gas permeabilities. It is noteworthy that the use of 40% [E1Py][NTf2]/CAct showed a seven-fold increase in CO2 permeability with CO2/N2 and CO2/CH4 permselectivities of 32 and 24, respectively.
Surveying cellulose modified materials, Lin’s research group102 doped cellulose triacetate (CTA, a polymer that is used industrially in membranes for the separation of CO2/CH4) with 1-ethyl-3-methylimidazolium tetrafluoroborate and 1-ethyl-3-methylimidazolium dicyanamide in order to enhance the cellulose affinity towards CO2. Upon doping, both the crystallinity and glass transition temperature decreased. An increase in CO2/CH4 selectivity and CO2/N2 permeability was observed.
A new type of hydrophilic microfibrillated cellulose (MFC) was investigated by Baschetti and co-workers103 who used pure MFC films and MFC/Lupamin® (a commercially-available polyvinylamine, BASF). The synthesized material showed good CO2/N2 and CO2/CH4 selectivity, in the order of 500 and 350 respectively, which was achieved by pure MFC films. The CO2 permeability was very low and never exceeded 25 Barrer, even at 90% relative humidity. The permeability of a 1:1 MFC/Lupamin® nanocomposite (the fabrication process is shown in Fig. 6) was better with a six-fold enhancement over that of the pure MFC and a permeability of 190 Barrer at the same humidity conditions.
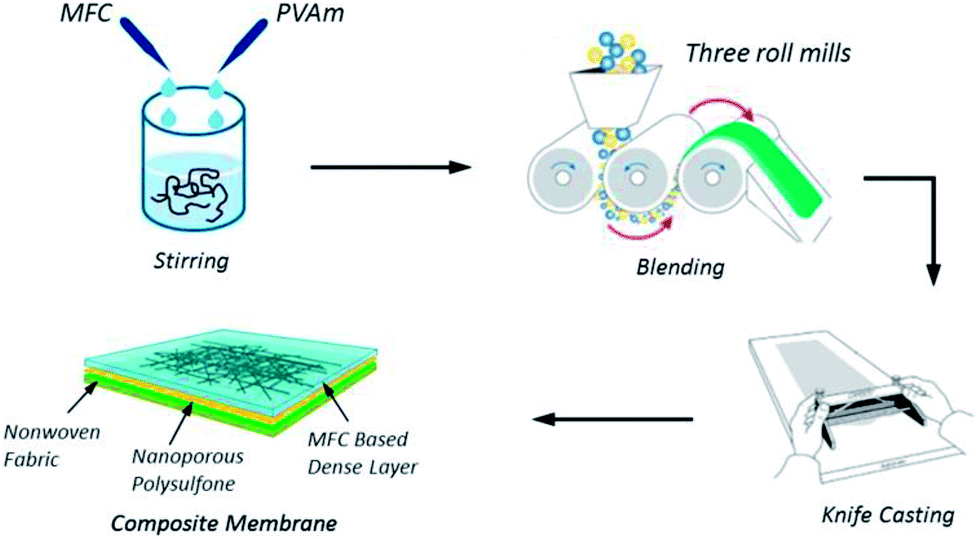 | ||
| Fig. 6 MFC/Lupamin® nanocomposite preparation. Reprinted with permission from ref. 102. | ||
In 2017, Dassanayake et al.104 reported an interesting study for a series of nanocrystalline cellulose mesoporous silica composite materials with tethered double-amidoxime functional groups (NCC-AO). The sorption capacities recorded for the NCC-AO were 3.30 and 5.54 mmol CO2 per g sorbent at 25 and 120 °C, respectively. The authors claimed that the sorption capacity at such elevated temperatures and atmospheric conditions of CO2 was the highest among other sorbents reported in the literature.
In the same year, Styring and co-workers105 synthesized a cellulose supported ionic liquid (SoILs) which showed improved CO2 uptake up to 39-fold in comparison with ILs. This means a reduction in the amount of IL used which can associate faster kinetics for sorption together with faster regeneration cycles. The optimal working ratio of cellulose to IL is 1:4, which might result in a reduction of operational costs and size of facilities.
Conclusions
Bio-renewable materials represented by the most abundant polysaccharides might be the next generation of sorbents that are introduced for the capturing of CO2, which was present heavily in the atmosphere due to the severe anthropogenic effect. In our consensus, several research groups have introduced mild to harsh conditions for the activation of the title materials, and what matters the most is the efficiency/commercial availability of the chosen sorbent. This is considered as one reason for the utilisation of water-immiscible polymers as solid supports for CO2 capturing purposes. In addition, another good advantage of such materials is their naturally benign-by-design architecture that provides a wide platform to capture CO2, ranging from physisorption to chemisorption based mechanisms, dry to wet scrubbing or even the introduction of hybrid materials that can work in parallel with the biopolymers to enhance the CO2 storage. In our opinion, a strong recommendation for future work is maintained throughout the introduction of oligomers,29 reported elsewhere as a first generation green sorbent, rather than polymers that can mimic the parent polysaccharides for wet scrubbing applications. “Supramolecular Chemisorption” is further facilitated by other hydrogen-bonding, future candidates of different functionalities. Furthermore, the implementation of a new fine named “green tax” (1% per successful fund taken from each accepted proposal submitted to any national/international funding agencies or world-wide universities to plant trees in arid areas and developing countries) might provide a good source of the title saccharides as well as cleansing the atmosphere from pollution while trying to save this green planet. As a matter of fact, what was damaged industrially and disrupted the carbon cycle can be fixed simply and in an eco-friendly manner by returning back to nature. Actually, Climeworks106 have utilised filters that are primarily composed of cellulose upon grafting with amines to capture CO2 from the air. Upon reaching the full capacity, the filter can be regenerated at 95 °C. A further recommendation is to find universal protocols to capture CO2 in terms of gas uptake measuring conditions (volumetric vs. gravimetric), as this may hinder a fair comparison among sorbents.29 Also, modification/functionalisation of the naturally-occurring polymers might provide better reactivities and performance characteristics.Conflicts of interest
There are no conflicts to declare.Acknowledgements
AFE acknowledges research support from the Deanship of Scientific Research at the Hashemite University, Jordan.Notes and references
- E. S. Poloczanska, C. J. Brown, W. J. Sydeman, W. Kiessling, D. S. Schoeman, P. J. Moore, K. Brander, J. F. Bruno, L. B. Buckley, M. T. Burrows, C. M. Duarte, B. S. Halpern, J. Holding, C. V. Kappel, M. I. O’Connor, J. M. Pandolfi, C. Parmesan, F. Schwing, S. A. Thompson and A. J. Richardson, Nat. Clim. Change, 2013, 3, 919–925 CrossRef.
- I. Nagelkerken and S. D. Connell, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 13272–13277 CrossRef CAS PubMed.
- A. W. R. Seddon, M. Macias-Fauria, P. R. Long, D. Benz and K. J. Willis, Nature, 2016, 531, 229–232 CrossRef CAS PubMed.
- UNFCCC, Adoption of the Paris Agreement., Report No. FCCC/CP/2015/L.9/Rev.1, http://unfccc.int/resource/docs/2015/cop21/eng/l09r01.pdf.
- P. Fragkos, N. Tasios, L. Paroussos, P. Capros and S. Tsani, Energy Policy, 2017, 100, 216–226 CrossRef.
- D. G. Victor, K. Akimoto, Y. Kaya, M. Yamaguchi, D. Cullenward and C. Hepburn, Nature, 2017, 548, 25–27 CrossRef PubMed.
- J. Rogelj, M. den Elzen, N. Höhne, T. Fransen, H. Fekete, H. Winkler, R. Schaeffer, F. Sha, K. Riahi and M. Meinshausen, Nature, 2016, 534, 631–639 CrossRef CAS PubMed.
- S. I. Seneviratne, M. G. Donat, A. J. Pitman, R. Knutti and R. L. Wilby, Nature, 2016, 529, 477–483 CrossRef CAS PubMed.
- L. Bretschger, Environ. Econ. Pol. Stud., 2017, 19, 1–14 CrossRef.
- D. M. D’Alessandro, B. Smit and J. R. Long, Angew. Chem., Int. Ed., 2010, 49, 6058–6082 CrossRef PubMed.
- J.-S. M. Lee, G. Rochelle, P. Styring, P. Fennell, G. Wilson, M. Trusler, P. Clough, J. Blamey, M. Dunstan, N. MacDowell, S. Lyth, J. Yao, T. Hills, M. Gazzani, P. Brandl, R. Anantharaman, S. Brandani, J. Stolaroff, M. Mazzotti, G. Maitland, C. Muller, G. Dowson, J. Gibbins, R. Ocone, K. Sedransk Campbell, M. Erans, L. Zheng, D. Sutter, A. Armutlulu and B. Smit, Faraday Discuss., 2016, 192, 125–151 RSC.
- H. A. Patel, J. Byun and C. T. Yavuz, ChemSusChem, 2017, 10, 1303–1317 CrossRef CAS PubMed.
- P. T. Anastas and M. M. Kirchhoff, Acc. Chem. Res., 2002, 35, 686–694 CrossRef CAS PubMed.
- M. Poliakoff, J. M. Fitzpatrick, T. R. Farren and P. T. Anastas, Science, 2002, 297, 807–810 CrossRef CAS PubMed.
- P. Anastas and N. Eghbali, Chem. Soc. Rev., 2010, 39, 301–312 RSC.
- E. D. Bates, R. D. Mayton, I. Ntai and J. H. Davis, J. Am. Chem. Soc., 2002, 124, 926–927 CrossRef CAS PubMed.
- D. J. Heldebrant, P. K. Koech, M. T. C. Ang, C. Liang, J. E. Rainbolt, C. R. Yonker and P. G. Jessop, Green Chem., 2010, 12, 713–721 RSC.
- A. Gandini, Macromolecules, 2008, 41, 9491–9504 CrossRef CAS.
- A. Gandini, T. M. Lacerda, A. J. F. Carvalho and E. Trovatti, Chem. Rev., 2016, 116, 1637–1669 CrossRef CAS PubMed.
- D. K. Schneiderman and M. A. Hillmyer, Macromolecules, 2017, 50, 3733–3749 CrossRef CAS.
- B. Medronho, A. Romano, M. G. Miguel, L. Stigsson and B. Lindman, Cellulose, 2012, 19, 581–587 CrossRef CAS.
- M. Rinaudo, Prog. Polym. Sci., 2006, 31, 603–632 CrossRef CAS.
- D. G. Ramirez-Wong, M. Ramirez-Cardona, R. J. Sanchez-Leija, A. Rugerio, R. A. Mauricio-Sanchez, M. A. Hernandez-Landaverde, A. Carranza, J. A. Pojman, A. M. Garay-Tapia, E. Prokhorov, J. D. Mota-Morales and G. Luna-Barcenas, Green Chem., 2016, 18, 4303–4311 RSC.
- H. Mahmood, M. Moniruzzaman, S. Yusup and T. Welton, Green Chem., 2017, 19, 2051–2075 RSC.
- R. P. Swatloski, S. K. Spear, J. D. Holbrey and R. D. Rogers, J. Am. Chem. Soc., 2002, 124, 4974–4975 CrossRef CAS PubMed.
- P. Styring, in Carbon Dioxide Utilisation, Elsevier, Amsterdam, 2015, pp. 19–32 Search PubMed.
- R. W. Flaig, T. M. Osborn Popp, A. M. Fracaroli, E. A. Kapustin, M. J. Kalmutzki, R. M. Altamimi, F. Fathieh, J. A. Reimer and O. M. Yaghi, J. Am. Chem. Soc., 2017, 139, 12125–12128 CrossRef CAS PubMed.
- A. F. Eftaiha, A. K. Qaroush, K. I. Assaf, F. Alsoubani, T. Markus Pehl, C. Troll and M. I. El-Barghouthi, New J. Chem., 2017, 41, 11941–11947 RSC.
- A. K. Qaroush, D. A. Castillo-Molina, C. Troll, M. A. Abu-Daabes, H. M. Alsyouri, A. S. Abu-Surrah and B. Rieger, ChemSusChem, 2015, 8, 1618–1626 CrossRef CAS PubMed.
- P. V. Kortunov, M. Siskin, L. S. Baugh and D. C. Calabro, Energy Fuels, 2015, 29, 5940–5966 CrossRef CAS.
- P. V. Kortunov, M. Siskin, L. S. Baugh and D. C. Calabro, Energy Fuels, 2015, 29, 5919–5939 CrossRef CAS.
- F. Inagaki, C. Matsumoto, T. Iwata and C. Mukai, J. Am. Chem. Soc., 2017, 139, 4639–4642 CrossRef CAS PubMed.
- Y. Park, K.-Y. A. Lin, A.-H. A. Park and C. Petit, Frontiers in Energy Research, 2015, 3, 42 CrossRef.
- Sigma-Aldrich Co., Ethanolamine ≥98%, http://www.sigmaaldrich.com/catalog/product/sial/e9508?lang=en%26region=JO, accessed on 15.Oct.2017.
- Alfa Aesar™, Ethanolamine, 98+%, https://www.fishersci.com/shop/products/ethanolamine-98-3/p-7081646#?keyword=Monoethanolamine, accessed on 15.Oct.2017.
- S. Seo, M. Quiroz-Guzman, M. A. DeSilva, T. B. Lee, Y. Huang, B. F. Goodrich, W. F. Schneider and J. F. Brennecke, J. Phys. Chem. B, 2014, 118, 5740–5751 CrossRef CAS PubMed.
- V. V. Chaban, RSC Adv., 2016, 6, 8906–8912 RSC.
- S. Zeng, X. Zhang, L. Bai, X. Zhang, H. Wang, J. Wang, D. Bao, M. Li, X. Liu and S. Zhang, Chem. Rev., 2017, 117, 9625–9673 CrossRef CAS PubMed.
- P. G. Jessop, D. J. Heldebrant, X. Li, C. A. Eckert and C. L. Liotta, Nature, 2005, 436, 1102 CrossRef CAS PubMed.
- P. Pollet, C. A. Eckert and C. L. Liotta, Chem. Sci., 2011, 2, 609–614 RSC.
- J. J. Gassensmith, H. Furukawa, R. A. Smaldone, R. S. Forgan, Y. Y. Botros, O. M. Yaghi and J. F. Stoddart, J. Am. Chem. Soc., 2011, 133, 15312–15315 CrossRef CAS PubMed.
- D. Wu, J. J. Gassensmith, D. Gouvêa, S. Ushakov, J. F. Stoddart and A. Navrotsky, J. Am. Chem. Soc., 2013, 135, 6790–6793 CrossRef CAS PubMed.
- J. J. Gassensmith, J. Y. Kim, J. M. Holcroft, O. K. Farha, J. F. Stoddart, J. T. Hupp and N. C. Jeong, J. Am. Chem. Soc., 2014, 136, 8277–8282 CrossRef CAS PubMed.
- H. A. Patel, T. Islamoglu, Z. Liu, S. K. M. Nalluri, A. Samanta, O. Anamimoghadam, C. D. Malliakas, O. K. Farha and J. F. Stoddart, J. Am. Chem. Soc., 2017, 139, 11020–11023 CrossRef CAS PubMed.
- R. Rajamanickam, H. Kim and J.-W. Park, Sci. Rep., 2015, 5, 10688 CrossRef CAS PubMed.
- A. K. Qaroush, K. I. Assaf, A. Al-Khateeb, F. Alsoubani, E. Nabih, C. Troll, B. Rieger and A. F. Eftaiha, Energy Fuels, 2017, 31, 8407–8414 CrossRef.
- K. I. Assaf, A. K. Qaroush and A. F. Eftaiha, Phys. Chem. Chem. Phys., 2017, 19, 15403–15411 RSC.
- A. C. O’Sullivan, Cellulose, 1997, 4, 173–207 CrossRef.
- I. Siró and D. Plackett, Cellulose, 2010, 17, 459–494 CrossRef.
- J. Cai and L. Zhang, Macromol. Biosci., 2005, 5, 539–548 CrossRef CAS PubMed.
- C. L. McCormick, P. A. Callais and B. H. Hutchinson, Macromolecules, 1985, 18, 2394–2401 CrossRef CAS.
- H. Qi, C. Chang and L. Zhang, Cellulose, 2008, 15, 779–787 CrossRef CAS.
- S. Zhu, Y. Wu, Q. Chen, Z. Yu, C. Wang, S. Jin, Y. Ding and G. Wu, Green Chem., 2006, 8, 325–327 RSC.
- M. Abe, Y. Fukaya and H. Ohno, Chem. Commun., 2012, 48, 1808–1810 RSC.
- D. L. Minnick and A. M. Scurto, Chem. Commun., 2015, 51, 12649–12652 RSC.
- X. Sun, Y. Chi and T. Mu, Green Chem., 2014, 16, 2736–2744 RSC.
- Q. Zhang, M. Benoit, K. De Oliveira Vigier, J. Barrault and F. Jérôme, Chem.–Eur. J., 2012, 18, 1043–1046 CrossRef CAS PubMed.
- Q.-P. Liu, X.-D. Hou, N. Li and M.-H. Zong, Green Chem., 2012, 14, 304–307 RSC.
- M. Francisco, A. van den Bruinhorst and M. C. Kroon, Green Chem., 2012, 14, 2153–2157 RSC.
- Q. Zhang, N. S. Oztekin, J. Barrault, K. De Oliveira Vigier and F. Jérôme, ChemSusChem, 2013, 6, 593–596 CrossRef CAS PubMed.
- Y. Yang, L. Song, C. Peng, E. Liu and H. Xie, Green Chem., 2015, 17, 2758–2763 RSC.
- J. Wang, Z. Xue, T. Yu, Z. Liu and T. Mu, Chem.–Asian J., 2017, 12, 1773–1779 CrossRef CAS PubMed.
- L. Song, Y. Yang, H. Xie and E. Liu, ChemSusChem, 2015, 8, 3217–3221 CrossRef CAS PubMed.
- J. Wang, Z. Xue, C. Yan, Z. Li and T. Mu, Phys. Chem. Chem. Phys., 2016, 18, 32772–32779 RSC.
- M. Gunnarsson, H. Theliander and M. Hasani, Cellulose, 2017, 24, 2427–2436 CrossRef CAS.
- S. Y. Oh, D. I. Yoo, Y. Shin and G. Seo, Carbohydr. Res., 2005, 340, 417–428 CrossRef CAS PubMed.
- H. Xie, X. Yu, Y. Yang and Z. K. Zhao, Green Chem., 2014, 16, 2422–2427 RSC.
- P. Nanta, W. Skolpap, K. Kasemwong and Y. Shimoyama, J. Supercrit. Fluids, 2017, 130, 84–90 CrossRef CAS.
- E. M. M. D. Valle, Process Biochem., 2004, 39, 1033–1046 CrossRef.
- Z. Liu, S. K. M. Nalluri and J. F. Stoddart, Chem. Soc. Rev., 2017, 46, 2459–2478 RSC.
- R. A. Smaldone, R. S. Forgan, H. Furukawa, J. J. Gassensmith, A. M. Z. Slawin, O. M. Yaghi and J. F. Stoddart, Angew. Chem., Int. Ed., 2010, 49, 8630–8634 CrossRef CAS PubMed.
- Note: Using diisopropyl ether (iPr2O) instead of MeOH resulted in the formation of a metal coordination framework (MCF). For more details please see the following reference: J. J. Gassensmith, R. A. Smaldone, R. S. Forgan, C. E. Wilmer, D. B. Cordes, Y. Y. Botros, A. M. Z. Slawin, R. Q. Snurr and J. F. Stoddart, Org. Lett., 2012, 14, 1460–1463 CrossRef CAS PubMed.
- R. S. Forgan, R. A. Smaldone, J. J. Gassensmith, H. Furukawa, D. B. Cordes, Q. Li, C. E. Wilmer, Y. Y. Botros, R. Q. Snurr, A. M. Z. Slawin and J. F. Stoddart, J. Am. Chem. Soc., 2012, 134, 406–417 CrossRef CAS PubMed.
- Note: Neither γ-cyclodextrin nor crushed CD-MOF-2 (an amorphous material as obtained from XRD measurements) were able to absorb CO2.
- D. L. Sivadas, K. P. Vijayalakshmi, R. Rajeev, K. Prabhakaran and K. N. Ninan, RSC Adv., 2013, 3, 24041–24045 RSC.
- Y.-C. Zhao, L. Zhao, L.-J. Mao and B.-H. Han, J Mater Chem A, 2013, 1, 9456–9461 CAS.
- T. Guo, A. H. Bedane, Y. Pan, B. Shirani, H. Xiao and M. Eić, Energy Fuels, 2017, 31, 4186–4192 CrossRef CAS.
- G. V. S. M. Carrera, N. Jordao, L. C. Branco and M. Nunes da Ponte, Faraday Discuss., 2015, 183, 429–444 RSC.
- P. R. Austin, in Chitin, Chitosan, and Related Enzymes, Academic Press, 1984, pp. 227–237 Search PubMed.
- P. R. Sivashankari and M. Prabaharan, in Chitosan Based Biomaterials Volume 1, Woodhead Publishing, 2017, pp. 117–133 Search PubMed.
- H. Xie, S. Zhang and S. Li, Green Chem., 2006, 8, 630–633 RSC.
- X. Sun, C. Huang, Z. Xue and T. Mu, Energy Fuels, 2015, 29, 1923–1930 CrossRef CAS.
- X. Sun, Z. Xue and T. Mu, Green Chem., 2014, 16, 2102–2106 RSC.
- G. E. Romanos, P. S. Schulz, M. Bahlmann, P. Wasserscheid, A. Sapalidis, F. K. Katsaros, C. P. Athanasekou, K. Beltsios and N. K. Kanellopoulos, J. Phys. Chem. C, 2014, 118, 24437–24451 CAS.
- K. Pohako-Esko, M. Bahlmann, P. S. Schulz and P. Wasserscheid, Ind. Eng. Chem. Res., 2016, 55, 7052–7059 CrossRef CAS.
- J. Fujiki and K. Yogo, Chem. Lett., 2013, 42, 1484–1486 CrossRef CAS.
- J. Fujiki and K. Yogo, Energy Fuels, 2014, 28, 6467–6474 CrossRef CAS.
- A. F. Eftaiha, F. Alsoubani, K. I. Assaf, W. M. Nau, C. Troll and A. K. Qaroush, RSC Adv., 2016, 6, 22090–22093 RSC.
- A. F. Eftaiha, F. Alsoubani, K. I. Assaf, C. Troll, B. Rieger, A. H. Khaled and A. K. Qaroush, Carbohydr. Polym., 2016, 152, 163–169 CrossRef CAS PubMed.
- A. K. Qaroush, K. I. Assaf, S. K. Bardaweel, A. Al-Khateeb, F. Alsoubani, E. Al-Ramahi, M. Masri, T. Bruck, C. Troll, B. Rieger and A. F. Eftaiha, Green Chem., 2017, 19, 4305–4314 RSC.
- C. Gebald, J. A. Wurzbacher, P. Tingaut, T. Zimmermann and A. Steinfeld, Environ. Sci. Technol., 2011, 45, 9101–9108 CrossRef CAS PubMed.
- C. Gebald, J. A. Wurzbacher, P. Tingaut and A. Steinfeld, Environ. Sci. Technol., 2013, 47, 10063–10070 CrossRef CAS PubMed.
- M. Sevilla and A. B. Fuertes, Energy Environ. Sci., 2011, 4, 1765–1771 CAS.
- D. M. Pacheco, J. R. Johnson and W. J. Koros, Ind. Eng. Chem. Res., 2012, 51, 503–514 CrossRef CAS.
- F. S. Li, R. P. Lively, J. S. Lee and W. J. Koros, Ind. Eng. Chem. Res., 2013, 52, 8928–8935 CrossRef CAS.
- Y.-J. Heo and S.-J. Park, Energy, 2015, 91, 142–150 CrossRef CAS.
- F. L. Bernard, D. M. Rodrigues, B. B. Polesso, A. J. Donato, M. Seferin, V. V. Chaban, F. D. Vecchia and S. Einloft, Fuel Process. Technol., 2016, 149, 131–138 CrossRef CAS.
- K. J. Shah and T. Imae, Biomacromolecules, 2016, 17, 1653–1661 CrossRef CAS PubMed.
- Y. Hu, X. Tong, H. Zhuo, L. Zhong, X. Peng, S. Wang and R. Sun, RSC Adv., 2016, 6, 15788–15795 RSC.
- L. Jiang and G. Hinrichsen, Angew. Makromol. Chem., 1997, 253, 193–200 CrossRef CAS.
- J. Deng, L. Bai, S. Zeng, X. Zhang, Y. Nie, L. Deng and S. Zhang, RSC Adv., 2016, 6, 45184–45192 RSC.
- B. Lam, M. Wei, L. Zhu, S. Luo, R. Guo, A. Morisato, P. Alexandridis and H. Lin, Polymer, 2016, 89, 1–11 CrossRef CAS.
- L. Ansaloni, J. Salas-Gay, S. Ligi and M. G. Baschetti, J. Membr. Sci., 2017, 522, 216–225 CrossRef CAS.
- R. S. Dassanayake, C. Gunathilake, A. C. Dassanayake, N. Abidi and M. Jaroniec, J Mater Chem A, 2017, 5, 7462–7473 CAS.
- D. G. Reed, G. R. M. Dowson and P. Styring, Frontiers in Energy Research, 2017, 5, 13 CrossRef.
- Business Opportunities in Carbon Capture and Utilisation, 56, 2017, http://www.climeworks.com/wp-content/uploads/2017/05/<?pdb_no 2017?>2017<?pdb END?>_03_30__Carbon-Captur-Journal_Climeworks-captures-CO2-directly-from-the-atmosphere.pdf.
| This journal is © The Royal Society of Chemistry 2018 |