
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIriomoteolides: novel chemical tools to study actin dynamics†
A.
Unzue‡
a,
R.
Cribiú§
a,
M. M.
Hoffman
a,
T.
Knehans‡
b,
K.
Lafleur¶
a,
A.
Caflisch
*b and
C.
Nevado
 *a
*a
aDepartment of Chemistry, University of Zürich, Winterthurerstrasse 190, CH-8057, Zürich, Switzerland. E-mail: cristina.nevado@chem.uzh.ch
bDepartment of Biochemistry, University of Zürich, Winterthurerstrasse 190, CH-8057, Zürich, Switzerland. E-mail: caflisch@bioc.uzh.ch
First published on 3rd April 2018
Abstract
Despite its promising biological profile, the cellular targets of iriomoteolide-3a, a novel 15-membered macrolide isolated from Amphidinium sp., have remained unknown. A small library of non-natural iriomoteolide-3a analogues is presented here as a result of a novel, highly convergent, catalysis-based scaffold-diversification campaign, which revealed the suitable sites for chemical editing in the original core. We provide compelling experimental evidence for actin as one of iriomoteolides' primary cellular targets, establishing the ability of these secondary metabolites to inhibit cell migration, induce severe morphological changes in cells and cause a reversible cytoplasmic retraction and reduction of F-actin fibers in a time and dose dependent manner. These results are interpreted in light of the ability of iriomoteolides to stabilize F-actin filaments. Molecular dynamics simulations provide evidence for iriomoteolide-3a binding to the barbed end of G-actin. These results showcase iriomoteolides as novel and easily tunable chemical probes for the in vitro study of actin dynamics in the context of cell motility processes including cell invasion and division.
Introduction
Screening of free-swimming dinoflagellates Amphidinium sp. via genetic and metabolomic analyses enabled the identification of the HYA024 strain as a source of unknown bioactive metabolites.1 Three new 20-membered ring cytotoxic lactones, iriomoteolides-1a-c, could be isolated together with a novel vinyl epoxide containing 15-membered ring macrolide named iriomoteolide-3a (1) and its 7,8-isopropylidene analogue (2).2 The carbon chain length as well as the C1- and oxygen distribution observed in 1 differ from that of most macrolides which possess an even-numbered lactone ring and a C1–Me/C2–O pattern as a result of the polyketide synthase machinery. The unique pattern found in 1 thus hints towards a non-successive incorporation of acetates or propionates in the assembly of the polyketide chain (Scheme 1, top left).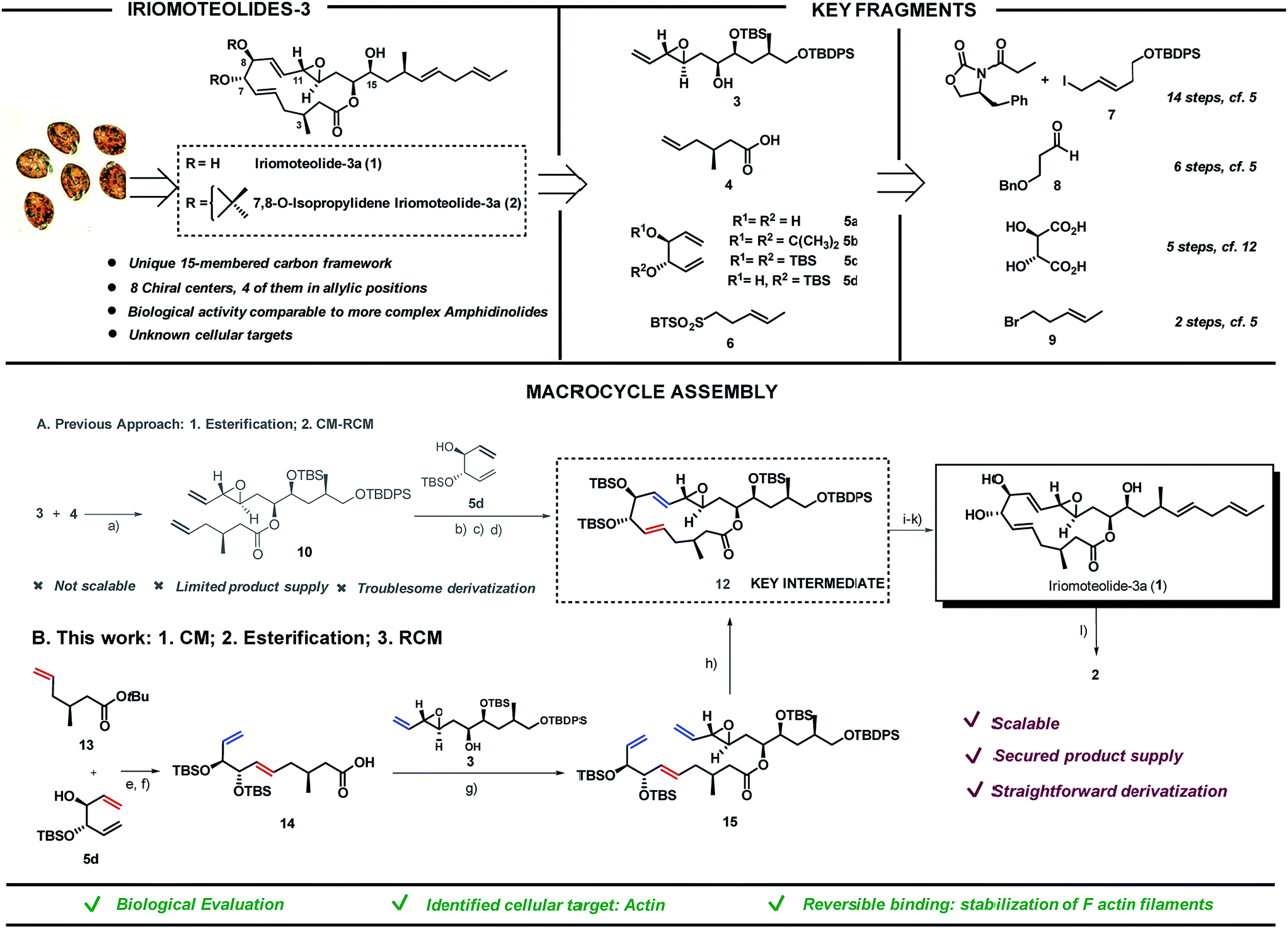 | ||
Scheme 1 Iriomoteolide-3a: key fragments (top) and different strategies for final assembly (bottom). (A) Original route via esterification/CM-RCM: (a) EDC·HCl, 4-pyrrolidinopyridine, CH2Cl2, 25 °C, 72%; (b) 5d (5 eq.), Grubbs II 11 (5 mol%), toluene, 25 to 50 °C; (c) TBSOTf, 2,6-lutidine, THF, 0 °C, 80%; (d) Grubbs II 11 (12 mol%), toluene, 25 °C, 76%. (B) Optimized route via CM/esterification/RCM: (e) Grubbs II 11 (5 mol%), toluene, 50 °C, 82%; (f) 2,6-lutidine, TBSOTf, CH2Cl2 followed by EtOAc/MeOH/water, Na2CO3, 78%; (g) 3, 4-pyrrolidinopyridine, EDC·HCl, CH2Cl2, 84%; (h) Grubbs II 11 (5 mol%), toluene, 25 °C, 81%; (i) NH4F, MeOH, 58%; (j) DMP, CH2Cl2, then 6, K[N(SiMe3)2], THF, 0 °C, 93![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :7 E/Z, 76%; (k) TBAF, THF, 86%. (l) 2,3-Dimethoxypropane, PPTS, CH2Cl2, 25 °C, 20%. EDC = 1-ethyl-3-(3-dimethylaminopropyl)-carbodiimide. DMP = Dess–Martin periodinane. :7 E/Z, 76%; (k) TBAF, THF, 86%. (l) 2,3-Dimethoxypropane, PPTS, CH2Cl2, 25 °C, 20%. EDC = 1-ethyl-3-(3-dimethylaminopropyl)-carbodiimide. DMP = Dess–Martin periodinane. | ||
The ability of iriomoteolides to impair cell growth on human B lymphocyte DG-75 and Epstein–Barr virus positive (EBV+) Raji cells was recognized early on.2 These results are in line with those reported for amphidinolides, polyketides also isolated from Amphidinium species, most of which exhibit respectable levels of cytotoxicity against standard murine and/or human tumor cell lines in vitro, in some cases with potencies that rival those of renowned anticancer agents. As it is typical for secondary metabolites with high level of molecular complexity, additional biological evaluation is hampered by the low yield in which they can be isolated from the natural source (0.015% of the wet weight of the dinoflagelate in the case of 1). Thus, in depth studies to unravel both the mode of action and primary cellular targets of these molecules must rely on chemical synthetic efforts. Both the challenging structural features and the potential of these natural products as biological probes have brought amphidinolides3–10 and iriomoteolides11–16 to the forefront of synthetic efforts in recent years. Our group reported the first total synthesis of iriomoteolide-3a (1), which enabled the confirmation of its proposed structure.17 However, even if access to the desired compound was granted, our initial strategy fell short to provide significant amounts of this secondary metabolite and derivatives thereof for further evaluation (Scheme 1A).
Herein we present a new synthetic approach towards iriomoteolide-3a which has enabled a scaffold-diversification campaign and solved the supply problem (Scheme 1B). The suitable sites for chemical edition of the original core have been identified and the impact of conformation in the bioactivity of these molecules has been proved.18 In addition, based on a varied array of cell-based and in vitro studies, we demonstrate that actin is one of the primary cellular receptors for iriomoteolide-3a and derivatives thereof. This study provides a comprehensive characterization of the mode of action of these metabolites as actin reversible stabilizers. The iriomoteolides therefore constitute a new class of chemical probes towards the reversible modulation of actin dynamics beyond their potential relevance as cytotoxic targets.
Results and discussion
Strategic considerations and new synthetic approach
Iriomoteolide-3a (1) was divided into four key fragments of comparable complexity (3–6),17 which were prepared from readily available starting materials (Scheme 1, top right).19 The north skeleton of the molecule, containing the sensitive vinyl epoxide moiety and the three additional stereocenters, was assembled from Evans oxazolidinone20 and (E)-tert-butyl((5-iodopent-3-en-1-yl)oxy)diphenylsilane (7)21,22 in a 14 steps sequence involving an asymmetric dihydroxylation, and in situ lactonization followed by functional group interconversion to produce fragment 3. The southern portion was produced in 6 steps from 3-(benzyloxy)propanal 823via catalytic asymmetric cyclocondensation with acetyl bromide24,25 followed by treatment with dimethyl cuprate to install the key C3–Me stereocenter in fragment 4. L-Tartaric acid was used as precursor for the 1,3-dienes 5a–d following previously reported procedures.26,27 Finally, sulfone 6 was prepared from (E)-5-bromo-2-pentene 928 in only two steps via halogen displacement and oxidation.29 The original approach to assemble the macrocycle (Scheme 1A) started with an intermolecular esterification between the secondary alcohol in 3 and acid 4 using EDC as activating agent in the presence of 4-pyrrolidinopyridine to produce ester 10 in 72% yield. To complete the macrolide we were inspired by the C2-symmetric nature of the C5,C10-portion of the molecule. A “capping” strategy via a highly E,E-stereoselective cross metathesis (CM)/-ring closing metathesis (RCM) sequence between 10 and C2-symmetric fragment 5 using Grubbs 2nd generation catalyst 1130 was sought.31 While the high reactivity of diol 5a and acetonide 5b resulted in complex reaction mixtures, bis-silylated olefin 5c delivered a contracted 11-membered ring lactone together with self-immolative CM products as a result of the steric hindrance imposed by the two bulky silyl groups flanking the double bonds in 5c (data not shown).27,32 We envisioned that the TBS group in 5d could prevent the coordination of the Ru-catalyst (11) and thus the CM reaction on its neighbouring double bond. As expected, the cross-metathesis between ester 10 and mono-TBS protected diol (5d) afforded a mixture of regioisomers in a ca. 1:1 ratio but, unfortunately, the RCM of this mixture of regioisomers did not afford the desired products but rather truncated lactones and dimeric 5d signalling the lack of reactivity of the olefin vicinal to the silyl group even in the case of a RCM event.33–35 Fortunately, after per-silylation of the mixture, a productive RCM was observed to give lactone 12 as a single isomer in 76% overall yield. The usefulness of Grubbs-type catalysts for the assembly of molecular complexity in challenging settings is once again portrayed here.36–40
While this approach secured access to the desired compound in four additional steps (namely, deprotection of the primary OTBDPS,41,42 oxidation of the primary alcohol with Dess–Martin periodinane followed by a Julia–Kocienski olefination with sulfone 6 and final TBAF deprotection of the remaining silyl groups), further chemical edition of the macrolide as well as access to a sufficient supply of the natural product itself for subsequent biological evaluation were hampered by the lack of scalability of this strategy. As a result, we embarked in the design of an alternative synthetic route to access key intermediate 12 in substantial amounts. The new approach encoded the previously described fragments (3–6) but aimed at a more productive assembly strategy both in terms of scalability and synthetic efficiency. In short, the southern and western part of the molecule exemplified by fragments 4 and 5, respectively, would be coupled first via cross metathesis reaction prior to the intermolecular esterification with northern fragment 3. Ring-closing metathesis would then construct the 15-membered lactone converging with our previously reported strategy. tert-Butyl ester 13 and 1,5-diene 5d were coupled via cross metathesis reaction in the presence of Grubb's 2nd generation catalyst 11 in excellent yield with complete E-stereocontrol (Scheme 1B). Silylation of the secondary alcohol under standard conditions followed by hydrolysis of the tert-butyl ester moiety produced fragment 14 in 78% yield. Intermolecular esterification with fragment 3 under the conditions previously reported for fragment 4 produced ester 15 in 84% yield. The strategic potential of RCM was again illustrated here with the use of 5 mol% of 11 to efficiently close the 15-membered lactone with complete E-stereoselectivity converging with our original synthesis. This approach afforded iriomoteolide-3a 1 in only 7 steps from the corresponding building blocks.19 7,8-O-Isopropylidene derivative 2 was prepared by treatment of 1 with 2,2-dimethoxypropane in the presence of pyridinium p-toluene sulfonate. Multi-mg quantities of these two molecules could be prepared with the new route, thus demonstrating the advantage of this strategy compared to the originally reported one.
Preparation of a focused library of iriomoteolide analogues
Next, a structural diversification campaign18,43,44 was designed to map the macrocyclic framework of the natural lead by straightforward modifications of the 3–6 subunits (Scheme 2). Our first goal was to explore the conformational landscape of the macrolide, as conformation is known to play a key role in the biological activity of drugs entitling medium or large ring sizes.19,45–48 The southern portion of the molecule, in particular the substitution pattern at C3 was studied first. This goal seemed easy to attain as it would only require the replacement of a single building block (i.e.4) in the entire sequence. Interestingly, we noticed that the 2JH2a/H2b and 3JH2a/H3 determined in our synthetic sample of 1 differed in ca. 4 and 2 Hz from the ones reported for the isolated sample, respectively.2 Since a sample of the natural material could not be made available to us, we focused on the preparation of the C3-epimer of 1 first. Applying the strategy described in Scheme 1B,19 persilylated derivative 16 could be prepared in multi-mg scale. To our surprise, and in striking contrast to the smooth removal of the silicon protecting groups observed en route to compound 1, the reaction of 16 in the presence of TBAF delivered two products. A careful purification of the reaction mixture revealed the formation of the desired irio-C3epimer 17 together with a ring-enlarged analogue 18 formed via transesterification between the C15–OH and the lactone moiety in 63 and 15% yields respectively (Scheme 2A). Both compounds displayed a completely different array of spectroscopic signals in 1H and 13C NMR compared to parent compound 1, qualitatively reflecting the population of a different conformational space elicited by the change in configuration at a single stereocenter (see ESI†). Although ring enlargements are not uncommon in macrolides under basic conditions,49–51 the result summarized in Scheme 2A was completely unexpected given the clean desilylation observed to produce 1 in both the original as well as the more recently developed total syntheses campaigns summarized in Scheme 1.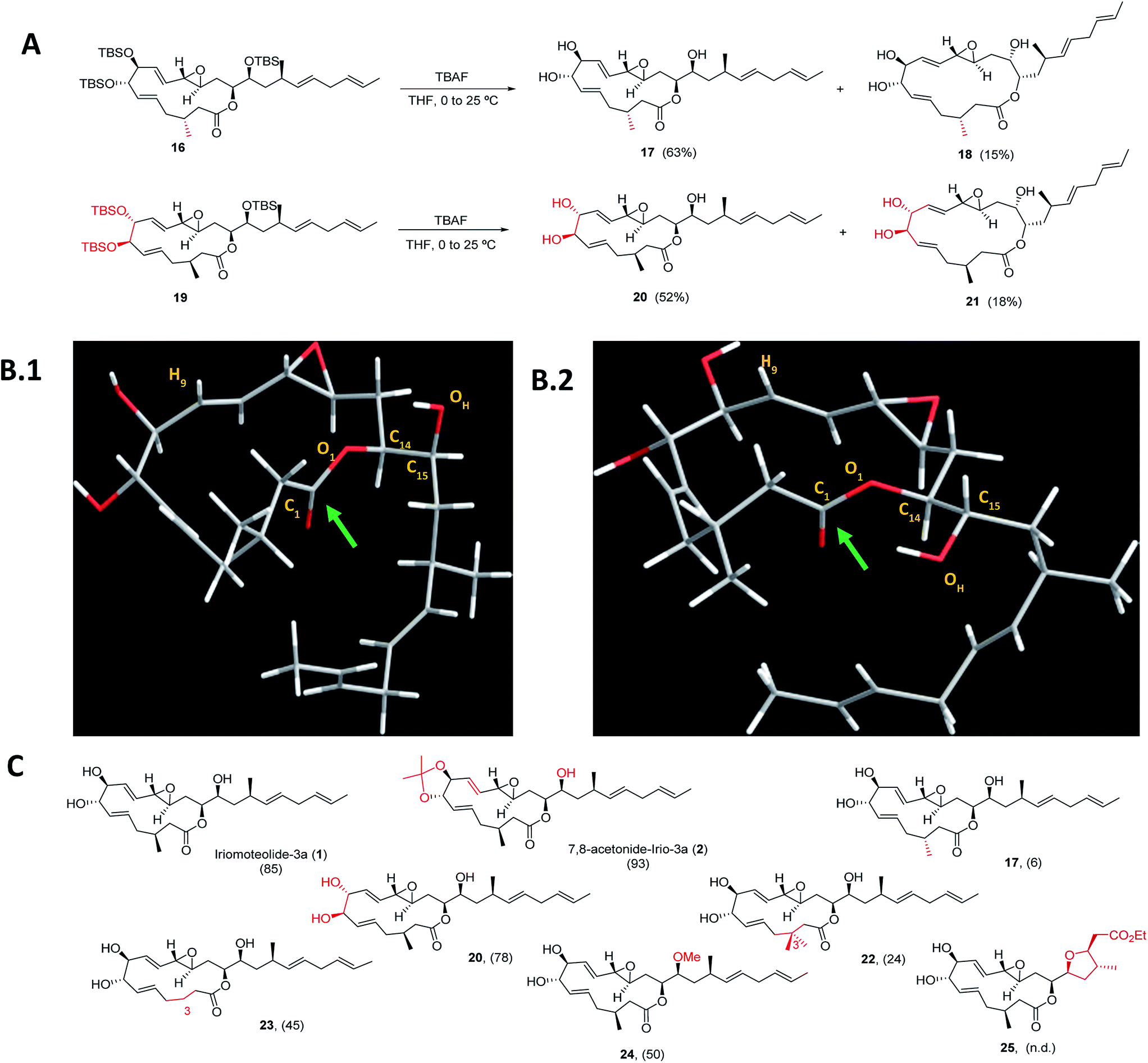 | ||
| Scheme 2 (A) Final deprotection towards C3-irio-3a epimer (17) and C7,C8-irio-3a diastereoisomer (20) in the presence of TBAF. (B) Blueprint of the most stable conformers for iriomoteolide-3a (1, B.1) and its C3-epimer (17, B.2). 1: O1–C14–C15–OH = −63°. 17: O1–C14–C15–OH = +62°. (C) Synthetic iriomoteolide derivatives using the general strategy reported in Scheme 1B and preliminary evaluation of their anti-proliferative activity. Values in brackets represent the % of growth inhibition of Daudi cells at 10 μM concentration of the compound. N.d.: not determined. | ||
Molecular dynamics simulations with the MMFF94 force field in the CHARMM program52,53 with implicit solvent were performed on 1 and 17 to find a qualitative explanation for this highly distinct behaviour (see molecular dynamics simulations section in the ESI† for further details).19 Two sets of clusters were found using a 10 kcal mol−1 maximum energy difference with the corresponding global minima as capping criteria. This restriction afforded 62 clusters for the natural product (1) and 31 for the C3-epimer (17).
For the two molecules investigated, the lower energy clusters (clusters 1–5 with a max. ΔE = 2 kcal mol−1 and comprising more than 1/3 of the total number of conformers) present a rather distinct conformational blueprint as depicted in Scheme 2B (irio-3a 1: Scheme 2B.1 and C3-epimer analogue 17: Scheme 2B.2). Especially remarkable is the influence that the methyl group at position C3 exerts on the dihedral angle O1–C14–C15–OH, which is shifted from −63° in the natural compound to +62° in 17. In turn, the π* C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O orbital in 17 is favorably aligned with the OH group explaining the observed transesterification reaction. The relative disposition of the lateral chain compared to the macrocycle is also strongly affected, with the dihedral C1–O1–C14–C15 going from −5° in 1 to −44° in the synthetic analogue 17. This effect also translates into a macrocycle “folding” in 17 compared to the relatively flat structure observed in the parent compound 1. Interestingly, these clusters present structural features that are in congruence with the recorded NMR data of these compounds in different organic solvents. Thus, the resonance for H9 appears at higher field for compound 17 than for the natural molecule, which reflects a shielding effect in the former case as this hydrogen is pointing towards the interior of the macrocyclic cavity.7 To further explore the conformational landscape of the macrocycle, the (7R,8R)-diastereoisomeric precursor 19 was easily obtained from D-tartaric acid following the route detailed in Scheme 1B. As in the case of the C3-epimer, formation of a one-carbon atom expanded lactone 21 could also be observed, together with the desired C7,C8-irio-3a diastereoisomer 20 in the final desilylation step (Scheme 2A). Aiming to further investigate the effect of C3 in the conformation and biological activity of these molecules, a derivative of 1 bearing two methyl groups at C3 (22) and a second one devoid of any substituent at this position (23) were also prepared following the general strategy outlined in Scheme 1B. An additional criterion in our design involved the modification of the hydrogen bond network of the molecule. We investigated whether the hydroxy group at C15 plays a key role in the activity of these macrolides, and thus the methyl ether derivative at this position (24) was also prepared. Along the same lines, a tetrahydrofuran derivative 25 was included as a result of the cyclization of a vinyl ester truncated analogue. The vinyl epoxide was conserved throughout the analogues as it has been recognized as a critical motif for covalent binding to protein targets in related macrolides.54 Finally, it is important to note that the abovementioned chemical edition could not have been probed by standard derivatization of the natural product (Scheme 2C).
O orbital in 17 is favorably aligned with the OH group explaining the observed transesterification reaction. The relative disposition of the lateral chain compared to the macrocycle is also strongly affected, with the dihedral C1–O1–C14–C15 going from −5° in 1 to −44° in the synthetic analogue 17. This effect also translates into a macrocycle “folding” in 17 compared to the relatively flat structure observed in the parent compound 1. Interestingly, these clusters present structural features that are in congruence with the recorded NMR data of these compounds in different organic solvents. Thus, the resonance for H9 appears at higher field for compound 17 than for the natural molecule, which reflects a shielding effect in the former case as this hydrogen is pointing towards the interior of the macrocyclic cavity.7 To further explore the conformational landscape of the macrocycle, the (7R,8R)-diastereoisomeric precursor 19 was easily obtained from D-tartaric acid following the route detailed in Scheme 1B. As in the case of the C3-epimer, formation of a one-carbon atom expanded lactone 21 could also be observed, together with the desired C7,C8-irio-3a diastereoisomer 20 in the final desilylation step (Scheme 2A). Aiming to further investigate the effect of C3 in the conformation and biological activity of these molecules, a derivative of 1 bearing two methyl groups at C3 (22) and a second one devoid of any substituent at this position (23) were also prepared following the general strategy outlined in Scheme 1B. An additional criterion in our design involved the modification of the hydrogen bond network of the molecule. We investigated whether the hydroxy group at C15 plays a key role in the activity of these macrolides, and thus the methyl ether derivative at this position (24) was also prepared. Along the same lines, a tetrahydrofuran derivative 25 was included as a result of the cyclization of a vinyl ester truncated analogue. The vinyl epoxide was conserved throughout the analogues as it has been recognized as a critical motif for covalent binding to protein targets in related macrolides.54 Finally, it is important to note that the abovementioned chemical edition could not have been probed by standard derivatization of the natural product (Scheme 2C).
As a way to compare the performance of the synthetic derivatives with that of the parent compound in an already established setting, a preliminary evaluation of cell viability on two different human lymphoma cell lines (Daudi and Akata) was carried out. Iriomoteolide-3a 1, C7,C8-acetonide 2 and derivatives 17, 20, 22–24 were tested at two different concentrations 10 and 2.5 μM (% of growth inhibition at 10 μM on Daudi are shown in Scheme 2C. For complete data set, see Fig. S1 in the ESI†). All compounds showed anti-proliferative activity at 10 μM except for C3-epimer 17, which was completely inactive. These results confirm the striking influence that a single-point mutation in the stereochemical composition of a molecule can have in its conformational landscape, and subsequently, in its biological activity.19,45–48 Along the same lines, both the addition and the elimination of a methyl group at C3 (analogues 22 and 23) resulted in a reduced anti-proliferative activity. Although the methylation of the hydroxy group at C15 (24) was tolerated, the cytotoxicity of this derivative substantially decreased compared to the parent compound, indicating that the presence of a hydrogen bond donor at this position is key for the activity.
Characterization of cellular targets
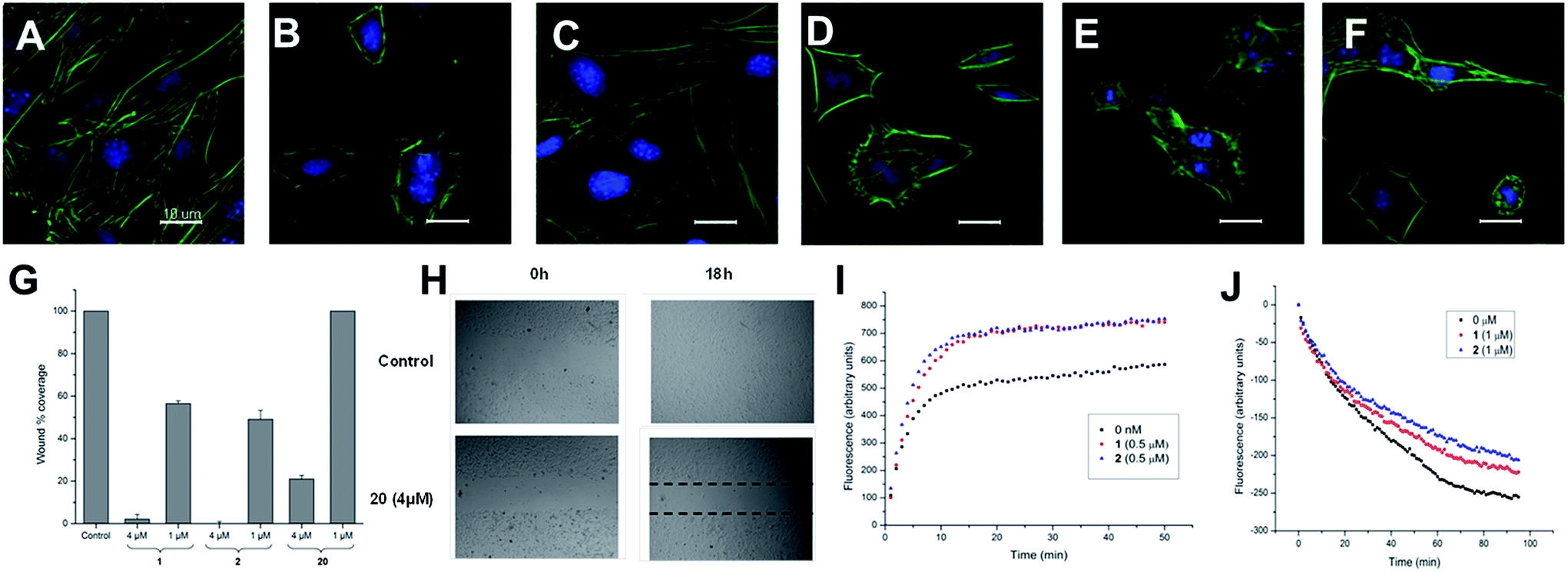 | ||
| Fig. 1 (A–F) Effect of iriomoteolides-3a on mouse fibroblast: fluorescence micrographs (40×) of the actin cytoskeleton of NIH/3T3 cells. Actin cytoskeleton is stained with FITC-phalloidin (green) and nuclei with 2-(4-amidinophenyl)-6-indolecarbamidine hydrochloride (DAPI) (blue). Cell viability >80% according to a fluorimetric assay with resazurin. (A) Control cells. (B) Cells incubated with irio-3a (1) at 250 nM for 2 hours and (C) after 8 hours showing complete recovery of their normal morphology and microfilament organization. (D and E) Cells incubated with 1 at 1 and 4 μM for 2 hours. (F) Cells incubated with synthetic analogue 20 (10 μM) for 8 hours. (G and H) Determination of the effects of irio-3a (1) and synthetic analogues 2 and 20 on cell migration by means of the scratch-wound assay on NIH/3T3 cells. (G) Percentage of surface area filled 18 hours after treatment with 1, 2 and 20 at two different concentrations (4 and 1 μM). The results are presented as mean ± SEM of three independent experiments. (H) Scratch wound carried out by a pipette tip and recolonized after 18 h under the effect of compound 20 (4 μM). (I and J) Effect of iriomoteolide 3a (1) and acetonide (2) on actin polymerization in vitro. (I) Effect of 1 and 2 (500 nM) on the copolymerization of actin and pyrenyl labeled G-actin. Actin/pyrenyl-labeled actin (4 μM) was incubated with 1 (red), 2 (blue) or without either (black). Polymerization was started by the addition of inducing salts (50 mM KCl, 2 mM MgCl2, 1 mM ATP) and drugs solved in DMSO at time = 0 (less than 5% DMSO). (J) Effects of 1 (1 μM, red) and 2 (1 μM, blue) on the rate of pyrenyl F-actin (2.3 μM, final concentration) depolymerization. Pyrenyl-labeled actin F-actin (23 μM) was diluted to 2.3 μM and the drugs were added. The samples were mixed to give a 1 μM final concentration of drug. | ||
These two sets of data indicate that iriomoteolides are likely to stabilize F-actin filaments, enhancing, as a consequence actin polymerization (Fig. 1I) and inhibiting F-actin depolymerization (Fig. 1J). Interestingly, amphidinolide H, which is isolated from the Amphidinium species as iriomoteolide-3a (1), has also been classified as an actin stabilizer whereas its effect is due to covalent binding to actin through its epoxide moiety.54,79 In contrast, no covalent adduct could be detected by MALDI-TOF upon incubation of 1 with G-actin, indicating that iriomoteolides, despite their epoxide moiety, do not form covalent adducts with actin, in line with the reversible effects seen in Fig. 1C (see Fig. S6 in the ESI†).
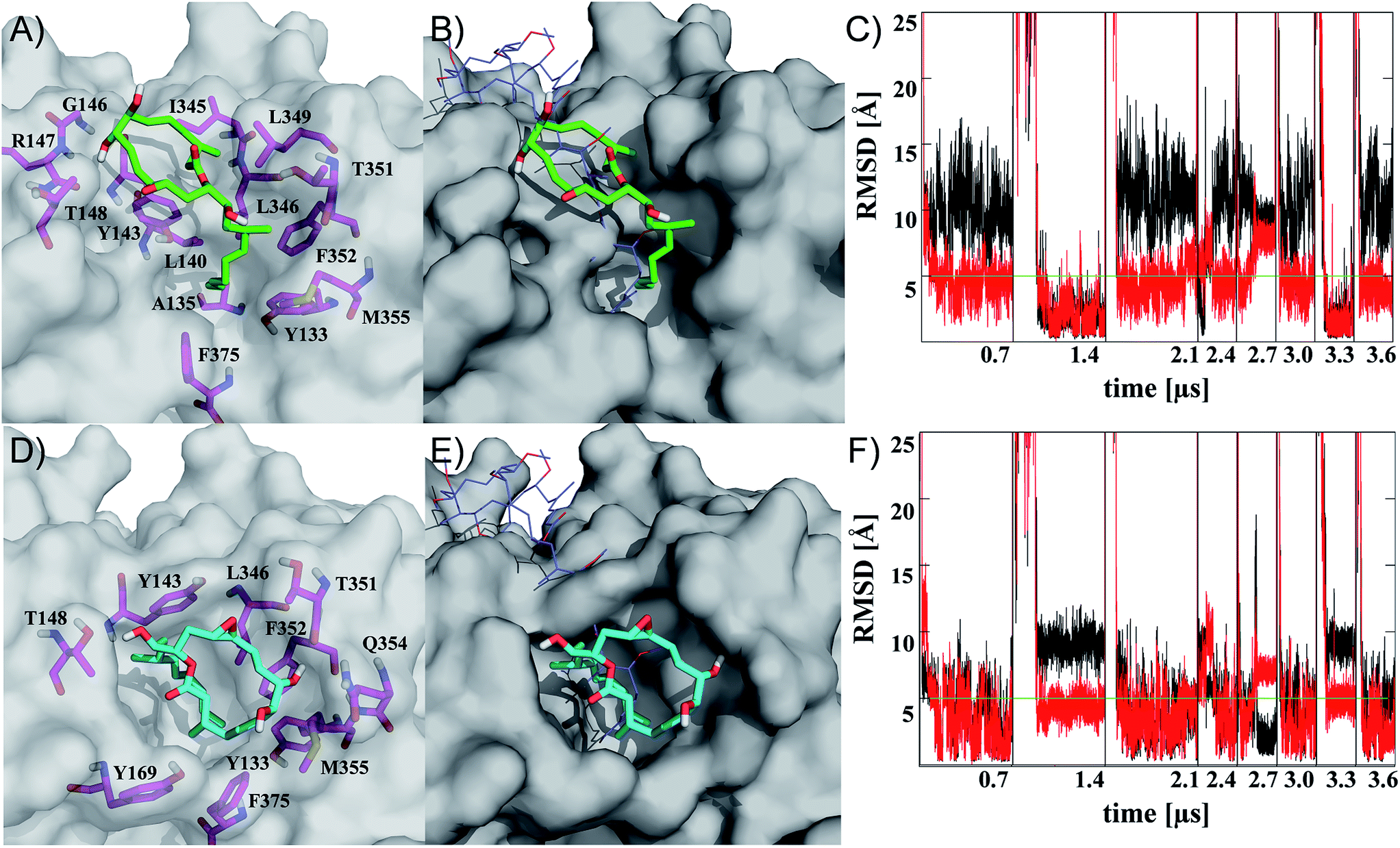 | ||
| Fig. 2 Representatives of the two most populated clusters sampled upon binding of iriomoteolide-3a (1) at the barbed end of monomeric G-actin (A, B cluster 1; D, E cluster 2). Binding modes are shown with residues within 4.5 Å highlighted (A, D magenta sticks) and overlayed with crystal structure coordinates from reidispongiolide A (B, E blue lines, PDB: 2asm). For the representative of cluster 1 (green sticks), a binding mode similar to natural products like reidispongiolide A is observed (B). The hydrophobic tail is inserted into the barbed and the macrocycle is located along the hydrophobic patch (A, B). The representative of cluster 2 (cyan sticks) shows a similar arrangement of the hydrophobic tail while, contrary to cluster 1, the macrolide resides on top of the tail section and not along the hydrophobic patch of the barbed end periphery (D, E). Time series of RMSD from the representative structures of clusters 1 (C) and 2 (F) for ring (black) or tail (red) moiety. Individual trajectories are concatenated in the time series, and starting points are marked (black vertical lines). Images rendered in Pymol http://www.pymol.org. | ||
Representatives of these two clusters are shown in Fig. 2 (for complete set of simulations, heat map analysis of contact interactions and clustering results, see Fig. S7–10 in the ESI†). The two binding modes are similar in their relative arrangement of the hydrophobic tail of iriomoteolide-3a (1). The tail is inserted into the barbed end interacting with mainly hydrophobic residues (Fig. 2A, D). The conformation of the tail section for the concatenated trajectories is similar for both binding modes (Fig. 2C, F red line). In contrast, the macrocycle is either extended from the tail in an orientation more similar to other natural products like reidispongiolide A,73 kabiramide C87 and trisoxasoles88 (cluster 1, Fig. 2A, B) or is located on top of the tail section (cluster 2, Fig. 2D, E). The time series of RMSD from the cluster representatives confirm the structural heterogeneity for the macrolide ring in both putative binding modes (Fig. 2C, F black) while the tail moiety remains structurally more homogeneous (Fig. 2C, F red).
Additionally, although the macrolide conformation for basin 2 has been sampled in more simulations than basin 1, once basin 1 was reached it was structurally more stable than basin 2 (Fig. 2C). The high sampling rate of the iriomoteolide-3a tail moiety in close contact with barbed end residues within the combined trajectories would be consistent with previous reports that suggested that interactions of actin binding macrolide tails at the barbed end of actin affect polymerization.61,73,89 The different binding sites of iriomoteolide-3a (within the barbed end) and amphidinolide H, which covalently binds to actin within subdomain IV,54 could explain the non-covalent interaction between iriomoteolide-3a and actin (see Fig. S6 in the ESI†). The simulation results thus indicate that iriomoteolide-3a (1) likely binds to the barbed end of monomeric actin although the macrocycle might be able to adopt several conformations.
Conclusions
Iriomoteolides are a novel family of cytotoxic macrolides, whose highly-convergent, catalysis-based synthesis has been accomplished attesting the usefulness of alkene cross- and ring-closing metathesis reactions.In this study, conformation has been unraveled as a key feature to enable the design of iriomoteolide-3a analogues able to at least equal the activity of the natural product, thus highlighting the power behind “total-synthesis-driven” campaigns for in depth studies of complex biologically active molecules. Small molecules that target the actin cytoskeleton have long been recognized as valuable molecular probes due to the essential role of actin in a wide range of cellular functions. In this work, we have established actin as a cellular target of these marine secondary metabolites. At submicromolar concentrations, iriomoteolide-3a and analogues are able to elicit profound morphological changes in the actin network in cells. Iriomoteolides appear to stabilize F-actin filaments by enhancing G-actin polymerization and inhibiting F-actin depolymerization. Contrary to amphidinolide H, these marine toxins interact with actin in a reversible, non-covalent fashion. Multiple molecular dynamics simulations with explicit solvent suggest that iriomoteolide 3a preferentially binds within the barbed end of G-actin. Our studies reveal the potential of iriomoteolide-3a and its non-natural analogues as new versatile chemical probes that can interfere with the highly complex dynamics of the actin cytoskeleton in order to understand cell motility processes including cell invasion and division.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Dr Navid Nadal for a generous loan of lymphoma cell lines and Dr Sabrina Sonda for helpful discussions and technical assistance. We also thank the Swiss National Science Foundation (200021_134686/1) the Legerlotz Stiftung (R. C. and M. H.) and the Forschungskredit of the University of Zürich (to K. L.) for financial support.Notes and references
- A. J. Calado and O. Moes, Phycologia, 2005, 44, 112–119 CrossRef.
- K. Oguchi, M. Tsuda, R. Iwamoto, Y. Okamoto, J. Kobayashi, E. Fukushi, J. Kawabata, T. Ozawa, A. Masuda, Y. Kitaya and K. Omasa, J. Org. Chem., 2008, 73, 1567–1570 CrossRef CAS PubMed.
- B. M. Trost, P. E. Harrington, J. D. Chisholm and S. T. Wrobleski, J. Am. Chem. Soc., 2005, 127, 13598–13610 CrossRef CAS PubMed.
- B. M. Trost, S. T. Wrobleski, J. D. Chisholm, P. E. Harrington and M. Jung, J. Am. Chem. Soc., 2005, 127, 13589–13597 CrossRef CAS PubMed.
- B. M. Trost and P. E. Harrington, J. Am. Chem. Soc., 2004, 126, 5028–5029 CrossRef CAS PubMed.
- H. W. Lam and G. Pattenden, Angew. Chem., Int. Ed. Engl., 2002, 41, 508–511 CrossRef CAS PubMed.
- P. Va and W. R. Roush, J. Am. Chem. Soc., 2006, 128, 15960–15961 CrossRef CAS PubMed.
- D. R. Williams and K. G. Meyer, J. Am. Chem. Soc., 2001, 123, 765–766 CrossRef CAS PubMed.
- A. K. Ghosh and C. Liu, J. Am. Chem. Soc., 2003, 125, 2374–2375 CrossRef CAS PubMed.
- A. Furstner, Isr. J. Chem., 2011, 51, 329–345 CrossRef.
- L. Fang, J. Yang and F. Yang, Org. Lett., 2010, 12, 3124–3127 CrossRef CAS PubMed.
- A. K. Ghosh and H. Yuan, Org. Lett., 2010, 12, 3120–3123 CrossRef CAS PubMed.
- J. Xie, Y. Ma and D. A. Horne, Chem. Commun., 2010, 46, 4770–4772 RSC.
- G. Zhao, Z. Q. Ye and T. Y. Gao, Tetrahedron, 2011, 67, 5979–5989 CrossRef.
- C. R. Reddy, G. Dharmapuri and N. N. Rao, Org. Lett., 2009, 11, 5730–5733 CrossRef CAS PubMed.
- Y. Zhang, L. Deng and G. Zhao, Org. Biomol. Chem., 2011, 9, 5730–5733 Search PubMed.
- R. Cribiu, C. Jager and C. Nevado, Angew. Chem., Int. Ed. Engl., 2009, 48, 8780–8783 CrossRef CAS PubMed.
- R. M. Wilson and S. J. Danishefsky, J. Org. Chem., 2006, 71, 8329–8351 CrossRef CAS PubMed.
- E. Moulin, V. Zoete, S. Barluenga, M. Karplus and N. Winssinger, J. Am. Chem. Soc., 2005, 127, 6999–7004 CrossRef CAS PubMed.
- D. A. Evans, J. Bartroli and T. L. Shih, J. Am. Chem. Soc., 1981, 103, 2127–2129 CrossRef CAS.
- X. T. Zhou and R. G. Carter, Chem. Commun., 2004, 2138–2140 RSC.
- K. Ohtsuki, K. Matsuo, T. Yoshikawa, C. Moriya, K. Tomita-Yokotani, K. Shishido and M. Shindo, Org. Lett., 2008, 10, 1247–1250 CrossRef CAS PubMed.
- P. Perlmutter, W. Selajerern and F. Vounatsos, Org. Biomol. Chem., 2004, 2, 2220–2228 CAS.
- S. G. Nelson, T. J. Peelen and Z. H. Wan, J. Am. Chem. Soc., 1999, 121, 9742–9743 CrossRef CAS.
- S. G. Nelson, Z. H. Wan and M. A. Stan, J. Org. Chem., 2002, 67, 4680–4683 CrossRef CAS PubMed.
- M. A. Robbins, P. N. Devine and T. Oh, Org. Synth., 1999, 76, 101–109 CrossRef CAS.
- S. Michaelis and S. Blechert, Org. Lett., 2005, 7, 5513–5516 CrossRef CAS PubMed.
- Y. C. Hwang and F. W. Fowler, J. Org. Chem., 1985, 50, 2719–2726 CrossRef CAS.
- A. B. Charette, C. Berthelette and D. St-Martin, Tetrahedron Lett., 2001, 42, 6619 CrossRef CAS.
- M. Scholl, S. Ding, C. W. Lee and R. H. Grubbs, Org. Lett., 1999, 1, 953–956 CrossRef CAS PubMed.
- S. Randl and S. Blechert, in Handbook of Metathesis, Wiley-VCH Verlag GmbH, 2003, pp. 151–175, DOI:10.1002/9783527619481.ch16.
- K. J. Quinn, A. K. Isaacs, B. A. DeChristopher, S. C. Szklarz and R. A. Arvary, Org. Lett., 2005, 7, 1243–1245 CrossRef CAS PubMed.
- A. Furstner and K. Langemann, J. Org. Chem., 1996, 61, 3942–3943 CrossRef PubMed.
- A. Furstner and K. Langemann, Synthesis, 1997, 792–803, DOI:10.1055/S-1997-4472.
- A. Furstner, O. R. Thiel and C. W. Lehmann, Organometallics, 2002, 21, 331–335 CrossRef.
- A. Deiters and S. F. Martin, Chem. Rev., 2004, 104, 2199–2238 CrossRef CAS PubMed.
- J. Cossy, S. Arseniyadis and C. Meyer, in Metathesis in Natural Product Synthesis, Wiley-VCH Verlag GmbH & Co. KGaA, 2010, DOI:10.1002/9783527629626.ch1.
- A. Furstner, Chem. Commun., 2011, 47, 6505–6511 RSC.
- K. C. Nicolaou, P. G. Bulger and D. Sarlah, Angew. Chem., Int. Ed., 2005, 44, 4490–4527 CrossRef CAS PubMed.
- A. H. Hoveyda and A. R. Zhugralin, Nature, 2007, 450, 243–251 CrossRef CAS PubMed.
- R. Lira and W. R. Roush, Org. Lett., 2007, 9, 533–536 CrossRef CAS PubMed.
- W. J. Zhang and M. J. Robins, Tetrahedron Lett., 1992, 33, 1177–1180 CrossRef CAS.
- S. Basu, B. Ellinger, S. Rizzo, C. Deraeve, M. Schürmann, H. Preut, H.-D. Arndt and H. Waldmann, Proc. Natl. Acad. Sci., 2011, 108, 6805–6810 CrossRef CAS PubMed.
- R. W. Huigens III, K. C. Morrison, R. W. Hicklin, T. A. Flood Jr, M. F. Richter and P. J. Hergenrother, Nat. Chem., 2013, 5, 195–202 CrossRef PubMed.
- D. L. Boger, T. M. Ramsey, H. Cai, S. T. Hoehn and J. Stubbe, J. Am. Chem. Soc., 1998, 120, 9149–9158 CrossRef CAS.
- R. W. Hoffmann, Angew. Chem., Int. Ed., 1992, 31, 1124–1134 CrossRef.
- R. W. Hoffmann, Angew. Chem., Int. Ed., 2000, 39, 2054–2070 CrossRef PubMed.
- A. Furstner, E. Moulin, C. Nevado, J. Gagnepain, G. Kelter and H. H. Fiebig, Tetrahedron, 2010, 66, 6421–6428 CrossRef.
- E. J. Corey, D. J. Brunelle and K. C. Nicolaou, J. Am. Chem. Soc., 1977, 99, 7359–7360 CrossRef CAS.
- S. E. Denmark and J. M. Muhuhi, J. Am. Chem. Soc., 2010, 132, 11768–11778 CrossRef CAS PubMed.
- F. Sarabia, M. Garcia-Castro and S. Chammaa, Tetrahedron Lett., 2005, 46, 7695–7699 CrossRef CAS.
- B. R. Brooks, C. L. Brooks III, A. D. MacKerell Jr, L. Nilsson, R. J. Petrella, B. Roux, Y. Won, G. Archontis, C. Bartels, S. Boresch, A. Caflisch, L. Caves, Q. Cui, A. R. Dinner, M. Feig, S. Fischer, J. Gao, M. Hodoscek, W. Im, K. Kuczera, T. Lazaridis, J. Ma, V. Ovchinnikov, E. Paci, R. W. Pastor, C. B. Post, J. Z. Pu, M. Schaefer, B. Tidor, R. M. Venable, H. L. Woodcock, X. Wu, W. Yang, D. M. York and M. Karplus, J. Comput. Chem., 2009, 30, 1545–1614 CrossRef CAS PubMed.
- B. R. Brooks, R. E. Bruccoleri, B. D. Olafson, D. J. States, S. Swaminathan and M. Karplus, J. Comput. Chem., 1983, 4, 187–217 CrossRef CAS.
- T. Usui, S. Kazami, N. Dohmae, Y. Mashimo, H. Kondo, M. Tsuda, A. G. Terasaki, K. Ohashi, J. Kobayashi and H. Osada, Chem. Biol., 2004, 11, 1269–1277 CrossRef CAS PubMed.
- K. C. Nicolaou, F. Roschangar and D. Vourloumis, Angew. Chem., Int. Ed. Engl., 1998, 37, 2015–2045 Search PubMed.
- A. Schmidt and M. N. Hall, Annu. Rev. Cell Dev. Biol., 1998, 14, 305–338 CrossRef CAS PubMed.
- T. D. Pollard, L. Blanchoin and R. D. Mullins, Annu. Rev. Biophys. Biomol. Struct., 2000, 29, 545–576 CrossRef CAS PubMed.
- K. S. Yeung and I. Paterson, Angew. Chem., Int. Ed. Engl., 2002, 41, 4632–4653 CrossRef CAS PubMed.
- G. Fenteany and S. Zhu, Curr. Top. Med. Chem., 2003, 3, 593–616 CrossRef CAS PubMed.
- M. Kita and H. Kigoshi, Nat. Prod. Rep., 2015, 32, 534–542 RSC.
- R. Ueoka, A. R. Uria, S. Reiter, T. Mori, P. Karbaum, E. E. Peters, E. J. N. Helfrich, B. I. Morinaka, M. Gugger, H. Takeyama, S. Matsunaga and J. Piel, Nat. Chem. Biol., 2015, 11, 705–712 CrossRef CAS PubMed.
- T. Usui, Biosci., Biotechnol., Biochem., 2007, 71, 300–308 CrossRef CAS PubMed.
- A. Furstner, D. De Souza, L. Turet, M. D. B. Fenster, L. Parra-Rapado, C. Wirtz, R. Mynott and C. W. Lehmann, Chem.–Eur. J., 2007, 13, 115–134 CrossRef PubMed.
- A. Furstner, D. Kirk, M. B. Fenster, C. Aissa, D. De Souza, C. Nevado, T. Tuttle, W. Thiel and O. Muller, Chem.–Eur. J., 2007, 13, 135–149 CrossRef PubMed.
- A. Furstner, T. Nagano, C. Muller, G. Seidel and O. Muller, Chem.–Eur. J., 2007, 13, 1452–1462 CrossRef PubMed.
- D. W. Goddette and C. Frieden, J. Biol. Chem., 1986, 261, 15974–15980 CAS.
- I. Spector, N. R. Shochet, Y. Kashman and A. Groweiss, Science, 1983, 219, 493–495 CAS.
- I. Spector, N. R. Shochet, D. Blasberger and Y. Kashman, Cell Motil. Cytoskeleton, 1989, 13, 127–144 CrossRef CAS PubMed.
- M. R. Bubb, I. Spector, A. D. Bershadsky and E. D. Korn, J. Biol. Chem., 1995, 270, 3463–3466 CrossRef CAS PubMed.
- A. V. Statsuk, R. Bai, J. L. Baryza, V. A. Verma, E. Hamel, P. A. Wender and S. A. Kozmin, Nat. Chem. Biol., 2005, 1, 383–388 CrossRef CAS PubMed.
- S. A. Rizvi, D. S. Courson, V. A. Keller, R. S. Rock and S. A. Kozmin, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 4088–4092 CrossRef CAS PubMed.
- S. A. Rizvi, S. Liu, Z. Chen, C. Skau, M. Pytynia, D. R. Kovar, S. J. Chmura and S. A. Kozmin, J. Am. Chem. Soc., 2010, 132, 7288–7290 CrossRef CAS PubMed.
- J. S. Allingham, A. Zampella, M. V. D'Auria and I. Rayment, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 14527–14532 CrossRef CAS PubMed.
- M. R. Bubb, A. M. Senderowicz, E. A. Sausville, K. L. Duncan and E. D. Korn, J. Biol. Chem., 1994, 269, 14869–14871 CAS.
- M. R. Bubb, I. Spector, B. B. Beyer and K. M. Fosen, J. Biol. Chem., 2000, 275, 5163–5170 CrossRef CAS PubMed.
- J. A. Barden, M. Miki, B. D. Hambly and C. G. Dos Remedios, Eur. J. Biochem., 1987, 162, 583–588 CrossRef CAS PubMed.
- J. Wehland, M. Osborn and K. Weber, Proc. Natl. Acad. Sci. U. S. A., 1977, 74, 5613–5617 CrossRef CAS.
- P. M. Sorensen, R. E. Iacob, M. Fritzsche, J. R. Engen, W. M. Brieher, G. Charras and U. S. Eggert, ACS Chem. Biol., 2012, 7, 1502–1508 CrossRef CAS PubMed.
- J. Kobayashi, H. Shigemori, M. Ishibashi, T. Yamasu, H. Hirota and T. Sasaki, J. Org. Chem., 1991, 56, 5221–5224 CrossRef CAS.
- C. Trigili, B. Pera, M. Barbazanges, J. Cossy, C. Meyer, O. Pineda, C. Rodriguez-Escrich, F. Urpi, J. Vilarrasa, J. F. Diaz and I. Barasoain, ChemBioChem, 2011, 12, 1027–1030 CrossRef CAS PubMed.
- M. A. Jordan and L. Wilson, Curr. Opin. Cell Biol., 1998, 10, 123–130 CrossRef CAS PubMed.
- C. Hayot, O. Debeir, P. Van Ham, M. Van Damme, R. Kiss and C. Decaestecker, Toxicol. Appl. Pharmacol., 2006, 211, 30–40 CrossRef CAS PubMed.
- K. Micoine and A. Furstner, J. Am. Chem. Soc., 2010, 132, 14064–14066 CrossRef CAS PubMed.
- J. H. Ju, S. R. Rajski, S. K. Lim, J. W. Seo, N. R. Peters, F. M. Hoffmann and B. Shen, J. Am. Chem. Soc., 2009, 131, 1370–1371 CrossRef CAS PubMed.
- C. Gaul, J. T. Njardarson, D. Shan, D. C. Dorn, K. D. Wu, W. P. Tong, X. Y. Huang, M. A. S. Moore and S. J. Danishefsky, J. Am. Chem. Soc., 2004, 126, 11326–11337 CrossRef CAS PubMed.
- K. Micoine, P. Persich, J. Llaveria, M. H. Lam, A. Maderna, F. Loganzo and A. Furstner, Chem. - Eur. J., 2013, 19, 7370–7383 CrossRef CAS PubMed.
- J. Tanaka, Y. L. Yan, J. Choi, J. Bai, V. A. Klenchin, I. Rayment and G. Marriott, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 13851–13856 CrossRef CAS PubMed.
- V. A. Klenchin, J. S. Allingham, R. King, J. Tanaka, G. Marriott and I. Rayment, Nat. Struct. Biol., 2003, 10, 1058–1063 CrossRef CAS PubMed.
- R. D. Perrins, G. Cecere, I. Paterson and G. Marriott, Chem. Biol., 2008, 15, 287–294 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Details for all biological assays as well as experimental and computational procedures and spectroscopic data. See DOI: 10.1039/c7sc04286h |
| ‡ Present Addresses: Discovery and Development Technologies, Merck KGaA, Frankfurter Str. 250, 64293 Darmstadt, Germany. |
| § Present Address: Biosynth AG, Sankt Gallen, Switzerland. |
| ¶ Present Address: Novartis, Fabrikstrasse 2, 4056 Basel, Switzerland. |
| This journal is © The Royal Society of Chemistry 2018 |