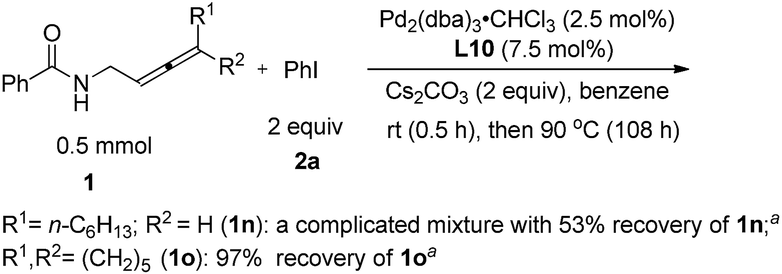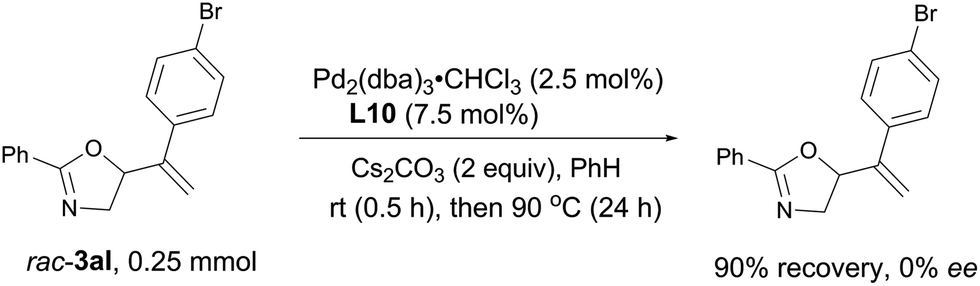DOI:
10.1039/C7SC04079B
(Edge Article)
Chem. Sci., 2018,
9, 1964-1969
A catalytic highly enantioselective allene approach to oxazolines†
Received
17th September 2017
, Accepted 2nd January 2018
First published on 5th January 2018
Abstract
Oxazolines are a very important class of heterocyclic compounds. However, catalytic enantioselective syntheses are very limited. Here, a highly enantioselective palladium-catalyzed coupling-cyclization of readily available N-(buta-2,3-dienyl) amides with aryl or 1-alkenyl iodides has been developed for the asymmetric construction of oxazoline derivatives. Many synthetically useful functional groups are tolerated in this reaction. The absolute configuration of the chiral center in the products has been established by X-ray diffraction study. A model for prediction of the absolute configuration of the chiral center in the products from this cyclic enantioselective nucleophilic allylation has been proposed. The synthetic potentials based on the unique structure of the products formed have also been demonstrated.
Introduction
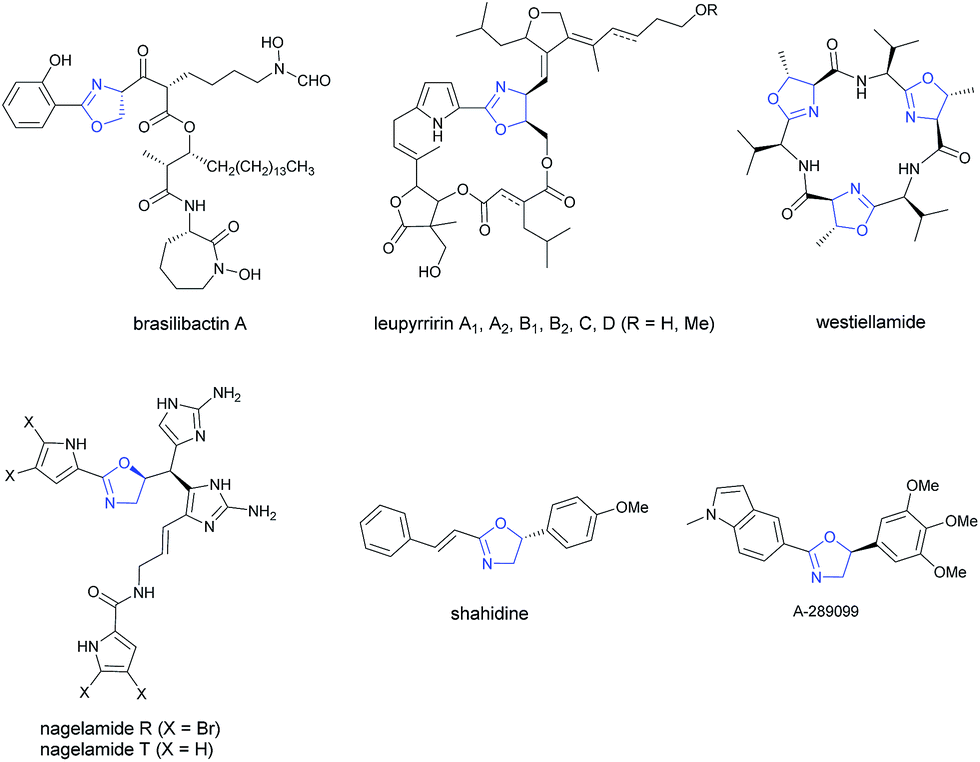
As a very important class of heterocyclic compounds, optically active oxazoline is not only present in many biologically active natural and unnatural compounds1–7 (Scheme 1), but also a very versatile functional group in organic synthesis.8–11
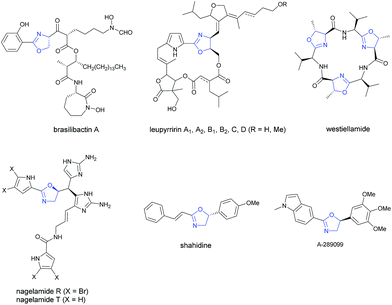 |
| | Scheme 1 Oxazoline natural products and drug. Selected examples of natural products and drugs containing oxazoline with chiral centers at 4- or 5-position. | |
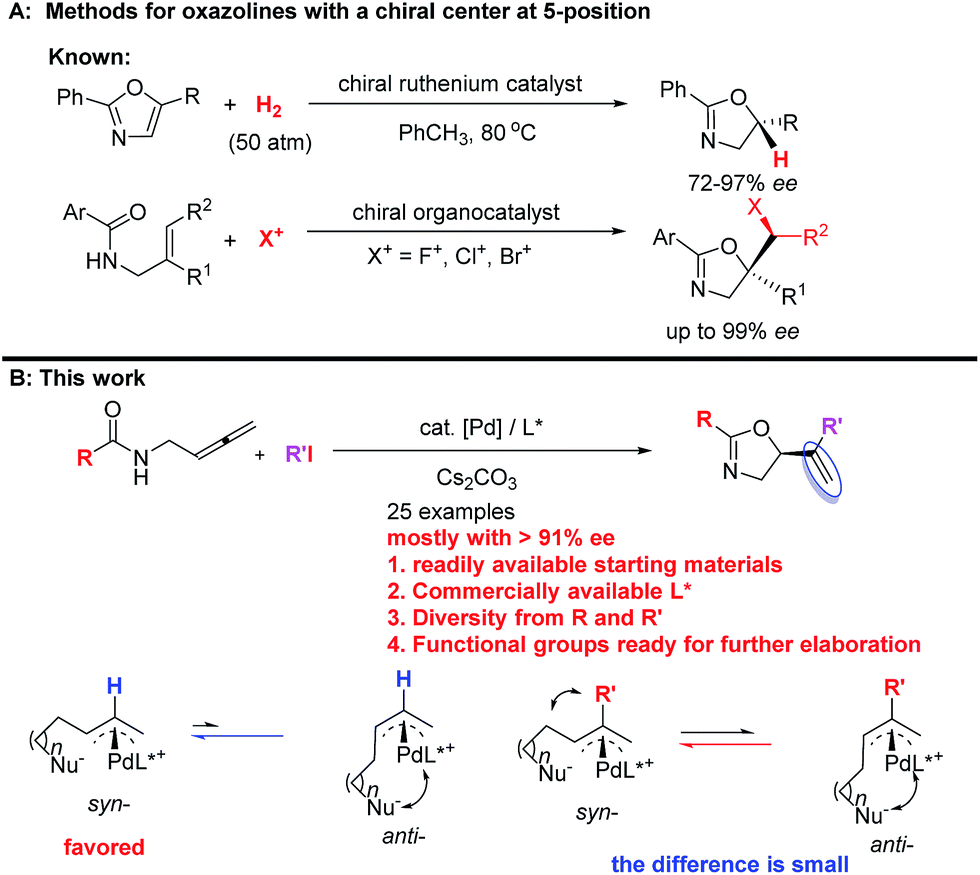
They are usually synthesized from stoichiometric amounts of optically active amino alcohols, which instead are prepared from optically active amino acids. However, catalytic enantioselective approaches to optically active oxazolines are still very limited. Enantioselective alkylation of oxazolines under the catalysis of optically active phase-transfer catalyst12–14 and desymmetrization of N-(1,3-dihydroxy-2-alkyl) amides15 have been developed for the synthesis of optically active oxazolines with a chiral center at the 4-position. Enantioselective reactions of oxazolines,16–18meso-aziridines,19 and 2-isocyanocarbozylates20–27 have been realized by applying different chiral catalysts for the synthesis of optically active oxazolines with chiral centers at both 4- and 5-positions. On the other hand, the ruthenium-catalyzed chemo- and enantio-selective hydrogenation of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond in pre-formed oxazoles with 50 atm of H2
C bond in pre-formed oxazoles with 50 atm of H2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 28 and enantioselective halocyclization of stereodefined N-(2-alkenyl) amides with an organocatalys29–33 have been reported for oxazoline derivatives with a chiral center at the 5-position (Scheme 2A). These two pioneering contributions suffer from limitation of the starting materials and products. Thus, due to the importance of oxazolines, development of new catalytic enantioselective protocols for the efficient preparation of optically active oxazolines from readily available common chemicals is of current interest. On the other hand, we have been interested in developing enantioselective cyclization of allenes with a nucleophilic functionality for the catalytic asymmetric synthesis of hetereocyclic compounds.34–39 Based on our recent development of straightforward method for the synthesis of allenes,41 we envisioned a catalytic enantioselective allene approach to this class of compounds via the in situ generation of 2-substituted π-allylic palladium intermediate and subsequent intramolecular allylic trapping by the oxygen atom in the amide functionality.40–42 Such an enantioselective allylation is challenging due to the presence of the 2-substituent (R′), which has greatly complicated the relative energy of the syn- and anti-π-allylic palladium intermediates (Scheme 2B). Herein, we wish to report our recent results for identifying a chiral ligand for the catalytic highly enantioselective synthesis of oxazoline derivatives with a chiral center at the 5-position. Such a strategy enjoys the diversity from R and R′ of both readily available starting materials and the construction of library for nagelamide R,5 shahidine,6 and the anticancer agents A-289099.7 In addition, the reactive functional groups in R and R′ and the CC bonds in the products make this method synthetically very attractive.
28 and enantioselective halocyclization of stereodefined N-(2-alkenyl) amides with an organocatalys29–33 have been reported for oxazoline derivatives with a chiral center at the 5-position (Scheme 2A). These two pioneering contributions suffer from limitation of the starting materials and products. Thus, due to the importance of oxazolines, development of new catalytic enantioselective protocols for the efficient preparation of optically active oxazolines from readily available common chemicals is of current interest. On the other hand, we have been interested in developing enantioselective cyclization of allenes with a nucleophilic functionality for the catalytic asymmetric synthesis of hetereocyclic compounds.34–39 Based on our recent development of straightforward method for the synthesis of allenes,41 we envisioned a catalytic enantioselective allene approach to this class of compounds via the in situ generation of 2-substituted π-allylic palladium intermediate and subsequent intramolecular allylic trapping by the oxygen atom in the amide functionality.40–42 Such an enantioselective allylation is challenging due to the presence of the 2-substituent (R′), which has greatly complicated the relative energy of the syn- and anti-π-allylic palladium intermediates (Scheme 2B). Herein, we wish to report our recent results for identifying a chiral ligand for the catalytic highly enantioselective synthesis of oxazoline derivatives with a chiral center at the 5-position. Such a strategy enjoys the diversity from R and R′ of both readily available starting materials and the construction of library for nagelamide R,5 shahidine,6 and the anticancer agents A-289099.7 In addition, the reactive functional groups in R and R′ and the CC bonds in the products make this method synthetically very attractive.
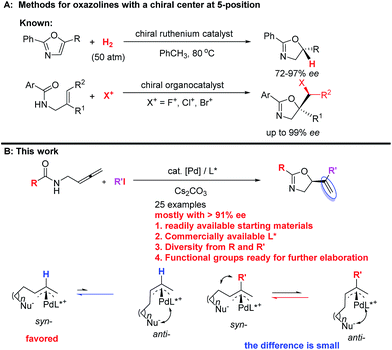 |
| | Scheme 2 Enantioselective approaches to oxazolines. (A) Known methods for oxazolines with a chiral center at 5-position. (B) This work: palladium-catalyzed asymmetric cyclization protocol from readily available common chemicals. | |
Results
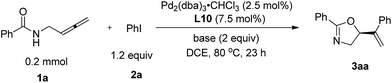
Optimization of the reaction
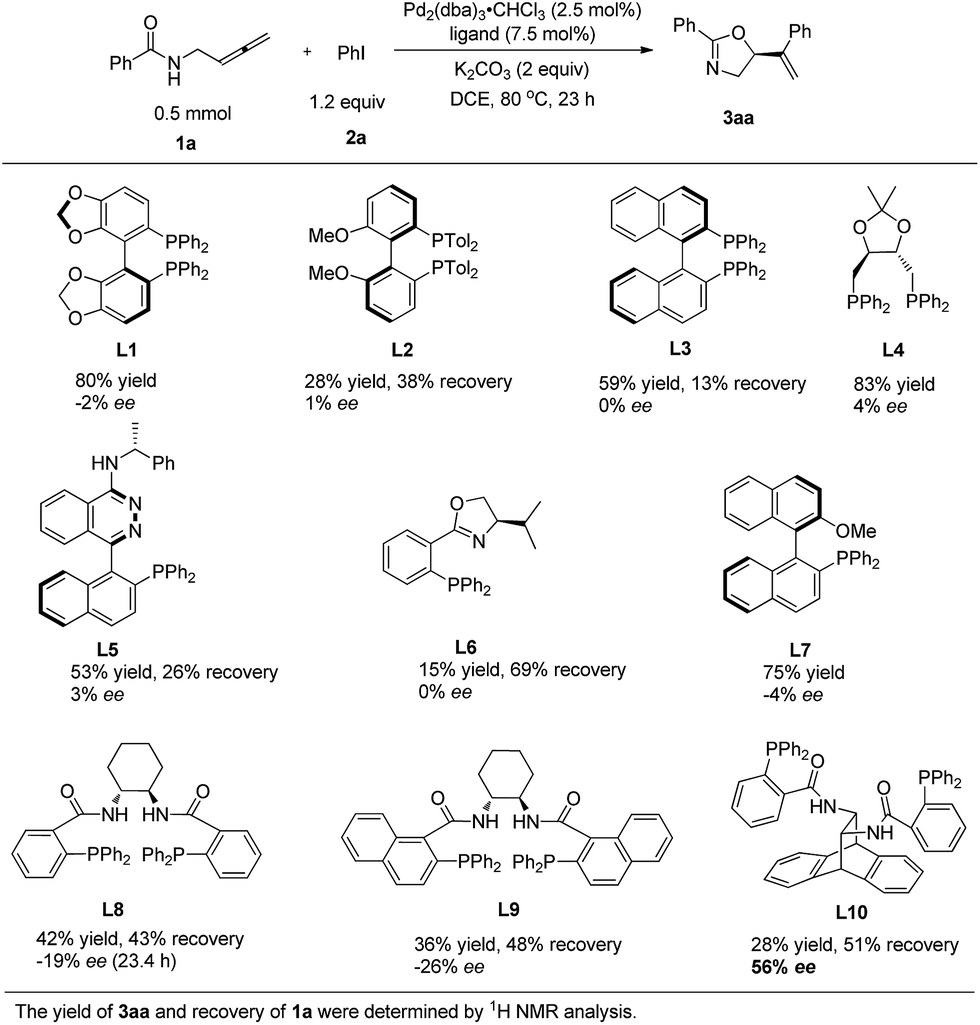
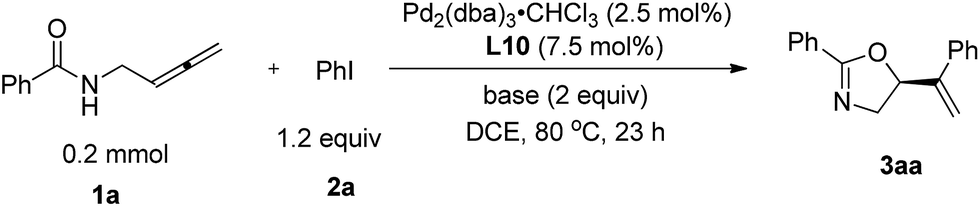
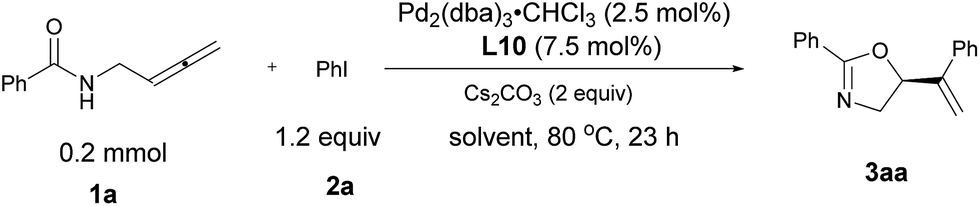
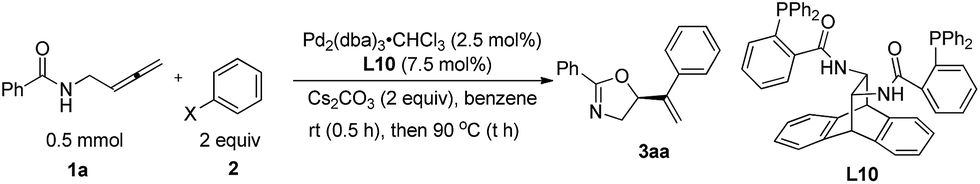
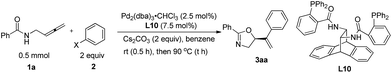
N-(Buta-2,3-dienyl)amide 1a and iodobenzene 2a were selected for our preliminary study on asymmetric cyclizative coupling reaction. However, with the biaryl-based phosphine ligand (S)-SEGPHOS (L1), the desired oxazoline 3aa was afforded in only 2% ee albeit with a high yield. The reaction with other ligands, such as P,P-ligands (L2–L4), P,N-ligands (L5–L6), and P,O-ligand (L7), still yielded almost a racemic product. To our delight, Trost ligands (L8–L10)43,44 performed better in this reaction, and L10 was found to be the best (Scheme 3, For the results with more ligands, see Scheme S1 in the ESI†).
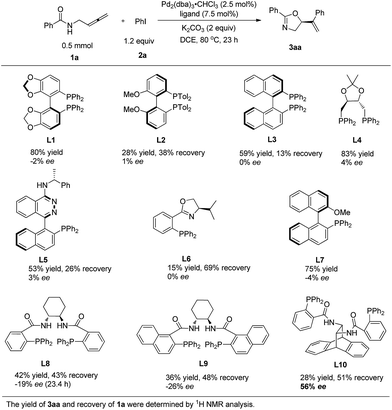 |
| | Scheme 3 The effect of ligands. L10 was found to be the best. | |
Further screening on the effect of bases led to the observation that Cs2CO3 is the best (Table 1, for the results with more bases, see Table S1 in the ESI†).
Table 1 The effect of basea
|
|
| Entry |
Base |
3aa
|
Recovery of 1ab (%) |
| Yieldb (%) |
eec (%) |
|
Unless indicated otherwise, the experiments were performed with 1a (0.2 mmol), 2a (0.24 mmol), Pd2(dba)3·CHCl3 (0.005 mmol), L10 (0.015 mmol), and base (0.4 mmol) in DCE (2 mL) at 80 °C under Ar atmosphere.
The yield and recovery were determined by 1H NMR analysis using 1,3,5-trimethylbenzene as the internal standard.
The ee values were determined by HPLC analysis.
Reaction time: 22.5 h.
n.d. = not determined.
|
| 1d |
Na2CO3 |
4 |
n.d.e |
85 |
| 2 |
K2CO3 |
42 |
51 |
43 |
| 3 |
Rb2CO3 |
67 |
66 |
27 |
|
4
|
Cs
2
CO
3
|
77
|
72
|
6
|
| 5 |
Na3PO4 |
47 |
58 |
37 |
| 6 |
K3PO4 |
66 |
71 |
16 |
| 7 |
LiOH |
36 |
42 |
47 |
| 8 |
NaOH |
64 |
45 |
19 |
| 9 |
KOH |
27 |
62 |
47 |
| 10 |
LiOtBu |
37 |
31 |
25 |
| 11 |
TEA |
3 |
n.d.e |
71 |
The striking effect of solvent
To further improve the enantioselectivity, we investigated the solvent effect (Table 2). Other halogen-containing solvents such as 1,1-dichloroethane and 1,1,1-trichloroethane failed to provide better results (Table 2, entries 1 and 2). Etheric solvents such as DME, THF, MTBE, and dixoane also failed to improve the enantioselectivity (Table 2, entries 3–6). Interestingly, when aromatic solvents were employed, higher enantioselectivity was constantly observed (Table 2, entries 7–11, 79–88% ee) albeit with some recovery of 1a. It is surprising that the reaction with a slightly higher equiv. of 2a and a low concentration in benzene at a higher temperature of 90 °C improved the ee to 94% (compare entry 11 with entry 12) (for more details, see Tables S2–S4 in the ESI†)! Thus, we defined that the reaction of N-(buta-2,3-dienyl)amide 1a (0.5 mmol) with iodobenzene 2a (1.0 mmol) in benzene (6 mL) at 90 °C as the standard conditions.
Table 2 The effect of solventa
|
|
| Entry |
Solvent |
3aa
|
Recovery of 1ab (%) |
| Yieldb (%) |
eec (%) |
|
Unless indicated otherwise, the experiments were performed with 1a (0.2 mmol), 2a (0.24 mmol), Pd2(dba)3·CHCl3 (0.005 mmol), L10 (0.015 mmol), and Cs2CO3 (0.4 mmol) in solvent (2 mL) for 23 h at 80 °C under Ar atmosphere.
The yield and recovery were determined by 1H NMR analysis using 1,3,5-trimethylbenzene as the internal standard.
The ee values were determined by HPLC analysis.
The reaction was conducted at 70 °C.
Reaction time: 22.5 h.
n.d. = not determined.
Reaction time: 21 h.
The reaction was conducted with 1a (0.5 mmol), 2a (1.0 mmol), Pd2(dba)3·CHCl3 (0.0125 mmol), L10 (0.0375 mmol), and Cs2CO3 (1.0 mmol) in benzene (6 mL) for 0.5 h at rt, then 39 h at 90 °C under Ar atmosphere.
Isolated yield.
|
| 1d |
CH3CHCl2 |
60 |
64 |
30 |
| 2e |
CH3CCl3 |
24 |
67 |
64 |
| 3d |
THF |
6 |
n.d.f |
94 |
| 4 |
DME |
80 |
10 |
3 |
| 5 |
MTBE |
59 |
52 |
— |
| 6 |
Dioxane |
54 |
46 |
34 |
| 7 |
PhCH3 |
68 |
79 |
5 |
| 8 |
o-Xylene |
69 |
87 |
25 |
| 9 |
m-Xylene |
66 |
88 |
27 |
| 10 |
p-Xylene |
71 |
87 |
24 |
| 11g |
Benzene |
69 |
85 |
27 |
|
12
|
Benzene
|
83
|
94
|
—
|
Substrate scope
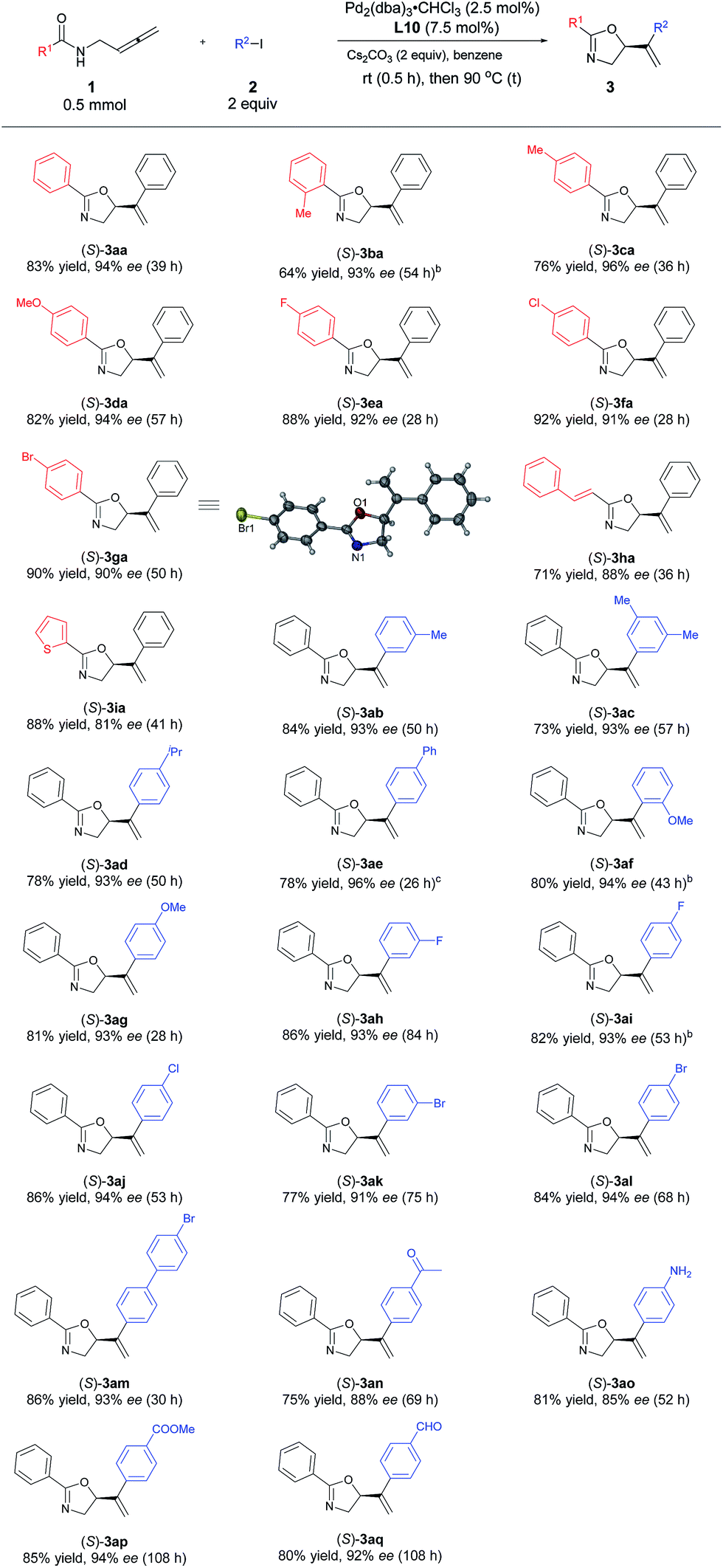
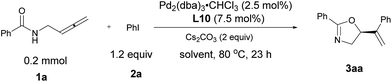
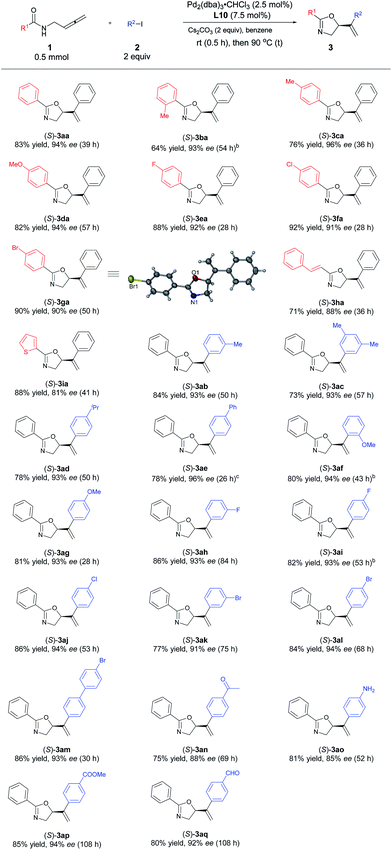
With the optimized protocol in hand, we investigated the generality of this transformation. As shown in Table 3, a variety of optically active oxazoline derivatives could be prepared: N-(buta-2,3-dienyl)amides 1 with R1 being electron-donating substituents, such as 2- or 4-methyl and 4-methoxy groups, performed well with iodobenzene, affording the desired oxazoline products 3ba–3da with 64–82% yields and 93–96% ees. Synthetically attractive halogen groups (F, Cl, Br) in substrates 1 were also tolerated in this reaction with high yields and ee values. The reaction of alkenyl or heteroaryl substituted amides proceeded smoothly to afford the corresponding products 3ha or 3ia in 71% and 88% yields, 88% and 81% ees, respectively. For aryl iodides, different electron-donating groups (Me, iPr, OMe) or Ph (2b–2g) were tolerated with very limited influence on the yields and enantioselectivity (73–84% yields, 93–96% ees). When 4-acylphenyl iodide 2n was subjected to the reaction, the corresponding oxazoline 3an was obtained in 75% yield and 88% ee. Halogen group (F, Cl, Br)-substituted aryl iodides 2 are also suitable for this transformation, affording oxazoline derivatives 3ah–3am in 77–86% yields and 91–94% ees. Sensitive functional groups such as primary amine (2o), ester (2p), and aldehyde group (2q) could be tolerated. The absolute configuration of 3ga with L10 was established as S by single-crystal X-ray diffraction study.45
Table 3 Scope of palladium-catalyzed asymmetric coupling cyclization reaction of N-(buta-2,3-dienyl)amides and aryl iodidesa
|
Unless indicated otherwise, the experiments were performed with 1 (0.5 mmol), 2 (1.0 mmol), Pd2(dba)3·CHCl3 (0.0125 mmol), L10 (0.0375 mmol), and Cs2CO3 (1.0 mmol) in benzene (6 mL) at rt (0.5 h) and 90 °C under Ar atmosphere.
The reaction was conducted in 4 mL of benzene.
With (S,S)-L10 purchased from Sigma-Aldrich, 90% ee of (R)-3ag was obtained.
|
|
We also tried the reaction of non-terminal allenes 1n and 1o with phenyl iodide, however, neither of them afforded the corresponding oxazoline product (Scheme 4).
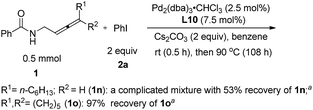 |
| | Scheme 4 The reaction of non-terminal allenes. aDetermined by 1H NMR analysis using 1,3,5-trimethylbenzene as the internal standard. | |
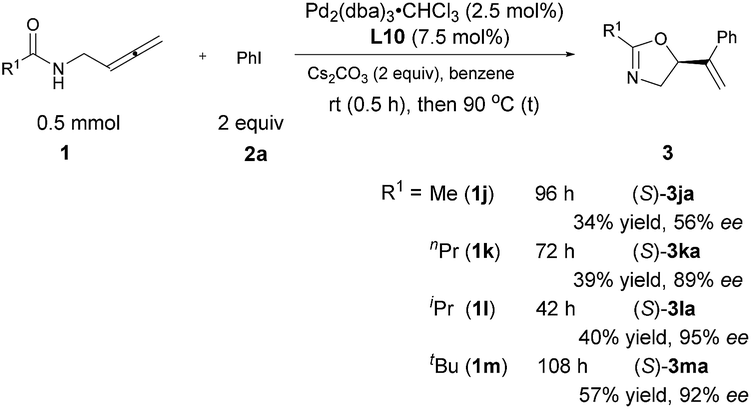
The steric effect of the R1 group
Interestingly, R1 may also be an alkyl group albeit with a low yield and the ee increased with its steric hindrance: with R1 being iPr, the ee reached 95%, indicating that the steric hindrance of the R1 group is also critical for enantioselectivity shown in Table 3. The reaction of t-butyl-substituted amide 1m is much slower with an enantioselectivity of 92% (Scheme 5).
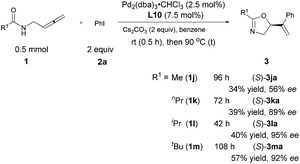 |
| | Scheme 5 Steric effect of the R1 group. | |
The effect of the leaving group X
Moreover, the reactivity of phenyl chloride is too low resulting in 97% recovery of starting material 1a; the reaction of phenyl bromide proceeded much slower (35% conversion in 65 hours); phenyl triflate afforded a complicated mixture (Table 4).
Table 4 The reaction of phenyl chloride, bromide, and triflatea
|
|
| Entry |
X |
Time (h) |
Resultsa |
|
Determined by 1H NMR analysis using 1,3,5-trimethylbenzene as the internal standard.
|
| 1 |
Cl |
65 |
97% recovery of 1a |
| 2 |
Br |
65 |
32% 3aa, 65% recovery of 1a |
| 3 |
OTf |
108 |
Complicated mixture |
Synthetic applications
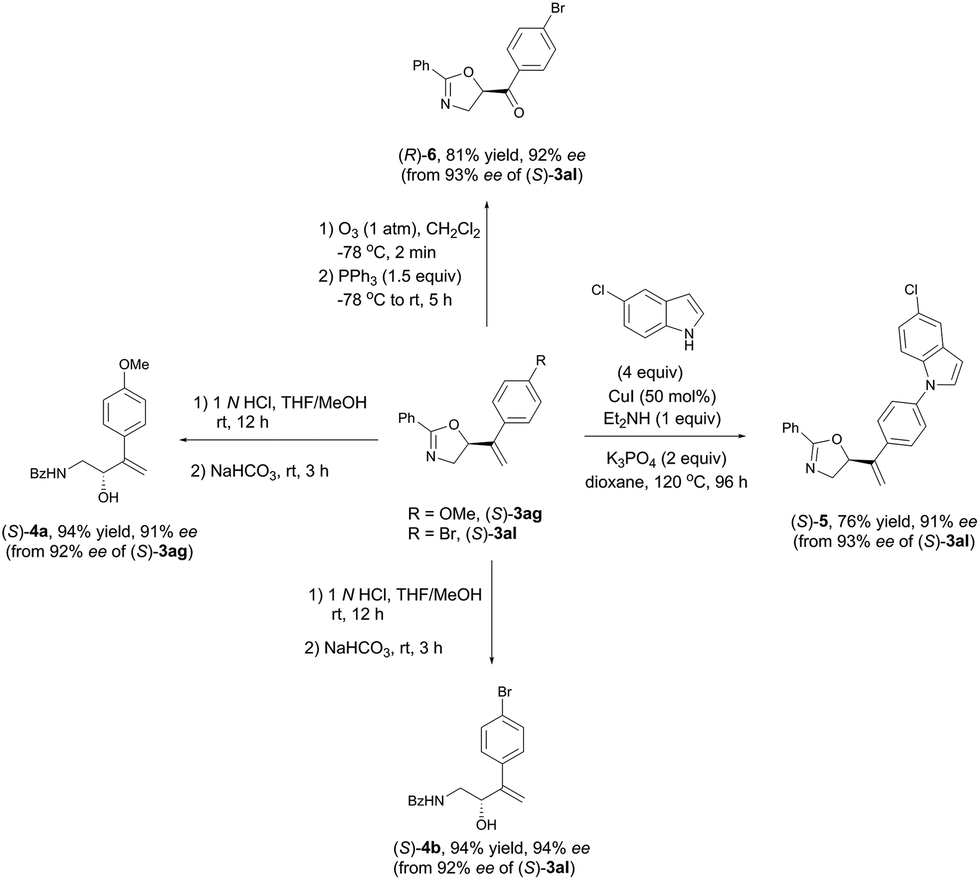
As shown in Table 3 and Scheme 5, these synthetically useful substituents such as Ar-OMe, Ar–Cl, Ar–Br, and Ar-acyl groups and the pre-installed (in (S)-3ha) or in situ formed CC bonds in every case make these chiral oxazolines ready for further transformations as demonstrated in Scheme 6. Hydrolysis of (S)-3ag and (S)-3al under acidic conditions readily afforded the useful optically active β-amino alcohols (S)-4a and (S)-4b in 91% and 94% ees, respectively.46 The C–N coupling of (S)-3al with 5-chloro-1H-indole was also realized using 50 mol% of CuI affording (S)-5 in 76% yield and 91% ee.47 Additionally, ozonolysis of the CC bond in (S)-3al afforded the rather sensitive product, 5-acyl-substituted oxazoline (R)-6, without racemization.48
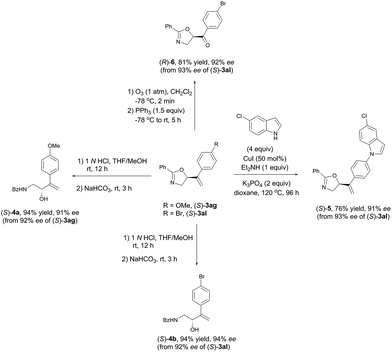 |
| | Scheme 6 Synthetic potentials of oxazoline products. | |
Discussion
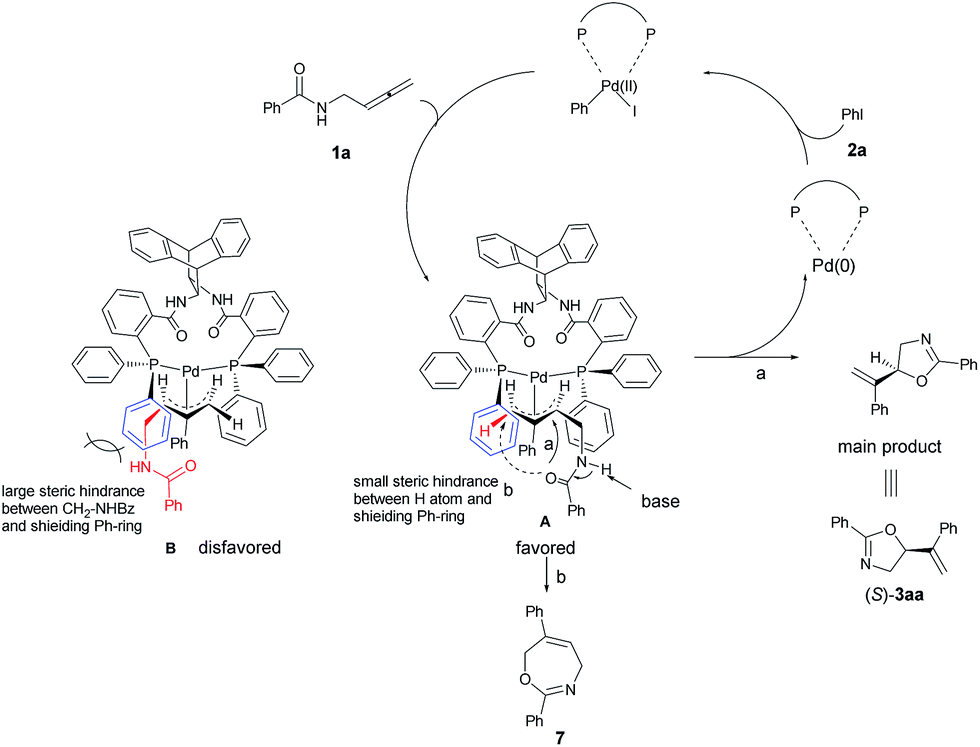
A model for the prediction of absolute configuration in the products with the Trost ligand L10 is shown below: oxidative addition of Pd(0) with iodobenzene 2a and subsequent carbopalladation with N-(buta-2,3-dienyl)amide 1a would form two π-allylic palladium intermediates A or B. The intermediates A is preferred over B due to the steric interaction of the amide group with the axial phenyl group, thus, subsequent nucleophilic O-attack in intermediate A (path a) leads to the product 3aa with the observed S-configuration (Scheme 7). In addition, the formation of the seven-membered product 7 is disfavored for the longer distance of nucleophilic O-centered nucleophile as compared to that for the formation the 5-membered 3aa (path a vs. path b).
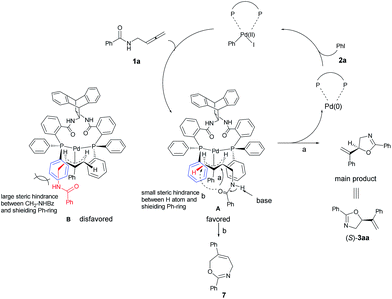 |
| | Scheme 7 A proposed mechanism and prediction of the absolute configuration of the product. Due to the steric interaction of the amide group with the axial phenyl group, the product 3aa was observed with the absolute S-configuration. | |
Stirring racemic 3al under the optimized reaction system for 24 hours, no ee was detected with the recovered oxazoline, which means that the allylic cleavage in rac-3al is not possible (Scheme 8).
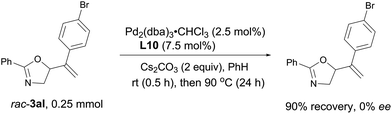 |
| | Scheme 8 Mechanistic study. No ee was detected with the recovered oxazoline, which means that the nucleophilic O-attack in intermediate A of the mechanistic proposal is irreversible. | |
Conclusion
In conclusion, we have developed a completely different enantioselective approach for the asymmetric construction of oxazoline derivatives with an excellent enantioselectivity via the palladium-catalyzed coupling-cyclization of N-(buta-2,3-dienyl)amides with aryl iodides. The easily accessible and diversified nature of both starting materials,40,49 the compatibility of useful functional groups, generality, and the synthetic potentials of the oxazoline and CC bond make this method of high interest to both organic and medicinal chemists. Further studies in this area are being actively pursued in our laboratory.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
Financial support from the National Science Foundation of China (Grant No. 21690063) is greatly appreciated. We thank Mr Shihua Song in this group for reproducing the results of (S)-3ca, (S)-3ea, and (S)-3af presented in this study.
Notes and references
- B. S. Davidson, Chem. Rev., 1993, 93, 1771 CrossRef CAS.
- M. Tsuda, M. Yamakawa, S. Oka, Y. Tanaka, Y. Hoshino, Y. Mikami, A. Sato, H. Fujiwara, Y. Ohizumi and J. Kobayashi, J. Nat. Prod., 2005, 68, 462 CrossRef CAS PubMed.
- H. B. Bode, H. Irschik, S. C. Wenzel, H. Reichenbach, R. Müller and G. Höfle, J. Nat. Prod., 2003, 66, 1203 CrossRef CAS PubMed.
- M. R. Prinsep, R. E. Moore, I. A. Levine and G. M. L. Patterson, J. Nat. Prod., 1992, 55, 140 CrossRef CAS.
- A. Araki, T. Kubota, K. Aoyama, Y. Mikami, J. Fromont and J. Kobayashi, Org. Lett., 2009, 11, 1785 CrossRef CAS PubMed.
- S. Faizi, F. Farooqi, S. Zikr-Ur-Rehman, A. Naz, F. Noor, F. Ansari, A. Ahmad and S. A. Khan, Tetrahedron, 2009, 65, 998 CrossRef CAS.
- S. K. Tahir, M. A. Nukkala, N. A. Z. Mozny, R. B. Credo, R. B. Warner, Q. Li, K. W. Woods, A. Claiborne, S. L. Gwaltney, D. J. Frost, H. L. Sham, S. H. Rosenberg and S.-C. Ng, Mol. Cancer Ther., 2003, 2, 227 CAS.
- R. H. Wiley and L. L. Bennett, Chem. Rev., 1949, 44, 447 CrossRef CAS.
- J. A. Frump, Chem. Rev., 1971, 71, 483 CrossRef CAS.
- M. Reuman and A. I. Meyers, Tetrahedron, 1985, 41, 837 CrossRef CAS.
- T. G. Gant and A. I. Meyers, Tetrahedron, 1994, 50, 2297 CrossRef CAS.
- S.-S. Jew, Y.-J. Lee, J. Lee, M. J. Kang, B.-S. Jeong, J. H. Lee, M.-S. Yoo, M.-J. Kim, S.-H. Choi, J.-M. Ku and H.-G. Park, Angew. Chem., Int. Ed., 2004, 43, 2382 CrossRef CAS PubMed.
- Y.-J. Lee, J. Lee, M.-J. Kim, T.-S. Kim, H.-G. Park and S.-S. Jew, Org. Lett., 2005, 7, 1557 CrossRef CAS PubMed.
- Y.-J. Lee, J. Lee, M.-J. Kim, B.-S. Jeong, J. H. Lee, T.-S. Kim, J. Lee, J.-M. Ku, S.-S. Jew and H.-G. Park, Org. Lett., 2005, 7, 3207 CrossRef CAS PubMed.
- Y. Tsuda, M. Kuriyama and O. Onomura, Chem.–Eur. J., 2012, 18, 2481 CrossRef CAS PubMed.
- H. Suga, K. Ikai and T. Ibata, Tetrahedron Lett., 1998, 39, 869 CrossRef CAS.
- H. Suga, K. Ikai and T. Ibata, J. Org. Chem., 1999, 64, 7040 CrossRef CAS.
- D. A. Evans, J. M. Janey, N. Magomedov and J. S. Tedrow, Angew. Chem., Int. Ed., 2001, 40, 1884 CrossRef CAS PubMed.
- M. Punk, C. Merkley, K. Kennedy and J. B. Morgan, ACS Catal., 2016, 6, 4694 CrossRef CAS PubMed.
- Y. Ito, M. Sawamura and T. Hayashi, J. Am. Chem. Soc., 1986, 108, 6405 CrossRef CAS.
- S. D. Pastor and A. Togni, J. Am. Chem. Soc., 1989, 111, 2333 CrossRef CAS.
- F. Gorla, A. Togni and L. M. Venanzi, Organometallics, 1994, 13, 1607 CrossRef CAS.
- J. M. Longmire and X. Zhang, Organometallics, 1998, 17, 4374 CrossRef CAS.
- Y. Motoyama, H. Kawakami, K. Shimozono, K. Aoki and H. Nishiyama, Organometallics, 2002, 21, 3408 CrossRef CAS.
- M.-X. Xue, C. Guo and L.-Z. Gong, Synlett, 2009, 13, 2191 Search PubMed.
- F. Sladojevich, A. Trabocchi, A. Guarna and D. J. Dixon, J. Am. Chem. Soc., 2011, 133, 1710 CrossRef CAS PubMed.
- H. Y. Kim and K. Oh, Org. Lett., 2011, 13, 1306 CrossRef CAS PubMed.
- R. Kuwano, N. Kameyama and R. Ikeda, J. Am. Chem. Soc., 2011, 133, 7312 CrossRef CAS PubMed.
- A. Jaganathan, A. Garzan, D. C. Whitehead, R. J. Staples and B. Borhan, Angew. Chem., Int. Ed., 2011, 50, 2593 CrossRef CAS PubMed.
- A. Jaganathan and B. Borhan, Org. Lett., 2014, 16, 3616 CrossRef CAS PubMed.
- V. Rauniyar, A. D. Lackner, G. L. Hamilton and F. D. Toste, Science, 2011, 334, 1681 CrossRef CAS PubMed.
- Y. Kawato, A. Kubota, H. Ono, H. Egami and Y. Hamashima, Org. Lett., 2015, 17, 1244 CrossRef CAS PubMed.
- Y. Kawato, H. Ono, A. Kubota, Y. Nagao, N. Morita, H. Egami and Y. Hamashima, Chem.–Eur. J., 2016, 22, 2127 CrossRef CAS PubMed.
- Q. Yang, X. Jiang and S. Ma, Chem.–Eur. J., 2007, 13, 9310 CrossRef CAS PubMed.
- W. Shu, Q. Yang, G. Jia and S. Ma, Tetrahedron, 2008, 64, 11159 CrossRef CAS.
- W. Shu and S. Ma, Chem. Commun., 2009, 6198 RSC.
- W. Shu and S. Ma, Tetrahedron, 2010, 66, 2869 CrossRef CAS.
- W. Shu, Q. Yu, G. Jia and S. Ma, Chem.–Eur. J., 2011, 17, 4720 CrossRef CAS PubMed.
- X. Xie and S. Ma, Chem. Commun., 2013, 49, 5693 RSC.
- For the racemic version of such a transformation, see:
(a) G. R. Cook and W. Xu, Heterocycles, 2006, 67, 215 CrossRef CAS;
(b) S. Inuki, Y. Yoshimitsu, S. Oishi, N. Fujii and H. Ohno, Org. Lett., 2009, 11, 4478 CrossRef CAS PubMed.
- For the synthetic application of this racemic transformation, see: B. Chen, N. Wang, W. Fan and S. Ma, Org. Biomol. Chem., 2012, 10, 8465 CAS.
- For 1,2-chiral induction of such a transformation, see: N. Wang, B. Chen and S. Ma, Asian J. Org. Chem., 2014, 3, 723 CrossRef CAS.
- B. M. Trost and D. L. Van Vranken, Angew. Chem., Int. Ed., 1992, 31, 228 CrossRef.
- B. M. Trost, D. L. Van Vranken and C. Bingel, J. Am. Chem. Soc., 1992, 114, 9327 CrossRef CAS.
- Crystal data for compound (S)-3ga: C17H14BrNO, MW = 328.20, orthorhombic, space group P2(1)2(1)2(1), final R indices [I > 2σ(I)], R1 = 0.0230, wR2 = 0.0559; R indices (all data), R1 = 0.0260, wR2 = 0.0569; a = 6.1624(6) Å, b = 7.7041(7) Å, c = 30.136(3) Å, α = 90.00°, β = 90.00°, γ = 90.00°, V = 1430.7(2) Å3, T = 296(2) K, Z = 4, reflections collected/unique 16442/2517 (Rint = 0.0340), number of observations [> 2σ(I)] 2337, parameters: 181. ESI, CCDC 1506167.†.
- J.-E. Joo, K.-Y. Lee, V.-T. Pham, Y.-S. Tian and W.-H. Ham, Org. Lett., 2007, 9, 3627 CrossRef CAS PubMed.
- N. Wang, B. Chen and S. Ma, Adv. Synth. Catal., 2014, 356, 485 CrossRef CAS.
- V. Komanduri and M. J. Krische, J. Am. Chem. Soc., 2006, 128, 16448 CrossRef CAS PubMed.
- H. Luo and S. Ma, Eur. J. Org. Chem., 2013, 3041 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental section, characterization of all compounds, and copies of 1H and 13C NMR spectra of selected compounds. CCDC 1506167. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7sc04079b |
| ‡ These three authors contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence c and
Shengming
Ma
c and
Shengming
Ma