
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Gold-catalyzed stereoselective dearomatization/metal-free aerobic oxidation: access to 3-substituted indolines/oxindoles†
Kai
Liu
,
Guangyang
Xu
and
Jiangtao
Sun
 *
*
Jiangsu Key Laboratory of Advanced Catalytic Materials & Technology, School of Petrochemical Engineering, Changzhou University, Changzhou 213164, P. R. China. E-mail: jtsun08@gmail.com; jtsun@cczu.edu.cn
First published on 6th November 2017
Abstract
An unprecedented dearomatization of indoles with diazoesters has been developed via cationic gold(I) catalysis. The functionalization selectively occurs at the C3-position to deliver methylene indole derivatives in good yields with excellent Z-selectivity, demonstrating unusual reactivity and selectivity compared with other noble metal catalysis. Importantly, simply followed by silica gel adsorption, an unprecedented metal-free aerobic oxidation occurs for indoles bearing N-electron donating substituents, providing a novel and efficient approach towards 3-substituted indolin-2-ones with a newly formed quaternary stereocenter in excellent stereoselectivity. Notably, these processes afford direct and selective access to a variety of valuable intermediates from abundant feedstock chemicals.
Introduction
Transition-metal-catalyzed carbene transfer from diazo compounds represents a powerful tool in modern organic synthesis, allowing the rapid assembly of a range of valuable structures which cannot be easily achieved by other methodologies.1 Particularly, different metal-carbenes often exhibit distinct reactivity and selectivity toward the same reaction precursors, which increases molecular complexity. A good example is the reaction of diazoesters with 2,3-nonsubstituted indoles, which often leads to two principal products, namely cyclopropane derivatives (Scheme 1a)2 and formal C(sp2)–H insertion products (Scheme 1b).3–7 Indeed, for the addition reaction, the use of rhodium,3 copper,4 iron5 and palladium6 complexes selectively afforded C3-alkylation products. In contrast, C2-alkylation has been observed for 1H-indoles upon exposure to a ruthenium catalyst.7 Additionally, annulation8 and the N–H insertion reaction9 have also been reported. | ||
| Scheme 1 Functionalization of indoles by metal-carbene transfer from diazo compounds: previous reports and our discovery. | ||
On the other hand, recent literature disclosed that the reactivity and chemoselectivity of gold-carbene highly depended on the electronic and steric properties of the ancillary ligand to the gold center,10,11 as well as the choice of counterion.12 Variation of the gold catalysts has recently been shown to even allow the formation of gold(III) intermediates from gold(I) precursors.13 In 2014, Shi and co-workers described a ligand-controlled gold-catalyzed addition of arenes to α-aryl diazoesters.14 They mentioned one example of indole C3-alkylation in the presence of an electron-deficient phosphite-gold catalyst, which exhibited similar chemo- and site-selectivity to rhodium and copper catalysis (Scheme 1b). However, we recently found that N-heterocyclic carbene (NHC) gold complexes displayed inverse chemoselectivity to phosphite gold catalysts even for the same substrates.15b Thus, we envisioned that, when exposed to different gold catalysts, the reaction of indoles with diazo compounds would probably result in distinct reaction pathways. In continuation with our interests in gold-carbene chemistry,15 as anticipated but not expected, herein we report the unprecedented stereoselective dearomatization of indoles with diazoesters under cationic gold(I) catalysis (Scheme 1c). Furthermore, when N-electron-donating substituted indoles are utilized, an unprecedented metal-free aerobic oxidation occurs after the initial dearomatization.
Results and discussion
At the outset, we employed N-boc indole 1a, N-benzyl indole 2a and phenyl diazoacetate 3a as model substrates to investigate the reaction (Table 1). The use of Ph3PAuCl/AgSbF6 (5 mol%) in dichloromethane at room temperature afforded C3-insertion product 5a in 70% yield (entry 1), while JohnPhosAuCl/AgSbF6 gave 16% yield of 5a (entry 2). When t-BuXPhosAuCl was used, 10% yield of 4a was obtained although 5a was still the major product (entry 3). Not surprisingly, the electron-deficient phosphite gold complex only gave 5a as a single product (entry 4). Gratifyingly, the use of IPrAuCl/AgSbF6 provided 4a in 70% yield (entry 5). We therefore started to survey other NHC gold complexes as well so as to try different counterions. To our delight, the yield of 4a was improved to 79% by IPrAu(PhCN)SbF6 (entry 6), and was further increased to 81% by IPrAu(PhCN)BArF16 (entry 7). A screen of the solvents revealed chloroform was the best one (entries 8 to 11), providing 4a in 87% yield (entry 9), and no significant side products were detected. The structure of 4a was confirmed with NMR spectroscopy and was further identified using X-ray analysis.17 Notably, when N-benzyl indole 2a was subjected to this reaction, the corresponding 3-methyleneindoline was not obtained and 3-substituted indolin-2-one 6a was obtained instead in 77% yield (entry 12, see ESI† for details), indicating that an aerobic oxidation occurred. Variation of the solvents did not improve the reaction (entries 13 to 15).|
|
|||||
|---|---|---|---|---|---|
| Entry | Indole | Catalyst | Solvent | Yieldb (%) | |
| 4a(5a) | 6a | ||||
| a Reaction conditions: 1a or 2a (0.2 mmol) and 3a (0.3 mmol) in 2 mL of the solvent were added to a solution of 5 mol% gold catalyst in 2 mL of the solvent via a syringe pump under argon for 2 h. The mixture was stirred at rt for another 2 h. For 6a, silica gel (5 g) adsorption of crude products was performed and was kept in air for 12 h at rt. b Isolated yields. (ArO) = (2,4-di-tert-butylphenyl). | |||||
| 1 | 1a | Ph3PAuCl/AgSbF6 | CH2Cl2 | 0(70) | — |
| 2 | 1a | JohnPhosAuCl/AgSbF6 | CH2Cl2 | 0(16) | — |
| 3 | 1a | t-BuXPhosAuCl/AgSbF6 | CH2Cl2 | 10(61) | — |
| 4 | 1a | (ArO)3PAuCl/AgSbF6 | CH2Cl2 | 0(75) | — |
| 5 | 1a | IPrAuCl/AgSbF6 | CH2Cl2 | 70(13) | — |
| 6 | 1a | IPrAu(PhCN)SbF6 | CH2Cl2 | 79(10) | — |
| 7 | 1a | IPrAu(PhCN)BArF | CH2Cl2 | 81(8) | — |
| 8 | 1a | IPrAu(PhCN)BArF | DCE | 81(9) | — |
| 9 | 1a | IPrAu(PhCN)BAr F | CHCl 3 | 87(<5) | — |
| 10 | 1a | IPrAu(PhCN)BArF | Toluene | 70(15) | — |
| 11 | 1a | IPrAu(PhCN)BArF | THF | 25(<5) | — |
| 12 | 2a | IPrAu(PhCN)BAr F | CHCl 3 | — | 77 |
| 13 | 2a | IPrAu(PhCN)BArF | CH2Cl2 | — | 70 |
| 14 | 2a | IPrAu(PhCN)BArF | DCE | — | 65 |
| 15 | 2a | IPrAu(PhCN)BArF | THF | — | 50 |

We then started to explore the substrate scope for the first dearomatization (Scheme 2). The reaction of indole 1a with a variety of diazoesters was firstly examined. It was observed that methyl phenyl diazoacetate gave the corresponding product in better yield than other esters (4a to 4d). Aromatic diazoesters with different substituents were employed, providing the corresponding 3-methyleneindolines in moderate to excellent yields (4e to 4k). In general, electron-deficient aromatic diazoesters furnished the products (4e to 4i) in higher yields than electron-rich substrates (4j to 4k). Next, the scope of indoles was investigated. Gratifyingly, C5-, C6- and C7-substituted N-boc indoles bearing either electron-withdrawing or electron-donating groups were all tolerated, furnishing the corresponding indolines in moderate to excellent yields (4l to 4p). Furthermore, different N-substituted indoles were also examined. The protecting groups such as tosyl (Ts), benzyloxycarbonyl (Cbz), benzoyl (Bz) and acetyl (Ac) were all amenable to the reaction and the corresponding products were obtained in acceptable yields (4q to 4t). This protocol was also amenable to pyrroles. The desired dearomatization products were isolated in good yields (4u to 4x). The structure of 4x was further confirmed by single-crystal X-ray crystallography.17 It should be noted that all of the methylene derivatives were isolated in the single Z-configuration.
 | ||
| Scheme 2 Substrate scope and reaction conditions: to a solution of 5 mol% of IPrAu(PhCN)BArF in CHCl3 (2 mL) a solution of 1 (0.2 mmol) and 3 (0.3 mmol) in 2 mL CHCl3 was added at rt for 2 h via a syringe pump under argon. The resulting mixture was stirred at rt for another 2 h. Isolated yields have been listed. 4 equiv. of the diazo substrate were used for the preparation of 4g. | ||
Next, we investigated the substrate scope of the tandem reaction towards the formation of 3-substituted indolin-2-ones (Scheme 3). Generally, the reaction of 2a with aromatic diazoesters either bearing electron-donating or electron-withdrawing substituents proceeded smoothly to afford the corresponding products in moderate to good yields (6a to 6h). A longer oxidation time (24 h) is needed for 6e to 6h. Afterwards, indoles bearing various substituents were examined. The use of N-benzyl 5-methoxy and 5-bromo indoles provided 6i and 6j in 81% and 74% yield, while 6-chloro and 6-methyl delivered the corresponding products in 75% and 78% yield, respectively. 7-Methoxy N-benzyl indole was also examined, and 6m was isolated in 53% yield. Finally, N-methyl and N-phenyl indoles were tested and the corresponding products (6n and 6o) were obtained in moderate yields. The structure of 6a and 6l was confirmed by X-ray analysis.
 | ||
| Scheme 3 Tandem reaction of the dearomatization and aerobic oxidation. Reaction conditions: IPrAu(PhCN)BArF (5 mol%), 2 (0.2 mmol) and 3 (0.3 mmol) in 4 mL CHCl3 at rt for 4 h. Then SiO2 (5 g) adsorption in air at rt for 12 to 24 h. Isolated yields have been listed. | ||
Deuterium labeling and control experiments were conducted to understand the reaction mechanism (Scheme 4). First, 4a cannot be converted to 5a under standard reaction conditions (Scheme 4a), which ruled out the possibility of preferential formation of 5 followed by isomerization and vice versa. Next, the reaction of D-1a with 3a yielded D-4a. The high deuterium incorporation at the 2-position might indicate that a 1,2-hydrogen shift is likely involved in the reaction (Scheme 4b). Since there is significant loss of deuterium, we suspected that the reaction may be interfered with by adventitious water present in the reaction mixture. Thus, a control reaction with 3 equivalents of D2O was run. Indeed, significant deuterium incorporation into the product was observed (Scheme 4c), suggesting that there is D/H exchange with the adventitious proton source during the reaction’s progress. A study on the kinetic isotope effect (KIE) using intermolecular competition between 1a and D-1a indicated a KIE value of 3.35 (Scheme 4d), which is consistent with the 1,2-hydrogen shift being the rate-determining step. Furthermore, compound 7 was separately prepared and subjected to standard reaction conditions. Unfortunately, no reaction was observed. The chemical incompetence of 7 rules out its possible role as an intermediate in this process (Scheme 4e).
 | ||
| Scheme 4 Mechanistic studies on the dearomatization reaction. | ||
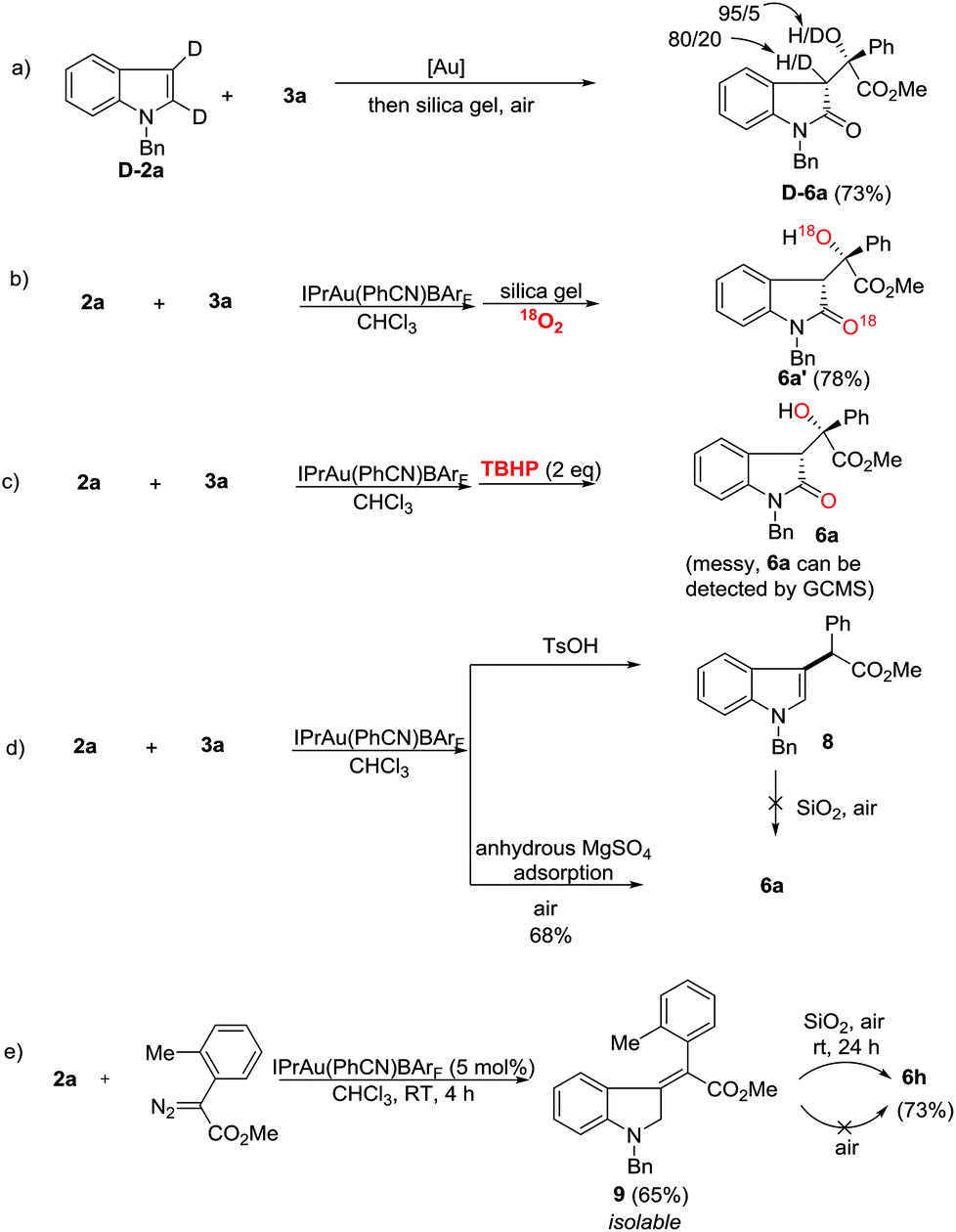
Next, mechanistic studies for the aerobic oxidation were carried out (Scheme 5). First, the reaction of D-2a with 3a gave D-6a in 73% yield with a low ratio of deuterium labeling (Scheme 5a). Moreover, the 18O-labeled product 6a′ was obtained in 78% yield under an 18O2 atmosphere, indicating that the oxygen atom in 6 came from the air (Scheme 5b). Using TBHP (tert-butyl hydroperoxide) instead of air as an oxidant, the reaction was messy although 6a could still be detected by GC/MS (Scheme 5c). To determine the role of the silica gel, p-toluene sulfonic acid was added and the C-3 alkylation product 8 was obtained in high yield (Scheme 5d). Moreover, by replacing the silica gel with anhydrous MgSO4, 6a was isolated in 68% yield upon exposure to air for 12 h (Scheme 5d). Therefore, the acidic property of the silica gel may not be critically important for this transformation. Its role is presumably to facilitate the aerobic oxidation by increasing the contact surface of the olefin intermediate with oxygen. Just recently, López and co-workers reported the gold-catalyzed formal insertion of aryl diazoesters into ferrocene to generate functionalized metallocenes.18 They also found that the silica gel promoted the aerobic oxidation leading to tertiary-substituted ferrocenyl alcohols. In that case, they believed the aerobic oxidation might proceed through initial electron-transfer from iron to molecular oxygen, which triggered the radical sequence to give the target product. Thus, to determine the reactive intermediate for this oxidation process, the C3-alkylation product 8 was subjected to silica gel in air, however, no reaction occurred (Scheme 5d). Gratifyingly, although the reaction intermediates are extremely unstable, we successfully isolated intermediate 9 after carefully checking all of the reactions listed in Scheme 3. Furthermore, the treatment of 9 with silica gel in air provided 6h in 73% yield (Scheme 5e). This result indicated that gold catalysts did not work during the aerobic oxidation. Without silica gel adsorption, 6h cannot be obtained either. Different from López’s report, this aerobic oxidation does not involve metal participation.
 | ||
| Scheme 5 Mechanistic studies on the aerobic oxidation. | ||
Until now, several reaction mechanisms have been proposed for indole functionalization with diazo compounds in the presence of rhodium, copper or other metal complexes.2–8 Clearly, this gold-catalyzed dearomatization is quite different from these known processes.19,20 Although the exact reaction mechanism is not clear at this moment, plausible ones have been proposed (Scheme 6). In view of the unique formation of Z-olefins, the carboxylate group may assist the olefin selectivity. Partially analogous to Fox’s description of rhodium-catalyzed C-3 alkylation of indoles with diazoesters3d and density functional theory (DFT) calculations reported by Xie et al.,3f the reaction of 3a with the cationic gold catalyst first generates gold carbene species IA or IB, which is followed by nucleophilic attack with indole to produce ylide IIIvia transition state II. Then this ylide intermediate would undergo a 1,2-hydrogen shift to give the final dearomatization intermediate IV, together with catalyst regeneration. The assistance of the ester carbonyl group explains the Z-configuration of the observed product. Next, silica gel-assisted aerobic oxidation occurs. The reaction of IV with molecular oxygen generates intermediate Vvia the Schenck ene reaction.21 Owing to the high oxidation ability of the peroxide motif and the electron-rich nature of the indole ring, subsequent internal epoxidation can rapidly take place via either VI or VII. Finally, semi-pinacol rearrangement of VI22 or rearrangement of the amino epoxide motif in VII23 leads to the observed amide 6. Considering the high reactivity of peroxide V, the epoxidation step occurs on the same face of the indole plane once it is formed (C–C bond rotation is thus discouraged). In this scenario, the stereochemistry integrity determines the high diastereoselectivity observed in the final product.
 | ||
| Scheme 6 Proposed reaction mechanisms. | ||
Conclusions
In summary, we have developed an unprecedented gold-catalyzed stereoselective dearomatization of indoles with diazoesters, providing 3-methyleneindolines in good to excellent yields with unique Z-configuration. Moreover, when N-donating substituent indoles were subjected to the reaction, a tandem reaction sequence occurred including the initial dearomatization and a sequential metal-free aerobic oxidation to produce 3-substituted indolin-2-ones. As a result, molecular oxygen has been successfully inlaid into the final structure. Notably, the use of the cationic gold(I) catalyst IPrAu(PhCN)BArF is crucial to the whole process.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the National Natural Science Foundation of China (21572024), the Natural Science Foundation of Jiangsu Province (BK20151184), the Jiangsu Key Laboratory of Advanced Catalytic Materials and Technology (BM2012110) and the Advanced Catalysis & Green Manufacturing Collaborative Innovation Center for their financial support.Notes and references
- For selected leading reviews, see: (a) H. M. L. Davies and S. J. Hedley, Chem. Soc. Rev., 2007, 36, 1109 RSC; (b) M. P. Doyle, R. Duffy, M. Ratnikov and L. Zhou, Chem. Rev., 2010, 110, 704 CrossRef CAS PubMed; (c) X. Guo and W. Hu, Acc. Chem. Res., 2013, 46, 2427 CrossRef CAS PubMed; (d) Y. Xia, Y. Zhang and J. Wang, ACS Catal., 2013, 3, 2586 CrossRef CAS; (e) X. Xu and M. P. Doyle, Acc. Chem. Res., 2014, 47, 1396 CrossRef CAS PubMed; (f) A. Ford, H. Miel, A. Ring, C. N. Slattery, A. R. Maguire and M. A. McKervey, Chem. Rev., 2015, 115, 9981 CrossRef CAS PubMed.
- (a) F. Gnad, M. Poleschak and O. Reiser, Tetrahedron Lett., 2004, 45, 4277 CrossRef CAS; (b) S. J. Hedley, D. L. Ventura, P. M. Dominiak, C. L. Nygren and H. M. L. Davies, J. Org. Chem., 2006, 71, 5349 CrossRef CAS PubMed; (c) M. Delgado-Rebollo, A. Prieto and P. J. Pérez, ChemCatChem, 2014, 6, 2047 CrossRef CAS and reference therein; (d) V. Lehner, H. M. L. Davies and O. Reiser, Org. Lett., 2017, 19, 4722 CrossRef CAS PubMed.
- (a) J. L. Wood, B. M. Stoltz, H.-J. Dietrich, D. A. Pflum and D. T. Petsch, J. Am. Chem. Soc., 1997, 119, 9641 CrossRef CAS; (b) R. Gibe and M. A. Kerr, J. Org. Chem., 2002, 67, 6247 CrossRef CAS PubMed; (c) Y. Lian and H. M. L. Davies, Org. Lett., 2010, 12, 924 CrossRef CAS PubMed; (d) A. DeAngelis, V. W. Shurtleff, O. Dmitrenko and J. M. Fox, J. Am. Chem. Soc., 2011, 133, 1650 CrossRef CAS PubMed; (e) Y. Lian and H. M. L. Davies, Org. Lett., 2012, 14, 1934 CrossRef CAS PubMed; (f) Q. Xie, X.-S. Song, D. Qu, L.-P. Guo and Z.-Z. Xie, Organometallics, 2015, 34, 3112 CrossRef CAS.
- M. B. Johansen and M. A. Kerr, Org. Lett., 2010, 12, 4956 CrossRef CAS PubMed.
- Y. Cai, S.-F. Zhu, G.-P. Wang and Q.-L. Zhou, Adv. Synth. Catal., 2011, 353, 2939 CrossRef CAS.
- X. Gao, B. Wu, W.-X. Huang, M.-W. Chen and Y.-G. Zhou, Angew. Chem., Int. Ed., 2015, 54, 11956 CrossRef CAS PubMed.
- W.-W. Chan, S.-H. Yeung, Z. Zhou, A. S. C. Chan and W.-Y. Yu, Org. Lett., 2010, 12, 604 CrossRef CAS PubMed.
- For selected examples, see: (a) Y. Lian and H. M. L. Davies, J. Am. Chem. Soc., 2010, 132, 440 CrossRef CAS PubMed; (b) C. Jing, Q.-Q. Cheng, Y. Deng, H. Arman and M. P. Doyle, Org. Lett., 2016, 18, 4550 CrossRef CAS PubMed; (c) M. Li, X. Guo, W. Jin, Q. Zheng, S. Liu and W. Hu, Chem. Commun., 2016, 52, 2736 RSC; (d) L. Jiang, W. Jin and W. Hu, ACS Catal., 2016, 6, 6146 CrossRef CAS; for intramolecular reactions, (e) A. R. Reddy, F. Hao, K. Wu, C.-Y. Zhou and C.-M. Che, Angew. Chem., Int. Ed., 2016, 55, 1810 CrossRef CAS PubMed; (f) M. J. James, P. O′Brien, R. J. K. Taylor and W. P. Unsworth, Angew. Chem., Int. Ed., 2016, 55, 9671 CrossRef CAS PubMed; (g) H. Xu, Y.-P. Li, Y. Cai, G.-P. Wang, S.-F. Zhu and Q.-L. Zhou, J. Am. Chem. Soc., 2017, 139, 7697 CrossRef CAS PubMed.
- V. Arredondo, S. C. Hiew, E. S. Gutman, I. D. U. A. Premachandra and D. L. Van Vranken, Angew. Chem., Int. Ed., 2017, 56, 4156 CrossRef CAS PubMed.
- For selected reviews, see: (a) A. S. K. Hashmi and G. J. Hutchings, Angew. Chem., Int. Ed., 2006, 45, 7896 CrossRef PubMed; (b) A. Fürstner and P. W. Davies, Angew. Chem., Int. Ed., 2007, 46, 3410 CrossRef PubMed; (c) D. J. Gorin, B. D. Sherry and F. D. Toste, Chem. Rev., 2008, 108, 3351 CrossRef CAS PubMed; (d) A. Corma, A. Leyva-Pérez and M. J. Sabater, Chem. Rev., 2011, 111, 1657 CrossRef CAS PubMed; (e) N. Krause and C. Winter, Chem. Rev., 2011, 111, 1994 CrossRef CAS PubMed; (f) R. Dorel and A. M. Echavarren, Chem. Rev., 2015, 115, 9028 CrossRef CAS PubMed; (g) Z. Zheng, Z. Wang, Y. Wang and L. Zhang, Chem. Soc. Rev., 2016, 45, 4448 RSC.
- For recent representative examples on gold-catalyzed diazo reactions, see: (a) V. V. Pagar, A. M. Jadhav and R.-S. Liu, J. Am. Chem. Soc., 2011, 133, 20728 CrossRef CAS PubMed; (b) G. Lonzi and L. A. López, Adv. Synth. Catal., 2013, 355, 1948 CrossRef CAS; (c) J. F. Briones and H. M. L. Davies, J. Am. Chem. Soc., 2012, 134, 11916 CrossRef CAS PubMed; (d) J. F. Briones and H. M. L. Davies, J. Am. Chem. Soc., 2013, 135, 13314 CrossRef CAS PubMed; (e) S. K. Pawar, C.-D. Wang, S. Bhunia, A. M. Jadhav and R.-S. Liu, Angew. Chem., Int. Ed., 2013, 52, 7559 CrossRef CAS PubMed; (f) Z.-Y. Cao, X. Wang, C. Tan, X.-L. Zhao, J. Zhou and K. Ding, J. Am. Chem. Soc., 2013, 135, 8197 CrossRef CAS PubMed; (g) Z. Yu, B. Ma, M. Chen, H.-H. Wu, L. Liu and J. Zhang, J. Am. Chem. Soc., 2014, 136, 6904 CrossRef CAS PubMed; (h) V. V. Pagar and R.-S. Liu, Angew. Chem., Int. Ed., 2015, 54, 4923 CrossRef CAS PubMed; (i) Z. Yu, H. Qiu, L. Liu and J. Zhang, Chem. Commun., 2016, 52, 2257 RSC; (j) F.-M. Liao, Z.-Y. Cao, J.-S. Yu and J. Zhou, Angew. Chem., Int. Ed., 2017, 56, 2459 CrossRef CAS PubMed; (k) B. Ma, Z. Chu, B. Huang, Z. Liu, L. Liu and J. Zhang, Angew. Chem., Int. Ed., 2017, 56, 2749 CrossRef CAS PubMed; (l) F. Zhao, N. Li, T. Zhang, Z.-Y. Han, S.-W. Luo and L.-Z. Gong, Angew. Chem., Int. Ed., 2017, 56, 3247 CrossRef CAS PubMed; (m) S. K. Pawar, M.-C. Yang, M.-D. Su and R.-S. Liu, Angew. Chem., Int. Ed., 2017, 56, 5035 CrossRef CAS PubMed. For reviews, see: (n) F. Wei, C. Song, Y. Ma, L. Zhou, C.-H. Tung and Z. Xu, Sci. Bull., 2015, 60, 1479 CrossRef CAS; (o) L. Liu and J. Zhang, Chem. Soc. Rev., 2016, 45, 506 RSC; (p) M. R. Fructos, M. M. Díaz-Requejo and P. J. Pérez, Chem. Commun., 2016, 52, 7326 RSC.
- M. Jia and M. Bandini, ACS Catal., 2015, 5, 1638 CrossRef CAS.
- (a) C.-Y. Wu, T. Horibe, C. B. Jacobsen and F. D. Toste, Nature, 2015, 517, 449 CrossRef CAS PubMed; (b) L. Huang, M. Rudolph, F. Rominger and A. S. K. Hashmi, Angew. Chem., Int. Ed., 2016, 55, 4808 CrossRef CAS PubMed; (c) L. Huang, F. Rominger, M. Rudolph and A. S. K. Hashmi, Chem. Commun., 2016, 52, 6435 RSC; (d) S. Witzel, J. Xie, M. Rudolph and A. S. K. Hashmi, Adv. Synth. Catal., 2017, 359, 1522 CrossRef CAS; (e) P. T. Bohan and F. D. Toste, J. Am. Chem. Soc., 2017, 139, 11016 CrossRef CAS PubMed.
- Y. Xi, Y. Su, Z. Yu, B. Dong, E. J. McClain, Y. Lan and X. Shi, Angew. Chem., Int. Ed., 2014, 53, 9817 CrossRef CAS PubMed.
- (a) D. Zhang, G. Xu, D. Ding, C. Zhu, J. Li and J. Sun, Angew. Chem., Int. Ed., 2014, 53, 11070 CrossRef CAS PubMed; (b) G. Xu, C. Zhu, W. Gu, J. Li and J. Sun, Angew. Chem., Int. Ed., 2015, 54, 883 CrossRef CAS PubMed; (c) C. Zhu, G. Xu and J. Sun, Angew. Chem., Int. Ed., 2016, 55, 11867 CrossRef CAS PubMed.
- N. Huguet, D. Lebœuf and A. M. Echavarren, Chem.–Eur. J., 2013, 19, 6581 CrossRef CAS PubMed.
- CCDC 1551068 (4a), 1551070 (4x), 1572912 (6a) and 1572913 (6l).†.
- When we prepared this work, López and co-workers reported a different iron-promoted aerobic oxidation of ferrocene derivatives: E. López, J. Borge and L. A. López, Chem.–Eur. J., 2017, 23, 3091 CrossRef PubMed.
- For DFT studies on gold-catalyzed direct arene C-H bond functionalization, see: (a) Y. Liu, Z. Yu, J. Z. Zhang, L. Liu, F. Xia and J. Zhang, Chem. Sci., 2016, 7, 1988 RSC; (b) M. R. Fructos, M. Besora, A. A. C. Braga, M. M. Díaz-Requejo, F. Maseras and P. J. Pérez, Organometallics, 2017, 36, 172 CrossRef CAS.
- For leading examples on the cyclopropanation reaction, see: (a) M. P. Doyle, J. H. Griffin, V. Bagheri and R. L. Dorow, Organometallics, 1984, 3, 53 CrossRef CAS; (b) M. P. Doyle, V. Bagheri, T. J. Wandless, N. K. Harn, D. A. Brinker, C. T. Eagle and K. L. Loh, J. Am. Chem. Soc., 1990, 112, 1906 CrossRef CAS; (c) M. P. Doyle, Q.-L. Zhou, A. B. Dyatkin and D. A. Ruppar, Tetrahedron Lett., 1995, 36, 7579 CrossRef CAS; (d) D. T. Nowlan III, T. M. Gregg, H. M. L. Davies and D. A. Singleton, J. Am. Chem. Soc., 2003, 125, 15902 CrossRef PubMed and reference therein.
- According to the reviewers’ suggestions, for selected examples, see: (a) M. Prein and W. Adam, Angew. Chem., Int. Ed., 1996, 35, 477 CrossRef CAS; (b) H. M. R. Hoffmann, Angew. Chem., Int. Ed., 1969, 8, 556 CrossRef CAS; (c) B. M. Snider, Acc. Chem. Res., 1980, 13, 426 CrossRef CAS.
- T. J. Snap, Chem. Soc. Rev., 2007, 36, 1823 RSC.
- Y. Zhang, Y. Zou, N. L. Brock, T. Huang, Y. Lan, X. Wang, Z. Deng, Y. Tang and S. Lin, J. Am. Chem. Soc., 2017, 139, 11887 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1551068, 1551070, 1572912 and 1572913. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7sc04086e |
| This journal is © The Royal Society of Chemistry 2018 |