
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis and kinetic resolution of substituted tetrahydroquinolines by lithiation then electrophilic quench†
Nicholas
Carter
,
Xiabing
Li
,
Lewis
Reavey
,
Anthony J. H. M.
Meijer
 and
Iain
Coldham
*
and
Iain
Coldham
*
Department of Chemistry, University of Sheffield, Brook Hill, Sheffield, S3 7HF, UK. E-mail: i.coldham@sheffield.ac.uk
First published on 14th December 2017
Abstract
Treatment of N-Boc-2-aryl-1,2,3,4-tetrahydroquinolines with n-butyllithium in THF at −78 °C resulted in efficient lithiation at the 2-position and the organolithiums were trapped with a variety of electrophiles to give substituted products. Variable temperature NMR spectroscopy gave kinetic data that showed that the rate of tert-butoxycarbonyl (Boc) rotation was fast (ΔG‡ ≈ 45 kJ mol−1 at −78 °C) and in situ ReactIR spectroscopy showed fast lithiation at −78 °C. By carrying out the lithiation in the presence of the chiral ligand sparteine, kinetic resolutions with very high levels of enantioselectivity were achieved. The resulting enantioenriched N-Boc-2-aryltetrahydroquinolines were converted to 2,2-disubstituted products without significant loss in enantiopurity. Most electrophiles add at the 2-position and the chemistry provides a way to access tetrahydroquinolines that are fully substituted alpha to the nitrogen atom. Notably, either enantiomer of the 2,2-disubstituted tetrahydroquinolines can be obtained with high selectivity from the same enantiomer of the chiral ligand. Unusually, when methyl cyanoformate was used as the electrophile, substitution occurred in the ortho position of the aryl ring attached at C-2. This change in regioselectivity on changing the electrophile was probed by deuterium isotope studies and by DFT calculations which suggested that the binding of the cyanoformate altered the structure of the intermediate organolithium. Secondary amine products can be prepared by removing the Boc group with acid or by inducing the Boc group to rearrange to the 2-position in the presence of triethylborane and this carbonyl N-to-C rearrangement occurs with retention of configuration from the intermediate enantiomerically enriched organolithium species.
Introduction
The tetrahydroquinoline ring system is of great importance in natural products and medicinal compounds.1 Substituted 1,2,3,4-tetrahydroquinolines are present in alkaloids such as angustureine, cuspareine, galipinine, martinellic acid, virantmycin, many with (for example) antiviral, antibacterial, antifungal, antimalarial, or antitumour activities.1 The majority of syntheses of tetrahydroquinolines involve the reduction of quinolines or dihydroquinolines,2 a Povarov type reaction,3 or a cyclization process to make one of the bonds in the partially saturated ring.4 This often leads to tetrahydroquinolines that are monosubstituted at positions in the partially saturated ring, for example at C-2, rather than 2,2-disubstituted products. The ability to prepare 2,2-disubstituted tetrahydroquinolines would be attractive, opening up a greater diversity of products and exploring more chemical space. However, there are few examples of their enantioselective preparation from achiral compounds. You and co-workers reported an isolated example of an asymmetric reduction of a substituted quinoline and intramolecular trapping with an indole (Scheme 1a).5 Zhao, Shi and co-workers developed an asymmetric Povarov reaction (Scheme 1b).6 Hopkins and Wolfe reported a palladium catalyzed carboamination to give 2,2-dialkyl tetrahydroquinolines (Scheme 1c).4d Enantioselective intramolecular N-arylation by Cai and co-workers was found with copper catalysis (Scheme 1d).7 Here we report the use of deprotonation followed by electrophile trapping as a convenient approach to 2,2-disubstituted tetrahydroquinolines (Scheme 1e). This strategy has found only limited use in a racemic sense with 2-cyanotetrahydroquinolines.8 Our research group has been studying the lithiation then electrophilic quench of N-Boc heterocycles, with a particular recent focus on piperidines,9 and tetrahydroisoquinolines.10,11 We show in this study that we can extend our lithiation chemistry with N-Boc-2-aryl-heterocycles to tetrahydroquinolines. These substrates have been found to undergo highly enantioselective kinetic resolution.12,13 The organolithium is configurationally stable at −78 °C and can be trapped to give 2,2-disubstituted tetrahydroquinolines with excellent enantioselectivities.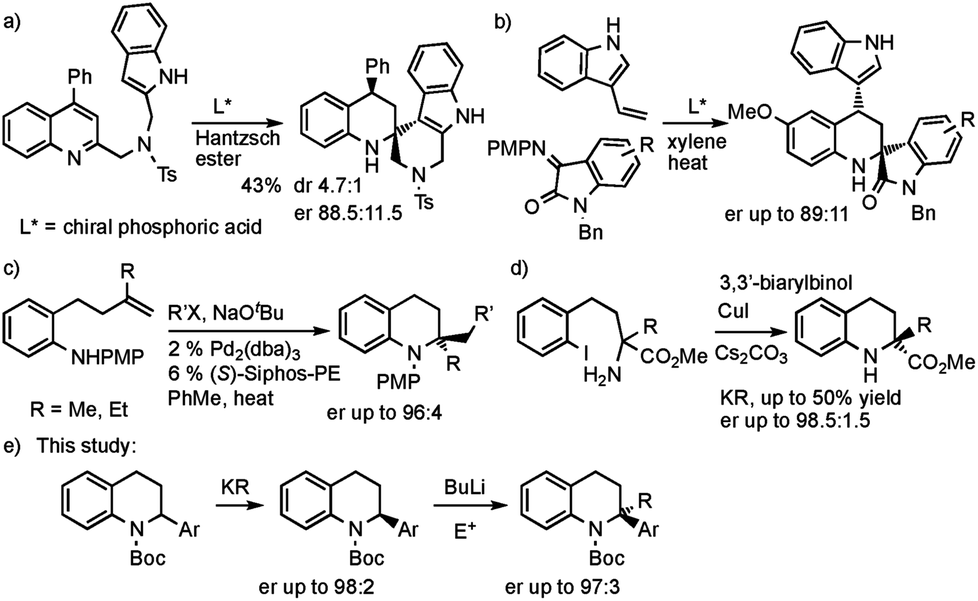 | ||
| Scheme 1 Asymmetric methods to 2,2-disubstituted tetrahydroquinolines. | ||
Results and discussion
We needed access to N-Boc-2-aryltetrahydroquinolines to test our lithiation chemistry. These were prepared from the quinolines 1a–e by reduction with sodium cyanoborohydride in acetic acid using a known method,14 followed by Boc protection of the amine to give the novel products 2a–e (Scheme 2).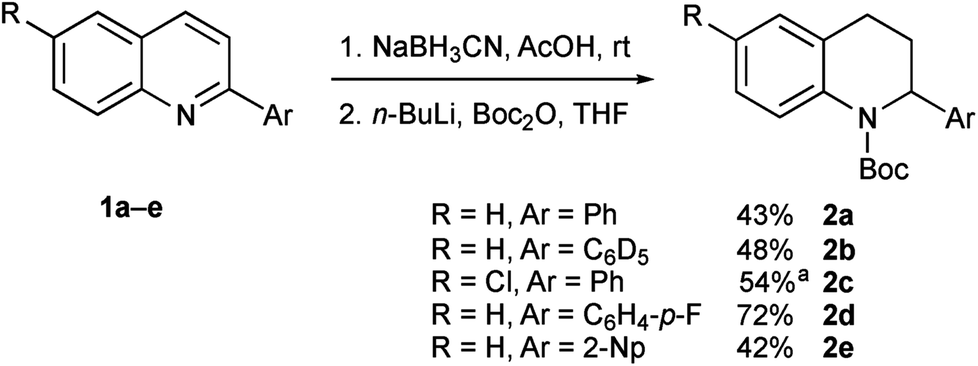 | ||
| Scheme 2 Preparation of tetrahydroquinolines 2a–e. aBoc-ON rather than Boc2O was used. | ||
We were aware from earlier studies that the rate of lithiation is dependent on the orientation of the Boc group.9d Therefore we probed the rate of rotation of the Boc group in compound 2a by using VT-NMR spectroscopy (see ESI†) and found activation parameters for Boc rotation, giving ΔG‡ ≈ 47 kJ mol−1 at −78 °C for each rotamer. This suggests that the Boc group rotates quickly (t1/2 ≈ 1 s) even at −78 °C. This was confirmed by ReactIR spectroscopy, which showed rapid lithiation (within a few minutes) at this temperature (Fig. 1 and ESI†).
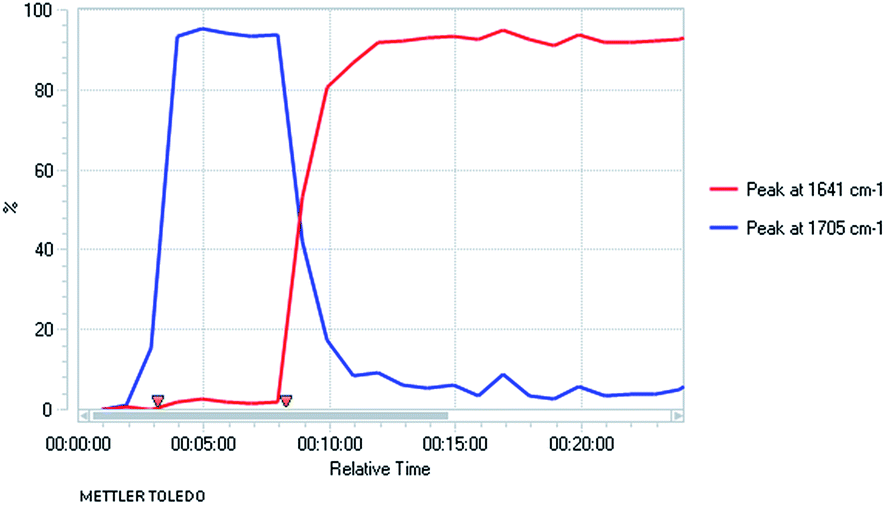 | ||
Fig. 1
In situ IR spectroscopy of the deprotonation of 2a with n-BuLi, THF at −78 °C with time in h:min:s with n-BuLi added at time 8 min (νC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O2a 1705 cm−1, νCO lithiated 2a 1641 cm−1). O2a 1705 cm−1, νCO lithiated 2a 1641 cm−1). | ||
The intermediate organolithium could be quenched with a selection of electrophiles to give the 2,2-disubstituted tetrahydroquinoline products 3a–i (Scheme 3). Generally good yields of the products were obtained. The only exception to this was the use of the electrophile methyl cyanoformate, which gave the product 4 rather than the expected product 3a. This is discussed further below.
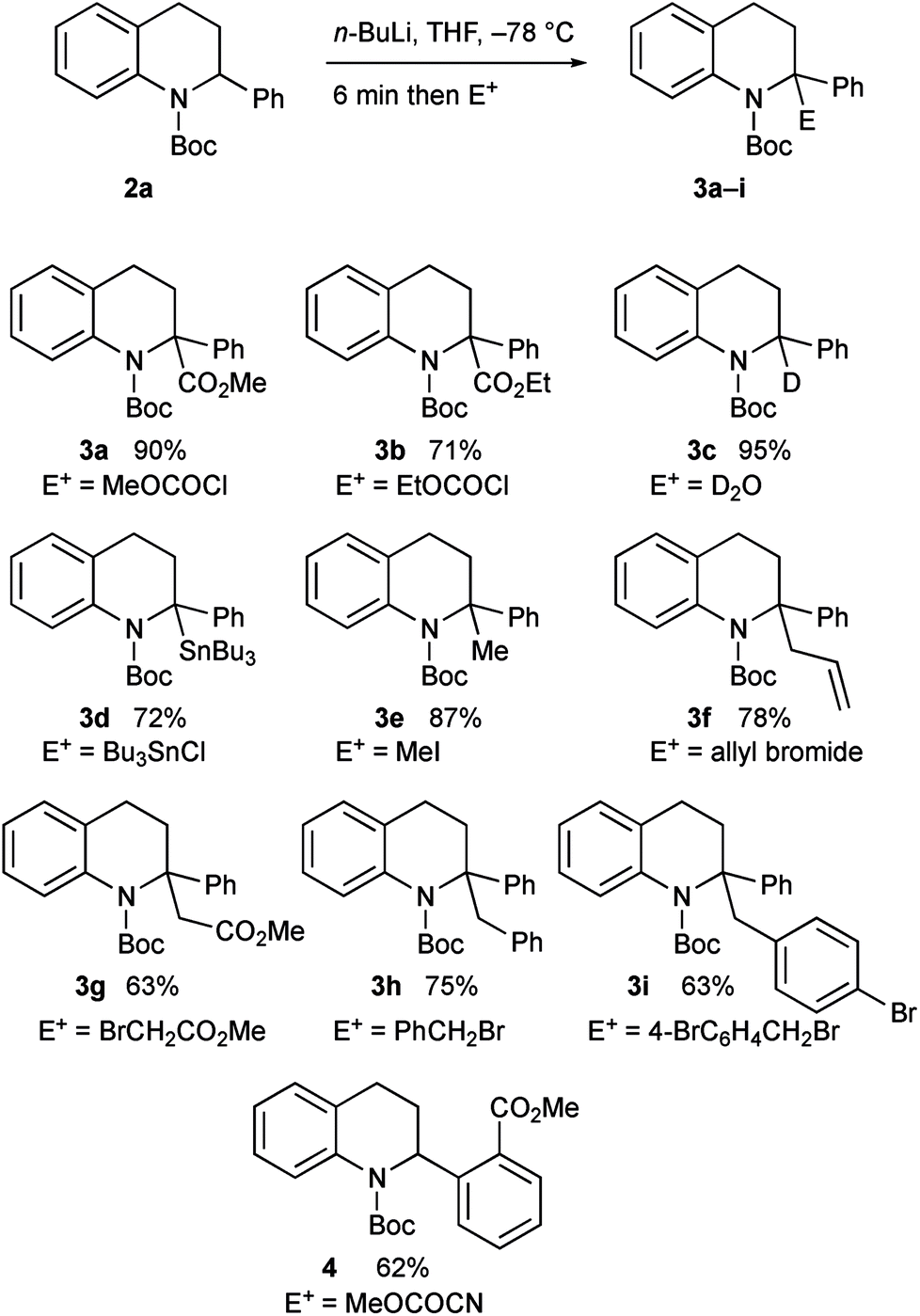 | ||
| Scheme 3 Lithiation–trapping of tetrahydroquinoline 2a. | ||
Kinetic resolution of the tetrahydroquinoline 2a was studied using (−)-sparteine as the chiral ligand in PhMe and adding n-BuLi to this mixture.15 Moderately good enantiomer ratios of the recovered tetrahydroquinoline 2a and the product 3a were obtained by this method. However improved results were found by pre-mixing the n-BuLi and (−)-sparteine before adding the tetrahydroquinoline 2a (Scheme 4). The deprotonation was relatively slow under these conditions and despite being a kinetic resolution, it was best to use 1.2 equiv. n-BuLi to achieve a suitable rate of reaction. The recovered tetrahydroquinoline 2a was isolated with high enantiomer ratio (er 97![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3) and this equates to a selectivity factor (krel) = 20.
:3) and this equates to a selectivity factor (krel) = 20.
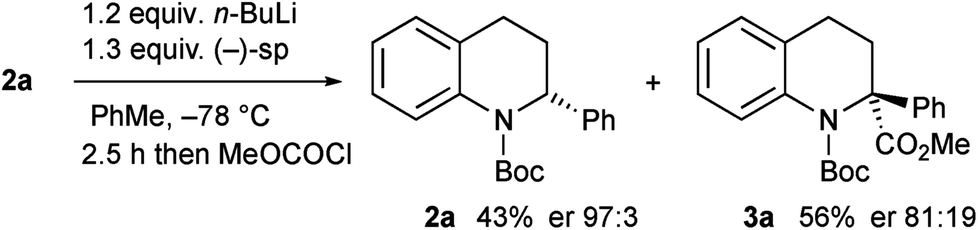 | ||
| Scheme 4 Kinetic resolution of tetrahydroquinoline 2a with (−)-sparteine. | ||
The opposite enantiomer of the recovered starting material 2a could be obtained by using (+)-sparteine in the kinetic resolution (Scheme 5). Several runs were conducted, all with very good enantioselectivities. Recrystallisation of the product 3a from run 1 gave material that was suitable for single crystal X-ray analysis. This confirmed the absolute configuration to be (R)-3a as indicated (see ESI†), and as expected based on previous findings of the stereoselectivity preference for BuLi·sparteine in the deprotonation of N-Boc-piperidines.15,16 The kinetic resolution was extended to the tetrahydroquinolines 2c–e (Scheme 6). High enantiomer ratios of the recovered tetrahydroquinolines were obtained in each case, particularly if more than just a slight excess of sparteine was added to the reaction mixture. The quenched products could be separated from the recovered starting material to give the desired tetrahydroquinolines (S)-2c–e.
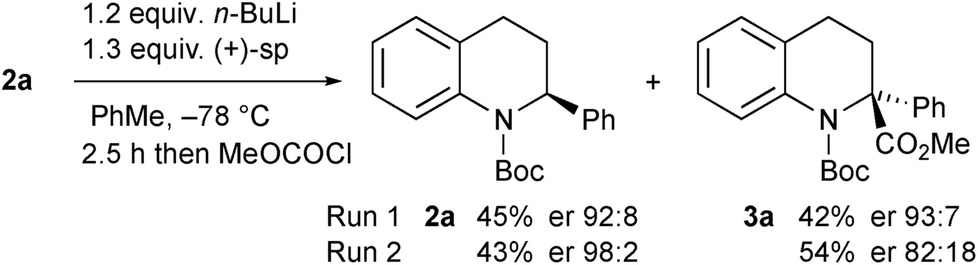 | ||
| Scheme 5 Kinetic resolution of tetrahydroquinoline 2a with (+)-sparteine. | ||
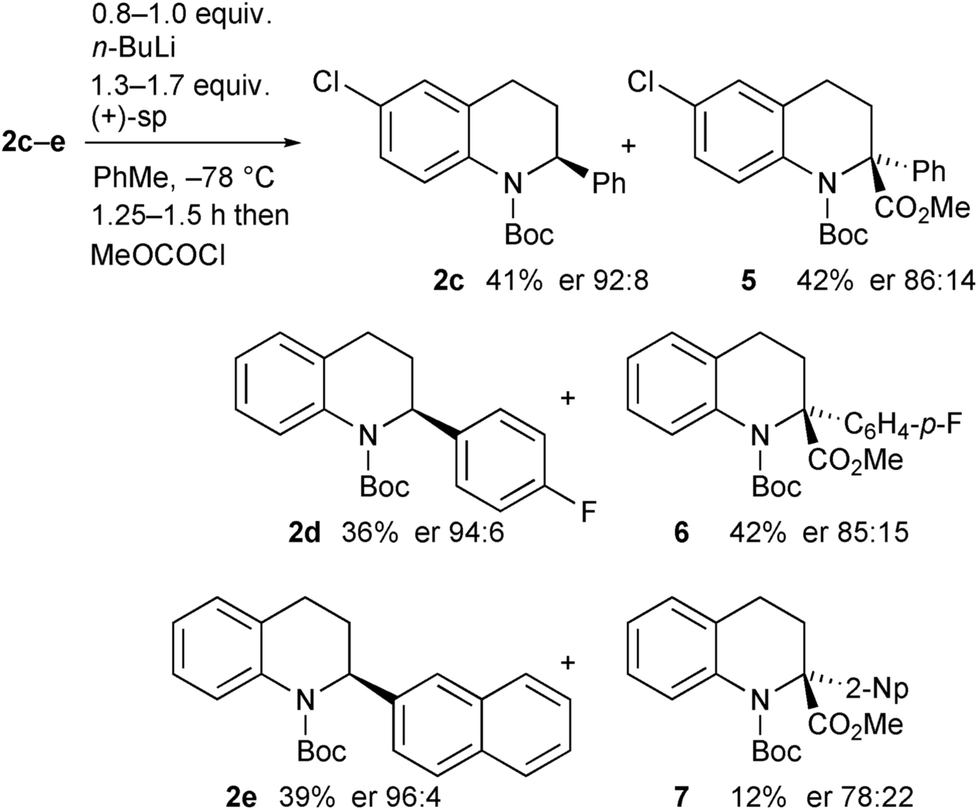 | ||
| Scheme 6 Kinetic resolution of tetrahydroquinolines 2c–e with (+)-sparteine. | ||
The enantioenriched 2-aryltetrahydroquinolines 2a and 2c–e were treated with n-BuLi in THF at −78 °C and the resulting organolithiums were found to be configurationally stable. After electrophilic quench, the products 3a–b, 3e, 3g, and 8–10 were obtained with high enantiomer ratios (Scheme 7). A slight loss of enantioselectivity was noticeable on using iodomethane as the electrophile, possibly as this reacts more slowly allowing some racemization on warming prior to quench.16b The tetrahydroquinoline 3g was recrystallised and its absolute configuration was confirmed by single crystal X-ray analysis. The major diastereomer of the oxazolidinones 10 was confirmed by X-ray analysis. For X-ray data, see ESI.†
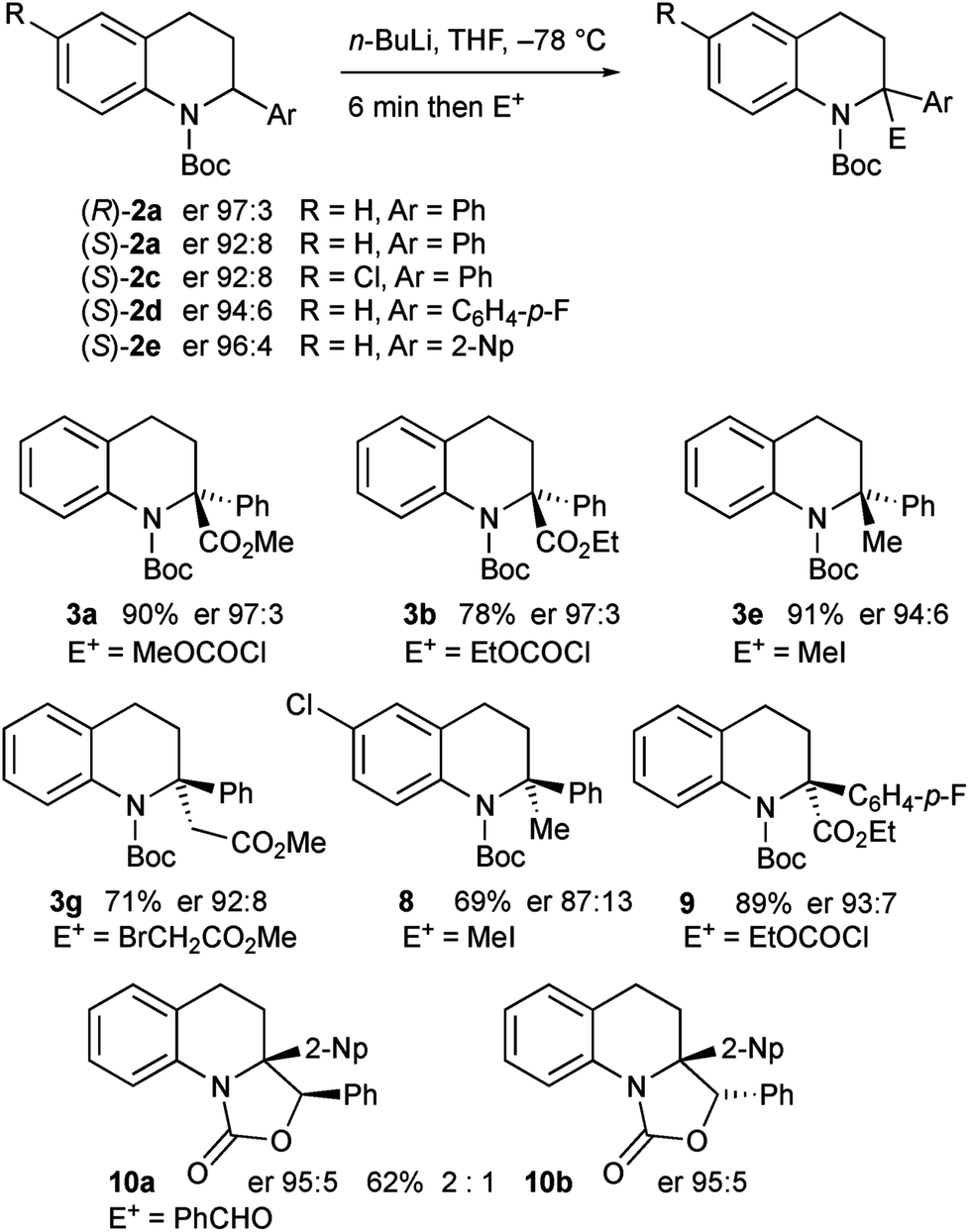 | ||
| Scheme 7 Lithiation–quench of enantioenriched tetrahydroquinolines 2a, 2c–e. | ||
Another electrophile that we were interested in testing was a trialkylborane, as this could result in a borate intermediate that should be prone to rearrange.17 However we found that, instead of quenching the organolithium, addition of triethylborane promoted Boc group migration to give the tetrahydroquinoline 11 (Scheme 8).18 The absolute configuration of the product 11 was confirmed by single crystal X-ray analysis showing that the rearrangement occurred with retention of configuration starting with the highly enantioenriched tetrahydroquinoline 2a. We speculate that the borane coordinates to the carbonyl oxygen atom to effect the migration and this must be preferable to direct reaction of the organolithium on the boron atom. The same reaction occurred with BEt3 as a catalyst (0.2 equiv. gave product 11, 59% yield), or even in the absence of any catalyst (42% yield of 11 on using BuLi then warming without any BEt3). We were able to remove the Boc group from the nitrogen atom in the products by using trifluoroacetic acid (TFA), for example from compound 3g to give the secondary amine 12 with only minimal loss of enantiopurity.
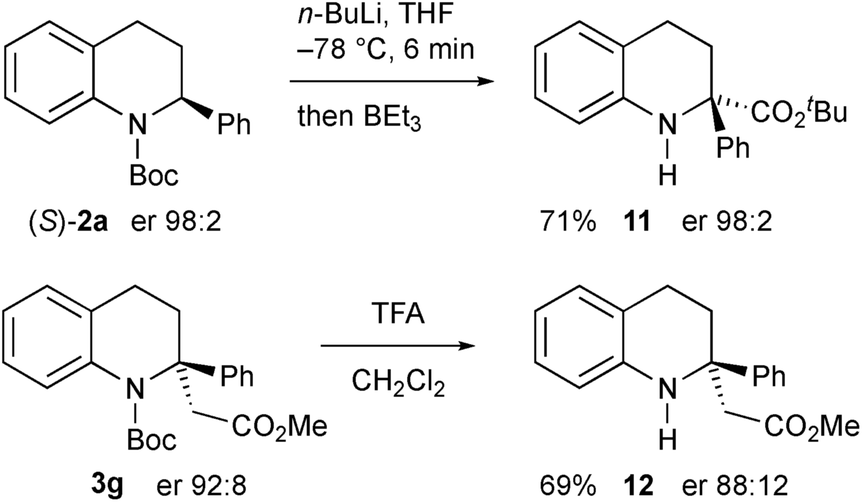 | ||
| Scheme 8 Formation of secondary amine products. | ||
We were surprised to find that the electrophile methyl cyanoformate gave the product 4 rather than the expected product 3a. We were concerned that there may have been competitive ortho lithiation,19 although this would be contrary to the formation of the products 3a–i. We recently uncovered an example of such an unusual change in regioselectivity with a benzylic organolithium on changing the electrophile,11c and wanted to probe this reactivity further. Treatment of the deuterated tetrahydroquinoline 3c under the same conditions (1.2 equiv. n-BuLi in THF) followed by addition of MeOCOCN returned recovered starting material 3c, indicating a large kinetic isotope effect.20 This suggests that the benzylic proton in 2a is indeed removed and not the ortho proton. Forcing the deprotonation with 3 equiv. n-BuLi for 1 h before addition of MeOCOCN still gave predominantly recovered 3c but did give a small amount (8% yield) of the product 4 (no deuterium present). Treatment of the tetrahydroquinoline 2b with n-BuLi then MeOCOCl gave the expected 2,2-disubstituted product 13 (Scheme 9). However, with MeOCOCN as the electrophile, the ortho substituted product 14 was obtained in moderate yield as a mixture in which the major product had deuterium at the 2-position. Hence the reaction must proceed by initial deprotonation alpha to the nitrogen atom. Most electrophiles react at C-2 to give the products 3a–i. With methyl cyanoformate, substitution occurs at the ortho position and then rearomatisation takes place (see ESI†). The transfer of the proton (or deuterium) is likely to occur non-selectively and indeed on using enantioenriched tetrahydroquinoline 2a (er 92:8, prepared as described in Scheme 5), the product 4 was formed with low selectivity (er 61:39).
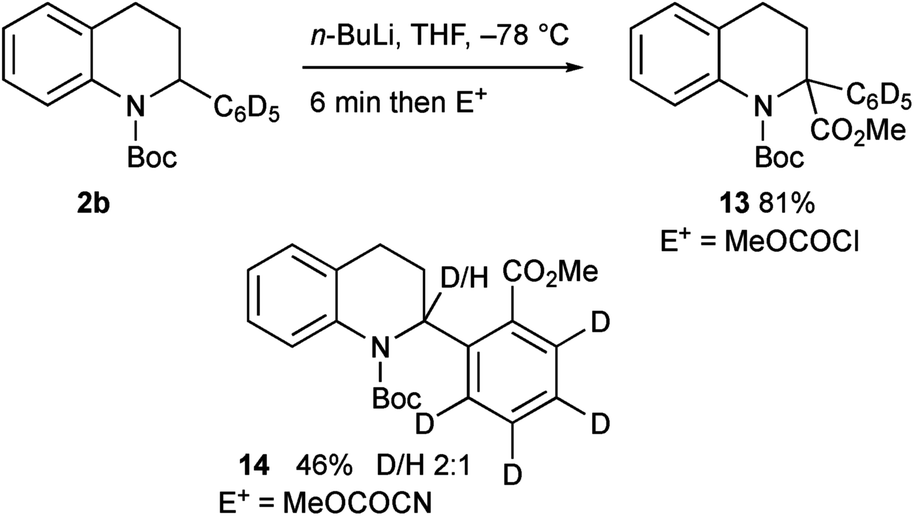 | ||
| Scheme 9 Lithiation–trapping of tetrahydroquinoline 2b. | ||
Calculations (using DFT/B3LYP-GD3BJ/6-311G**; see Computational methods below) initially focused on Boc rotation of tetrahydroquinoline 2a, for which we found an activation Gibbs energy of 48.7 kJ mol−1 in fair agreement with the experimental value of about 44 kJ mol−1 at 298 K. Subsequent calculations focused on the structures of the intermediate organolithium. In particular, we studied the complexation of the intermediate lithiated species with MeOCOCl and MeOCOCN. This gave insight into the potential reason for the change in regioselectivity. The minimised structures had the lithium atom coordinated to the carbonyl oxygen atom and close to C-2 when coordinated to THF or MeOCOCl (Fig. 2a and b). On the other hand, there was clearly an η3 co-ordination of the lithium atom when MeOCOCN was bound (Fig. 2c). An alternative explanation could be that released cyanide could affect the regiochemistry, however an experiment in which MeOCOCl was added to the organolithium after addition of one equivalent of NaCN returned only the alpha-substituted product 3a. Thus, we surmise that a change in structure of the organolithium on complexing the different electrophiles (MeOCOCl or MeOCOCN) must be influencing the regiochemistry on reaction with the electrophile, although the precise way that this happens will be subject to further study.21
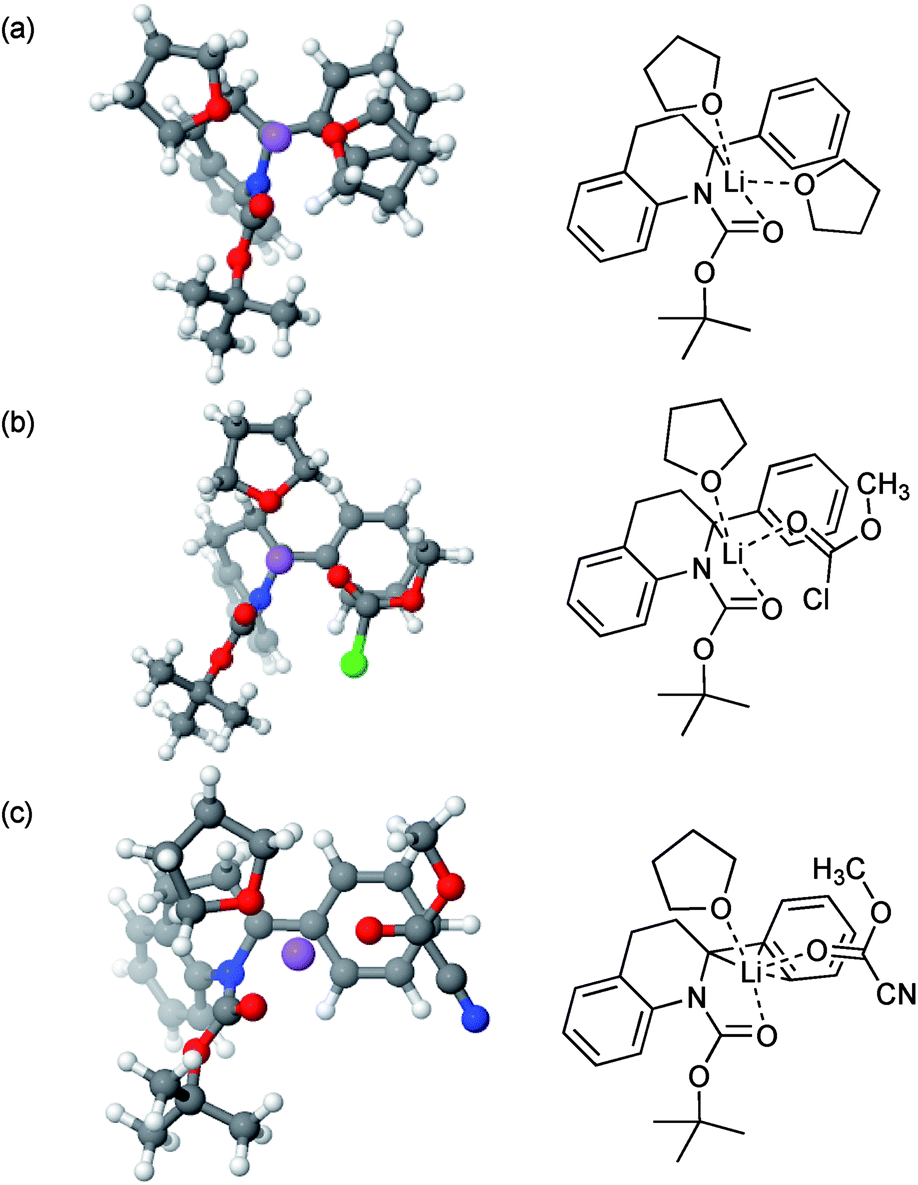 | ||
| Fig. 2 Lithiated intermediates by DFT [6-311G(d,p) basis set with B3LYP functional] and their ChemDraw representations. (a) In THF; (b) with MeOCOCl; (c) with MeOCOCN. | ||
Experimental
A representative method for the kinetic resolution of the tetrahydroquinoline 2a is given below. For further details and all data, see ESI.†
n-BuLi (0.6 mL, 0.39 mmol, 2.5 M in hexane) was added to freshly distilled (+)-sparteine (106 mg, 0.45 mmol) in PhMe (4 mL) at −78 °C. After 30 min, tetrahydroquinoline 2a (101 mg, 0.33 mmol, 0.3 M solution in toluene) was added. After 2.5 h, methyl chloroformate (0.09 mL, 1.15 mmol) was added. The mixture was allowed to warm to room temperature over 16 h and then MeOH (1 mL) was added. Purification by column chromatography on silica gel, eluting with petrol–EtOAc (95:5), gave the recovered tetrahydroquinoline (S)-2a (44 mg, 43%) as an amorphous solid; m.p. 60–61 °C; er 98:2, determined by CSP HPLC with a Cellulose-2 column; [α]23D −103.9 (0.6, CHCl3); and in addition the tetrahydroquinoline (R)-3a (65 mg, 54%) as an amorphous solid; m.p. 71–74 °C; er 82:18, determined by CSP HPLC.
Computational methods
All calculations were performed using density functional theory, employing the B3LYP22 functional as implemented in the D.01 version of Gaussian 09.23 Calculations included dispersion corrections using the GD3-BJ24 method. All calculations used the 6-311G(d,p)25 basis set. Solvent was included via the PCM method26 as implemented in Gaussian with the default parameters for THF.The starting positions of coordinated solvent and electrophile molecules were varied to obtain the lowest energy structures. Frequency calculations were performed on all optimized structures to confirm that these were true minima. One transition state calculation was performed, for which a single imaginary frequency was found, as expected. For the calculations on 2a no imaginary frequencies were found. The two complexes presented in Fig. 2a and b also showed no imaginary frequencies. The complex presented in Fig. 2c showed a single imaginary frequency of −16.5 cm−1. Inspection shows this mode is largely a torsional mode of the ligands around the lithium–ligand bond. Re-running the calculation with a finer integration grid and tighter optimization convergence led to a structure without imaginary frequencies. This latter structure is reported in the ESI.† All Gibbs energies reported were evaluated at 298.15 K and standard pressure. For the precise keywords used in each of the calculations see the ESI.†
Conclusions
In conclusion, we have developed a rapid access to 2,2-disubstituted tetrahydroquinolines from 2-aryltetrahydroquinolines by deprotonation with n-butyllithium followed by electrophilic quench. The reaction proceeds with retention of configuration on using enantiomerically enriched starting materials. Surprisingly, methyl cyanoformate reacted at the ortho position of the 2-aryl substituent and this change in regioselectivity on change in the electrophile is proposed, on the basis of DFT studies, to result from a small change in the structure of the organolithium intermediate. Excellent enantioselectivities in the kinetic resolution were obtained in the presence of the chiral ligand sparteine. This chemistry therefore provides a new method to prepare highly enantiomerically enriched 2-aryltetrahydroquinolines and tetrahydroquinolines that are fully substituted at the 2 position.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We acknowledge support from the EPSRC, the China Scholarship Council (UK-China Scholarships for Excellence), and the University of Sheffield. We thank Harry Adams and Craig Robertson for the single crystal X-ray analyses, Sandra van Meurs and Craig Robertson for help with NMR spectroscopy, and Ashraf el-Tunsi for further experiments.Notes and references
- V. Sridharan, P. A. Suryavanshi and J. C. Menéndez, Chem. Rev., 2011, 111, 7157 CrossRef CAS PubMed.
- For example, see: (a) Z. Zhang and H. Du, Org. Lett., 2015, 17, 2816 CrossRef CAS PubMed; (b) Y.-L. Du, Y. Hu, Y.-F. Zhu, X.-F. Tu, Z.-Y. Han and L.-Z. Gong, J. Org. Chem., 2015, 80, 4754 CrossRef CAS PubMed; (c) J. Wen, R. Tan, S. Liu, Q. Zhao and X. Zhang, Chem. Sci., 2016, 7, 3047 RSC; (d) Y. Wang, Y. Liu, D. Zhang, H. Wei, M. Shi and F. Wang, Angew. Chem., Int. Ed., 2016, 55, 3776 CrossRef CAS PubMed; (e) C. S. Lim, T. T. Quach and Y. Zhao, Angew. Chem., Int. Ed., 2017, 56, 7176 CrossRef CAS PubMed.
- For some reviews, see: (a) M. Fochi, L. Caruana and L. Bernardi, Synthesis, 2014, 46, 135 CrossRef; (b) J. S. B. Forero, J. Jones and F. M. da Silva, Curr. Org. Synth., 2016, 13, 157 CrossRef.
- For example, see: (a) Y. K. Kang, S. M. Kim and D. Y. Kim, J. Am. Chem. Soc., 2010, 132, 11847 CrossRef CAS PubMed; (b) K. Mori, K. Ehara, K. Kurihara and T. Akiyama, J. Am. Chem. Soc., 2011, 133, 6166 CrossRef CAS PubMed; (c) J. C. Anderson, J. P. Barham and C. D. Rundell, Org. Lett., 2015, 17, 4090 CrossRef CAS PubMed; (d) B. A. Hopkins and J. P. Wolfe, Chem. Sci., 2015, 5, 4840 RSC; (e) R.-R. Liu, B.-L. Li, J. Lu, C. Shen, J.-R. Gao and Y.-X. Jia, J. Am. Chem. Soc., 2016, 138, 5198 CrossRef CAS PubMed; (f) L. A. Leth, F. Glaus, M. Meazza, L. Fu, M. K. Thøgersen, E. A. Bitsch and K. A. Jørgensen, Angew. Chem., Int. Ed., 2016, 55, 15272 CrossRef CAS PubMed; (g) Y. Zhu, B. Li, C. Wang, Z. Dong, X. Zhong, K. Wang, W. Yan and R. Wang, Org. Biomol. Chem., 2017, 15, 4544 RSC.
- S.-G. Wang, W. Zhang and S.-L. You, Org. Lett., 2013, 15, 1488 CrossRef CAS PubMed.
- H.-H. Zhang, X.-X. Sun, J. Liang, Y.-M. Wang, C.-C. Zhao and F. Shi, Org. Biomol. Chem., 2014, 12, 9539 CAS.
- (a) W. Yang, Y. Long, S. Zhang, Y. Zeng and Q. Cai, Org. Lett., 2013, 15, 3598 CrossRef CAS PubMed; (b) F. Zhou, J. Guo, J. Liu, K. Ding, S. Yu and Q. Cai, J. Am. Chem. Soc., 2012, 134, 14326 CrossRef CAS PubMed.
- S. Shahane, F. Louafi, J. Moreau, J.-P. Hurvois, J.-L. Renaud, P. van de Weghe and T. Roisnel, Eur. J. Org. Chem., 2008, 4622 CrossRef CAS.
- (a) I. Coldham, S. Raimbault, P. T. Chovatia, J. J. Patel, D. Leonori, N. S. Sheikh and D. T. E. Whittaker, Chem. Commun., 2008, 4174 RSC; (b) I. Coldham, S. Raimbault, D. T. E. Whittaker, P. T. Chovatia, D. Leonori, J. J. Patel and N. S. Sheikh, Chem.–Eur. J., 2010, 16, 4082 CrossRef CAS PubMed; (c) I. Coldham and D. Leonori, J. Org. Chem., 2010, 75, 4069 CrossRef CAS PubMed; (d) N. S. Sheikh, D. Leonori, G. Barker, J. D. Firth, K. R. Campos, A. J. H. M. Meijer, P. O'Brien and I. Coldham, J. Am. Chem. Soc., 2012, 134, 5300 CrossRef CAS PubMed.
- (a) X. Li, D. Leonori, N. S. Sheikh and I. Coldham, Chem.–Eur. J., 2013, 19, 7724 CrossRef CAS PubMed; (b) X. Li and I. Coldham, J. Am. Chem. Soc., 2014, 136, 5551 CrossRef CAS PubMed; (c) R. A. Talk, A. Duperray, X. Li and I. Coldham, Org. Biomol. Chem., 2016, 14, 4908 RSC.
- For other ring systems, see: (a) S. P. Robinson, N. S. Sheikh, C. A. Baxter and I. Coldham, Tetrahedron Lett., 2010, 51, 3642 CrossRef CAS; (b) E. J. Cochrane, L. A. Hassall and I. Coldham, J. Org. Chem., 2015, 80, 5964 CrossRef CAS PubMed; (c) T. Aeyad, J. D. Williams, A. J. H. M. Meyer and I. Coldham, Synlett, 2017, 28, 2765 CrossRef CAS.
- For examples of kinetic resolutions to give tetrahydroquinolines, see: (a) M.-W. Chen, X.-F. Cai, Z.-P. Chen, L. Shi and Y.-G. Zhou, Chem. Commun., 2014, 50, 12526 RSC; (b) K. Saito, H. Miyashita and T. Akiyama, Chem. Commun., 2015, 51, 16648 RSC; (c) D. Kong, S. Han, R. Wang, M. Li, G. Zi and G. Hou, Chem. Sci., 2017, 8, 4558 RSC.
- For examples of kinetic resolutions of amines, see: (a) I. Kreituss and J. W. Bode, Acc. Chem. Res., 2016, 49, 2807 CrossRef CAS PubMed; (b) I. Kreituss and J. W. Bode, Nat. Chem., 2017, 9, 446 CrossRef CAS; (c) S. Das, N. Majumdar, C. K. De, D. S. Kundu, A. Döhring, A. Garczynski and B. List, J. Am. Chem. Soc., 2017, 139, 1357 CrossRef CAS PubMed; (d) J. I. Murray, N. J. Flodén, A. Bauer, N. D. Fessner, D. L. Dunklemann, O. Bob-Egbe, H. S. Rzepa, T. Bürgi, J. Richardson and A. C. Spivey, Angew. Chem., Int. Ed., 2017, 56, 5760 CrossRef CAS PubMed.
- A. D. Lackner, A. V. Samant and F. D. Toste, J. Am. Chem. Soc., 2013, 135, 14090 CrossRef CAS PubMed.
- E. J. Cochrane, D. Leonori, L. A. Hassall and I. Coldham, Chem. Commun., 2014, 50, 9910 RSC.
- (a) W. F. Bailey, P. Beak, S. T. Kerrick, S. Ma and K. B. Wiberg, J. Am. Chem. Soc., 2002, 124, 1889 CrossRef CAS PubMed; (b) D. Stead, G. Carbone, P. O'Brien, K. R. Campos, I. Coldham and A. Sanderson, J. Am. Chem. Soc., 2010, 132, 7260 CrossRef CAS PubMed.
- I. Coldham, J. J. Patel, S. Raimbault, D. T. E. Whittaker, H. Adams, G. Y. Fang and V. K. Aggarwal, Org. Lett., 2008, 10, 141 CrossRef CAS PubMed.
- For examples of N to C migration of carbonyl groups, see: (a) N. Kise, H. Ozaki, H. Terui, K. Ohya and N. Ueda, Tetrahedron Lett., 2001, 42, 7637 CAS; (b) K. W. Kells, A. Ncube and J. M. Chong, Tetrahedron, 2004, 60, 2247 CrossRef CAS; (c) N. Dieltiens, C. V. Stevens, K. G. R. Masschelein and T. Rammeloo, Tetrahedron, 2005, 61, 6749 CrossRef CAS; (d) V. Capriati, S. Florio, R. Luisi and B. Musio, Org. Lett., 2005, 7, 3749 CrossRef CAS PubMed; (e) D. M. Hodgson, P. G. Humphreys, Z. Xu and J. G. Ward, Angew. Chem., Int. Ed., 2007, 46, 2245 CrossRef CAS PubMed.
- (a) F. Affortunato, S. Florio, R. Luisi and B. Musio, J. Org. Chem., 2008, 73, 9214 CrossRef CAS PubMed; (b) V. Capriati, S. Florio and R. Luisi, Eur. J. Org. Chem., 2014, 5397 CrossRef CAS.
- For kinetic isotope effects on deprotonation, see D. Hoppe, M. Paetow and F. Hintze, Angew. Chem., Int. Ed., 1993, 32, 394 CrossRef.
- For a change in regioselectivity between alpha and para substitution on reaction of a benzyllithium based on a change in structure, see: (a) U. Kroesen, L. Knauer and C. Strohmann, Angew. Chem., Int. Ed., 2017, 56, 6232 CrossRef CAS PubMed; (b) J. G. Peters, M. Seppi, R. Fröhlich, B. Wibbeling and D. Hoppe, Synthesis, 2002, 381 CrossRef CAS. For examples of structures of benzyllithiums, see: (c) H. Ahlbrecht, J. Harbach, T. Hauck and H.-O. Kalinowski, Chem. Ber., 1992, 125, 1753 CrossRef CAS; (d) G. Boche, M. Marsch, J. Harbach, K. Harms, B. Ledig, F. Schubert, J. C. W. Lohrenz and H. Ahlbrecht, Chem. Ber., 1993, 126, 1887 CrossRef CAS; (e) T. Tatic, S. Hermann, M. John, A. Loquet, A. Lange and D. Stalke, Angew. Chem., Int. Ed., 2011, 50, 6666 CrossRef CAS PubMed; (f) M. G. Davidson, D. Garcia-Vivo, A. R. Kennedy, R. E. Mulvey and S. D. Robertson, Chem.–Eur. J., 2011, 17, 3364 CrossRef CAS PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456 CrossRef CAS PubMed.
- (a) A. D. McLean and G. S. Chandler, J. Chem. Phys., 1980, 72, 5639 CrossRef CAS; (b) K. Raghavachari, J. S. Binkley, R. Seeger and J. A. Pople, J. Chem. Phys., 1980, 72, 650 CrossRef.
- (a) G. Scalmani and M. J. Frisch, J. Chem. Phys., 2010, 132, 114110 CrossRef PubMed; (b) M. Cossi, N. Rega, G. Scalmani and V. Barone, J. Comput. Chem., 2003, 24, 669 CrossRef CAS PubMed and references therein.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, spectroscopic data, ReactIR, X-ray and DFT data, and NMR spectra. For products 3a, 3g, 10a, and 11. CCDC 1578266–1578269. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7sc04435f |
| This journal is © The Royal Society of Chemistry 2018 |