DOI:
10.1039/C7SC03617E
(Edge Article)
Chem. Sci., 2018,
9, 1902-1911
A simple multi-responsive system based on aldehyde functionalized amino-boranes†
Received
17th August 2017
, Accepted 2nd January 2018
First published on 4th January 2018
Abstract
A simple aldehyde functionalized amino-triarylborane donor–acceptor system (BO-1) was found to display distinct responses toward multiple external stimuli including solvent, temperature and pressure, with emission colours changing from blue to red. The operating mechanism most likely involves reversible switching between closed and open structures based on an intramolecular B ← O bond. Imbedded donor–acceptor charge transfer transitions played a key role in the multi-coloured fluorescent response of this new boron system to external stimuli. Multi-coloured and erasable fluorescent images on solid substrates based BO-1's “turn-on” response toward solvents, particularly water, are demonstrated.
Introduction
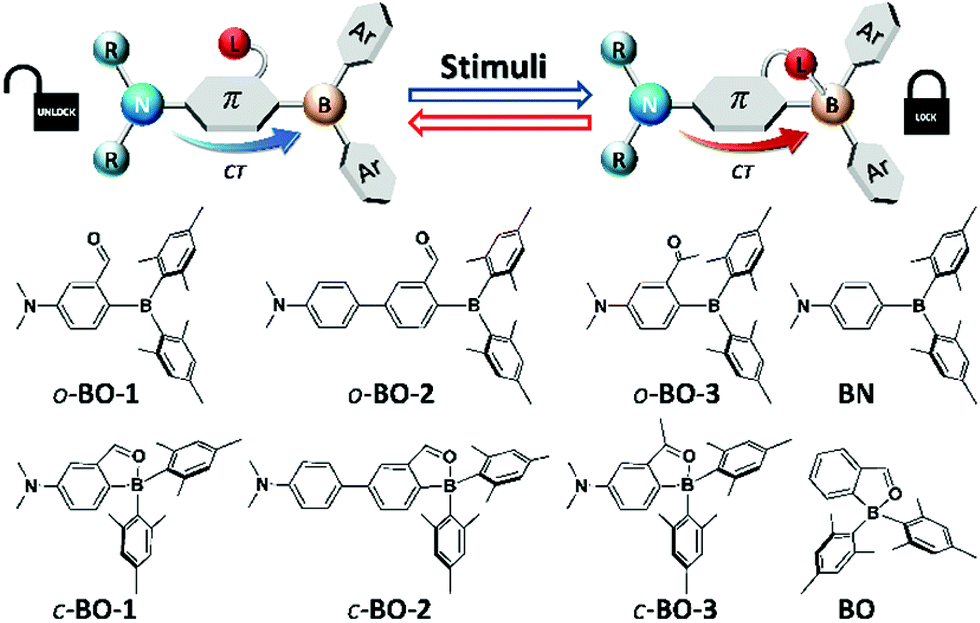
Multi-responsive systems are highly attractive and important because of their potential applications in a variety of sensing and monitoring/tracking technologies.1 Many previously reported stimuli-responsive systems are based on transition-metal compounds, some of which display highly sensitive and robust responses toward external stimuli such as light,1a,2 pressure,1a–d,3 temperature,1a,e,4 and oxygen,1a,5etc. Main-group based responsive systems are more appealing because of their low cost. As a result, main-group systems that respond to external stimuli have attracted much attention recently and many interesting examples have been reported.6–8 Nonetheless, multi-responsive main-group based systems remain rare and underexplored. Organoboron-based systems have recently been demonstrated to be very promising as responsive materials.6,7 Although the operating mechanisms of some of the boron-based responsive systems are often similar to those observed in other organic π-conjugated materials or transition metal based systems such as changes in molecular aggregation/intermolecular interactions or fluorescence to phosphorescence switching,7,8 recent studies have shown that boron-based responsive systems do have a few unique features for achieving responses to external stimuli. For example, the responsive properties of boron compounds could be modulated by the following: (a) reversible B ← X bond formation/cleavage,6a–d (b) donor–acceptor charge transfer (CT) transition modulation,6e,f (c) reversible B–C/C–C bond formation/cleavage, etc.,6g–k and (d) visible light induced E/Z isomerization.6c,d,l Recently reported organoboron-based responsive systems typically rely on/address one of these features. Donor–acceptor systems have a special place in organoboron chemistry and organoboron-based optoelectronic materials.9 Manipulating donor–acceptor interactions in boron-based systems has led to non-linear optical materials,10 anion sensors,11 charge transport materials for solar cells,12 emitters for organic light emitting devices (OLEDs),13 and molecular thermometers.6e,f Combining a switchable internal donor–acceptor bond with an internal donor–acceptor CT system could allow the creation of new and versatile multi-responsive materials. However, to the best of our knowledge, this strategy has not yet been explored for multi-responsive systems. Furthermore, for practical applications, it is always desirable to have a simple system that is easy and cheap to produce. Based on these considerations and inspired by the recent advances in boron-based responsive materials, we designed and investigated simple responsive boron systems, depicted in Scheme 1.
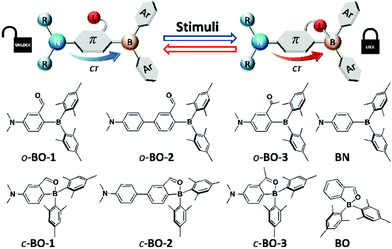 |
| | Scheme 1 The main operating mechanism and the structures of the newly designed donor–acceptor boron compounds and control compounds for the study of multi-responsive systems. | |
Two key features are implemented in the new molecules we designed. The first feature is the inclusion of a potentially switchable B ← O unit. In order to achieve reversible boron–donor (L) bond switching, the B–L bond must be relatively weak, which could be achieved by either enhancing steric congestion around the boron centre, introducing a weak donor, or reducing the electrophilicity of the boron atom. Thus, to ensure a weak B–L bond, the bulky mesityl groups were chosen for the aryl substituents on boron, which also protect the boron centre from nucleophilic attack by external donors such as water or solvent molecules. In addition, an aldehyde group was chosen as the donor for binding to boron because it is a relatively weak donor, as indicated by the 11B chemical shift of the previously reported compound BO at 25 ppm (compared to the corresponding imine analogue that has a 11B chemical shift of 5 ppm).14 The second feature is the installation of an NMe2 donor unit, which is well known to produce strong intramolecular charge transfer (ICT) transitions when combined with a triarylboron.9,10 Furthermore, the amino group allows the modulation of the electron density/electrophilicity of the boron centre. Two different linkers, phenyl and biphenyl, were included in the structural design of molecules BO-1 and BO-2 to further modulate the ICT and the B–O bond strength. The previously reported compounds, BO and10a,bBN, and a new compound BO-3 in which the aldehyde group is replaced by a ketone group, were also examined as control compounds for this work. We have found that molecule BO-1 responds to both pressure and temperature, while BO-2 and BO-3 either respond only to temperature, or have no response to temperature and pressure, due to their distinct difference in structures. In addition, BO-1 displays distinct “turn-on” fluorescence colour changes on solid substrates upon exposure to different solvent molecules, including water, enabling the use of this molecule for simple fluorescent patterning/writing with solvents. The details of this work are presented herein.
Results and discussion
Synthesis and crystal structures
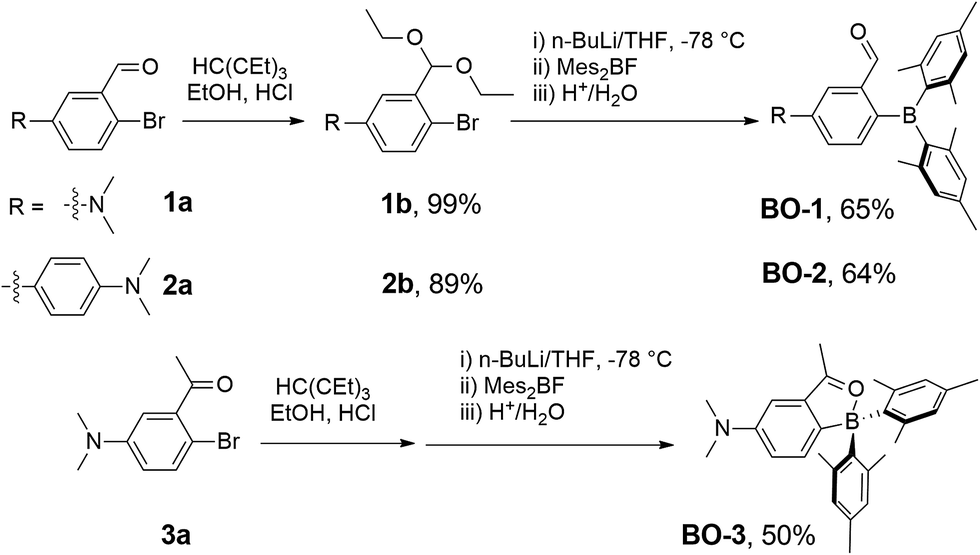
Compounds BO-1, BO-2 and BO-3 were obtained according to the procedure shown in Scheme 2. The starting materials 1a and 3a were prepared according to a literature procedure15 and the synthetic details for 2a are provided in the ESI.† After protecting the carbonyl group in 1a–3a with the ethoxy group, the BMes2 unit can be attached to the benzene ring via lithiation and substitution, producing BO-1–BO-3 in good yields. Because the intermediate 3b for BO-3 is unstable, it was not isolated. All three compounds were fully characterized by NMR, HRMS and single-crystal X-ray diffraction analyses. 11B{1H} NMR spectra indicated that these three compounds have distinct structures. In C6D6, the 11B chemical shift of BO-1 appears at 60 ppm, while that of BO-2 and BO-3 is at 30 ppm and 16 ppm, respectively. Compared to BO that has a 11B signal at 25 ppm, BO-2 and BO-3 likely have the closed structures shown in Scheme 1, while BO-1 likely has the open structure. The much upfield shifted 11B signal of BO-3 indicates that it has a stronger B–O bond than that of BO-2.
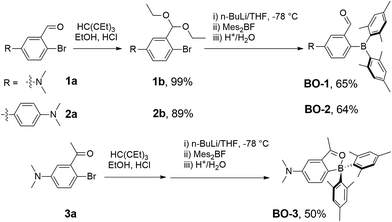 |
| | Scheme 2 Synthetic procedures for BO1, BO-2 and BO-3. | |
This was confirmed by crystallographic analyses. As shown in Fig. 1, BO-1 has a typical three-coordinate boron centre, while BO-2 and BO-3 have a tetrahedral boron centre with B–O bond length of 1.645(2) Å, and 1.626(3) Å, respectively (the B–O bond length in BO is 1.641(2) Å). The B–CMes bond lengths in BO-2/BO-3 (1.636(3), 1.637(3) Å for BO-2; 1.640(4), 1.642(3) Å for BO-3) are much longer than those in BO-1 (1.574(2), 1.570(2) Å), consistent with the more congested boron centre in BO-2/BO-3. The crystals/crystalline powders of BO-1 and BO-2/BO-3 have distinct colours in the crystalline state – yellow and red-orange, respectively, as shown in Fig. 1, which is clearly a consequence of their distinct structures. BO-1 forms an extended 1D structure in the crystal lattice via H bonds between the oxygen atom and the H atoms of the NMe2 unit. The fact that BO-1 has the open form can be attributed to the zwitterionic resonance structure shown in Fig. 1, which greatly reduces the electrophilicity of the boron center toward the oxygen atom. For the biphenyl linked BO-2 molecule, the zwitterion resonance structure is less important since it involves the dearomatization of two phenyl rings. As a result, the B center in BO-2 is more electrophilic and capable of forming the chelate structure. The electron donating and bulky methyl group in BO-3 is clearly responsible for the formation of the closed form. Efforts to obtain the closed form of BO-1 by using/mixing different solvents such as CH2Cl2, CH3CN, methanol, water, etc. in the crystallization process inevitably led to the formation of the o-BO-1 crystals that consistently display the same colour and the same unit cell parameters. All three compounds are stable for months under ambient conditions in solution and in the solid state (see Fig. S35†).
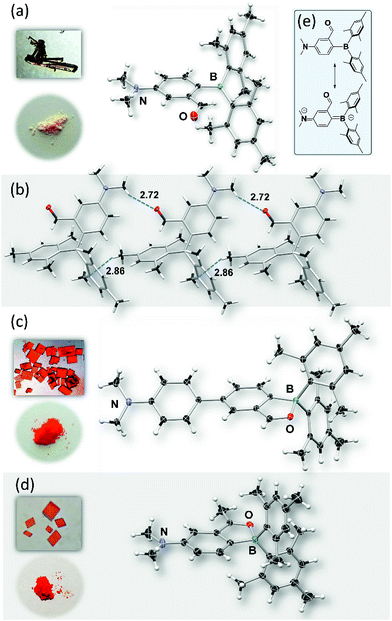 |
| | Fig. 1 Crystal structures of BO-1 (a), BO-2 (c) and BO-3 (d) with 35% thermal ellipsoids, the inset photographs show the colours of crystals and powders of BO-1, BO-2 and BO-3, respectively. (b) The packing diagram of BO-1 showing intermolecular H-bonds. (e) The two resonance forms of BO-1. | |
Distinct photophysical properties of BO-1–BO-3
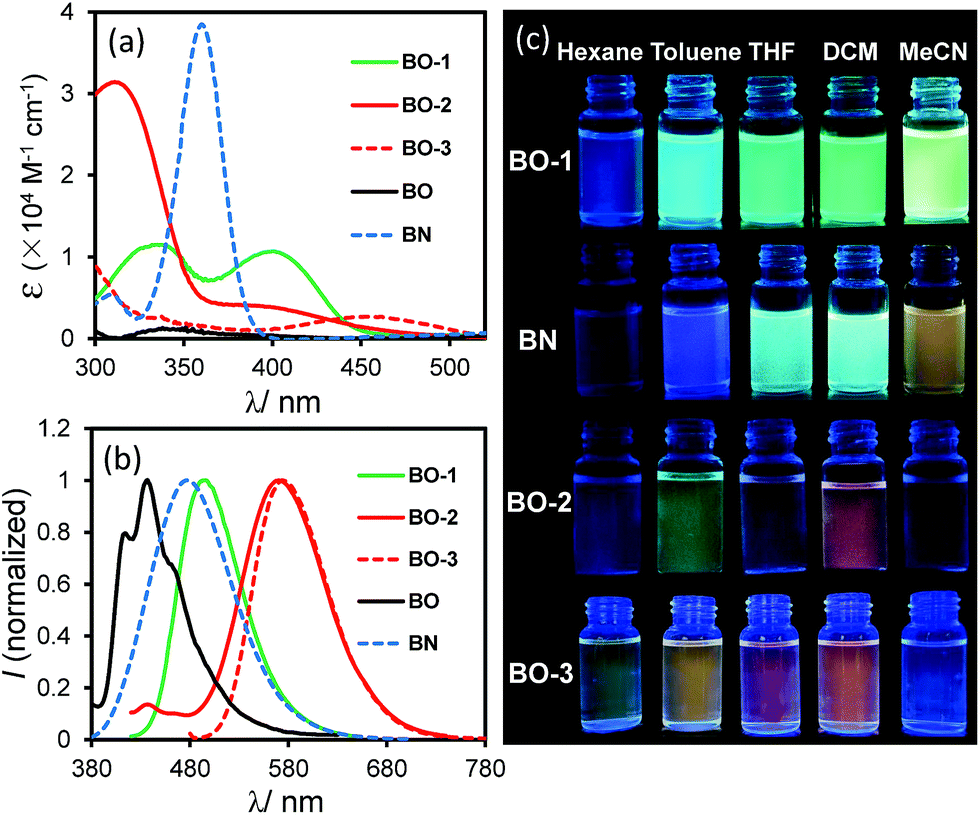
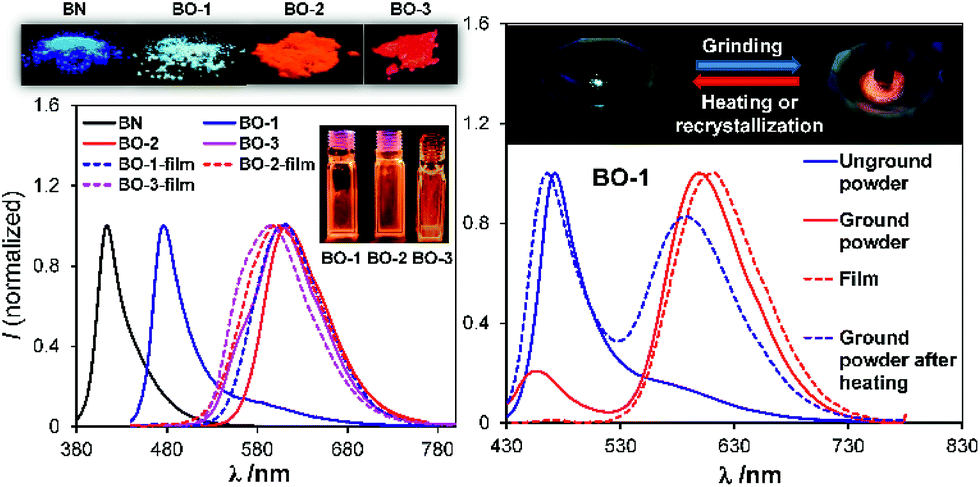
As shown in Fig. 2, the absorption spectrum of BO-1 is distinctly different from those of BO-2/BO-3, with a strong absorption band at ∼400 nm that could be assigned to the amino to boron/carbonyl ICT transition. A weak absorption band at ∼400 nm in the spectrum of BO-2 agrees with its closed structure shown by the crystal structure, which diminishes the ICT transitions. In addition, the absorption tail of BO-2 stretches further to the longer wavelength than that of BO-1 (CH2Cl2, 0.01 mM). BO-3 has a distinct absorption band at 450 nm. At 0.10 mM, BO-1 has a weak, but distinct low energy band at about 450 nm, resembling that of BO-3 (see Fig. 2 and ESI†). For comparison, the absorption spectra of BN and BO compounds are also included in Fig. 2. The ICT transition band of BN appears at a much higher energy and is more intense than those of BO-1 and BO-2/BO-3, while BO has only very weak absorption bands in the near UV region. This illustrates the dramatic impact on the electronic properties of the boron compounds induced by a subtle change in the structure or the introduction of a 2nd donor group. In the fluorescence spectra (CH2Cl2, 0.01 mM, 298 K), the emission peak of BO-2/BO-3 appears at 584 nm/591 nm, nearly 100 nm bathochromically shifted relative to that of BO-1 (490 nm). As a result, BO-1 displays a blue-green emission colour, while BO-2 and BO-3 have a pinkish red/red-orange colour in CH2Cl2. In addition to the emission colour difference, the emission quantum efficiency also varies sharply, with ΦFL = 0.61, 0.25, 0.45 in CH2Cl2, for BO-1, BO-2, and BO-3, respectively (see Table 1). Furthermore, the emission λmax and colour of BO-1, BO-2 and BO-3 have a distinct dependence on solvent polarity as shown in Fig. 2 (see ESI†). For BO-1, its solvent-dependent emission resembles that of BN, which, along with its high ΦFL, further supports the strong N to B/C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O ICT character in BO-1. BO-2 and BO-3 also display solvent dependent emission, although the emission spectral shift with solvent polarity is much less for BO-3. BO-2 is essentially non-emissive in DMF/CH3CN.
O ICT character in BO-1. BO-2 and BO-3 also display solvent dependent emission, although the emission spectral shift with solvent polarity is much less for BO-3. BO-2 is essentially non-emissive in DMF/CH3CN.
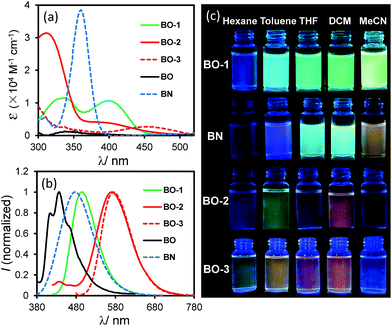 |
| | Fig. 2 Absorption (a) and fluorescence spectra (b) (recorded with λex = λmax of the first low energy absorption band) of BO-1, BO-2, BO-3, BO and BN in CH2Cl2 (0.01 mM). (c) Photographs showing fluorescent colours of BO-1, BO-2, BO-3 and BN in selected solvents (0.01 mM). Photos of BO can be found in ESI.† | |
Table 1 Photophysical data for compounds BO-1–BO-3
| Compounds |
Absorption (nm) |
Emission, 298 K |
Emission, 77 K |
| CH2Cl2 (0.1 mM) |
CH2Cl2 (0.1 mM) |
Unground powder |
Ground powder |
CH2Cl2 (0.1 mM) |
|
λ
max [nm], τ (ns), ΦFLa |
λ
max [nm], τ (ns), ΦFLa |
λ
max [nm], τ (ns), ΦFLa |
λ
max [nm], τ (ns), ΦFLa |
|
Determined using an absolute QY spectrometer.
|
|
BO-1
|
400 |
499, 14.1, 0.61 |
473, 2.9, 0.07 |
465, 1.5, 0.001 |
468, 8.9, 0.04 |
| 600, 9.1, 0.06 |
620, 6.1, 0.05 |
604, 13.5, 0.09 |
|
BO-2
|
400 |
584, 6.3, 0.25 |
611, 8.7, 0.10 |
N/A |
606, 28.2, 0.11 |
|
BO-3
|
454 |
591, 30, 0.45 |
628, 8.0, 0.11 |
N/A |
574, 44, 0.56 |
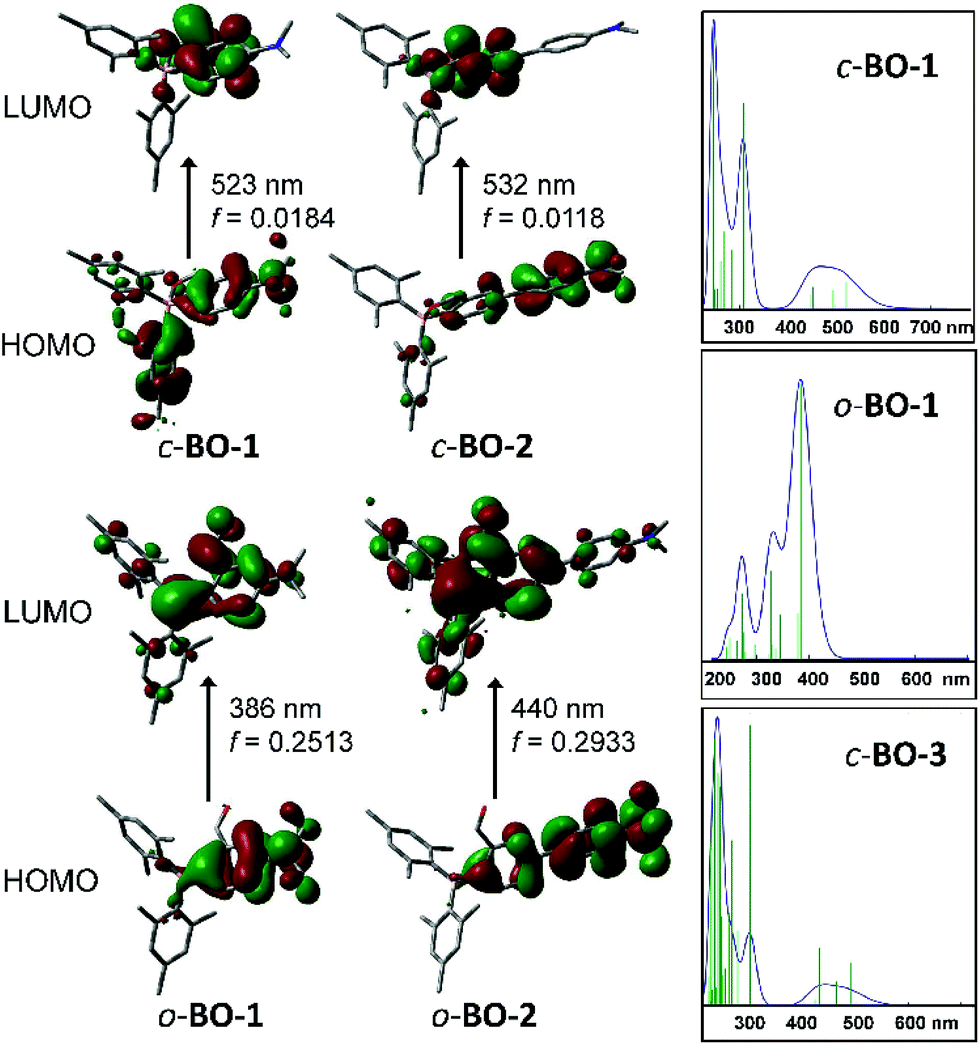
To elucidate the origin of electronic transitions that are responsible for the distinct photophysical properties of molecules BO-1 and BO-2/BO-3, TD-DFT computational studies at the B3LYP/6-31g(d) level of theory were performed for both open and closed forms of BO-1/BO-2, and the closed form of BO-3, and the following findings were obtained. For the closed forms, the S0 → S1 vertical excitation involves mainly HOMO to LUMO transitions (92% and 82%, for BO-1 and BO-2, respectively) at 523 nm (BO-1) and 532 nm (BO-2) with weak oscillator strength. The HOMO orbital of closed BO-1 and BO-2/BO-3 involves mainly a Mes and NMe2-Ph (BO-1/BO-3) or NMe2-Ph (BO-2), while the LUMO orbital involves the B,O-Ph chelate (see Fig. 3). Therefore, the S0 → S1 transitions for the closed molecules have some CT character. The emission peak at 584 nm/591 nm for BO-2/BO-3 can be attributed to the S1 → S0 transition of the closed form. For the open form, the S0 → S1 transition for both BO-1 and BO-2 is a CT transition involving HOMO and LUMO orbitals, located on the NMe2 and the Ar linker, and the boron-benzaldehyde, respectively, with very high oscillator strength as shown in Fig. 3. The weak absorption band of BO-1 at 450 nm is clearly not from the open form, based on TD-DFT data. The S0 → S1 transition energy of o-BO-1 is blue shifted about 100 nm, compared to that of c-BO-1. Based on the experimentally observed and the TD-DFT calculated absorption spectra (see ESI†), the distinct emission colour of BO-1 and BO-2/BO-3 is most likely caused by their preferential adoption of the open and the closed forms, respectively. The strongly solvent-dependent fluorescence of BO-1 agrees with the strong ICT character of the open form. The low emission quantum efficiency of BO-2 can be attributed to the low oscillator strength of the S0 → S1 transition in c-BO-2. For c-BO-3, the S0 → S1 transition oscillator strength (0.0207) is much greater than that of c-BO-2, thus leading to a brighter emission. Electrochemical analysis shows that the reduction potential of the aldehyde group in BO-1 is 0.25 V more positive than that of BO-2, which agrees with the DFT calculated LUMO energies of the o-BO-1 and the c-BO-2. Similarly, the oxidation potential of the amino group in BO-1 is ∼0.29 V more positive than that of BO-2, which also agrees with the calculated HOMO energy difference of o-BO-1 and the c-BO-2 (see Fig. S46, Tables S8 and S9†). DFT data indicate that in the gas phase, the o-BO-1 is slightly more stable than c-BO-1 (by about 0.8 kcal mol−1) and the activation barrier from the o-BO-1 to c-BO-1 is less than 7 kcal mol−1 (see Fig. S50†). It is therefore reasonable to suggest that in solution and at ambient temperature, o-BO-1 and c-BO-1 are likely in equilibrium, with o-BO-1 being the dominating form.
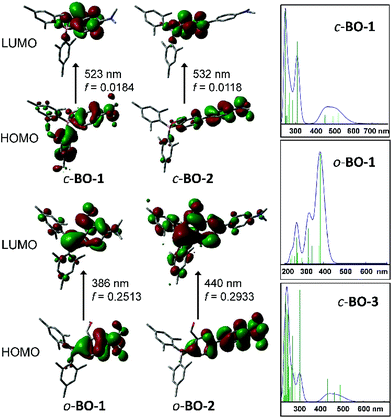 |
| | Fig. 3 HOMO and LUMO diagrams of BO-1 and BO-2 and TD-DFT (B3LYP/6-31g(d)) calculated absorption spectra of c-BO-1/o-BO-1 and c-BO-3. | |
Distinct impact of water on the emission colour of BO-1 and creating fluorescent paintings with solvents as ink
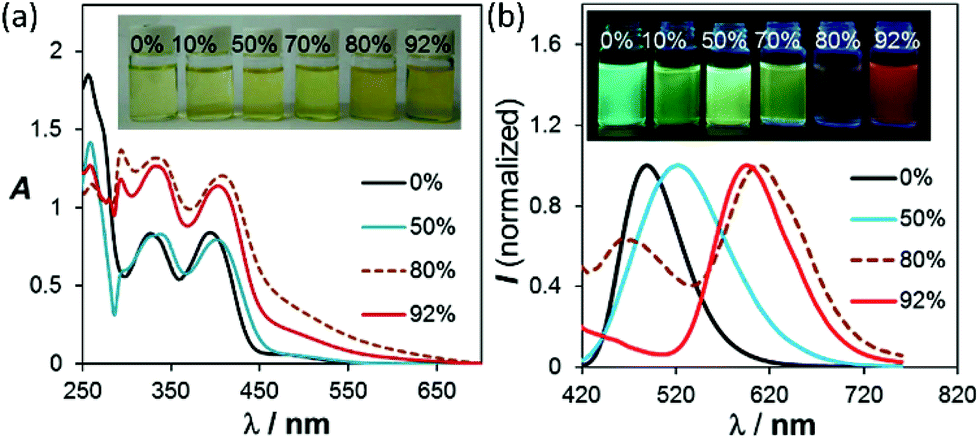
Among all the solvents we examined for BO-1, water was found to have a unique impact on the emission colour of BO-1. In either CH3CN or THF (see Fig. S26–S28†), the emission colour of the BO-1 solution changes gradually from blue green to green in THF, and green to whitish yellow in CH3CN with increasing water content, accompanied by a sharp decrease in ΦFL (e.g. from 0.45 in THF to 0.02 in a 70% water solution). When the water content exceeds 70%, the emission colour of the solution experiences a sharp change to red-orange, with an emission peak appearing at ∼605 nm, as shown in Fig. 4. Accompanying this is the distinct increasing of an absorption band at about 500 nm, which decreases a bit at 92% water level, due perhaps to a small amount of precipitate formation. The red emission peak of BO-1 in a mostly water solution resembles that of c-BO-2 (see Fig. S17†), except that the c-BO-2 emission in such a solution is extremely weak (ΦFL < 0.001) (c-BO-3 becomes insoluble). The emission peak of BO-2 experiences about a 35 nm bathochromic shift from THF to water (∼95%), while that of BO-1 has about 105 nm bathochromic shift. In contrast, the addition of water to the THF or CH3CN solution of BN shifts the emission band hypsochromically (∼60 nm from THF to 92% water, ∼90 nm from CH3CN to 92% water) with either increased ΦFL or little ΦFL change (see Fig. S25b†), which may be caused by aggregation induced structural rigidification and the highly hydrophobic nature of BN. TD-DFT calculations for o-BO-1 using water as the dielectric medium show that the amino group becomes pyramidal and the dihedral angle between the planes of phenyl and NMe2 increases from 11° to 27°, accompanied by a 30 nm red shift of the S0 → S1 vertical excitation energy (30 nm) with a very high oscillator strength (0.1990), compared to that in the gas phase (see ESI†), owing to the twisted ICT (TICT) effect.16 Because of the very low absorbance of the absorption band of BO-1 at 450 nm, it cannot be attributed to o-BO-1 in water. Water molecules likely interact with the amino group in o-BO-1, reducing its donating ability to the boron centre via TICT, thus increasing the electrophilicity of the boron center and facilitating the formation of the closed structure. The oxygen atom of the water molecules may also interact with the carbonyl group in o-BO-1, enhancing the nucleophilicity of the oxygen atom,17 allowing it to form a B–O bond. Addition of fluoride ions to BO-1 led to a distinct fluorescence quenching without colour change (see ESI†). Hence, the red emission colour of BO-1 in the presence of water is not caused by water binding to the boron centre. Definitive evidence for the formation of c-BO-1 in the presence of water was obtained from the 11B NMR spectra, which showed that the 11B peak of BO-1 shifts from 58 ppm in THF to ∼0 ppm in the 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 solvent mixture of water and THF (see Fig. S16a†), thus unequivocally establishing that BO-1 has a closed structure in the presence of water. The water induced change in BO-1 is fully reversible upon the addition of organic solvents.
:1 solvent mixture of water and THF (see Fig. S16a†), thus unequivocally establishing that BO-1 has a closed structure in the presence of water. The water induced change in BO-1 is fully reversible upon the addition of organic solvents.
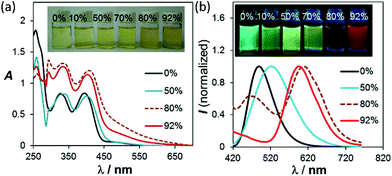 |
| | Fig. 4 (a) Changes in the absorption and (b) fluorescence spectra of BO-1 in THF (1 × 10−4 M), with increasing water content. For the complete spectra, see ESI.† | |
Although ΦFL (0.03 in 92% water + THF and 0.04 in 98% water + CH3CN) of BO-1 in water is much lower than that in THF (0.45) or CH3CN (0.58), the emission colour is readily visible. Most interesting is the finding that the solvent/water-dependent emission colour of BO-1 can also be visualized on solid substrates such as silica gel thin-layer chromatography (TLC) plates. One example is shown in Fig. 5(a). In this demonstration, three letters were written on a TLC plate using 1.3 mM CH2Cl2 solution of BO-1. After the solvent is evaporated, under UV light illumination, the three letters display the characteristic blue-green emission colour of BO-1. As the letters are wet with drops of water, the emission colour switches immediately to red-orange. After the TLC plate is dried to remove the water, the emission colour reverts to blue-green. Fluorescence spectra indicate that this is a fully reversible process (see Fig. S27†), supporting that the water induced structural transformation of BO-1 can occur reversibly on silica gel support. Addition of water to BO-1 crystals/powder does not change the emission colour, perhaps due to the high crystal lattice energy. The second example is shown in Fig. 5(b) and (d). In this example, the TLC plate was first treated with a 0.1 mM CH2Cl2 solution of BO-1 and then dried. Under UV illumination (365 nm), such treated TLC plates appear dark with very weak blue emission. Next, a small cotton swab, soaked with a selected solvent (pure solvent) was used to draw pictures on the plate. In order to provide sufficient time for the camera to capture the images created, high boiling point solvents such as DMF, toluene and water were used. These three solvents also provide distinct emission colours to the image – DMF: yellow-green, toluene: blue, water: red. Images created in such manner vanish as the ink/solvent evaporates, as shown by Fig. 5(c) and (e). The TLC plates can be reused for creating new images. This “solvent-turn on” emission effect of BO-1 on a TLC plate may be attributed to the large emission quantum efficiency difference of BO-1 in solution and in the solid state (e.g. ΦFL = 0.61 in CH2Cl2; 0.45 in 2-methoxyethyl ether (MOE); 0.15 in crystalline powder). Similar effects cannot be achieved for BO-2/BO-3 because of the similar and low ΦFL in both solution and solid states (e.g. ΦFL = 0.25 in CH2Cl2; 0.09 in MOE, 0.10 as crystalline powder) for BO-2, and the small emission colour change of BO-3 in different solvents. For the BN compound, it has a very high ΦFL in both solution and the solid state (e.g. ΦFL = 0.71 in CH2Cl2; 0.57 in MOE; 0.37 in the solid state). As a result, the background emission (blue) of BN is always dominant, making BN ineffective for creating multi-coloured images on a solid substrate using solvents as ink (see Fig. S45b and c†). In fact, BO-1 is the only molecule that displays full colours on the TLC plates. The highly visible red-orange emission colour of BO-1 generated by water on a silica gel substrate is certainly unique among the compounds we examined and critical for creating full-coloured paintings on TLC plates. The same approach can also be applied to non-fluorescent paper substrates. However, on filter papers, the dispersed BO-1 compound emits a weak red colour, which can be switched to bright blue, green or yellow emission colours with writing using solvents such as toluene or DMF, but cannot produce the red colour by writing with water (see Fig. S45d†). The cellulose of the filter paper likely behaves like water and stabilizes the closed form of BO-1.
 |
| | Fig. 5 (a) Photographs showing the reversible emission colour change of letters written on a pristine TLC plate (with a 1.3 mM CH2Cl2 solution of BO-1) with water and heating. (b and d) Photographs showing paintings created using a cotton tip and selected solvents as inks on a TLC plate treated with a BO-1 solution (CH2Cl2, 0.1 mM). Water: red; DMF: yellow; toluene: blue. A handheld UV lamp (365 nm) was used to illuminate the images created. (c and e) Photographs showing the disappearance of the images after the TLC plate is dried at rt. | |
Distinct temperature-dependent emission of BO-1
Both BO-1 and BO-2 display temperature-dependent emission spectral changes, but with a distinct difference. At 77 K the emission spectrum of BO-1 is dominated by a red-orange peak, with a notable presence of a blue emission peak at about 450 nm, which could be attributed to the closed and the open form of BO-1, respectively. The ratio of the yellow/orange peak versus the blue peak is dependent on the solvent and the concentration of BO-1 at 77 K. Increasing concentration greatly enhances the relative intensity of the yellow/orange peak of BO-1. For BO-2, its 77 K emission spectrum is almost entirely from the red-orange emission peak. From 77 K to near the melting point of the solvent, the emission peak of BO-2 loses intensity rapidly (e.g. ΦFL = ∼0.02 from 202 K to 157 K in CH2Cl2) and the emission colour shifts from yellow-orange to sky-blue with erratic changes in the CIE coordinates (see Fig. S43†). Above m.p., the emission colour of BO-2 gradually shifts back to yellow-green with increasing T, accompanied by a gradual increase in emission intensity. Increased ICT character of the c-BO-2 molecule with T may account for its emission intensity gain and bathochromic shift.
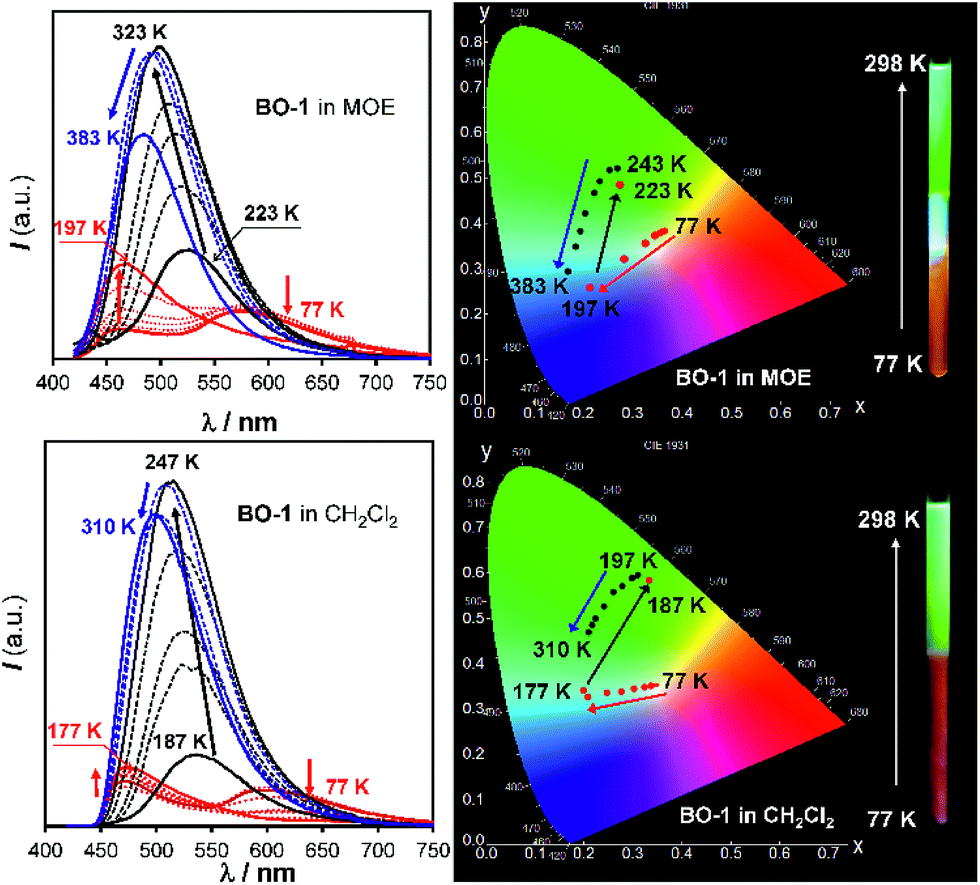
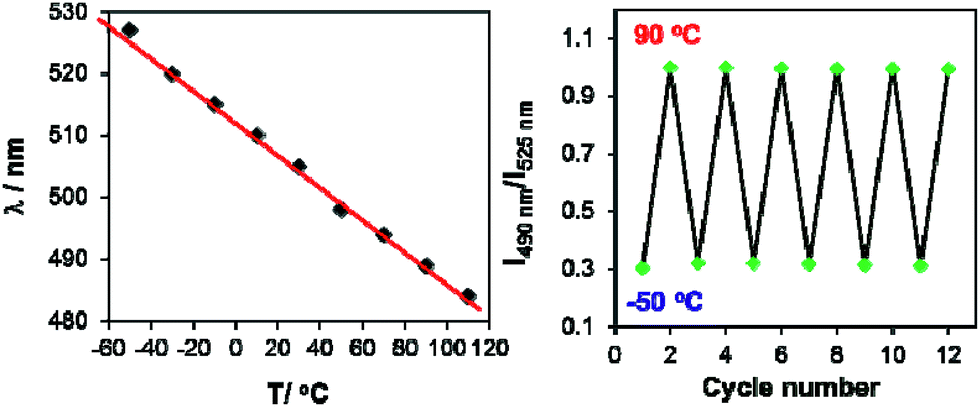
In contrast, for BO-1, in CH2Cl2 (m.p. = 176 K) or MOE (2-methoxyethyl ether, m.p. = 205 K, 0.1 mM), there are two well separated regions in the CIE diagram that display approximately linear changes in CIE coordinates (see Fig. S38(b)†) with T, one below the m.p. and one above the m.p. of the solvent (Fig. 6). Below m.p. (the red region of the emission spectra) the emission colour of BO-1 changes with increasing T from yellow-orange to blue. In CH2Cl2 or MOE, at or near the m.p., the CIE coordinates of BO-1 undergo a sharp transition from blue to green, which is clearly caused by the phase change of the solvent. Above m.p. (the black region of the emission spectra), the emission peak moves back toward blue and increases intensity with increasing T. The switching of the closed form to the open form is likely a main cause for the emission colour change in the low T region. Variable temperature 11B NMR experiments revealed that the 11B chemical shift of BO-1 in CD2Cl2 is about ∼38 ppm at 243 K, near that of BO-2 (see Fig. S36†), an indication that at 243 K and below, the closed form likely has a major presence in solution. The emission λmax or the ratio of I490 nm/I525 nm (see Fig. S38(a)†) of BO-1 changes linearly and reversibly with T in the region of 223 K to 383 K in MOE, as shown in Fig. 7, an indication that BO-1 is an effective temperature sensor over a wide temperature range. Because the T-dependent change of the emission spectrum of BO-1 in MOE at 223–383 K closely resembles that of BN (except that BN has a steeper slope, compared to that shown in Fig. 7, see Fig. S40–S42 and S44(a)†), the emission colour change of BO-1 in this temperature region is likely dominated by the donor (amino)–acceptor ICT fluorescence change with T. In solvents such as benzene and hexane (see Fig. S39a and b†), c-BO-1 is more dominant than o-BO-1 at low T. Interestingly, the sharp solvent-phase transition-caused changes in the CIE diagram of BO-1 in CH2Cl2 or MOE were not observed in hexane. Nonetheless, the CIE diagram of BO-1 in hexane also displays two distinct regions of change, 77 K to 157 K, and 167 K to 298 K. The T-dependent emission colour change of BO-1 has an excellent reversibility at 223–383 K in MOE, as shown by Fig. 7. In contrast, the emission of BN shows a relatively poor reversibility with T under the same conditions (see Fig. S42†).
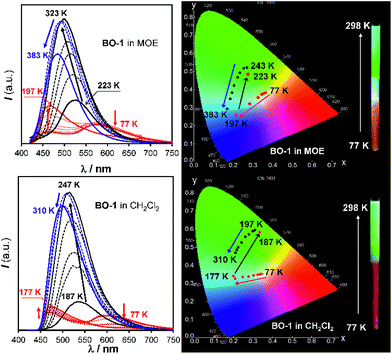 |
| | Fig. 6 Emission spectra (left) and CIE coordinate diagrams (right) of BO-1 in MOE (top) and CH2Cl2 (bottom) at various temperatures. Inset: photographs showing the emission colour change of BO-1 solutions with T in an NMR tube. The bottom half of the NMR tube was cooled to 77 K in liquid nitrogen and the photos were taken immediately after the NMR tube was pulled out of the liquid nitrogen. MOE represents 2-methoxyethyl ether. | |
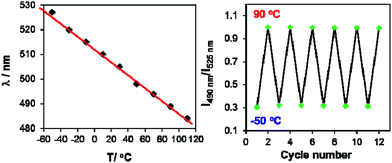 |
| | Fig. 7 Left: diagram showing the linear dependence of the emission λmax of BO-1 fluorescence in MOE on T from 223 K (−50 °C) to 383 K (110 °C). Right: diagram showing the reversible cycling of BO-1 emission between −50 °C and 90 °C, plotted with the intensity ratio of I490 nm/I525 nmvs. T. | |
The high contrast and reversible emission colour change from red/orange to blue/green of BO-1 over a wide temperature range is unique among the boron compounds examined in this work (BN has a deep blue emission colour at low T). Such a sharp contrasting colour change with T is rare for organic dyes. In a recently reported example,1a a multidentate Pt(II) chelate compound was shown to switch between monomer phosphorescence (sky-blue, low T) and excimer phosphorescence (red, high T), a trend opposite to that observed for BO-1. Compared to the Pt(II) based system, the BO-1 based system is certainly much easier and cheaper to synthesize, and simpler for incorporation into other supporting/functional systems such as polymers for practical temperature sensing applications, which is being investigated in our laboratory.
Emission colour change of BO-1 with pressure
In addition to temperature dependence, the emission spectrum of BO-1 also displays dependence on pressure. As shown in Fig. 8, upon grinding, the emission peak of BO-1 powder at 475 nm decreases in intensity and a new peak at ∼620 nm appears, which becomes the dominating emission peak with increasing grinding time. As a result, the emission colour of BO-1 powder changes from whitish blue to red-orange. The red emission band of BO-1 is in fact similar to those of BO-2 and BO-3 in the solid state (Fig. 8). The visual colour of BO-1 powder also changes from light yellow to orange with grinding. In addition, the amorphous film of BO-1 produced by casting the BO-1 solution in organic solvent such as CH2Cl2 on a glass substrate also has a red-orange emission band similar to that of the ground powder. Both the ground powder and the film of BO-1 have a distinct weak absorption band at ∼500 nm (see Fig. S30†), resembling those of BO-2 film/BO-3 solution/film, and BO-1 in THF:water solution (1:9). IR spectra showed the appearance of a distinct C–O stretching band at 1517 cm−1 for the partially ground powder of BO-1, which is identical to the strong C–O stretching band of BO-3 and BO powder (see Fig. S48†). Based on these observations, we suggest that the red-orange emission peak of the ground powder and the film of BO-1 likely originates from c-BO-1. The ground powder and the amorphous film of BO-1 can return to the crystalline state at ambient temperature, which can be accelerated with heating, either fully or partially restoring the blue emission peak (Fig. 8). It is possible to create blue and red emission patterns using a spatula to scratch the powder of BO-1 on a glass surface (see Fig. S45a†).
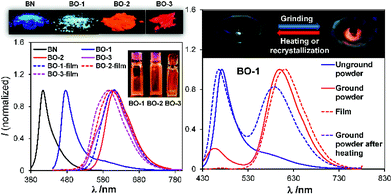 |
| | Fig. 8 Left: photographs showing the emission colours of BN, BO-1–BO-3 in the crystalline state and as amorphous films, and the corresponding emission spectra. Right, top: photographs showing the emission colour change of BO-1 powder in a mortar with grinding; bottom: emission spectra of BO-1 in various states. | |
Force-responsive systems based on boron compounds are very rare. To the best of our knowledge, the only well-established examples are those reported by Fraser and coworkers,7 which are based on BF2-chelated α,β-diketonate derivatives and do not involve structural change. Wakamiya and coworkers also recently reported a boron system that changes colour from yellow to green in the solid state upon grinding, and attributed this phenomenon to the change of the dye molecule from a crystalline state to a partially amorphous state.6aBO-1 is a new example of boron-based systems that display a drastic and reversible colour change in response to temperature and pressure.
Conclusions
We have established that the introduction of an ortho aldehyde group in proximity to a boron centre in an amino-triarylborane donor–acceptor system allows the achievement of a simple multi-stimuli responsive system, in which the reversible switching of a B ← O bond likely plays a key role. In addition, the imbedded intramolecular donor (amino)–acceptor (boron–aldehyde) charge transfer transition in this system also plays a critical role in modulating the structural switching and the emission colour. This finding introduces new guiding principles in molecular structure design/materials engineering for achieving simple and highly effective multi-responsive systems based on organoboron compounds.
Experimental
General
The THF solvent used for the synthesis of BO-1–BO-3 was freshly distilled over sodium metal and stored under nitrogen prior to use. All other solvents and commercial chemicals were used without further purification. UV-visible absorption spectra were recorded on a Cary 300 UV-Vis spectrophotometer. Fluorescence quantum efficiencies were determined using a Hamamatsu C11347-11 Quantaurus-QY spectrometer. 1H, 11B and 13C NMR spectra were recorded on a Bruker Avance 400 MHz or 700 MHz spectrometer. Luminescence spectra were recorded on an Edinburgh Instruments FLS980 or a Lengguang Tech F97 Pro spectrophotometer. Electrochemical measurements were conducted on an AUTOLAB-CV-75W analyzer with a scan rate of 100 mV s−1. The electrochemical cell was a standard three-compartment cell composed of a Pt working electrode, a Pt auxiliary electrode, and a Pt wire reference electrode. All measurements were performed using 0.10 M of NBu4PF6 in DMF as the supporting electrolyte. The potentials are reported relative to the ferrocene/ferrocenium couple. Powder X-ray diffraction patterns were recorded on a Bruker D8 Advance diffractometer. High resolution mass spectral data were obtained via ESI on an Agilent (Q-TOF 6520) analyzer. Column chromatography was carried out on silica gel (300–400 mesh). Analytical thin-layer chromatography was performed on glass plates of Silica Gel GF-254. Compounds BN10a and BO14 were prepared using methods described in the literature. 5-(N,N-Dimethylamino)-2-bromobenzaldehyde (1a) was also prepared according to a literature procedure.15
Synthesis of BO-1
Compound 1a (0.50 g, 2.19 mmol) and triethyl orthoformate (1.2 mL, 7.23 mmol) were dissolved in ethanol (10 mL), and then a catalytic amount of concentrated HCl (31 μL) was added. The reaction mixture was refluxed for 4 h. After compound 1a was completely consumed, the reaction mixture was brought to room temperature, then washed and extracted with water and ethyl acetate, respectively. The combined organic layers were washed with brine and dried over anhydrous Na2SO4. After removing the solvents under reduced pressure, compound 1b was obtained as a colourless liquid (0.65 g, 99% yield). 1H NMR (400 MHz, CDCl3): δ 7.33 (d, J = 8.8 Hz, 1H), 7.01 (d, J = 3.1 Hz, 1H), 6.55 (dd, J = 8.8, 3.1 Hz, 1H), 5.59 (s, 1H), 3.76–3.54 (m, 4H), 2.94 (s, 6H), 1.26 (t, J = 7.0 Hz, 6H) ppm. To an oven-dried Schlenk flask was added 1b (0.70 g, 2.63 mmol) and the flask was evacuated and back-filled with N2 three times. THF (10 mL) was then added and the mixture was cooled to −78 °C using an acetone and liquid nitrogen bath. n-BuLi (1.8 mL of 1.6 M solution in hexane, 2.89 mmol) was added over 30 min. After 1 h, a solution of Mes2BF (0.82 g, 2.73 mmol) in 4 mL of THF was added over 10 min. The reaction mixture was warmed to room temperature, and stirring was continued for 12 h. HCl (8 mL, 1 N) was added and stirring was continued for another 4 h. The reaction mixture was then extracted with diethyl ether. The combined organic layers were washed with a brine solution and dried over anhydrous Na2SO4. After removing the solvents at reduced pressure, the crude product was purified by silica gel chromatography using petroleum ether and ethyl acetate (20:1) as the eluent. Compound BO-1 was obtained as a yellow solid in 65% yield (0.67 g, 1.70 mmol). 1H NMR (400 MHz, C6D6): δ 9.86 (s, 1H), 7.55 (d, J = 8.4 Hz, 1H), 7.24 (d, J = 2.4 Hz, 1H), 6.79 (s, 4H), 6.45 (dd, J = 8.4, 2.4 Hz, 1H), 2.34 (s, 6H), 2.20 (s, 12H), 2.18 (s, 6H) ppm. 13C NMR (176 MHz, CD3CN): δ 197.19, 152.28, 142.75, 141.32, 137.77, 135.83, 129.55, 119.68, 111.01, 40.39, 23.89, 20.95 ppm. 11B NMR (128 MHz, C6D6): δ 60 ppm. HRMS (ESI, m/z): [M + H]+ calcd for C27H33BNO, 398.2650; found 398.2662.
Synthesis of BO-2
Compound 2b was prepared in the same manner as described above for compound 1b, using compound 2a (0.25 g, 0.82 mmol), triethyl orthoformate (0.45 mL, 2.71 mmol), ethanol (6 mL), and concentrated HCl (11.89 μL). Compound 2b was isolated as a white solid (0.28 g, 0.73 mmol, 89%). 1H NMR (400 MHz, CDCl3): δ 7.84 (d, J = 2.4 Hz, 1H), 7.53 (dd, J = 15.6, 8.6 Hz, 3H), 7.37 (dd, J = 8.3, 2.4 Hz, 1H), 6.79 (d, J = 8.8 Hz, 2H), 5.68 (s, 1H), 3.76–3.58 (m, 4H), 3.00 (s, 6H), 1.27 (t, J = 7.1 Hz, 6H). Using 2b as the starting material (0.25 g, 0.66 mmol), THF (10 mL), n-BuLi (0.45 mL of 1.6 M solution in hexane, 0.73 mmol), Mes2BF (0.22 g, 0.73 mmol), and 2 mL of 1 N HCl, compound BO-2 was synthesized according to the same procedure as described for BO-1. The crude product was purified by silica gel chromatography using CH2Cl2 and petroleum ether as the eluent (1:1). Compound BO-2 was obtained as red solid in 64% yield (0.20 g, 0.42 mmol). 1H NMR (400 MHz, C6D6): δ 8.59 (s, 1H), 7.88 (d, J = 7.8 Hz, 1H), 7.67 (d, J = 1.2 Hz, 1H), 7.58 (dd, J = 7.8, 1.6 Hz, 1H), 7.46 (d, J = 8.8 Hz, 2H), 6.85 (s, 4H), 6.65 (d, J = 8.8 Hz, 2H), 2.54 (s, 6H), 2.28 (s, 12H), 2.22 (s, 6H) ppm. 13C NMR (176 MHz, CDCl3): δ 196.47, 141.82, 141.01, 139.67, 136.16, 134.36, 133.19, 129.37, 127.92, 125.45, 24.40, 21.03 ppm. 11B NMR (128 MHz, C6D6): δ 30 ppm. HRMS (ESI, m/z): [M + H]+ calcd for C33H37BNO: 474.2963; found: 474.2966.
Synthesis of BO-3
A Schlenk flask was charged with 3a (354 mg; 1.46 mmol) and EtOH (7.0 mL) under air, and concentrated HCl (21 μL) was added. The solution was stirred at 100 °C for 4 h. Water (20 mL) and ethyl acetate (60 mL) were added, and the organic phase was separated, washed with water (3 × 20 mL), brine (30 mL), and dried with MgSO4. After the removal of the solvent, the product was dissolved in THF (20 mL). n-BuLi (1.6 M solution in hexane, 0.75 mL, 1.2 mmol) was added dropwise at −78 °C. The mixture was stirred for 1 h at −78 °C and BMes2F in THF (10 mL) was added, the solution was stirred at −78 °C for 1 h, then warmed to room temperature and stirred for 12 h. The HCl (1 mL, 1 M) was added to the mixture, and the solution was stirred at room temperature for 10 min. After the removal of the solvent, purification of the crude product by column chromatography (ethyl acetate/petroleum ether = 1/10) afforded the product as a red solid, 0.30 g, yield: 50%. 1H NMR (400 MHz, C6D6): δ 7.87 (d, J = 8.3 Hz, 1H), 7.15 (s, 1H), 6.87 (s, 4H), 6.70 (d, J = 8.3 Hz, 1H), 6.61 (s, 1H), 2.44 (s, 6H), 2.35 (s, 12H), 2.23 (s, 6H), 1.84 (s, 3H) ppm. 13C NMR (176 MHz, C6D6): δ 206.81, 149.05, 140.62, 138.43, 133.98, 132.60, 129.73, 127.98, 122.48, 106.38, 39.79, 24.85, 20.69, 19.23 ppm. 11B NMR (225 MHz, C6D6): δ 15.61 ppm. HRMS (ESI, m/z): [M + H]+ calcd for C28H34BNO, 412.2812; found 412.2804.
Computational study
DFT and TD-DFT calculations were performed using the Gaussian 09 suite of programs18 on the High Performance Computing Virtual Laboratory (HPCVL) at Queen's University. Geometry optimizations and vertical excitations of all compounds were obtained at the B3LYP19/6-31g(d)20 level of theory and the resulting structures were confirmed to be stationary points through vibrational frequency analysis. Calculated UV/Vis and IR spectra were generated by GaussSum V3.0.21 The transition state involved in the interconversion of the open BO-1 and the closed BO-1 structures was located by scanning the potential energy surface with the ModRedundant option in Gaussian along the boron–oxygen bond length, followed by an optimization. Vibrational frequency analysis contained only one true imaginary frequency, indicating that the transition state optimization was successful. For details, see ESI.†
Single-crystal X-ray diffraction analysis
The crystal data of BO, BO-1, BO-2 and BO-3 were collected on a Bruker D8-Venture diffractometer with Mo-target (λ = 0.71073 Å) at 180 K. Data were processed on a PC with the aid of the Bruker SHELXTL software package22 and corrected for absorption effects. The crystals of BO-1, BO-2 and BO-3 belong to the monoclinic space group P21/n, C2/c, and P21/c, respectively, while the crystals of BO belong to the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) . All non-hydrogen atoms were refined anisotropically. The positions of hydrogen atoms were calculated and their contributions in structural factors were included. The details of crystal data, collection parameters and results of analyses are provided in the ESI.† The crystal data of BO, BO-1, BO-2 and BO-3 were deposited to the Cambridge Crystallographic Data Center with deposition numbers of CCDC 1568989 (BO-1), 1568988 (BO-2), 1585930 (BO-3), 1569175 (BO).
. All non-hydrogen atoms were refined anisotropically. The positions of hydrogen atoms were calculated and their contributions in structural factors were included. The details of crystal data, collection parameters and results of analyses are provided in the ESI.† The crystal data of BO, BO-1, BO-2 and BO-3 were deposited to the Cambridge Crystallographic Data Center with deposition numbers of CCDC 1568989 (BO-1), 1568988 (BO-2), 1585930 (BO-3), 1569175 (BO).
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
Y. G. S., G. F. H., X. L., N. W., T. P. and S. W. thank the National Natural Science Foundation of China for financial support (grant no. 201501011, 201571017 and 21701011). H. J. L. thanks the Chinese Scholarship Council for graduate scholarship. H. J. L., S. K. M, and S. W. thank the Natural Sciences and Engineering Research Council of Canada for financial support (RGPIN 1193993-2013) and the High-Performance Computing Virtual Laboratory (HPCVL) at Queen's University for the computing facility.
Notes and references
-
(a) L. Liu, X. Wang, N. Wang, T. Peng and S. Wang, Angew. Chem., Int. Ed., 2017, 56, 9160 CrossRef CAS PubMed;
(b) P. Chen, Q. Li, S. Grindy and N. Holten-Andersen, J. Am. Chem. Soc., 2015, 137, 11590 CrossRef CAS PubMed;
(c) H.-Y. Ku, B. Tong, Y. Chi, H.-C. Kao, C.-C. Yeh, C.-H. Chang and G.-H. Lee, Dalton Trans., 2015, 44, 8552 RSC;
(d) X. Zhang, J. Y. Wang, J. Ni, L. Y. Zhang and Z. N. Chen, Inorg. Chem., 2012, 51, 5569 CrossRef CAS PubMed;
(e) D. S. Achilleos and M. Vamvakaki, Macromolecules, 2010, 43, 7073 CrossRef CAS.
- J. C. Chan, W. H. Lam, H. L. Wong, N. Zhu, W. T. Wong and V. W. Yam, J. Am. Chem. Soc., 2011, 133, 12690 CrossRef CAS PubMed.
-
(a) P. Xue, J. Ding, P. Wang and R. Lu, J. Mater. Chem. C, 2016, 4, 6688 RSC;
(b) L. M. Huang, G. M. Tu, Y. Chi, W. Y. Hung, Y. C. Song, M. R. Tseng, P. T. Chou, G. H. Lee, K. T. Wong, S. H. Cheng and W. S. Tsai, J. Mater. Chem. C, 2013, 1, 7582 RSC.
-
(a) A. Y.-Y. Tam, K. M.-C. Wong and V. W.-W. Yam, J. Am. Chem. Soc., 2009, 131, 6253 CrossRef CAS PubMed;
(b) K. Nishino, H. Yamamoto, K. Tanaka and Y. Chujo, Org. Lett., 2016, 18, 4064 CrossRef CAS PubMed.
- Y. Xing, C. Liu, J. H. Xiu and J. Y. Li, Inorg. Chem., 2015, 54, 7783 CrossRef CAS PubMed.
-
(a) H. Shimogawa, O. Yoshikawa, Y. Aramaki, M. Murata, A. Wakamiya and Y. Murata, Chem.–Eur. J., 2017, 23, 3784 CrossRef CAS PubMed;
(b) Y. Cao, J. K. Nagle, M. O. Wolf and B. O. Patrick, J. Am. Chem. Soc., 2015, 137, 4888 CrossRef CAS PubMed;
(c) N. Kano, J. Yoshino and T. Kawashima, Org. Lett., 2005, 7, 3909 CrossRef CAS PubMed;
(d) J. Yoshino, N. Kano and T. Kawashima, Tetrahedron, 2008, 64, 7774 CrossRef CAS;
(e) J. Feng, L. Xiong, S. Wang, S. Li, Y. Li and G. Yang, Adv. Funct. Mater., 2013, 23, 340 CrossRef CAS;
(f) J. Feng, K. Tian, D. Hu, S. Wang, S. Li, Y. Zeng, Y. Li and G. Yang, Angew. Chem., Int. Ed., 2011, 50, 8072 CrossRef CAS PubMed;
(g) Y. L. Rao, H. Amarne, S. B. Zhao, T. M. McCormick, S. Martic, Y. Sun, R. Y. Wang and S. Wang, J. Am. Chem. Soc., 2008, 130, 12898 CrossRef CAS PubMed;
(h) Y. L. Rao, H. Amarne and S. N. Wang, Coord. Chem. Rev., 2012, 256, 759 CrossRef CAS;
(i) J. Wang, B. Jin, N. Wang, T. Peng, X. Li, Y. Luo and S. Wang, Macromolecules, 2017, 50, 4629 CrossRef CAS;
(j) A. Iida, S. Saito, T. Sasamori and S. Yamaguchi, Angew. Chem., Int. Ed., 2013, 52, 3760 CrossRef CAS PubMed;
(k) H. Wang, J. Zhang and Z. Xie, Angew. Chem., Int. Ed., 2017, 56, 9198 CrossRef CAS PubMed;
(l) H. Qian, Y. Y. Wang, D. S. Guo and I. Aprahamian, J. Am. Chem. Soc., 2017, 139, 1037 CrossRef CAS PubMed;
(m) K. Dhanunjayarao, V. Mukundam and K. Venkatasubbaiah, Inorg. Chem., 2016, 55, 11153 CrossRef CAS PubMed.
-
(a) G. Zhang, J. Lu, M. Sabat and C. L. Fraser, J. Am. Chem. Soc., 2010, 132, 2160 CrossRef CAS PubMed;
(b) G. Zhang, J. P. Singer, S. E. Kooi, R. E. Evans, E. L. Thomas and C. L. Fraser, J. Mater. Chem., 2011, 21, 8295 RSC;
(c) H. Naito, K. Nishino, Y. Morisaki, K. Tanaka and Y. Chujo, Angew. Chem., Int. Ed., 2017, 56, 254 CrossRef CAS PubMed.
-
(a) H. Sohn, M. J. Sailor, D. Magde and W. C. Trogler, J. Am. Chem. Soc., 2003, 125, 3821 CrossRef CAS PubMed;
(b) Y. Liu, Y. Tang, N. N. Barashkov, I. S. Irgibaeva, J. W. Y. Lam, R. Hu, D. Birimzhanova, Y. Yu and B. Z. Tang, J. Am. Chem. Soc., 2010, 132, 13951 CrossRef CAS PubMed;
(c) Z. J. Zhao, B. Hea and B. Z. Tang, Chem. Sci., 2015, 6, 5347 RSC;
(d) J. N. Zhang, H. Kang, N. Li, S. M. Zhou, H. M. Sun, S. W. Yin, N. Zhao and B. Z. Tang, Chem. Sci., 2017, 8, 577 RSC.
-
(a) L. Ji, S. Griesbeck and T. B. Marder, Chem. Sci., 2017, 8, 846 RSC;
(b) F. Jäkle, Chem. Rev., 2010, 110, 3985 CrossRef PubMed;
(c) C. R. Wade, A. E. J. Broomsgrove, S. Aldridge and F. P. Gabbaï, Chem. Rev., 2010, 110, 3958 CrossRef CAS PubMed;
(d) Z. M. Hudson and S. Wang, Acc. Chem. Res., 2009, 42, 1584 CrossRef CAS PubMed;
(e) Y. Shirota and H. Kageyama, Chem. Rev., 2007, 107, 953 CrossRef CAS PubMed;
(f) C. Reus, S. Weidlich, M. Bolte, H.-W. Lerner and M. Wagner, J. Am. Chem. Soc., 2013, 135, 12892 CrossRef CAS PubMed.
-
(a) Z. Yuan, C. D. Entwistle, J. C. Collings, D. Albesa-Jov, A. S. Batsanov, J. A. K. Howard, N. J. Taylor, H. M. Kaiser, D. E. Kaufmann, S.-Y. Poon, W.-Y. Wong, C. Jardin, S. Fathallah, A. Boucekkine, J.-F. Halet and T. B. Marder, Chem.–Eur. J., 2006, 12, 2758 CrossRef CAS PubMed;
(b) Z. Yuan, J. C. Collings, N. J. Taylor, T. B. Marder, C. Jardin and J.-F. Halet, J. Solid State Chem., 2000, 154, 5 CrossRef CAS;
(c) Z. Yuan, N. J. Taylor, Y. Sun, T. B. Marder, I. D. Williams and C. Lap-Tak, J. Organomet. Chem., 1993, 449, 27 CrossRef CAS;
(d) Z. Yuan, N. J. Taylor, T. B. Marder, I. D. Williams, S. K. Kurtz and L.-T. Cheng, J. Chem. Soc., Chem. Commun., 1990, 1489 RSC.
-
(a) P. Chen, R. A. Lalancette and F. Jäkle, Angew. Chem., Int. Ed., 2012, 51, 7994 CrossRef CAS PubMed;
(b) Y. Kim and F. P. Gabbaï, J. Am. Chem. Soc., 2009, 131, 3363 CrossRef CAS PubMed;
(c) S. Yamaguchi, S. Akiyama and K. Tamao, J. Am. Chem. Soc., 2001, 123, 11372 CrossRef CAS PubMed;
(d) X. D. Yin, K. L. Liu, Y. Ren, R. A. Lalancette, Y.-L. Loo and F. Jäkle, Chem. Sci., 2017, 8, 5497 RSC.
-
(a) X. Long, Z. Ding, C. Dou, J. Zhang, J. Liu and L. Wang, Adv. Mater., 2016, 28, 6504 CrossRef CAS PubMed;
(b) C. Dou, X. Long, Z. Ding, Z. Xie, J. Liu and L. Wang, Angew. Chem., Int. Ed., 2016, 55, 1436 CrossRef CAS PubMed;
(c) R. Zhao, C. Dou, Z. Xie, J. Liu and L. Wang, Angew. Chem., Int. Ed., 2016, 55, 5313 CrossRef CAS PubMed;
(d) C. Dou, Z. Ding, Z. Zhang, Z. Xie, J. Liu and L. Wang, Angew. Chem., Int. Ed., 2015, 54, 3648 CrossRef CAS PubMed.
-
(a) Y. J. Shiu, Y. C. Cheng, W. L. Tsai, C. C. Wu, C. T. Chao, C. W. Lu, Y. Chi, Y. T. Chen, S. H. Liu and P. T. Chou, Angew. Chem., Int. Ed., 2016, 55, 3017 CrossRef CAS PubMed;
(b) K. Suzuki, S. Kubo, K. Shizu, T. Fukushima, A. Wakamiya, Y. Murata, C. Adachi and H. Kaji, Angew. Chem., Int. Ed., 2015, 54, 15231 CrossRef CAS PubMed;
(c) H. Hirai, K. Nakajima, S. Nakatsuka, K. Shiren, J. Ni, S. Nomura, T. Ikuta and T. Hatakeyama, Angew. Chem., Int. Ed., 2015, 54, 13581 CrossRef CAS PubMed;
(d) F. H. Li, W. L. Jia, S. Wang, Y. Q. Zhao and Z. H. Lu, J. Appl. Phys., 2008, 103, 034509 CrossRef;
(e) W. L. Jia, X. D. Feng, D. R. Bai, Z. H. Lu, S. Wang and G. Vamvounis, Chem. Mater., 2005, 17, 164 CrossRef CAS;
(f) D. L. Crossley, I. A. Cade, E. R. Clark, A. Escande, M. J. Humphries, S. M. King, I. Vitorica-Yrezabal, M. J. Ingleson and M. L. Turner, Chem. Sci., 2015, 6, 5144 RSC.
-
(a) B. Neue, R. Fröhlich, B. Wibbeling, A. Fukazawa, A. Wakamiya, S. Yamaguchi and E.-U. Würthwein, J. Org. Chem., 2012, 77, 2176 CrossRef CAS PubMed;
(b) Z. García-Hernández and F. P. Gabbaï, Z. Naturforsch., 2009, 64b, 1381 Search PubMed.
- M. Barbasiewicz, M. Michalak and K. Grela, Chem.–Eur. J., 2012, 18, 14237 CrossRef CAS PubMed.
-
(a) X. G. Liu, Q. L. Qiao, W. M. Tian, W. J. Liu, J. Chen, M. J. Lang and Z. C. Xu, J. Am. Chem. Soc., 2016, 138, 6960 CrossRef CAS PubMed;
(b) G. Balkowski, A. Szemik-Hojniak, I. H. M. van Stokkum, H. Zhang and W. J. Buma, J. Phys. Chem. A, 2005, 109, 3535 CrossRef CAS PubMed;
(c) X. G. Liu, J. M. Cole and Z. C. Xu, J. Phys. Chem. C, 2017, 121, 13274 CrossRef CAS.
-
F. A. Carey and R. J. Sundberg, Advanced Organic Chemistry, Part A: Structure and Mechanism, Plenum Press, New York, 3rd edn, 1990 Search PubMed.
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. H. Petersson, M. Nakatsuji, X. Caricato, H. P. F. Li, A. Hratchian, J. Izmaylov, G. Bloino, J. L. Zheng, M. Sonnenberg, M. Hada, K. Ehara, R. Toyota, J. Fukuda, M. Hasegawa, T. Ishida, Y. Nakajima, O. Honda, H. Kitao, T. Nakai, J. A. Vreven, J. E. Montgomery Jr, F. Peralta, M. Ogliaro, J. J. Bearpark, E. Heyd, K. N. Brothers, V. N. Kudin, T. Staroverov, R. Keith, J. Kobayashi, K. Normand, A. Raghavachari, J. C. Rendell, S. S. Burant, J. Iyengar, M. Tomasi, N. Cossi, J. M. Rega, M. Millam, J. E. Klene, J. B. Knox, V. Cross, C. Bakken, J. Adamo, R. Jaramillo, R. E. Gomperts, O. Stratmann, A. J. Yazyev, R. Austin, C. Cammi, J. W. Pomelli, R. L. Ochterski, K. Martin, V. G. Morokuma, G. A. Zakrzewski, P. Voth, J. J. Salvador, S. Dannenberg, A. D. Dapprich, O. Daniels, J. B. Farkas, J. V. Foresman, J. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09 Revision C.01, 2010 Search PubMed.
- T. Yanai, D. Tew and N. Handy, Chem. Phys. Lett., 2004, 393, 51 CrossRef CAS.
-
(a) R. Ditchfield, W. J. Hehre and J. A. Pople, J. Chem. Phys., 1971, 54, 724 CrossRef CAS;
(b) A. D. McLean and G. S. Chandler, J. Chem. Phys., 1980, 72, 5639 CrossRef CAS.
- N. M. O'Boyle, A. L. Tenderholt and K. M. Langner, J. Comput. Chem., 2008, 29, 839 CrossRef PubMed.
-
SHELXTL, version 6.14; Bruker AXS, Madison, WI, 2000–2003 Search PubMed.
|
| This journal is © The Royal Society of Chemistry 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *ab
*ab