
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA simple route to highly active single-enzyme nanogels†
Ana
Beloqui‡
 *ab,
Andrei Yu
Kobitski
c,
Gerd Ulrich
Nienhaus
acde and
Guillaume
Delaittre
*ab
*ab,
Andrei Yu
Kobitski
c,
Gerd Ulrich
Nienhaus
acde and
Guillaume
Delaittre
*ab
aInstitute of Toxicology and Genetics, Karlsruhe Institute of Technology (KIT), Hermann-von-Helmholtz-Platz 1, 76344 Eggenstein-Leopoldshafen, Germany. E-mail: guillaume.delaittre@kit.edu; a.beloqui@nanogune.eu
bPreparative Macromolecular Chemistry, Institute for Technical Chemistry and Polymer Chemistry, Karlsruhe Institute of Technology (KIT), Engesserstrasse 15, 76131 Karlsruhe, Germany
cInstitute of Applied Physics, Karlsruhe Institute of Technology (KIT), Wolfgang-Gaede-Strasse 1, 76131 Karlsruhe, Germany
dInstitute of Nanotechnology, Karlsruhe Institute of Technology (KIT), Hermann-von-Helmholtz-Platz 1, 76344 Eggenstein-Leopoldshafen, Germany
eDepartment of Physics, University of Illinois at Urbana-Champaign, Urbana, Illinois 61801, USA
First published on 1st December 2017
Abstract
We have established a simple one-step synthesis of single-enzyme nanogels (SENs), i.e., nanobiocatalysts consisting of an enzyme molecule embedded in a hydrophilic, polymeric crosslinked nanostructure, as a most attractive approach to enhance the stability of enzymes. In contrast to earlier protocols, we demonstrate here that the addition of a small amount of sucrose makes the nanogel formation equally effective as earlier two-step protocols requiring enzyme pre-modification. This provides the dual advantage of skipping a synthetic step and preserving the surface chemistry of the enzymes, hence their native structure. Enzymes encapsulated in this way exhibit a high catalytic activity, similar to that of the free enzymes, in a markedly widened pH range. With our method, the thickness of the hydrogel layer can be finely tuned by careful adjustment of reaction parameters. This is most important because the shell thickness strongly affects both enzyme activity and stability, as we observe for a wide selection of proteins. Finally, a single-molecule analysis by means of two-color confocal fluorescence coincidence analysis confirms that our encapsulation method is highly efficient and suppresses the occurrence of nanoparticles lacking an enzyme molecule. The proposed method is therefore highly attractive for biocatalysis applications, ensuring a high activity and stability of the enzymes.
1. Introduction
The encapsulation of individual enzymes within a thin hydrogel mantle has proven extremely effective for increasing their stability under denaturing conditions, such as elevated temperatures or solutions with a high content of organic solvent.1–4 In contrast to classic encapsulation protocols, one of the particular features of this method is that the dimensions of the final catalysts are similar to those of the enzyme molecules. For (bio-)nanotechnology applications,5,6 this is a key advantage over other protein stabilization methods that involve crosslinking and/or aggregation.7 A second advantage is the presence of reactive motifs displayed by the hydrogel coating, through which the nanobiocatalysts can be immobilized on a variety of surfaces while preserving the protective effect provided by the encapsulation.8–11 Therefore, this approach is most attractive for a broad number of applications in biotechnology.Since the first description of single-enzyme nanogels (SENs) in 2006,12 most ensuing reports employed essentially the same encapsulation protocol. It entails a two-step procedure, in which the side chains of lysine residues are first modified to introduce vinyl groups. Subsequently, these groups covalently anchor to the enzyme surface a polyacrylamide layer formed by crosslinking polymerization during the second step (Scheme 1i and ii), which results in a putative core–shell enzyme-polymer nanohybrid. This pre-functionalization step, which was considered to be a sine qua non to synthesize SENs,13 constitutes a serious complication and limits the applicability of the method. In fact, while the modification of lysine residues with activated N-succinimidyl esters takes place with reasonable yields at basic pH (8.0–8.5),14 the removal of positively charged groups from the protein surface at these pH conditions may result in a drastic loss of enzyme stability and could also lead to inactivation.15,16 In the present study, we sought to extend the range of applicability of SENs by simplifying the synthesis to a one-step encapsulation procedure, while preserving the shielding effect of the polymeric network. With this approach, we succeeded in the encapsulation of a range of proteins with widely different structures, catalytic activities, and sizes, including redox enzymes (horseradish peroxidase, glucose oxidase, catalase, laccase, and alcohol oxidase), hydrolases (esterase, β-glucosidase, and lipase), as well as a non-catalytic protein (bovine serum albumin).
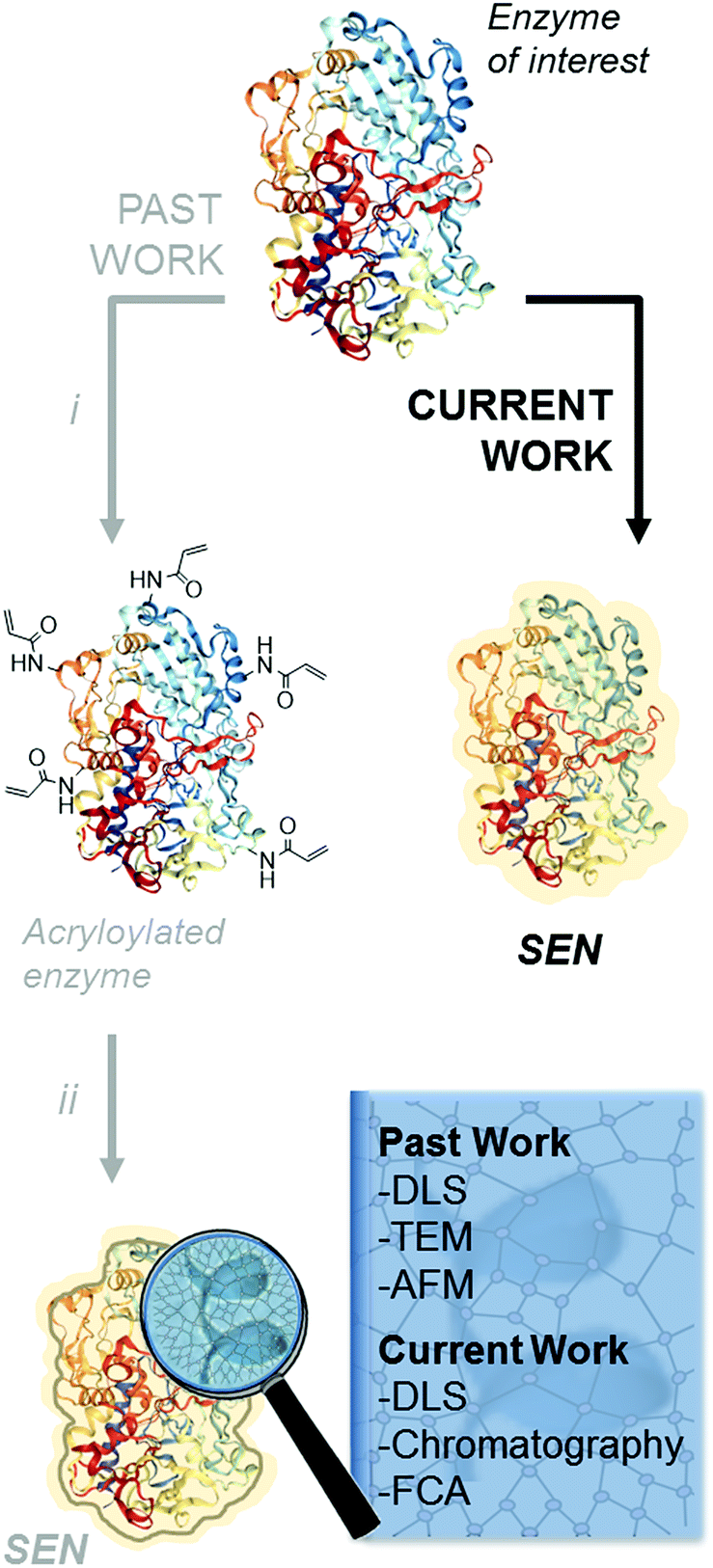 | ||
| Scheme 1 Synthetic protocols to produce SENs. Past work: (i) lysine modification using N-acryloylsuccinimide; (ii) nanogel formation using acrylamide, N,N′-methylenebisacrylamide, ammonium persulfate, tetra-methylethylenediamine. Current work: the enzyme acryloylation step is skipped and encapsulation is directly performed with added carbohydrate. | ||
Single-molecule encapsulation of small non-catalytic proteins without prior chemical modification was recently reported for drug delivery purposes, yet using specific charged monomers.17–19 Here, we focus on the encapsulation of a range of commercially available enzymes.
By variation of the encapsulation parameters, we have identified optimal SEN synthesis conditions and studied the effects of encapsulation on enzymatic stability and activity. Importantly, by using additives during the crosslinking polymerization, we show that native enzyme molecules can be encapsulated as efficiently as their lysine-modified variants. The hydrogel layer thickness can be easily controlled, and the degree of stabilization conferred to the enzyme increases with the hydrogel thickness, whereas the enzymatic activity decreases after a given thickness. In our quest for highly stable nanobiocatalysts, we found conditions under which SENs may exhibit constant, high activity over a wide pH range.
Finally, we have tackled an issue which has not yet been addressed in the literature, i.e., the question of whether there are enzyme-free nanogels in the final product of SEN synthesis. For some advanced applications involving immobilization of these nanobiocatalysts, the presence of such species could indeed be detrimental. Two-color fluorescence coincidence analysis (FCA) of photon bursts from freely diffusing particles permits the analysis of co-diffusion in a quantitative fashion at the single particle level.20–23 In this technique, confocal optical microscopy provides a tiny observation volume of typically one femtoliter,24,25 from which the fluorescence of two different emitters is detected in two separate spectral channels. By labeling the protein and the polymer nanogel with fluorescent dyes, we have utilized this powerful technique to examine the presence of protein and polymeric network in the same nanoparticle in order to provide, for the first time, robust and quantitative evidence of co-localization of both components at the molecular level.
2. Results and discussion
We first examined whether pre-acryloylation of lysine side chains on the surface of the enzymes was indeed necessary for encapsulation by using horseradish peroxidase (HRP) as a model enzyme. Acrylamide (AAm) and N,N′-methylenebisacrylamide (MBAAm) were employed to form the hydrogel coating, with a redox radical forming system, i.e., ammonium persulfate and tetramethylethylenediamine (APS/TEMED). Of note, this preparation mixture resembles that of a polyacrylamide electrophoretic gel. Non-modified, native HRP and its acryloylated counterpart (HRPa) were subjected to encapsulation in parallel experiments at two distinct protein concentrations (11.3 and 28.4 μM), with all other molar ratios kept identical ([HRP]/[AAm]/[MBAAm]/[APS]/[TEMED] = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :6000:1000:500:500). After purification, dynamic light scattering (DLS) measurements revealed that SENs originating from the acryloylated sample (HRPa_SEN) had a diameter approx. 15% greater than those obtained from native HRP (HRP_SEN) (Fig. 1A), indicating the beneficial effect of the lysine modification for nanogel formation. However, even without this modification, a sizeable nanogel growth was observed for native HRP at 28.4 μM, whereas only minimal growth was measured at 11.3 μM, presumably due to inefficient polymer wrapping on the protein surface. In the following, we show that the impaired encapsulation at low protein concentration can be remedied by adding a small fraction of stabilizing carbohydrate to the polymerization reaction.
:6000:1000:500:500). After purification, dynamic light scattering (DLS) measurements revealed that SENs originating from the acryloylated sample (HRPa_SEN) had a diameter approx. 15% greater than those obtained from native HRP (HRP_SEN) (Fig. 1A), indicating the beneficial effect of the lysine modification for nanogel formation. However, even without this modification, a sizeable nanogel growth was observed for native HRP at 28.4 μM, whereas only minimal growth was measured at 11.3 μM, presumably due to inefficient polymer wrapping on the protein surface. In the following, we show that the impaired encapsulation at low protein concentration can be remedied by adding a small fraction of stabilizing carbohydrate to the polymerization reaction.
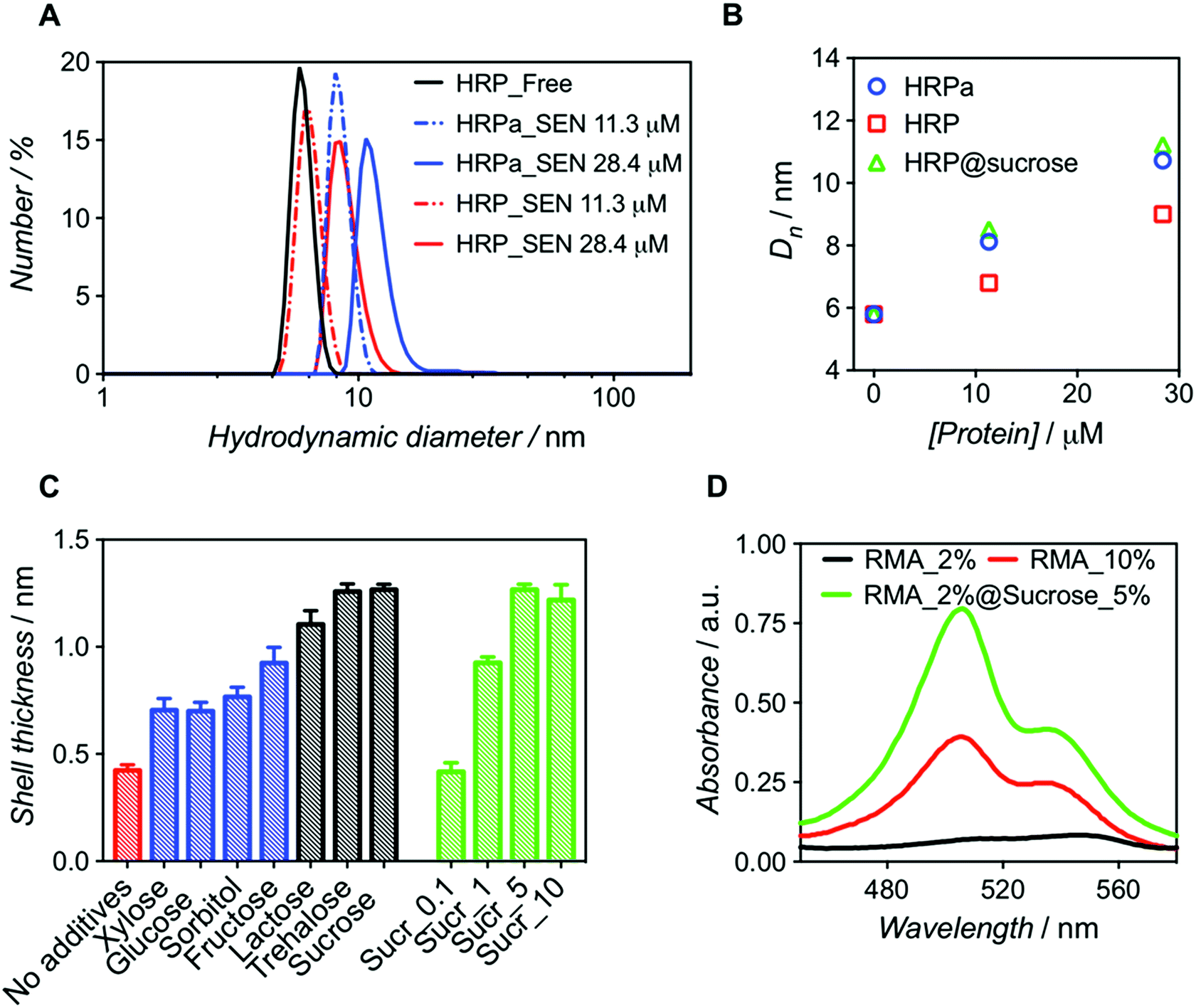 | ||
| Fig. 1 Comparative analysis of nanogels obtained upon encapsulation of acryloylated HRP (HRPa_SEN) and native HRP (HRP_SEN) and evaluation of the influence of sucrose (5% w/v) on the final diameter of SENs. (A) Hydrodynamic diameter number distributions of HRPa_SEN, HRP_SEN, and non-modified HRP at two protein seeding concentrations. (B) Evolution of the number-average diameter (Dn) after encapsulation of HRPa, HRP, and sucrose-doped HRP at 0, 11.3, and 28.4 μM protein seeding concentrations. See Fig. S1A† for the corresponding DLS spectra. (C) Nanogel thickness values after encapsulation of non-modified protein in the absence of additives (red) or in the presence of various carbohydrates (5.0% w/v): monosaccharides (in blue), disaccharides (in grey). Different sucrose concentrations (0.1, 1.0, 5.0, and 10.0% w/v) were also assayed (green). Protein concentration: 10 μM. (D) UV-vis spectra of rhodamine 6G labeled fluorescent SENs (5.3 μM), obtained by copolymerization with rhodamine 6G methacrylate (RMA) at different conditions (RMA percentages denote the molar ratio of RMA to acrylamide). | ||
Molecular dynamics simulations have previously suggested that single-enzyme nanogel formation was enhanced by pre-organization of acrylamide monomers at the protein surface via hydrophobic interactions and H-bonding between the monomer amide moieties and amino acid residues.13 This led us to hypothesize that the encapsulation of non-acryloylated enzymes can be enhanced by boosting these interactions using small carbohydrates, which are known to improve protein stability in complex mixtures.26 As a first survey, a series of monosaccharides (glucose, sorbitol, xylose, and fructose) and disaccharides (lactose, sucrose, and trehalose) were screened as polymerization enhancers (Fig. 1C). Note that, assuming a core–shell structure, we calculate the hydrogel shell thickness by relating the size of the enzyme nanogels to that of the corresponding free enzyme (see Table S1†). For a protein concentration of 10 μM, we obtained significantly thicker shells when adding disaccharides in the polymerization mixture; trehalose and sucrose showing the best performance. For sucrose, the influence of carbohydrate concentration was also assessed (Fig. 1C), leading to an optimal value of 5% w/v. Small carbohydrates, particularly disaccharides, may strengthen the monomer/protein interaction through reduction of hydrophobic/hydrophilic repulsion forces and, therefore, provide a surface microenvironment in which monomers accumulate. This may result in thicker polymeric shells and eventually lead to higher enzyme stabilization. On the basis of our survey (Fig. 1C), we selected sucrose (5% w/v) as an encapsulation promoter for the remainder of our study. Indeed, the original hydrogel layer thickness obtained for pre-modified HRPa could be fully restored for non-modified HRP upon addition of 5% w/v sucrose to the polymerization mixture (Fig. 1B), resulting in HRP_SENs with slightly higher activity than acryloylated HRPa_SENs (Fig. S1B†), presumably due to the fact that the protein remained in its native state, without alteration of its primary structure.
In order to further quantify the effect of sucrose on the polymerization, we used a chromophoric monomer, namely rhodamine 6G methacrylate (RMA), whose incorporation into the polymer network can easily be detected and quantified by UV-vis spectroscopy. Prior to these measurements, a RMA concentration calibration was established at 506 and 534 nm (Fig. S2†). Protein concentration was calculated using the characteristic heme Soret peak of HRP at 402 nm. Interestingly, when RMA was added at 2 mol% ([RMA])/([AAm] + [RMA]) in the presence of sucrose, we obtained SENs with a 6-fold higher incorporation of RMA than in the absence of the carbohydrate. Strikingly, in the absence of sucrose, even with 10 mol% of RMA in the initial feed, such a degree of incorporation could not be achieved (Table 1). Therefore, we propose that sucrose addition may open the door for the insertion of more complex and functional monomers, which may have been difficult to incorporate under the classical two-step conditions, resulting in SENs with entirely new properties. This effect is not only supported by the enhanced incorporation of RMA, but also by a diameter increase of 17% (from 9.9 to 11.6 nm), equivalent to 60% increase in volume and a shell thickness growth of 40%, in the presence of sucrose (Fig. 1D and S3,† and Table 1).
| Sample name | x RMA | [Sucrose] (w/v) | X RMA | D n (nm) |
|---|---|---|---|---|
| a x RMA = [RMA]/([AAm] + [RMA]); XRMA = number of RMA molecules incorporated per protein molecule. | ||||
| HRP_SEN_RMA2% | 0.02 | — | 0.44 | 9.9 |
| HRP_SEN_RMA2%@Sucrose5% | 0.02 | 5% | 2.63 | 11.6 |
| HRP_SEN_RMA10% | 0.10 | — | 1.38 | 9.3 |
Our new SEN preparation protocol was further investigated over an extended concentration range (3.3–33.5 μM) with glucose oxidase (GOx) as a model enzyme, leading to GOx_SENs. All other parameters were kept constant, i.e., [GOx]/[AAm]/[MBAAm]/[APS]/[TEMED] = 1:6000:1000:500:500 and 5% w/v sucrose (see Table S1†). After elution through a Sephadex G-75 column, UV-vis spectra of the collected fractions were recorded to determine the amount of residual, non-encapsulated protein. We constructed chromatograms at three different wavelengths – 230, 280, and 450 nm – for monitoring (i) both polymer and protein, (ii) mostly protein, and (iii) exclusively protein, respectively (Fig. S4 and S5†). Fractions eluting between 2.5 and 4.0 mL can be attributed to SENs, while the later ones contain essentially only free proteins as well as not or only poorly crosslinked polyacrylamide. Clear indication of poor encapsulation at low protein concentration is found with 45% of free GOx eluting at [GOx] = 3.3 μM (GOx_SEN1). Acceptable encapsulation yields (>90%) were achieved only at protein seeding concentrations greater than 10 μM (Fig. S6†). The relatively low value of this threshold contrasts with the experiments carried out without sucrose, where 43% GOx remained free at a protein concentration of 8 μM, and >90% encapsulation was only reached for [GOx] > 20 μM. The hydrodynamic diameters for this concentration series obtained in the presence of sucrose were measured by DLS (Table S1†). A clear linear correlation between the protein seeding concentration and the final shell thickness was observed (Fig. 2).
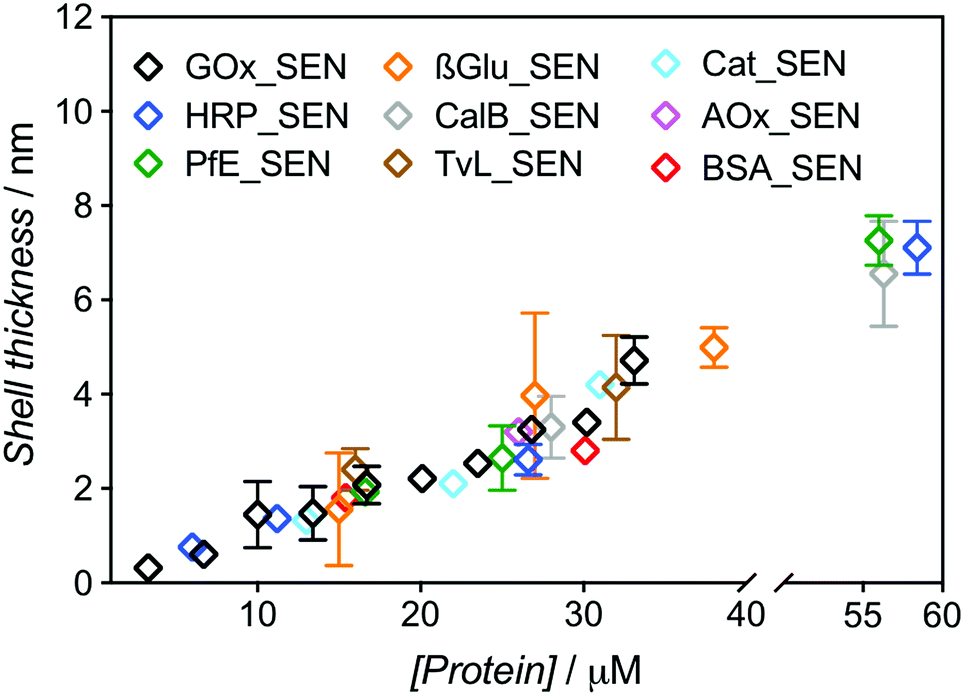 | ||
| Fig. 2 Dependence of the shell thickness on the protein seeding concentration for SENs synthesized from nine different proteins. Hydrodynamic diameters were measured by DLS (Fig. S7†). The shell thickness was calculated as half of the difference between overall particle diameter and the respective enzyme diameter. | ||
The same experimental setup was used to create single-biomolecule nanogels for a wider range of proteins (see ESI† for synthesis and purification procedure): HRP, an esterase (PfE), β-glucosidase (β-Glu), a lipase (CalB), a laccase (TvL), a catalase (Cat), an alcohol oxidase (AOx), and bovine serum albumin (BSA). Remarkably, all assayed proteins behaved in the same manner and followed the GOx trend extrapolated to about 60 μM, as depicted in Fig. 2. Above this concentration, gelation due to macroscopic crosslinking occurred.
These experiments demonstrate that, with our one-step synthesis, SENs can efficiently be synthesized from free, commercial, non-modified enzymes, with the shell thickness precisely adjustable through simple variation of the overall medium concentration (as protein concentrations varies, all concentration ratios remain constant). The process therefore appears as protein-independent and relies purely on the polymerization conditions.
Next, we enquired whether SENs would still conserve their ability to protect the enzymes from denaturation under damaging conditions, despite the lack of covalent anchoring points between the protein surface and the polymeric network. To this end, we examined the pH stability because it is well known that enzyme activity depends sensitively on pH. For GOx from Aspergillus niger, the pH optimum of 5.5 was confirmed here, with the activity rapidly dropping to either side of this point (Fig. 3A, blue). Remarkably, as the GOx_SEN shell thickness increased, the optimal pH range became broader. Indeed, with a shell thickness of about 2 nm, GOx_SEN displayed >80% of the maximum activity between pH 4 and 8 (Fig. 3A, pink). Furthermore, in analogy to previously prepared SENs, hybrids prepared with our protocol have an enhanced thermostability (80% of initial activity after incubation of GOx_SEN5 at 65 °C for 30 min – see Fig. S9A†), stable conversion rates in the presence of the protease trypsin (Fig. S9B†), and high storage stability at room temperature and through freeze-dry/freeze-thaw cycles. As another interesting example, we assayed SENs with a core consisting of laccase from Trametes versicolor (TvL), under (human) physiological conditions (37 °C, pH 7.0) at which the stability of laccase is drastically reduced. In fact, attempts have previously been made to increase laccase stability through mutagenesis to render it active in human blood.27 Using our encapsulation method, the enzyme showed impressive, 142- and 148-fold (TvL_SEN1 and TvL_SEN2, respectively) higher oxidation rates than the native enzyme (see ESI, Table S2†).
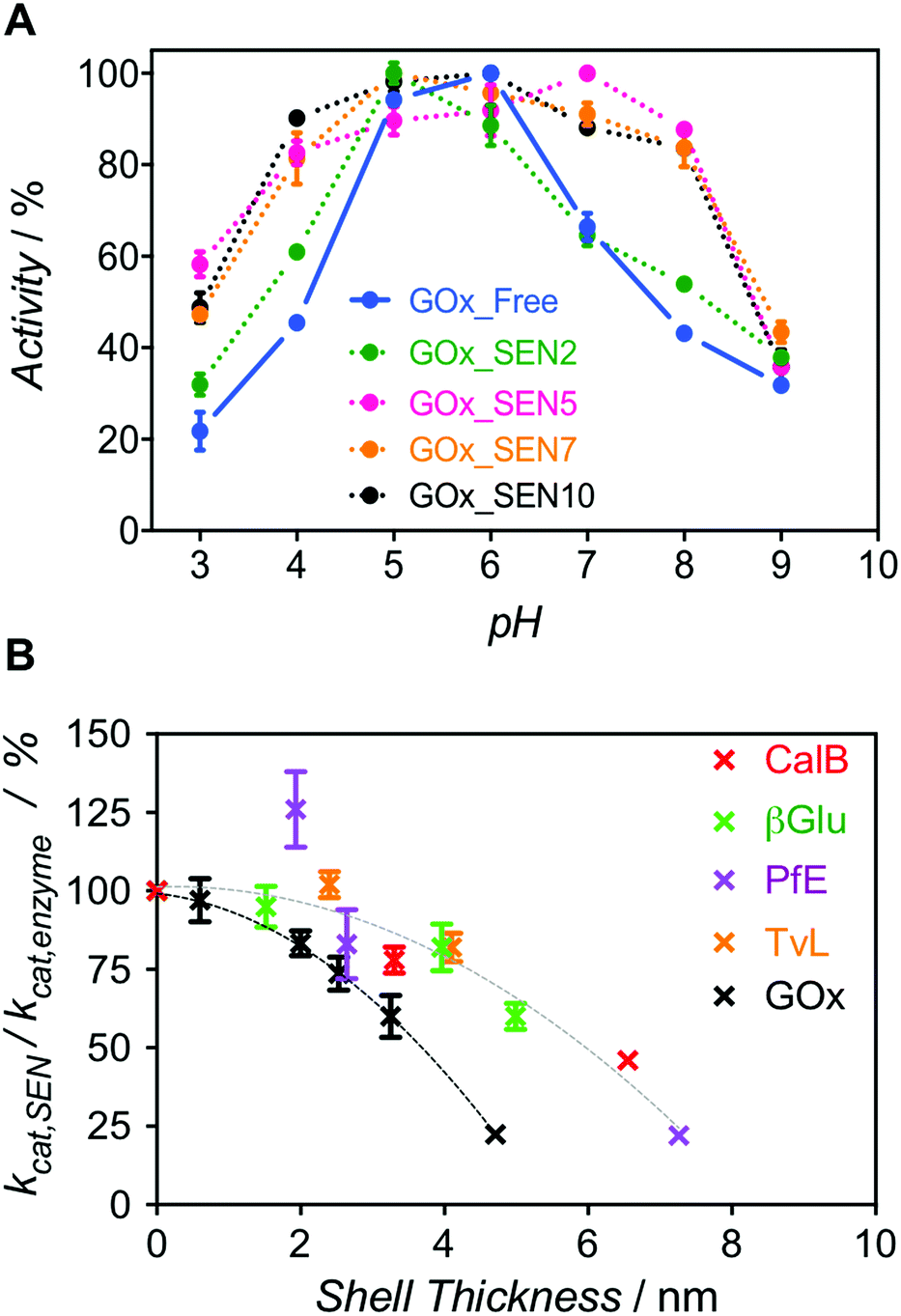 | ||
| Fig. 3 (A) Relative activity profiles over the pH range 3.0 to 9.0 for GOx_SENs with various shell thicknesses: GOx_SEN2 (0.6 nm), GOx_SEN5 (2.1 nm), GOx_SEN7 (2.5 nm), and GOx_SEN10 (4.7 nm). (B) Dependence of kcat,SEN/kcat,enzyme on the gel thickness, as determined for nine different proteins. | ||
However, thicker layers significantly compromised the enzymatic activity, as seen from the lower kcat values (Fig. S8 and Table S3†). Presumably, the thicker polymer barrier may hinder substrate diffusion. In order to further explore the hydrogel thickness effect, kcat values were calculated for several enzymes and their respective SENs (Fig. S10 and Table S4†). The ratio kcat,SEN/kcat,enzyme, plotted against the calculated gel thickness (Fig. 3B), shows a clear decrease with increasing gel thickness. Interestingly, the activity reduction effect seemed to be more pronounced for GOx_SENs, as shown by a decrease of 78% in kcat for a gel thickness of 4.7 nm. This may result from the catalytic assay chosen for GOx. For all enzymes but GOx, catalysis involves only one diffusion step before detection, i.e., penetration of the substrate through the hydrogel shell. However, in the case of GOx, a cascade reaction takes place: glucose diffuses in and one of the products, namely hydrogen peroxide, must diffuse out to convert an external chromogenic substrate by means of a second enzymatic reaction with non-encapsulated HRP. Nevertheless, it is also possible that the turnover rate reduction induced by thicker hydrogel layers is due to the restriction of protein internal movements. Indeed, we found a meaningful increase in KM values for the thickest shell (KM = 123 mM), in contrast to samples with a shell thickness of 2–3 nm (KM = 5–7 mM), presumably due to conformational constraints imposed by the increased stiffness of the thick polymer network. Yet, further studies are necessary to draw more definitive conclusions, e.g., by employing promiscuous enzymes and substrates of various sizes. All in all, for all studied proteins except GOx, we have observed a decrease of only up to ca. 10% in enzyme velocity for gel thicknesses <3 nm, while the polymer coating of similar thicknesses provides substantial protection to the enzymes, as demonstrated by the pH stability experiment.
Because formation of thicker nanogels with concomitant stronger stabilization could not be achieved with higher protein seeding concentrations due to macrogelation, we decided to explore the synthesis of a more densely crosslinked network. Hence, we increased the crosslinker amount from [AAm]/[MBAAm] = 6:1 to 2:1. Unfortunately, these conditions triggered polymer precipitation, presumably due to an excess of water-insoluble MBAAm in the reaction. However, we succeeded in preparing nanogels with a higher crosslinking density through a 6-fold decrease of the total monomer concentration ([AAm]/[protein] = [1000]) and [AAm]/[MBAAm] = 2:1 (GOx_SEN11, see ESI and Table S1†). At an identical protein concentration as for GOx_SEN8, smaller nanogels were obtained (13.9 nm for GOx_SEN8, 9.0 nm for GOx_SEN11) (see Fig. 4A). Most importantly, practically constant activity was observed over the pH range 3–9 with these SENs (Fig. 4B). However, similar to what we observed with thicker layers, the kcat value at pH 6.0 for the highly crosslinked GOx_SEN11 dropped to 32.2 ± 3.8 s−1, as compared to 98.1 ± 10.9 s−1 for GOx_SEN8, which possesses a thicker yet less crosslinked hydrogel protection layer. Presumably, in parallel to the aforementioned conformational constraints, the significantly reduced pore size may hinder the diffusion of substrate molecules to the enzyme.
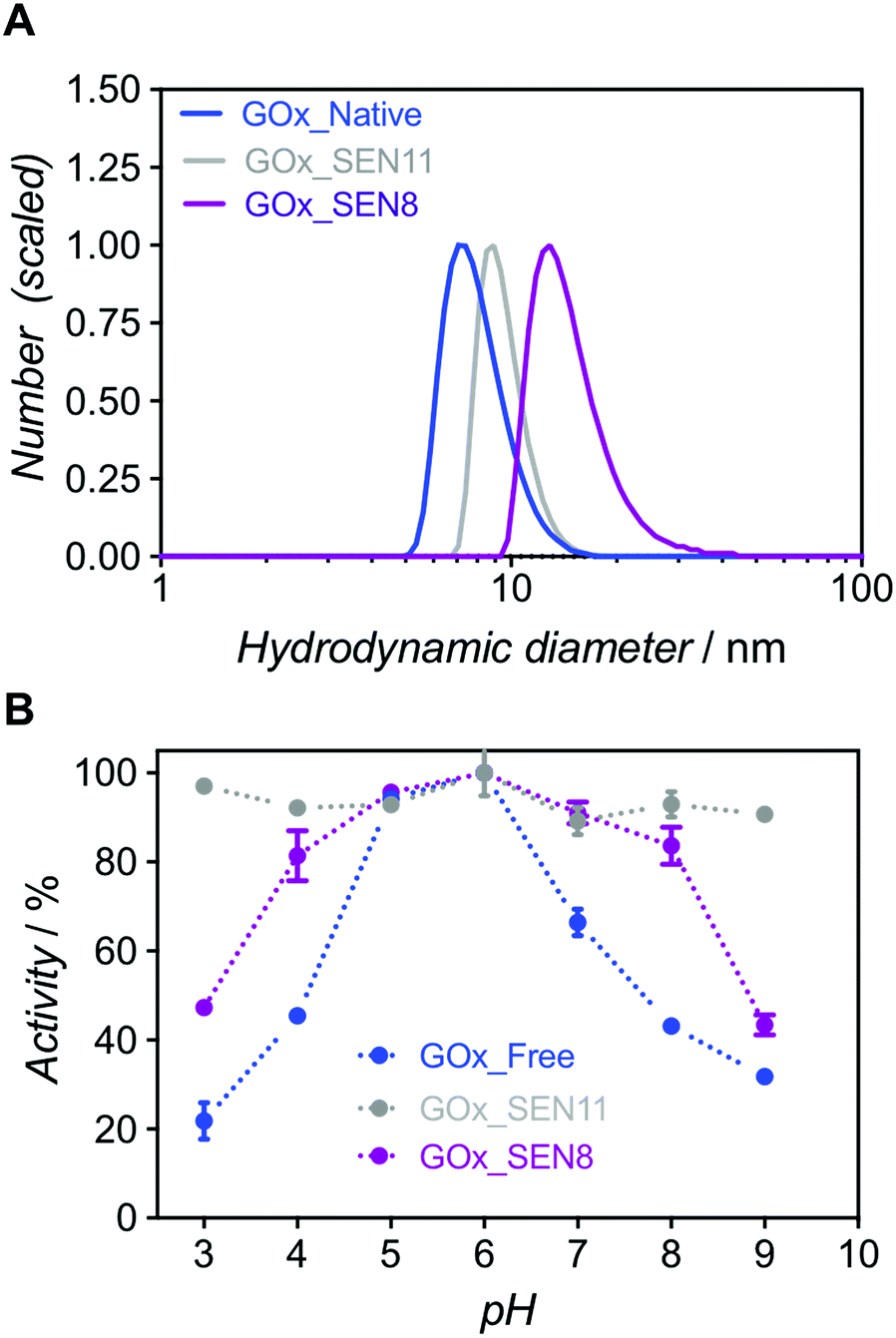 | ||
| Fig. 4 (A) Hydrodynamic diameter number distributions for GOx_SEN8 and GOx_SEN11, and native GOx as a reference. (B) Relative activity profiles over a range of pH values (from 3.0 to 9.0) for GOx_SENs made with different [AAm]/[MBAAm]: 6:1 for GOx_SEN8 (shell thickness 3.2 nm), and 2:1 for GOx_SEN11 (0.8 nm). | ||
At this stage, we can state with confidence that increased protein concentration or crosslinker ratio both lead to higher stabilization – within the presently explored range – through the formation of thicker or stiffer hydrogel layers. Such layers may provide a near-quantitative protection at extreme pH and perhaps also high temperature, accompanied by a reduced activity at some stage (Scheme 2).28 Thus, a balance must be found between stability and activity, depending on the intended type of application.
 | ||
| Scheme 2 Effect of the polyacrylamide layer thickness and porosity on the activity and stability of SENs. | ||
Finally, we aimed at measuring the degree of encapsulation at the single particle level. As yet, SENs have been structurally characterized by atomic force microscopy, transmission electron microscopy (TEM), or DLS. These methods provide size or shape information but characterize neither the composition nor the nanostructure of the SENs. It is, however, important to ensure that the synthesized nanoparticles are indeed all filled with a protein molecule. In fact, as suggested by classical crosslinking polymerization studies and observed here with blank experiments (Fig. S11†),29 hydrogel nanoparticles with diameters similar to those of SENs are produced in the absence of proteins under identical synthesis conditions. The protein molecules present in the medium during the SEN synthesis may be able to suppress the formation of “empty” nanogels due to the forces that attract the monomers to the protein surface.13 Still, the potential presence of “empty” nanogels could be a critical issue, particularly, if the SENs are to be immobilized.2,8,10,11 In that case, the real loading of protein on the support material would be overestimated.
To probe the presence of a protein molecule in each nanogel, we used single-particle two-color FCA, employing independent dye labeling of both the enzyme and the polyacrylamide hydrogel. Prior to encapsulation, native HRP was labeled with the Alexa Fluor 647 dye through attachment to lysine residues (see ESI†) and the polymer was labeled after encapsulation by thio-Michael addition on the residual double bonds7 using a thiolated rhodamine derivative (Fig. 5A). For encapsulation, we used our sucrose-based protocol with subsequent dialysis to remove low molar mass polymer. Note that the pre-encapsulation fluorescent labelling through lysines is especially enabled by our one-step encapsulation protocol. After polymer labeling, free rhodamine reagent was removed by sequential dialysis and centrifugal filtration steps. A careful sample labeling protocol is crucial for an accurate two-color single-particle measurement and, ideally, the two fluorophores should be present in comparable numbers.
 | ||
| Fig. 5 (A) Experimental workflow used for sample preparation in single-particle analysis experiments. HRP was labeled with Alexa Fluor 647 (i) and then encapsulated (ii.a); the polymeric shell was further labeled with rhodamine-PEG-thiol fluorophore (iii.a). Incomplete encapsulation events (ii.b) would lead to polyacrylamide (PA) particles excitable at 550 nm after the shell labeling step (iii.b). (B) UV-vis spectrum of doubly labeled HRP_SEN, from which the relative amounts of fluorescent labels in the sample are determined. (C) Density map of intensities in emission bursts from individual SENs upon green (532 nm) and red (637 nm) excitation. The color code (from green to red) depicts the average value of the apparent FRET efficiency, E = IA/(IA + ID), with ID and IA denoting the intensities emitted by donor (rhodamine) and acceptor (Alexa Fluor 647) of individual particles upon green excitation, respectively. SENs with burst intensities of <10 counts (grey-shaded regions) in either color channel under green and red excitation are assigned to particles without labeled HRP or without labeled nanogel component, respectively. | ||
Optimization experiments (Michael addition catalyst type, dye stoichiometry) were thus carried out with HRP as model protein, eventually leading to the double-labeled sample HRP@647_SEN@550 (Fig. S13–S16†). The corresponding UV-vis spectrum depicted in Fig. 5B displays a maximum at λ = 402 nm corresponding to the characteristic Soret peak of HRP, as well as two peaks at λ = 560 and 650 nm matching the rhodamine (Rho) and Alexa Fluor 647 (Alexa) absorption maxima. Based on previously established calibration curves (Fig. S12†), overall labeling ratios of 2.2 molecules of Alexa Fluor 647 per protein and 2.0 molecules of rhodamine per nanogel particle were determined. Assuming random labeling governed by a Poisson distribution, this implies that 11% and 13% of proteins and nanogels, respectively, do not carry a label for statistical reasons (Fig. S17†). With the chosen dye pair, precise colocalization can be evaluated not only by detecting co-diffusing molecules, but also by Förster resonance energy transfer (FRET), which reports on the proximity of two fluorophores in the 1–10 nm range, matching the SEN dimension.
Fluorescence burst analysis (Fig. 5C and S18†) shows nanoparticles with co-localized rhodamine and Alexa Fluor 647 emission, most of them with high FRET efficiency, indicating efficient encapsulation. Indeed, for a highly dilute sample (approx. 100 pM), co-localization is observed only if two molecules are detected as a single entity. The density map in Fig. 5C indicates that a fraction of ca. 30% of enzymes lacks the nanogel label. These events appear close to the y-axis. Considering the fraction of statistically non-labeled nanogels (ca. 13%, vide supra), 17% of enzymes were not encapsulated, which is in reasonable agreement with chromatographic experiments (see Fig. S6,† [GOx] = 10 μM). More importantly, the density map displays a fraction of approx. 10% of rhodamine-labeled nanogels lacking Alexa Fluor 647. This result matches the aforementioned 11% of proteins statistically lacking a label, leading to the conclusion that essentially all polymer species are linked to an enzyme. Therefore, our work provides strong evidence for the single-enzyme nanogel nature of our particles and their purity, as well as indirect evidence supporting the possible formation of core–shell (protein–polymer) structured nanoparticles, based on the previously reported molecular simulations13 and on the following observations collected in the present work: (1) after encapsulation, the enzyme stability is greatly enhanced against pH and temperature changes as well as proteases, the latter aspect suggesting a (near-)quantitative shielding phenomenon through encapsulation/wrapping, which could not be possible through simple, partial adsorption; (2) DLS distributions are monomodal, with average sizes increasing steadily and being small enough to preclude the presence of two enzymes within one particle; (3) FCA and FRET results prove the quantitative colocalization of both polymer and enzyme within the same particle.
3. Conclusions and outlook
In this work, we have shown that modification of lysine residues to introduce polymerizable groups prior to protein encapsulation is not needed for preparation of stable SENs, particularly if additives such as sucrose are added to the polymerization mixture. Using a facile one-step synthesis, we have achieved comparable nanogel size and protein encapsulation efficiency as with the classical method for a broad range of proteins. There remains a challenging task which cannot be tackled with currently available characterization methods: directly proving the yet-assumed core–shell structure by direct visualization. Indeed, TEM is unable to provide contrast between the protein and the polymer due to too-similar chemical compositions. Similarly, small-angle neutron scattering would require drastic changes to the system in order to provide contrast between the dispersant (aqueous solution) and the swollen polymer network. High-resolution optical microscopy is not yet able to reach the sub-10 nm resolution. Nevertheless, as indicated above, we provide additional indirect evidence supporting the core–shell assumption. Importantly, we showed for the first time that the polyacrylamide shell thickness can be precisely adjusted by varying the initial protein concentration in the polymerization reaction. This is crucial because we found that the activity and the stability of the SENs depends on the shell thickness. Thicker layers provide a higher stability, as inferred from the widening of the pH range of optimal enzyme performance. Such enhanced stability is most welcome in applications involving multi-enzyme processes, for which pH conditions at which all enzymes perform satisfactorily are not easily found. However, we also observed that thicker shells result in a decreased activity (lower kcat), presumably due to diffusion barriers or decreased conformational flexibility of the enzymes. Higher crosslinking of the shell will result in similar effects. Therefore, a suitable balance between activity and stability has to be found for a specific application.It is also important to confirm the purity of the SEN samples, particularly, whether all nanogels contain an enzyme molecule. Our in-depth chromatographic studies suggest that encapsulation performed at protein concentrations below 10 μM, as in most previously published studies, may not be quantitative. At higher concentrations, both chromatography and high-resolution fluorescence spectroscopy confirmed the absence of empty, enzyme-free nanogels. Therefore, the new SEN synthesis protocol presented here appears most advantageous for applications with immobilized SENs (e.g., on flat surfaces or micro-/nanocarriers), where attachment of catalytically non-functional nanogels would decrease the overall activity of the final biohybrid.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
G. D. would like to thank the German Federal Ministry of Education and Research (BMBF) for current funding in the frame of the Molecular Interaction Engineering program (Biotechnologie 2020+). A. B. would like to thank Diputación de Gipuzkoa for current funding in the frame of Gipuzkoa Fellows program. G. U. N. acknowledges support by the KIT within the context of the Helmholtz Program STN and by the Deutsche Forschungsgemeinschaft (DFG) grant GRK 2039. Dr Pavel Levkin (ITG, KIT) is thanked for providing access to the DLS instrument and Prof. Christopher Barner-Kowollik (ITCP, KIT) for constant support.References
- J. Ge, D. Lu, J. Wang and Z. Liu, Biomacromolecules, 2009, 10, 1612–1618 CrossRef CAS PubMed.
- L. Liu, W. Yu, D. Luo, Z. Xue, X. Qin, X. Sun, J. Zhao, J. Wang and T. Wang, Adv. Funct. Mater., 2015, 25, 5159–5165 CrossRef CAS.
- Y. Liu, J. Du, M. Yan, M. Y. Lau, J. Hu, H. Han, O. O. Yang, S. Liang, W. Wei, H. Wang, J. Li, X. Zhu, L. Shi, W. Chen, C. Ji and Y. Lu, Nat. Nanotechnol., 2013, 8, 187–192 CrossRef CAS PubMed.
- M. Yan, Z. Liu, D. Lu and Z. Liu, Biomacromolecules, 2007, 8, 560–565 CrossRef CAS PubMed.
- C.-M. Tîlmaciu and M. C. Morris, Front. Chem., 2015, 3, 59 Search PubMed.
- N. Dey and T. Devasena, Graphene, 2015, 3, 1–5 CrossRef.
- D. Brady and J. Jordaan, Biotechnol. Lett., 2009, 31, 1639–1650 CrossRef CAS PubMed.
- A. Beloqui, S. Baur, V. Trouillet, A. Welle, J. Madsen, M. Bastmeyer and G. Delaittre, Small, 2016, 12, 1716–1722 CrossRef CAS PubMed.
- J. Li, X. Jin, Y. Liu, F. Li, L. Zhang, X. Zhu and Y. Lu, Chem. Commun., 2015, 51, 9628–9631 RSC.
- M. Lin, D. Lu, J. Zhu, C. Yang, Y. Zhang and Z. Liu, Chem. Commun., 2012, 48, 3315 RSC.
- W. Wei, J. Du, J. Li, M. Yan, Q. Zhu, X. Jin, X. Zhu, Z. Hu, Y. Tang and Y. Lu, Adv. Mater., 2013, 25, 2212–2218 CrossRef CAS PubMed.
- M. Yan, J. Ge, Z. Liu and P. Ouyang, J. Am. Chem. Soc., 2006, 128, 11008–11009 CrossRef CAS PubMed.
- J. Ge, D. Lu, J. Wang, M. Yan, Y. Lu and Z. Liu, J. Phys. Chem. B, 2008, 112, 14319–14324 CrossRef CAS PubMed.
- N. Xia, Y. Xing, G. Wang, Q. Feng, Q. Chen, H. Feng, X. Sun and L. Liu, Int. J. Electrochem. Sci., 2013, 8, 2459–2467 CAS.
- P. P. Batra, M. A. Roebuck and D. Uetrecht, J. Protein Chem., 1990, 9, 37–44 CrossRef CAS PubMed.
- T. Masuda, N. Ide and N. Kitabatake, Chem. Senses, 2005, 30, 253–264 CrossRef CAS PubMed.
- H. Tian, J. Du, J. Wen, Y. Liu, S. R. Montgomery, T. P. Scott, B. Aghdasi, C. Xiong, A. Suzuki, T. Hayashi, M. Ruangchainikom, K. Phan, G. Weintraub, A. Raed, S. S. Murray, M. D. Daubs, X. Yang, X. B. Yuan, J. C. Wang and Y. Lu, ACS Nano, 2016, 10, 7362–7369 CrossRef CAS PubMed.
- M. Yan, J. Du, Z. Gu, M. Liang, Y. Hu, W. Zhang, S. Priceman, L. Wu, Z. H. Zhou, Z. Liu, T. Segura, Y. Tang and Y. Lu, Nat. Nanotechnol., 2010, 5, 48–53 CrossRef CAS PubMed.
- M. Zhao, Y. Liu, R. S. Hsieh, N. Wang, W. Tai, K. Il Joo, P. Wang, Z. Gu and Y. Tang, J. Am. Chem. Soc., 2014, 136, 15319–15325 CrossRef CAS PubMed.
- T. Winkler, U. Kettling, A. Koltermann and M. Eigen, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 1375–1378 CrossRef CAS.
- H. Li, L. Ying, J. J. Green, S. Balasubramanian and D. Klenerman, Anal. Chem., 2003, 75, 1664–1670 CrossRef CAS PubMed.
- K. Bacia and P. Schwille, Nat. Protoc., 2007, 2, 2842–2856 CrossRef CAS PubMed.
- R. M. Dörlich, Q. Chen, P. N. Hedde, V. Schuster, M. Hippler, J. Wesslowski, G. Davidson and G. U. Nienhaus, Sci. Rep., 2015, 5, 10149 CrossRef PubMed.
- K. Nienhaus and G. U. Nienhaus, J. Mol. Biol., 2016, 428, 308–322 CrossRef CAS PubMed.
- L. Zemanová, A. Schenk, M. J. Valler, G. U. Nienhaus and R. Heilker, Drug Discovery Today, 2003, 8, 1085–1093 CrossRef.
- J. C. Lee and S. N. Timasheff, J. Biol. Chem., 1981, 256, 7193–7201 CAS.
- D. M. Mate, D. Gonzalez-Perez, R. Kittl, R. Ludwig and M. Alcalde, BMC Biotechnol., 2013, 13, 38 CrossRef CAS PubMed.
- A. Buthe, Methods Mol. Biol., 2011, 743, 213–222 Search PubMed.
- N. Sanson and J. Rieger, Polym. Chem., 2010, 1, 965 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Materials, methods, synthesis procedure, enzymatic activity assays and single particle analysis. See DOI: 10.1039/c7sc04438k |
| ‡ Present address: Nanomaterials group, CICnanoGUNE, Avenida Tolosa 76, 20018 Donostia-San Sebastián, Spain. |
| This journal is © The Royal Society of Chemistry 2018 |