
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Mechanistic insights into boron-catalysed direct amidation reactions†
Sergey
Arkhipenko
a,
Marco T.
Sabatini
b,
Andrei S.
Batsanov
c,
Valerija
Karaluka
b,
Tom D.
Sheppard
 b,
Henry S.
Rzepa
d and
Andrew
Whiting
*a
b,
Henry S.
Rzepa
d and
Andrew
Whiting
*a
aCentre for Sustainable Chemical Processes, Department of Chemistry, Durham University, Science Site, South Road, Durham, DH1 3LE, UK. E-mail: andy.whiting@durham.ac.uk
bDepartment of Chemistry, University College London, 20 Gordon Street, London, WC1H 0AJ, UK
cDepartment of Chemistry, Durham University, Science Site, South Road, Durham, DH1 3LE, UK
dDepartment of Chemistry, Imperial College London, South Kensington Campus, London, SW7 2AZ, UK
First published on 2nd January 2018
Abstract
The generally accepted monoacyloxyboron mechanism of boron-catalysed direct amidation is brought into question in this study, and new alternatives are proposed. We have carried out a detailed investigation of boron-catalysed amidation reactions, through study of the interaction between amines/carboxylic acids and borinic acids, boronic acids and boric acid, and have isolated and characterised by NMR/X-ray crystallography many of the likely intermediates present in catalytic amidation reactions. Rapid reaction between amines and boron compounds was observed in all cases, and it is proposed that such boron–nitrogen interactions are highly likely to take place in catalytic amidation reactions. These studies also clearly show that borinic acids are not competent catalysts for amidation, as they either form unreactive amino-carboxylate complexes, or undergo protodeboronation to give boronic acids. It therefore seems that at least three free coordination sites on the boron atom are necessary for amidation catalysis to occur. However, these observations are not consistent with the currently accepted ‘mechanism’ for boron-mediated amidation reactions involving nucleophilic attack of an amine onto a monomeric acyloxyboron intermediate, and as a result of our observations and theoretical modelling, alternative proposed mechanisms are presented for boron-mediated amidation reactions. These are likely to proceed via the formation of a dimeric B–X–B motif (X = O, NR), which is uniquely able to provide activation of the carboxylic acid, whilst orchestrating the delivery of the amine nucleophile to the carbonyl group. Quantum mechanical calculations of catalytic cycles at the B3LYP+D3/Def2-TZVPP level (solvent = CH2Cl2) support the proposal of several closely related potential pathways for amidation, all of which are likely to be lower in energy than the currently accepted mechanism.
Introduction
Boron compounds have a rich, fascinating and complex structural chemistry, especially related to clusters, cages, complexes and anions associated with B–O1 and B–N2 containing systems. Indeed, such systems are well known to “have a distinct tendency to disproportionate, apparently more so than those of any other nonmetallic elements”.1a This observation means that identifying and controlling the role played by boron in reactions and catalytic processes is particularly challenging and has proved so on many occasions.Elegant single and cooperative solutions, exemplified in Scheme 1, have enabled the precise reactivity control of boron in different reactions. For example, in the catalytic asymmetric CBS reduction system3 both nitrogen and oxygen ligands control boron coordination to tune an external boron to behave cooperatively to control ketone Lewis-acid binding, activation and hence, reduction. Similarly, precise boron centre control can be employed to achieve highly efficient hydroxynaphthaquinone chiral Lewis-acid binding, activation and subsequent asymmetric Diels–Alder cycloaddition;4 the boron control and prevention of diborate-disproportionation deriving from use of hindered BINOL ligands, assembled by careful manipulation of unstable acyloxyboron species. Indeed, the model for these boron complexes was derived from ‘triacetoxyborate’ complexes of chiral juglone derivatives. However, triacetoxyborate is not stable at ambient temperatures and it rearranges to give tetra-acetyl diborate, as confirmed by X-ray crystallography.5 These seminal works led us to conclude that reaction mechanisms involving boron are non-trivial, because the structural chemistry of boron is itself non-trivial.
 | ||
| Scheme 1 General structural role of boron in different catalytic processes. | ||
It is generally the case that in the absence of a catalyst, amines and carboxylic acids do not usually react to form amides at ambient temperatures, and forcing conditions are often required to promote this condensation reaction to access amides.6 Although a direct thermally accessible reaction is possible, direct amidation is better facilitated at lower temperatures by using catalysts,7 with boron-based systems being particularly effective.8 Useful catalysts for direct amidation include boric acid,9 borate esters10 and boronic acids.11–13 Borinic acids have also been claimed to promote direct amidation reactions under relatively mild conditions.14 Clearly, an understanding of the mechanism of these boron-mediated amidations is crucial for designing catalysts with improved activity, but more importantly, a fundamental understanding of the role of the boron in such systems is required. To this end, we have been interested in the structural and mechanistic chemistry associated with boron compounds employed in direct amidation for several years.12a The diversity of the catalytic systems reported to date, from simple boric acid9 or boronic acids11–13 to carefully designed cooperative catalysts,15 including poly-boron-based systems,16 poses a challenge in terms of identifying key structural motifs which are necessary for effective catalysis. Even considering boronic acid catalysts alone, bifunctional systems containing nitrogen and boron,12 or iodine/bromine and boron13 and electron deficient aryl boronic acids11 have all been demonstrated to be highly effective. Currently, a comprehensive understanding of how the catalyst structure affects the activity is clearly lacking.
Previously, an acyloxyboron-based intermediate was proposed as the reactive catalytic species,11a with computational studies suggesting that it was energetically plausible (Scheme 2A).17 As shown in Scheme 2, this mechanism involved the formation of the acyloxyboron derivative which then had to undergo nucleophilic attack by an amine to give the amide, after loss of water. However, despite the fact that DFT calculations17 suggest that acyloxyboron species are feasible intermediates in direct amidation catalysis, actual definitive physical evidence of the mechanistic role of boron in such reactions is sparse and could be misleading due to the potential for numerous possible boron species to be present in such reactions.12a Indeed, it should be noted that acyloxyboron species are particularly prone to dimerization/oligomerization, as mentioned above with regard to B(OAc)3 (vide supra and Scheme 1).
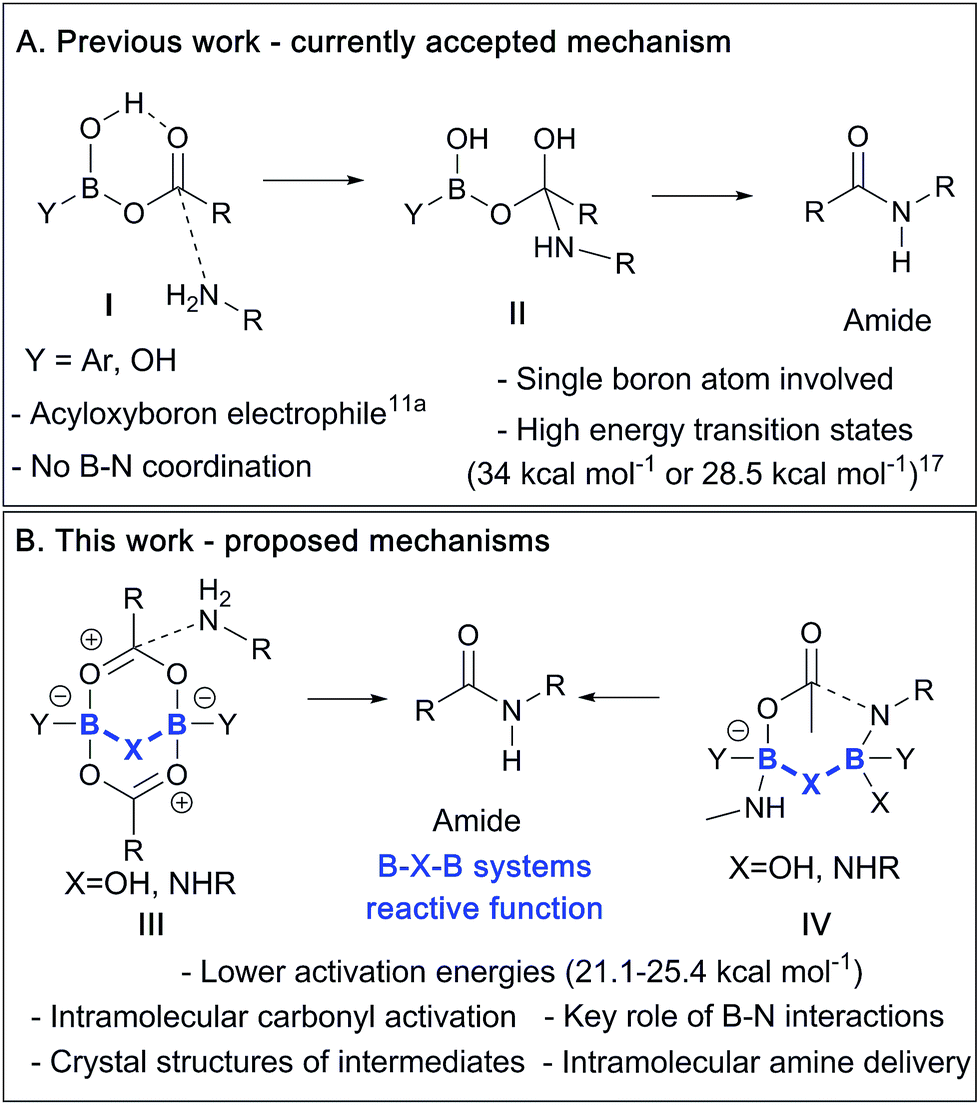 | ||
| Scheme 2 (A) Currently proposed mechanism via a monomeric acyloxyboron intermediate. (B) Summary of mechanisms proposed in this paper involving dimeric B–X–B motifs. | ||
In this work, we report recent studies in which we have probed the types of species potentially involved in boron-catalysed amidation reactions using a combination of experimental, structural and theoretical approaches. As a result, we postulate that acyloxyboron species like I (Scheme 2A) are unlikely to be sufficiently reactive as acylating agents, and we propose new mechanistic pathways for boron-mediated amidation which proceed through B–X–B systems, as shown in Scheme 2B. We also demonstrate their feasibility experimentally, supported by computational methods. Indeed, several routes for catalytic amidation are suggested, all of which were calculated to have lower energy barriers than the previously postulated single boron atom mechanisms.17 Importantly, they involve intermediates with two boron centres, and take account of the possible role of both boron–nitrogen and boron–oxygen interactions, which can be readily observed between amines and boronic or borinic acids, and between these boron compounds and carboxylic acids. These observations point to a generic structural motif which can potentially explain the success of cooperative,15 bifunctional12a and polyboron amidation catalysts,16 and they may even explain catalysis in other, related reactions.18
Results and discussion
Observations on the acyloxy mechanism
In recent computational studies,17 both tetrahedral17a and trigonal17b acyloxyboron species were considered as possible active acylating agents, with the latter leading to a lower energy barrier for the reaction with an amine. There is little experimental evidence for these proposed intermediates,11a and only a few classes of well-characterised acyloxyboron compounds have been reported.4,5,18 It is also not clear whether acyloxyboron compounds are effective acylating agents for amines; certainly, trigonal acyloxyboron compounds are likely to be Lewis acidic, and therefore, reaction of an amine at the boron atom should have a much lower energy barrier than the alternative nucleophilic attack at the carbonyl leading to amide formation. Indeed, readily accessible acyloxyboron compound 1a19a reacted cleanly with benzylamine to give the ‘ate’-complex 2 (Scheme 3). Even in the presence of excess amine, no conversion to amide was observed. An investigation of previously reported tetrahedral acyloxyboron species had suggested that they were also ineffective as acylating agents for amines. For example, borinate 1b, generated by reaction of triethylborane with glycine, was recovered unchanged after heating at reflux in toluene with benzylamine,19b and preliminary work in our laboratories found that MIDA-boronate 1c also showed no reactivity towards amines under mild conditions (others have reported related observations20) which clearly demonstrates that such species are not effective acylating agents. These results contrast with the mechanism examined computationally by Marcelli in which a tetrahedral acyloxyboronate was proposed to undergo attack by an amine in a reaction that typically takes place at room temperature.17a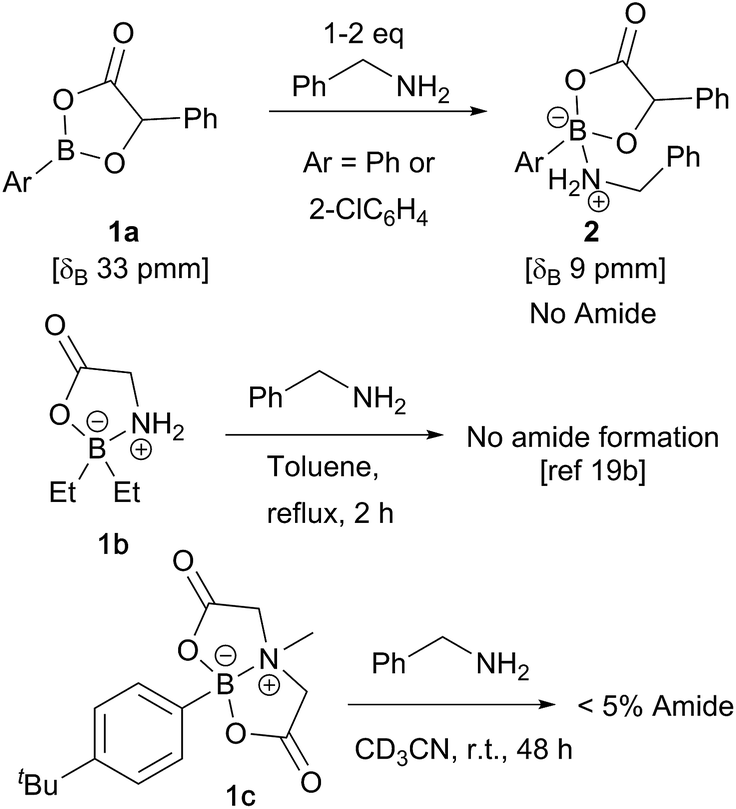 | ||
| Scheme 3 Monomeric acyloxyboron species typically show low reactivity towards amines. | ||
Borinic acids in direct amide formation
Analysis of carboxylic acid/amine/boronic acid reaction systems is a complicated task because multiple equilibria coexist in such reaction mixtures,12a leading to the presence of many different species. Hence, in order to better understand the potential catalytic role of boronic acids in direct amidation, we examined related diarylborinic acids as model systems. Interestingly, in our hands,14 these compounds were found to be ineffective, unreactive direct amidation catalysts, and therefore, ideal for probing the structural effects of coordination versus reaction with both carboxylic acids and amines.Our first finding was that amines (e.g. benzylamine and ethylenediamine) readily coordinate to borinic acids 3a–c (freshly obtained from their corresponding ethanolamine complexes, see ESI†) with formation of the corresponding “ate”-complexes 4 (see Scheme 4). Complexes of type 4 were observed by 11B NMR via a signal shift from δ 40–45 ppm for the borinic acids 3 to a peak in the “tetrahedral” region around 2–6 ppm. Importantly, this diagnostic shift was observed in all cases, and interestingly, when bis-3,4,5-trifluorophenylborinic acid 3a was employed, the adducts 4a and 4b crystallised directly from the reaction mixtures allowing unambiguous structural verification by X-ray crystallography (see Fig. 1).
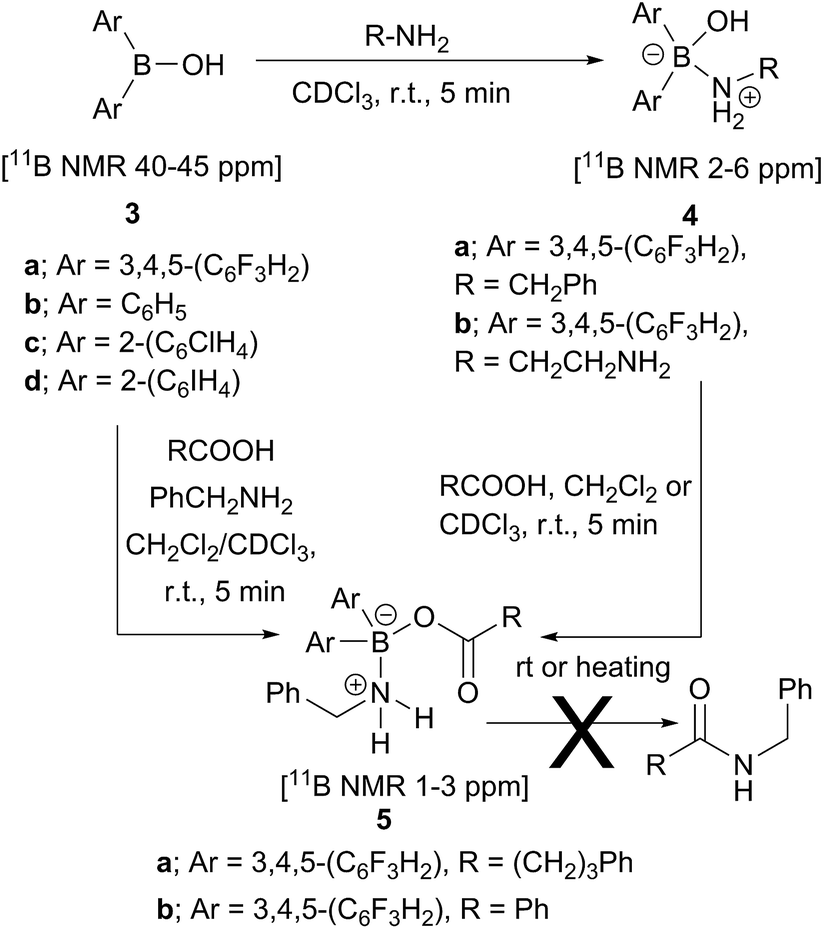 | ||
| Scheme 4 Reactions between borinic acids 3 and amines to give complexes 4, and subsequent conversion into aminocarboxylate complexes 5, which are not readily converted into amides. | ||
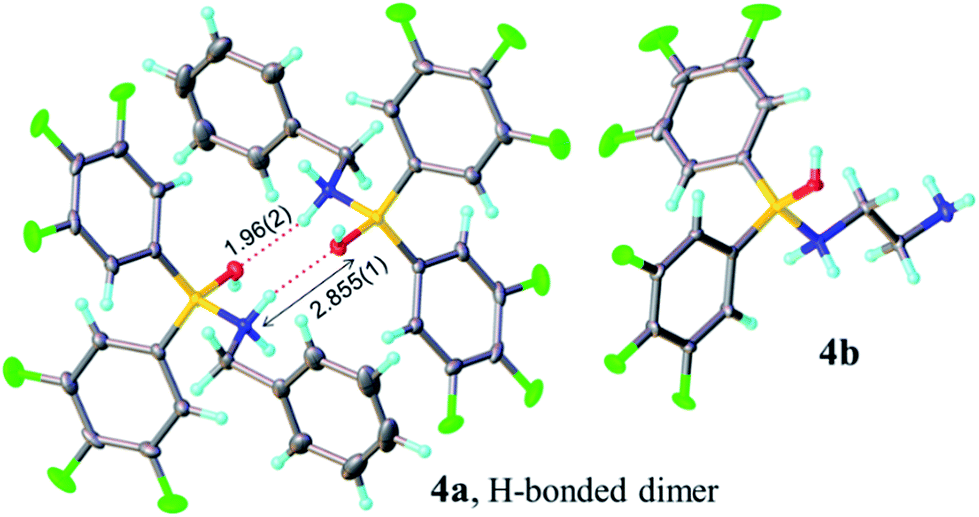 | ||
| Fig. 1 X-ray structures of Lewis adducts 4a (H-bonded dimer) and 4b. | ||
Borinic acid–amine complexes 4 were found to react with carboxylic acids (e.g. benzoic acid and 4-phenylbutyric acid) resulting in amine–carboxylic acid complexes 5 (Scheme 4). The same complexes 5 were also formed if all three components (borinic acid, amine and carboxylic acid) were mixed together simultaneously (see ESI† for related reactions and structures from the reactions of 3b and 3c). In many cases, these closely related products were also unambiguously characterised by X-ray crystallography (see Fig. 2). It should be noted that complexes 5 are structurally closely related to the acyloxyboron intermediate proposed for direct amidation reactions (I, Scheme 2A), with the NH2 unit potentially able to hydrogen bond to the carbonyl oxygen to provide activation. Most importantly, however, complexes 5 could not be converted to amides either by heating, or by addition of further aliquots of acid, amine or base; a result which strongly corroborates our hypothesis that, in general, single-boron acyloxy compounds are not competent acylating agents for amines.
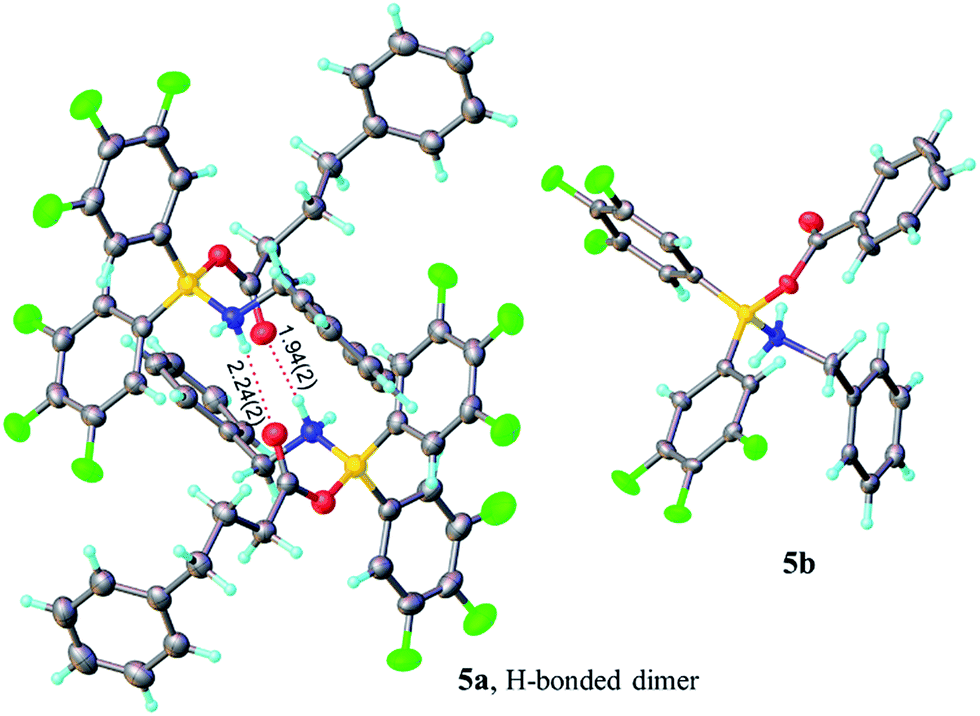 | ||
| Fig. 2 X-ray structures of amino-carboxylate borinic complexes 5a (showing a hydrogen-bonded dimer of two independent molecules) and 5b (disorder of phenyl ring is omitted for clarity). For 5c and 5d see ESI.† | ||
In contrast, when bis-3,4,5-trifluorophenylborinic acid 3a was reacted with carboxylic acids alone, immediate formation of a boron “ate”-complex was observed according to 11B NMR (Fig. 3), as shown in Scheme 5, and a subsequent slow protodeboronation reaction occurred which generated a boronic species (see Fig. 3). This was followed, in this case, by slow crystallisation of the corresponding boroxine 7a (Fig. 4). Hence, we propose that the “ate”-complex observed by 11B NMR was 6 (Scheme 5 and Fig. 3) based on systems of this type which have been isolated.21 In addition, the mass spectrum of the mixture showed an m/z 725.1340 (calcd m/z for 6a [M − H] 725.1343). Complex 6a underwent protodeboronation–dehydration to give the notably uncomplexed boroxine 7a, and its corresponding boronic acid 8a. The fact that such reactions occur readily at room temperature shows that formation of B–O–B species is extremely facile. Both borinic and boronic acids can thus readily form bridged B–O–B systems such as 6 and 7 (and 11, vide infra) which demonstrates that such motifs play an important role, both in boron chemistry in general, and likely also in direct amidation catalysis. It also exemplifies the ease with which a borinic acid undergoes protodeboronation in the presence of a carboxylic acid.
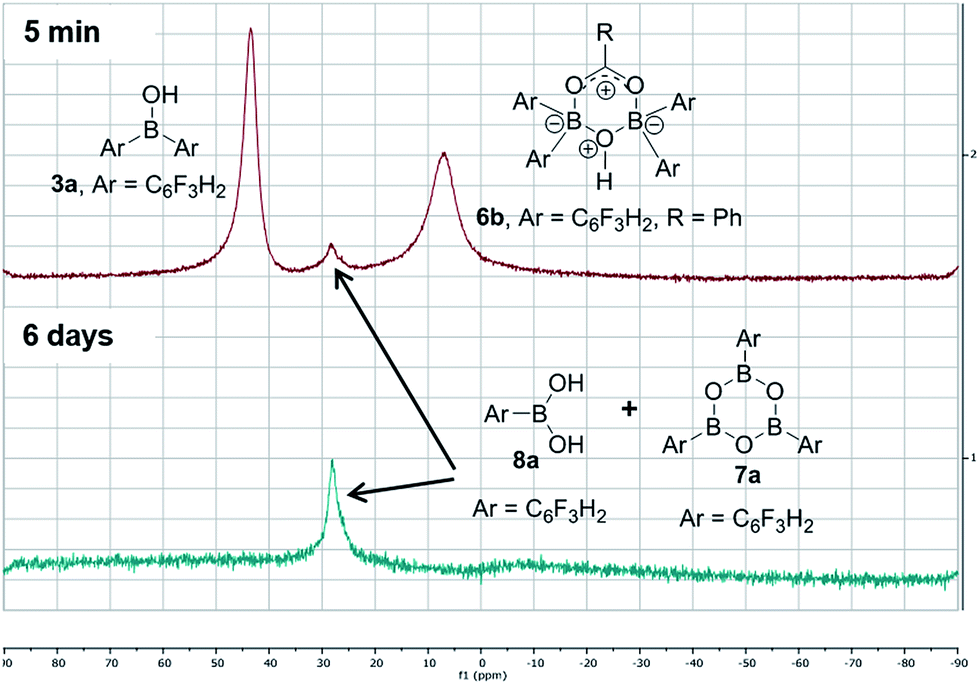 | ||
| Fig. 3 11B NMRs of a mixture of bis-3,4,5-trifluorophenylborinic acid 3a with benzoic acid after 5 min and after 6 days upon mixing (Ar = 3,4,5-trifluorophenyl). | ||
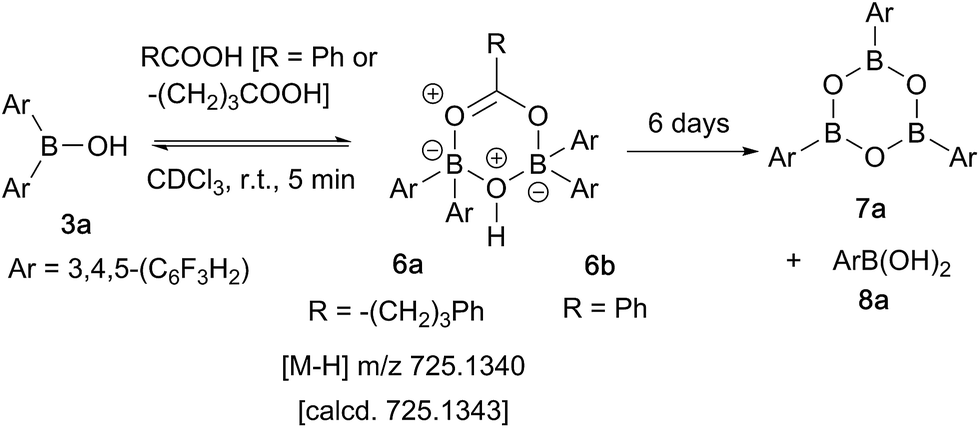 | ||
| Scheme 5 Reaction of 3,4,5-trifluorophenylborinic acid 3a with 4-phenylbutyric or benzoic acid, resulting in boroxine formation. | ||
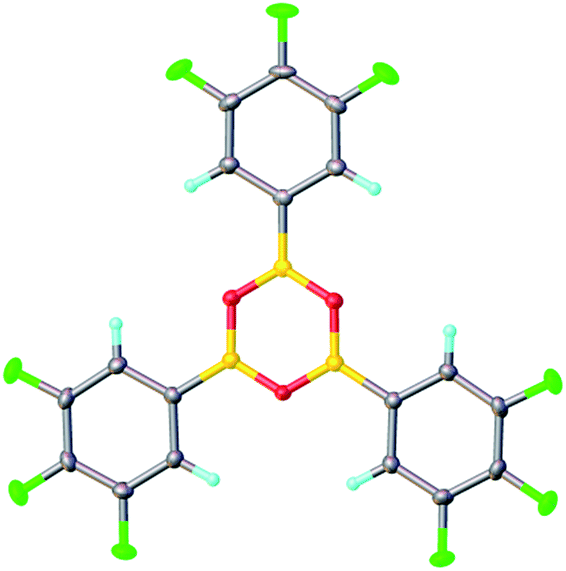 | ||
| Fig. 4 X-ray structure of 3,4,5-trifluorophenylboroxine 7a in orthorhombic β-form at 120 K. | ||
Having observed that borinic acids do not mediate amide bond formation, we were surprised by a recent report describing borinic acid-catalysed amide formation.14 However, when we attempted amidations under exactly the same conditions to those recently reported,14 amides could indeed be obtained in good yield; an explanation for which was required.
After further investigations, we discovered that the key to catalytic reactivity was the 15 minute “prestir” period, as explained in Scheme 6. When the claimed catalyst, bis-2-chlorophenylborinic acid 3c, was exposed to a carboxylic acid in the presence of activated 5 Å molecular sieves (MS), catalytic activity was observed. Without this “prestir” period, no amide formation occurred. The significant finding was that the 11B and 1H NMR spectra of the crude reaction mixtures were different depending on whether the “prestir” period was applied or not (Scheme 6A and B). This data indicated that if borinic acid 3c, phenylacetic acid and benzylamine were mixed simultaneously without the “prestir” period, aminocarboxylate complex 5c was formed and remained stable over at least one week in solution (vide supra, Scheme 4 and 6A). When analyzing the product mixtures obtained after the “prestir” period, significant amounts of boronic species were observed (Scheme 6B). We propose, therefore, that borinic acid 3c acts as a pre-catalyst, and its protodeboronation occurs rapidly during the “prestir” period to give 2-chlorophenylboronic acid 8c, which is well known to be an effective catalyst for direct amidation, as reported by Hall et al.13a This finding was further verified by a separate experiment in which bis-2-chlorophenylborinic acid 3c and phenylacetic acid were stirred for 15 minutes with 5 Å MS, leading to a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :6 ratio of borinic to boronic species (Scheme 6C). Interestingly, the process of protodeboronation occurred much more slowly when activated 4 Å MS were used compared to 5 Å MS, so the particular sieves used play an important role in assisting the deboronation reaction (it is worth noting that generally either 3 or 4 Å MS are used for achieving dehydration in most amidation reactions as an alternative to azeotropic water removal. See ref. 13b for a relevant discussion).
:6 ratio of borinic to boronic species (Scheme 6C). Interestingly, the process of protodeboronation occurred much more slowly when activated 4 Å MS were used compared to 5 Å MS, so the particular sieves used play an important role in assisting the deboronation reaction (it is worth noting that generally either 3 or 4 Å MS are used for achieving dehydration in most amidation reactions as an alternative to azeotropic water removal. See ref. 13b for a relevant discussion).
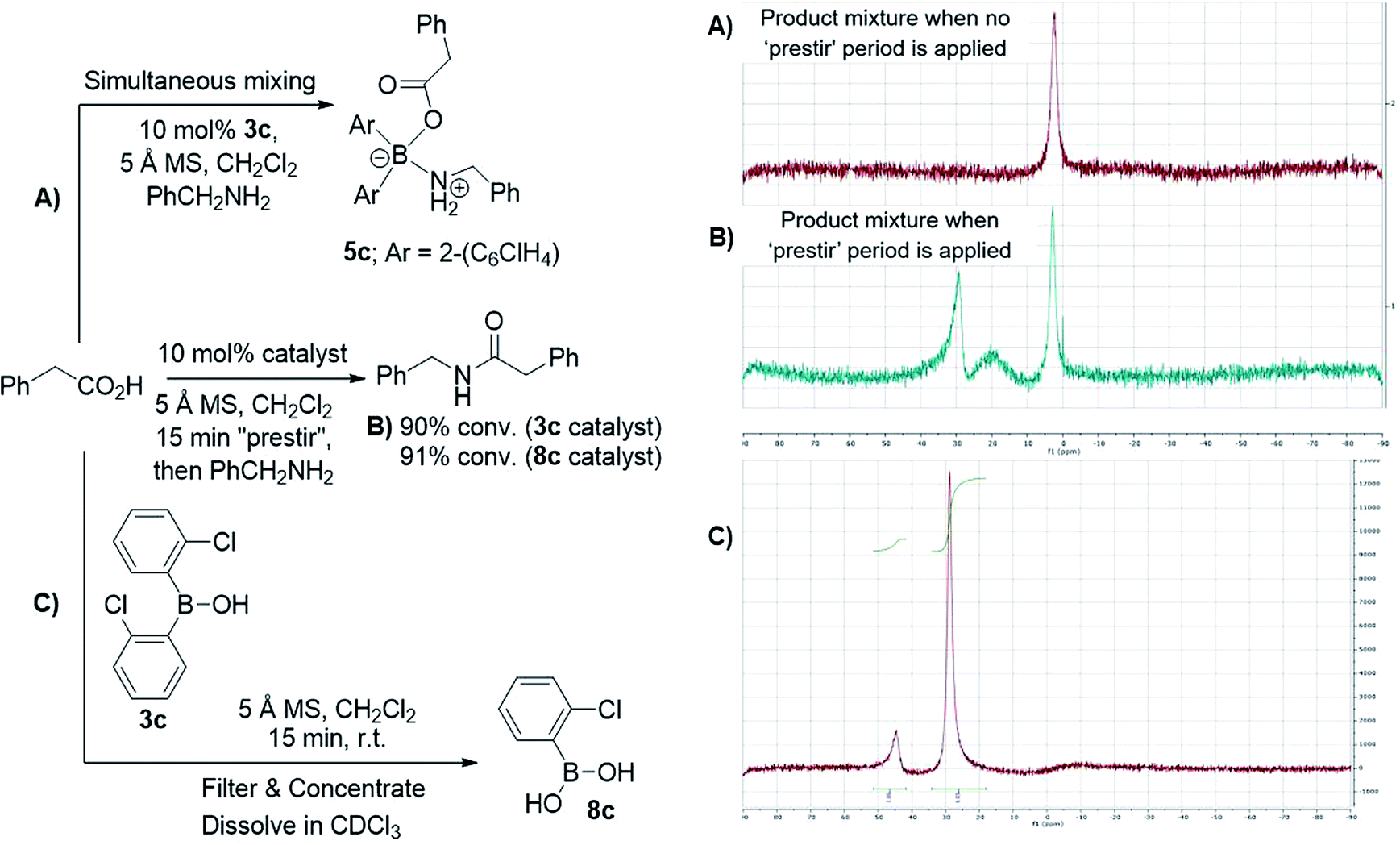 | ||
| Scheme 6 Examining bis-2-chlorophenylborinic acid 3c as a catalyst for direct amide formation and 11B NMR spectra of product mixtures obtained in 3 examined cases. | ||
To further support the fact that boronic acid 8c is actually formed from borinic acid 3cin situ under the “prestir” conditions and that 8c is the active catalyst, we compared 3c and commercial 2-chlorophenylboronic acid 8c side-by-side under the same conditions (Scheme 6B). Similar amide conversions were observed in the two reactions with 3c and 8c (91% and 90% respectively), which supports our proposal that the boronic acid is the actual catalyst. It can, therefore, be concluded that borinic acids do not catalyse amidations directly, and can only do so through protodeboronation to the boronic acid.
The fact that aminocarboxylate complexes 5 could not be converted into amides seems to be rather inconsistent with the commonly accepted mechanism for boron-mediated amidation (Scheme 2A). Since borinic acids are stronger Lewis acids than boronic acids, it might be expected that acyloxyborinic species 5 should actually be stronger acylating agents than the acyloxyboronic analogues, and these complexes also have the ability to form internal hydrogen bonds between the protonated amine and the carbonyl. Since both boronic acids and boric acid catalyse direct amidation,8 but borinic acids do not, it implies that at least three free coordination sites are needed on the boron atom in order for amidation catalysis to be possible. This conclusion led us to consider, therefore, whether B–X–B dimer formation might be essential for the observed catalytic activity, and that a bridging X-group (i.e. O or N) could be essential to orchestrate the catalytic process. We therefore looked more closely at the speciation that might be occurring in boronic acid-mediated direct amidation catalysis, using both experimental and theoretical methods.
Boronic acids in direct amide formation
Due to the complexity of carboxylic acid/amine/boronic acid systems, the reactivity of boronic acids with amines and carboxylic acids was initially explored separately. When treated with an amine, boronic acids appear to form complexes of general structures 9 and 10.20 3,4,5-Trifluorophenyl boroxine–benzylamine complex 10a (Fig. 5) and the adduct with ethylenediamine 10b (see ESI† for X-ray) were formed at room temperature and crystallised (Scheme 7), while phenylboroxine complexes 10c and 10d with two or four benzylamine molecules (Scheme 8 and Fig. 5) formed upon reaction of 8a with two equivalents of benzylamine at reflux in toluene (either with reflux condenser or with Dean–Stark water removal apparatus). An important observation was that the amine promotes dissolution of the boronic acid in organic solvent by formation of these boroxine complexes (see ESI†).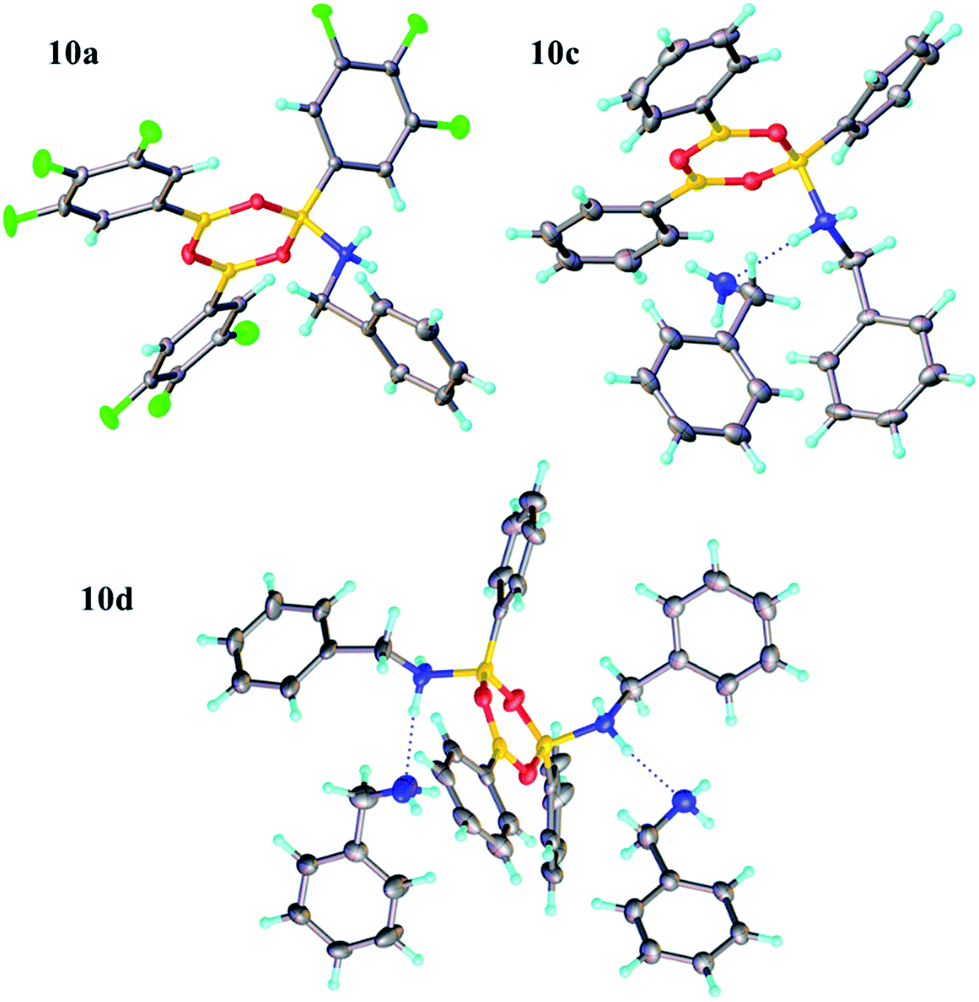 | ||
| Fig. 5 X-ray structures of 3,4,5-trifluorophenylboroxine-benzylamine adduct 10a and phenylboronic acid derived boroxine–amine adducts 10c and 10d. | ||
 | ||
| Scheme 7 Reactions between boronic acids 8a–d and amines at r.t. | ||
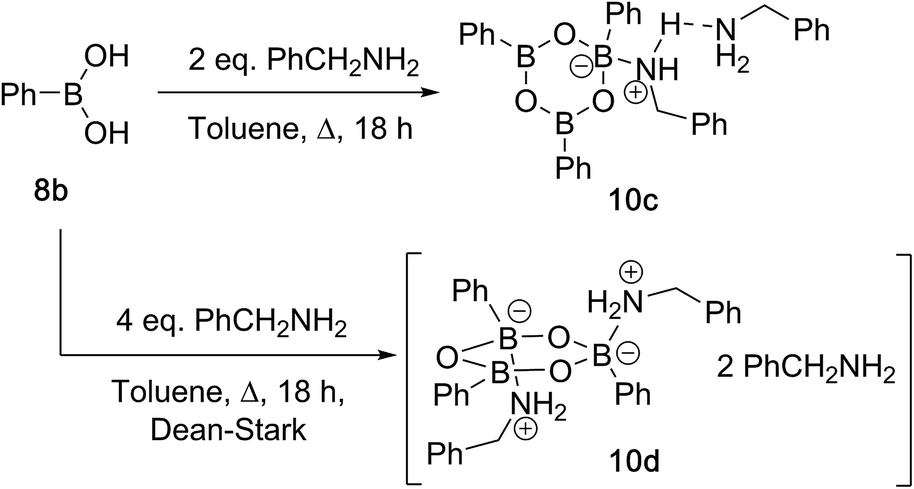 | ||
| Scheme 8 Reactions between phenylboronic acid 8a and benzylamine at reflux. | ||
These examples further underlined (also observed for borinic acids, vide supra) the importance of B–N interactions, which should readily occur between the amine and any trigonal boron species present in catalytic amidation systems. This is also consistent with our hypothesis that any trigonal acyloxyboron species is likely to react at the boron atom with an amine and indeed, such interactions may facilitate interconversion to and from boroxine species.
Having investigated the various interactions between amines and boronic acids, we next examined reactions between carboxylic acids and boronic acids. It was observed that boronic acid 8c did not react with phenylacetic acid unless 5 Å MS were introduced into the reaction mixture. In the presence of 5 Å MS alone, boronic acid 8c was converted into the corresponding boroxine 7c (Fig. 7A and B). With carboxylic acid present, initial formation of boroxine 7c occurred (Fig. 7C), and as more MS were added as well as phenylacetic acid, a new “ate”-complex (δ 5 ppm, Fig. 7D and E) was formed, and the equilibrium between it and boroxine 7c was pushed further towards the new species. Careful crystallisation from this heterogeneous system (containing solid 5 Å MS), allowed crystals to be separated which were suitable for X-ray characterisation. The product was unambiguously identified as the “ate”-complex 11c (see Fig. 6) which is structurally related to analogous complexes reported elsewhere.22 Interestingly, in a separate NMR reaction, addition of an excess of phenylacetic acid to boronic acid 8c, in the presence of 5 Å MS shifted the equilibrium even further towards the “ate”-complex as seen by 11B NMR (see further examples in the ESI†).
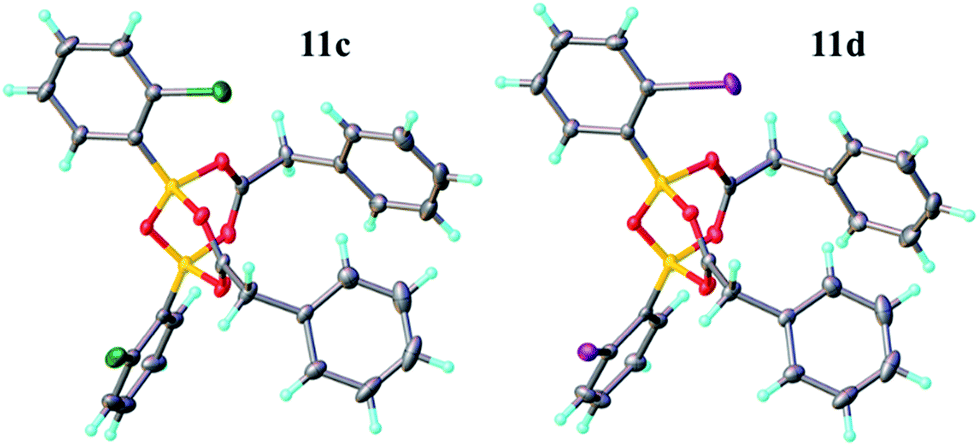 | ||
| Fig. 6 X-ray structures of B–O–B “ate”-complexes 11c and d. | ||
The amount of MS used also had a profound effect, and with a large excess of MS it was possible to generate >85% of the “ate”-complex in the mixture (calculated by 11B NMR, see Fig. 7E and ESI† for further examples). The resulting species 11c is analogous to “ate”-complexes 6 in the borinic acid series, which undergo subsequent protodeboronation to give boronic acid derivatives rather than act as amidation catalysts. This again highlights the need for the correct boron oxidation state, so that there are sufficient free coordination sites on the boron centre: borinic acids being unreactive but boronic acids/boric acid being catalytically active.
 | ||
| Fig. 7 11B NMR spectra showing interactions between 2-chlorophenylboronic acid 8c, phenylacetic acid and 5 Å MS. (A) 2-Chlorophenylboronic acid; (B) addition of 5 Å MS to (A), leading to boroxine formation; (C) addition of a tiny amount of 5 Å MS to a mixture of boronic and carboxylic acid; (D) addition of more 5 Å MS; (E) addition of even more 5 Å MS. | ||
It should be noted that without 5 Å MS the formation of the diacyloxy B–O–B bridged complexes 11 did not occur to a significant extent; even when 4 Å MS were used, less than 5% of the diacyloxy B–O–B complex 11c was observed. Interestingly though, a similar diacyloxy B–O–B bridged complex 11d could be isolated by reaction of 2-iodophenylboronic acid 8d and phenylacetic acid. In contrast, when boronic acids without an ortho-substituent, such as 3,4,5-trifluorophenylboronic acid 8a, were reacted with phenylacetic acid, the “ate”-complex was formed, as shown by 11B NMR, but did not crystallise and only boroxine 7a crystallised from the reaction mixture. In the case of ortho-substituted boronic acids, the boroxines are likely to be destabilized due to steric effects, and thus, the formation of dimeric “ate”-complexes 11 may be more favourable, enabling the formation of catalytically competent species.
Such sterically-induced destabilization of boroxines was indeed confirmed by relative free energy calculations29 (B3LYP+D3/Def2-TZVPP/solvent = dichloromethane) in which cyclisation of o-chlorophenyl boronic acid 8c to give 7c + 3H2O was found to be 2.55 kcal mol−1 more endoenergic than formation of the boroxine 7e from p-chlorophenyl boronic acid 8e (Scheme 9). For the former, this is due to reduced conjugation between the aryl rings and the boroxine ring as a result of twisting of ∼11°. At 298 K, this translates into a ∼105-fold decrease in the equilibrium concentration of the boroxine 7. Indeed, the high catalytic activity of such ortho-substituted systems may be due to these key reactivity effects which can destabilise the boroxine, a species which is likely to be an off-cycle resting state of the catalyst.
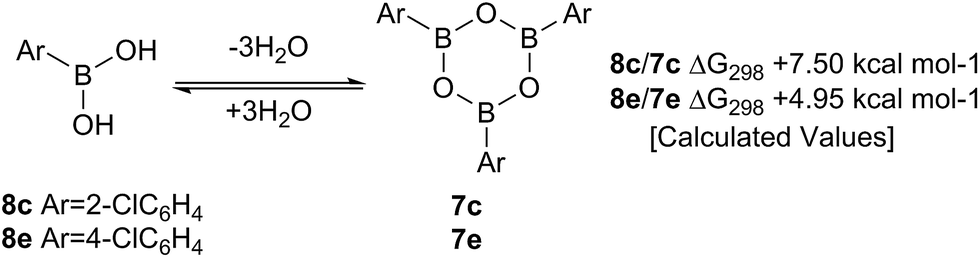 | ||
| Scheme 9 Calculated ΔG298 values for the formation of boroxines from o-chlorophenylboronic 8c acid and p-chlorophenylboronic acid 8e. | ||
Interestingly, when we attempted to reproduce the reported10 generation of an “acyloxy” boron derivative as described by Ishihara et al. using 3,4,5-trifluorophenylboronic acid 8a heated at reflux with phenylacetic acid in toluene (Dean–Stark water removal), there was no obvious reaction by either 11B and 1H NMR or ReactIR.
Having isolated the diacyloxy B–O–B complexes 11, we attempted to react these systems with benzylamine (Fig. 8). Surprisingly, when 1 equivalent of amine per dicarboxylate complex 11c was added, the 11B NMR showed the appearance of a new species 12 with a signal at 3 ppm, but 1H NMR showed no amide formation even after 2.5 h (see ESI†). However, when a second equivalent of amine was added, amide formation was seen within 20 minutes. This reaction of the diacyloxy B–O–B bridged complex 11c with two equivalents of benzylamine did not reach completion, and the signals we assign to the new species 12 remained in the 1H NMR even after 48 h. It is possible that this is due to the fact that there is an excess of boron compound (in total) with respect to amine in the reaction mixture, which indicates that an excess of amine and/or carboxylic acid per catalyst molecule is required for catalytic turnover and efficient reactivity.
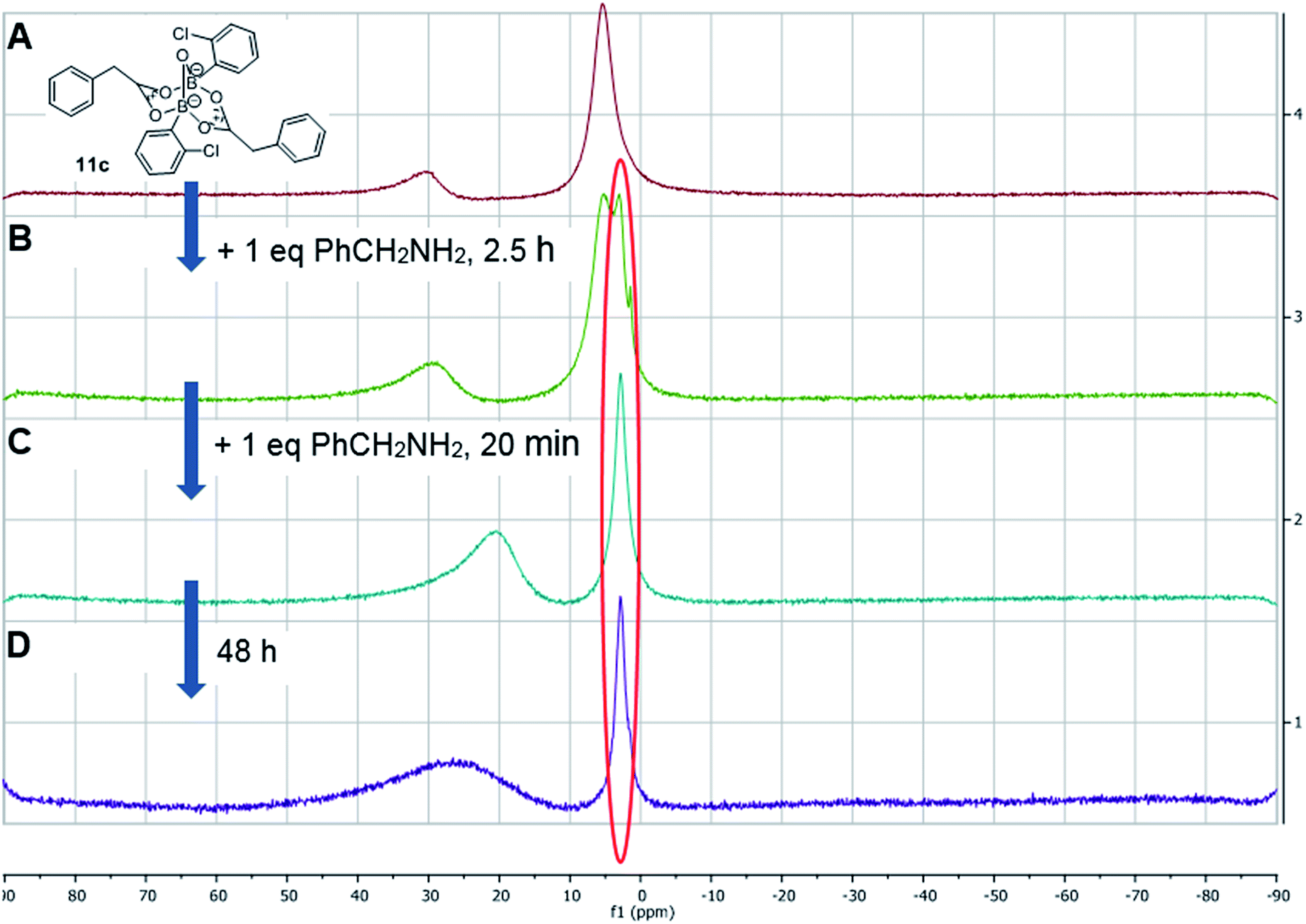 | ||
| Fig. 8 11B NMRs of (A) mixture of 2-chlorophenylboronic acid 8c and phenylacetic acid in the presence of 5 Å MS resulting in equilibrium between B–O–B dicarboxylate 11c (δ 5 ppm) and boronic species 7c (δ 30 ppm); (B) addition of 1 equivalent (per “ate”-complex 11c) of benzylamine, 2.5 h, showing formation of a new complex 12; (C) addition of 2nd equivalent of benzylamine, 20 min; (D) same mixture over 48 h. | ||
We then went on to investigate the species present in catalytic reaction mixtures containing boronic acid, amine and carboxylic acid (Scheme 10). Interestingly, the behavior of these systems was dependent on the order of reagent addition. When the carboxylic acid was premixed with boronic acid 8c in the presence of 5 Å molecular sieves, rapid formation of the “ate”-complex 11c was observed (Scheme 10A); subsequent addition of amine led to complete formation of amide within 2 h. During the amidation reaction, small quantities of boroxine–amine complex 10e were also observed, along with a signal consistent with the unknown species 12 (δB 3 ppm). The amine complex 10e became the dominant species after the amidation was complete. In contrast, when boronic acid 8c was mixed with the amine in the presence of 5 Å molecular sieves (Scheme 10B), rapid formation of a boroxine amine complex (e.g.10e) was observed, though a lower 11B NMR shift of 15 ppm suggests that 10e was likely in equilibrium with a compound analogous to 10d, in which two amine molecules are coordinated to the boroxine. Addition of carboxylic acid led to amide formation, but after 2 h the reaction had only reached 50% conversion. Again, during the amidation reaction both 10d and 12 were observed, with the latter being the major species. Finally, a reaction was carried out in which amine and carboxylic acid were pre-mixed to give the ammonium carboxylate salt and boronic acid 8c was then added (Fig. 10C). This exhibited similar behavior to the reaction shown in Fig. 10B, giving 53% conversion to amide after 2 h.
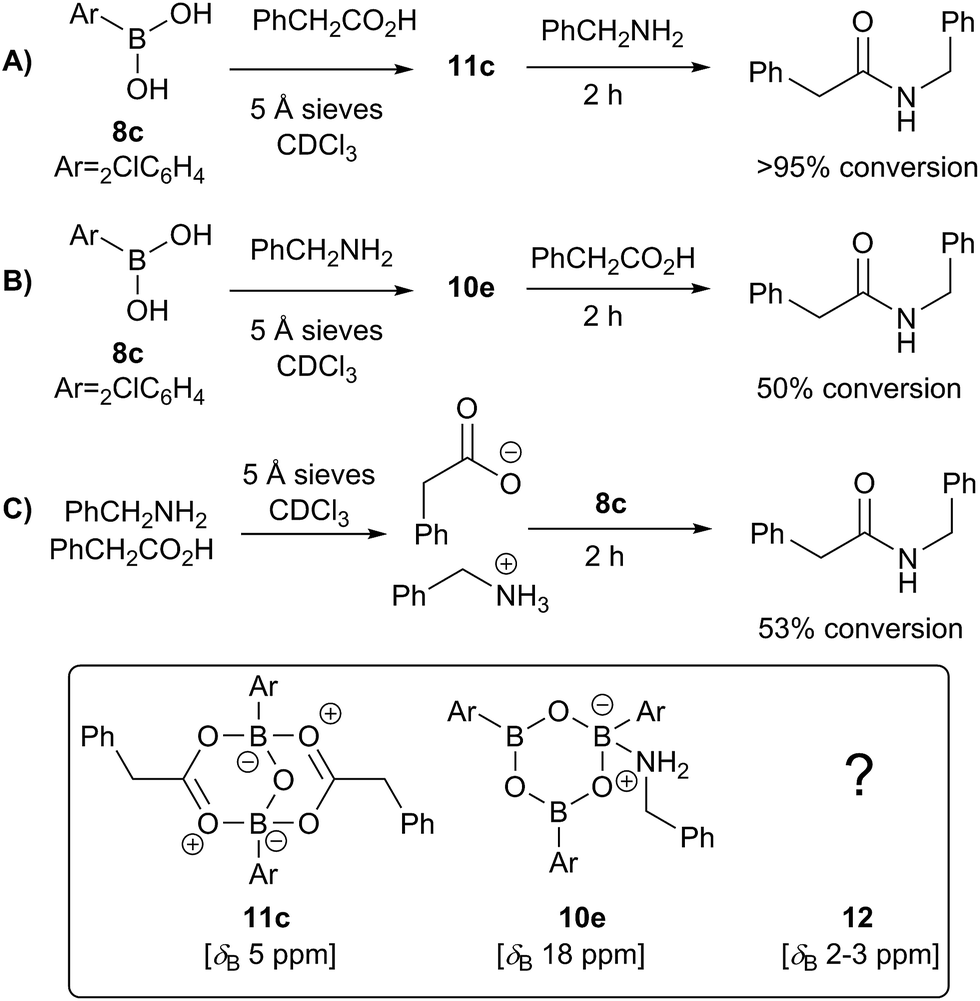 | ||
| Scheme 10 Observed boron-containing species in catalytic amidation reactions containing boronic acid 8c, carboxylic acid and amine. | ||
Finally, when either B2O3 or boric acid were mixed with benzylamine and phenylacetic acid (both insoluble in CDCl3), some dissolution was observed and the resulting 11B NMR showed the appearance of an “ate”-complex (see ESI†), suggesting that these boron compounds can be pulled into solution via complexation.
Summary of observations
A full summary of possible intermediates generated from borinic/boronic acids by reaction with carboxylic acids and amines is shown in Scheme 11, along with likely pathways for their formation. Thus, borinic acids 3 form inert complexes 5 (via4 or postulated intermediate A) in the presence of amine and carboxylic acid, but in the presence of carboxylic acid alone, they can undergo protodeboronation (probably via observed intermediate 6), to enter the boronic acid cycle. Boronic acids 8 undergo rapid complexation with amines to give intermediates of type 9, which are rapidly converted through to boroxine–amine complexes 10via postulated intermediate B, facilitating dissolution of the boronic acid in organic solvents. Direct condensation of a boronic acid 8 with a carboxylic acid should lead initially to C, the previously proposed reactive acylating agent.11a However, as we have shown above, C is likely to undergo self-condensation under the dehydrating conditions to give “ate”-complexes of type 11 in a similar fashion to the dimerization of B(OAc)3 (vide supra, Scheme 1). This latter complex could also potentially be formed from B, through condensation with two molecules of carboxylic acid. We tentatively propose that intermediate 12 may be the amine-bridged dimeric “ate”-complex shown, which is closely related to 11. This is based upon the fact that 12 is formed by reaction of 11 with amine under dehydrating conditions, and the fact that the NMR signals for 12 are similar in line shape to those for complex 5 (see ESI†). Under catalytic conditions, a similar boron NMR peak is observed that may be 12, or a related compound derived from it.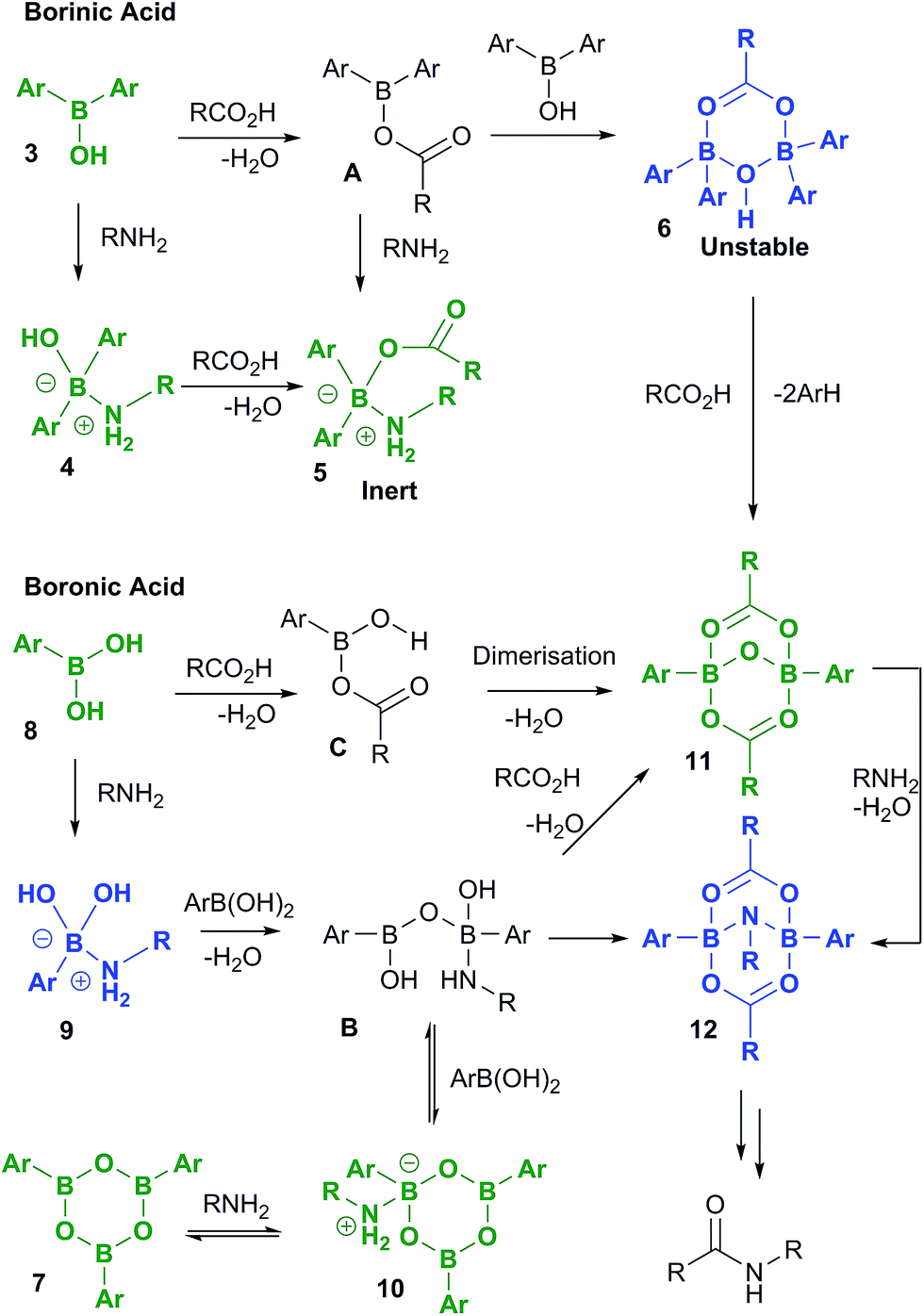 | ||
| Scheme 11 Boron species potentially generated from reaction of both borinic acids and boronic acids with carboxylic acids and amines. Compounds which have been fully characterized in this study by X-ray crystallography are shown in green, and those which have been observed by NMR and/or mass spectrometry are shown in blue. Proposed intermediates during the formation of the observed boron species are shown in black. | ||
Overall, these results lead to the conclusion that formation of complexes from reaction of boron centres with both carboxylic acids and amines (e.g.5 and 12) is not only observed at all three investigated boron oxidation levels (borinic, boronic and boric) but is also indicative that these types of species are likely to be generated in catalytic amidation reactions. It is also clear that B–O–B linkages and B–N interactions are vitally important, and likely to be present in all catalytically active boron-based systems, i.e. particularly boronic acids, and likely boric acid by extrapolation. As it is not possible to be certain what other types of B–X–B systems can potentially form, to more deeply understand the role of cooperativity resulting in direct amidation catalytic activity between two boron atoms via a B–X–B system, we probed a number of putative mechanistic pathways using quantum mechanical modelling, all of which involve dimeric species potentially derived from 11 and/or 12.
Mechanistic insights and theoretical calculations
We decided to formulate different diboron systems, linked by oxygens or nitrogens and with different complexes, including amine and carboxylate and their corresponding salts, in order to have a better chance of locating the types of systems that might be responsible for direct amidation catalysis. Preliminary exploration of transition states involving monomeric B–N species resulted in high overall energy barriers (>30.0 kcal mol−1). Hence, pathfinder explorations of the potential energy surfaces were then carried out for the different putative B–X–B-mediated catalytic cycles, organised around five possible routes based on the number of amines involved (Fig. 9–13), in order to explore the mechanistic diversity of these systems. Key transition state structures in each cycle are shown together in Fig. 14. To enable a high quality basis set to be employed (Def2-TZVPP), substituents were all modelled at this stage using methyl groups. Each cycle included the reaction free energies of the pre-step involving species used to assemble the reactant used as the start point in the cycle, and the energies of all subsequent steps in the cycle, ending in the final products. A final reaction free energy is included to re-generate the starting reactant ready for a second catalytic cycle.The cycle involving one amine is illustrated in Fig. 9. The pre-assembly condensation step (blue) indicates a modestly endoenergic step (ΔΔG298 +9.4 kcal mol−1) producing water, which agrees with the need for water removal using molecular sieves for the reaction to proceed. With the reactant in the cycle set to a relative energy of 0.0, the high point is reached with C–O cleavage (TS3) corresponding to ΔG298 +23.4 kcal mol−1, corresponding to a relatively slow ambient temperature reaction. The product in this cycle is +0.3 kcal mol−1 relative to the reactant, but −4.2 kcal mol−1 is recovered in the next regeneration step (red). Such a computational exploration can only demonstrate the viability of a single pathway to product; it does not preclude, of course, that lower energy pathways might not be discovered upon a more complete exploration of the potential energy surface. These variants might include lower energy conformations of the various species explored. Rather more certain is that the free energy barrier surmounted during the cycle is probably an upper calculated bound to any true value. It should also be noted that the model used for the substituents (methyl) may introduce further uncertainty, but since the purpose here was a broad exploration of various mechanistic alternatives, we did not undertake the much more computationally resource-intensive recalculation using larger substituents.
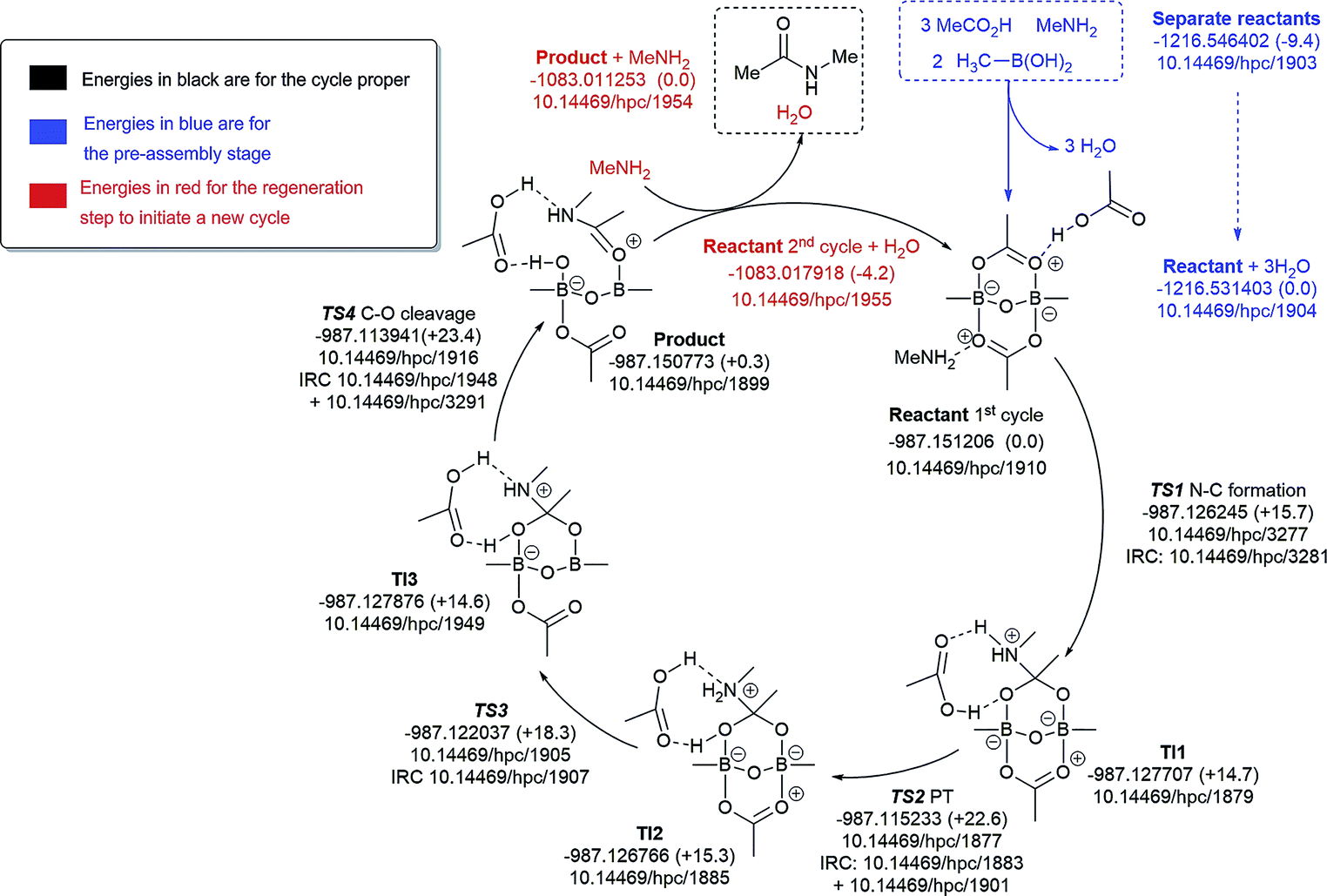 | ||
| Fig. 9 Catalytic cycle involving one amine (data29 sub-collection DOI: 10.14469/hpc/1854). The total computed free energy for each stationary point is shown in Hartree, with the relative energies shown in parentheses in kcal mol−1. The DOI for data repository entries for individual species are shown in the form e.g.10.14469/hpc/1885. Energies in blue are for the pre-assembly stage, in black for the cycle proper and in red for the regeneration step to initiate a new cycle. All DOIs in this figure are available as clickable links (final paginated PDF only). | ||
A cycle involving two amine species is illustrated in Fig. 10. The pre-assembly step (blue) is rather more endoenergic (ΔG298 +25.4 kcal mol−1) than the previous cycle, but the energetic maximum in the cycle (TS3, ΔG298 +24.8 kcal mol−1) relative to the assembled reactants is very similar. The product of the first cycle is exoenergic by ΔG298 22.3 kcal mol−1, offset in part by the regeneration step for the second cycle being ΔG298 +16.5 endoenergic.
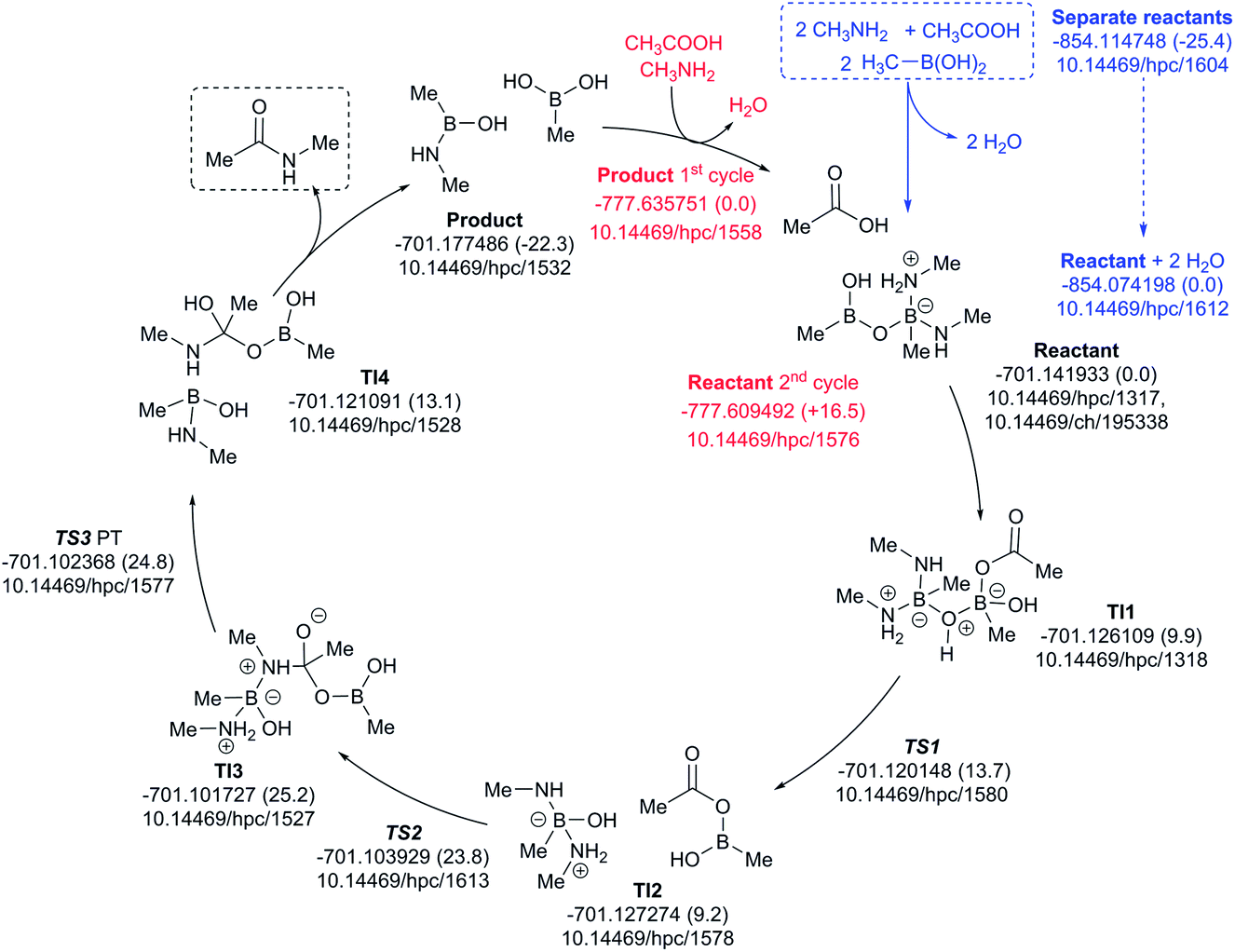 | ||
| Fig. 10 Catalytic cycle involving two amines (data29 sub-collection DOI: 10.14469/hpc/1622; the labels have the same meaning as in Fig. 9). All DOIs in this figure are available as clickable links (final paginated PDF only). | ||
The three-amine cycle (Fig. 11) again shows similar overall behavior. The condensation reaction resulting in the initial cycle reactant is endoenergic by ΔG298 +24.4 kcal mol−1, matched by the exoenergic products (ΔG298 −24.8 kcal mol−1), and with a regenerative step for the start of the second cycle being endoenergic by ΔG298 +14.1 kcal mol−1. The rate limiting TS3 has ΔG298 21.1 kcal mol−1, slightly more favourable than the previous two cycles.
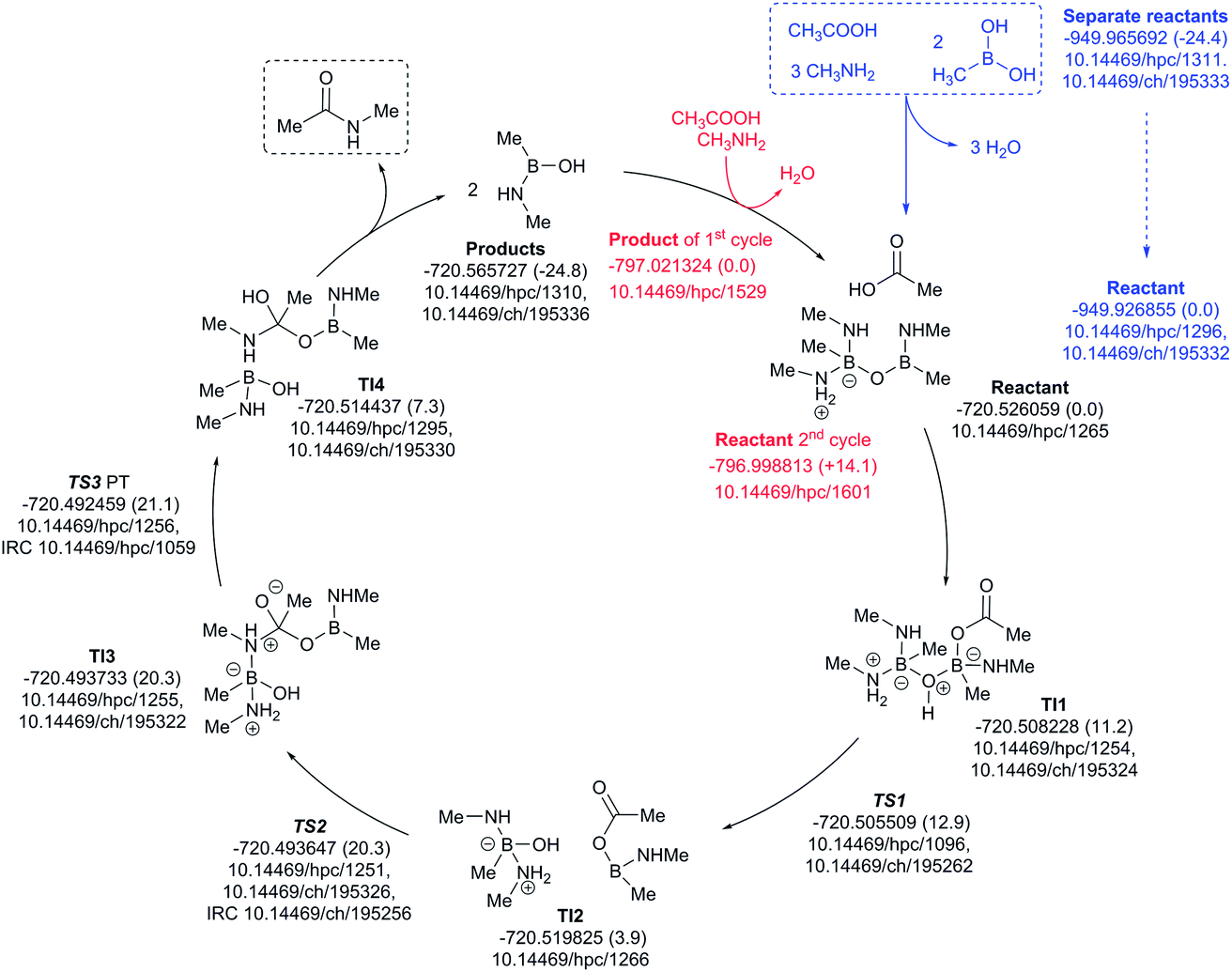 | ||
| Fig. 11 Catalytic cycle involving three amines (data29 sub-collection DOI: 10.14469/hpc/1621; the labels have the same meaning as in Fig. 9). All DOIs in this figure are available as clickable links (final paginated PDF only). | ||
The four-amine cycle (Fig. 12) has a highly endoenergic condensation step (ΔG298 +37.9 kcal mol−1), again offset by the products being exoenergic by 32.4 kcal mol−1. The regeneration step for the second cycle is endoenergic by +24.9 kcal mol−1 and the overall barrier originating from TS1 is 25.4 kcal mol−1. This makes this model cycle less favourable.
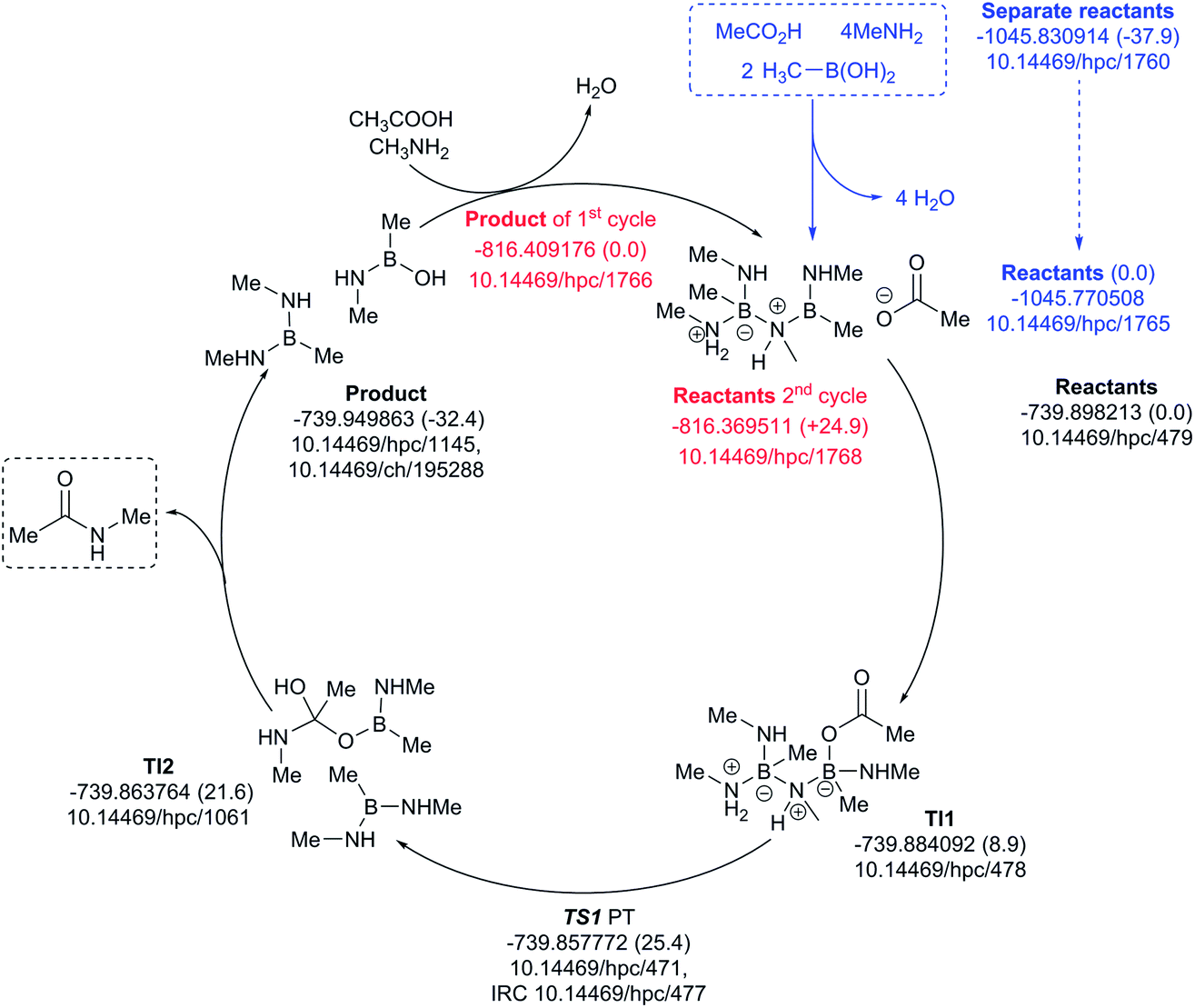 | ||
| Fig. 12 Catalytic cycle involving four amines (data29 sub-collection DOI: 10.14469/hpc/1728; the labels have the same meaning as in Fig. 9). All DOIs in this figure are available as clickable links (final paginated PDF only). | ||
Finally, a variation of the one amine cycle involving utilization of a second amine to form a B–N–B catalytic intermediate was examined (Fig. 13), for which the catalyst assembly step is similarly endoenergic (ΔG298 +26.0 kcal mol−1) to the B–O–B bridged two amine route above. The highest free energy point in the catalytic cycle (TS2, ΔG298 +23.2 kcal mol−1) is now relative to the initial tetrahedral intermediate (TI1), which forms exoenergically from the bicyclic reactant related to the previous cycles, but with a transition state of presumed low barrier corresponding to reactant reorganization which we were unable to locate. This suggests that the barriers resulting from species with B–N–B bridges are similar to those with B–O–B bridges.
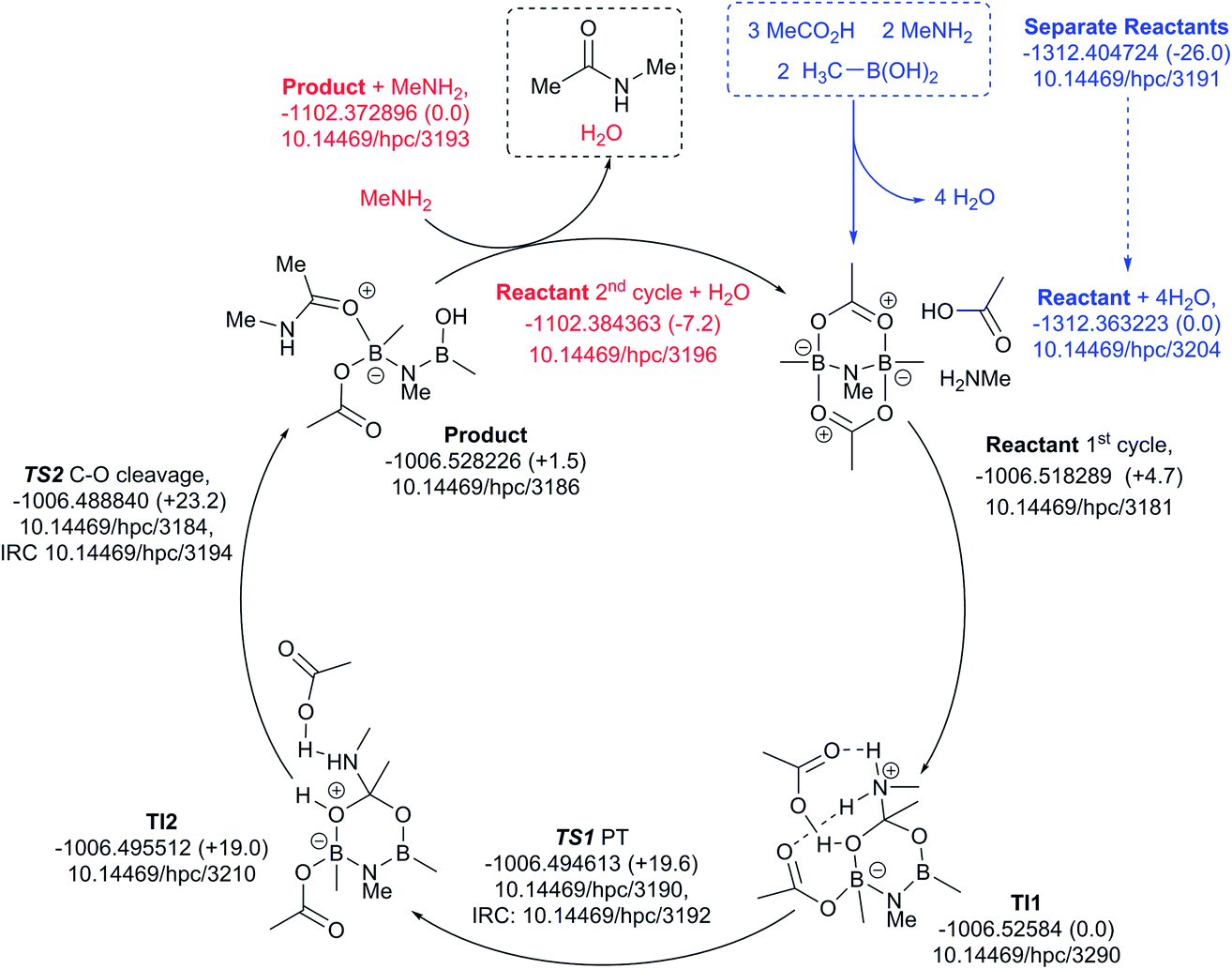 | ||
| Fig. 13 Catalytic cycle involving B–N–N bridging group (data29 sub-collection DOI: 10.14469/hpc/3188; the labels have the same meaning as in Fig. 9). All DOIs in this figure are available as clickable links (final paginated PDF only). | ||
These five pathfinding explorations have thus identified a model corresponding to a thermal reaction at 298 K, with the caveat that further optimization of these very complex catalytic combinations may always reveal even lower energy routes. In the future, we plan to exploit these core models to study substituent effects on the energies.
All five of the catalytic cycles investigated have reasonable thermal barriers for the catalytic mechanism (Fig. 14). It is difficult, however, at least purely on the basis of the computed energetics of such catalytic processes, to confidently discriminate between these. Importantly, all these systems have lower overall energetics than the monomeric acyloxyboron pathway previously proposed11a and computationally examined.17 Finally, on the basis of these calculations, the resting state of the catalyst would be expected to be a bridged (B–O–B or B–N–B) system containing tetrahedral boron atoms (e.g. TI2 in Fig. 13), consistent with the signals observed from NMR analysis of catalytic reaction mixtures (δB 3 ppm).
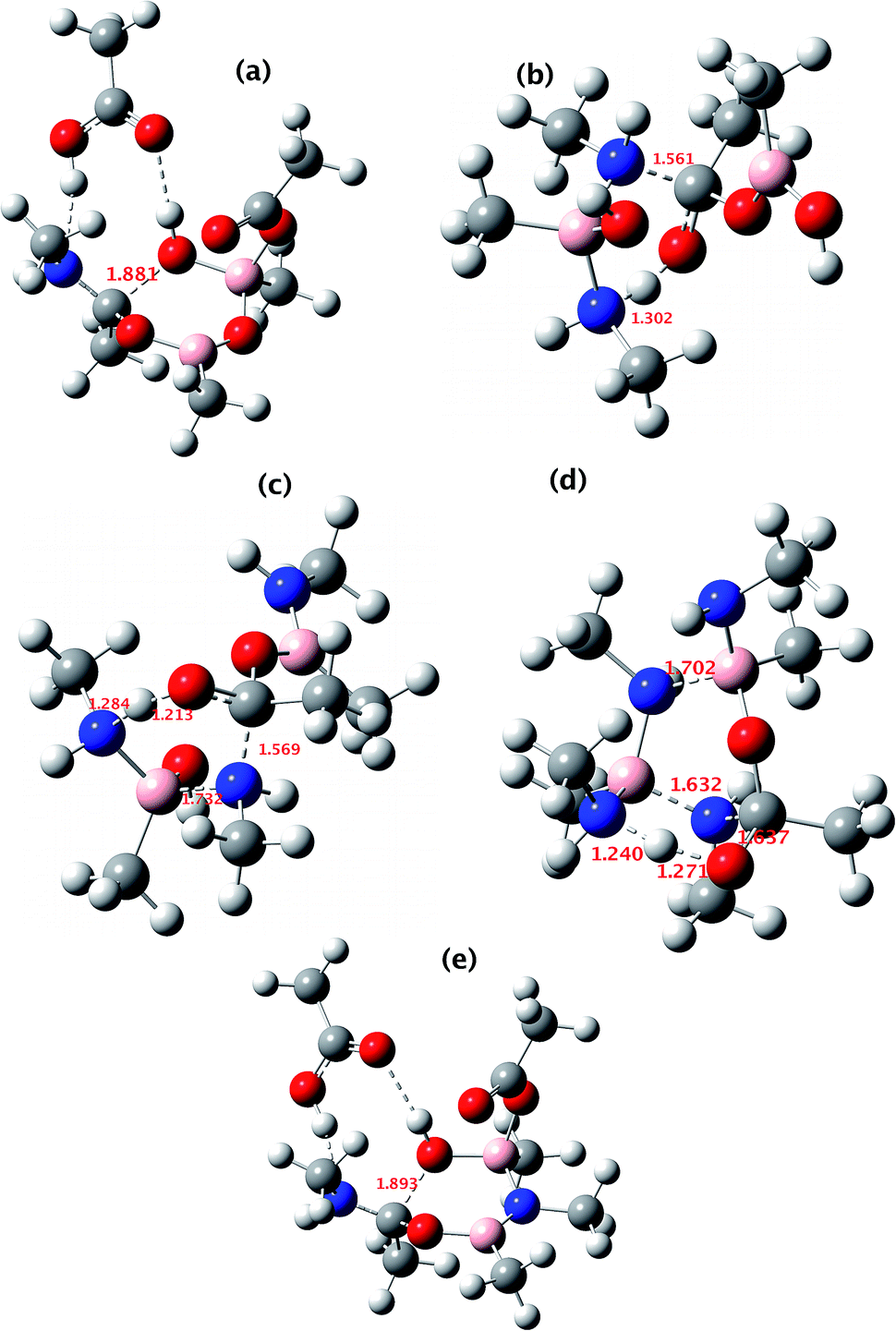 | ||
| Fig. 14 The computed structure for: (a) TS3 shown in Fig. 9; (b) TS3 shown in Fig. 10; (c) TS3 shown in Fig. 11; (d) TS1 shown in Fig. 12 and (e) TS2 shown in Fig. 13. Indicated bond length are in Å. The activation free energies ΔG298 for the five transition states are respectively 23.4, 25.2, 21.1, 25.4 and 23.2 kcal mol−1. | ||
Kinetic analysis of a catalytic amidation reaction
Preliminary studies of the reaction kinetics for a boronic acid catalyzed amidation reaction revealed some interesting findings (Scheme 12 and ESI†). The selection of reaction conditions and amine/carboxylic acid reactants was guided by the need for a homogenous reaction mixture in order to obtain useful kinetic data. Reaction of 4-phenylbutylamine with benzoic acid12b was carried out in tert-amyl methyl ether (TAME) under Dean–Stark conditions10f using 8c as the amidation catalyst. Analysis of reaction profiles was carried out using the Burés method (see ESI†).23 The reaction was first order in catalyst up to a loading of 5 mol%, but a catalytic loading of 10 mol% did not provide a significant rate acceleration over 5 mol%. Interesting, the first order dependence on catalyst was observed up to 10 mol% loading, when 1.4 equivalents of amine were present. This may suggest that the amine is important for generating the active catalytic species, perhaps related to dimer 12. A positive order of reaction with respect to the amine concentration was also observed at 3.5 mol% catalyst loading. Unexpectedly, increasing the concentration of carboxylic acid had a negative effect on the reaction rate, and even a slight excess of 1.2 equivalents of carboxylic acid led to almost complete inhibition of the reaction at 3.5 mol% catalyst loading; the effect was less pronounced at higher catalyst loadings.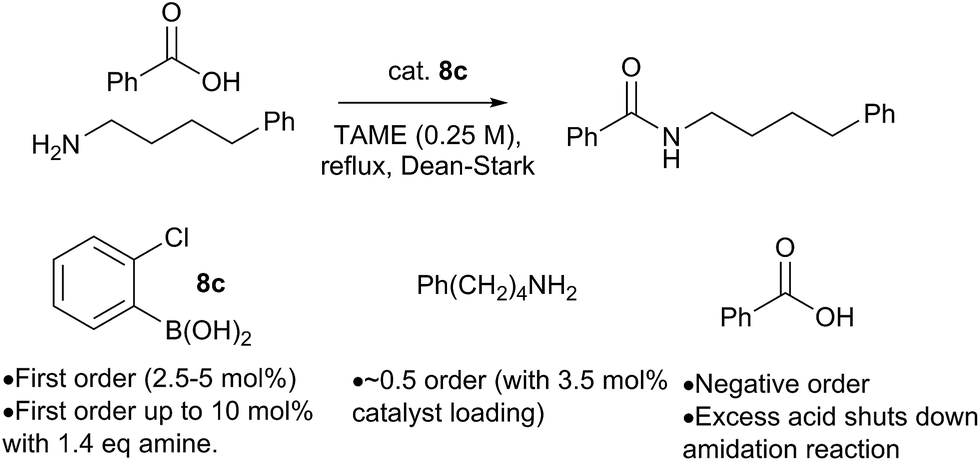 | ||
| Scheme 12 Preliminary kinetics studies on a boronic acid catalyzed amidation reaction. | ||
At present, this observation is difficult to explain, but it appears that excess carboxylic acid prevents formation of the active catalytic species, e.g. via formation of off-cycle species such as the ammonium carboxylate salt or dimer 11. Similar observations regarding catalyst inhibition by carboxylic acid were noted in Zr-catalysed amidation reactions.24
All the mechanisms outlined above, as well as those previously proposed in the literature, involve an essentially unimolecular turnover limiting step, so reaction kinetics do not provide a useful tool for distinguishing between them. The fact that the reaction kinetics show a first order dependence on the catalyst concentration is consistent with either a monomeric or dimeric active catalyst, provided it remains as a monomer/dimer throughout the catalytic cycle.23c,24 The positive interaction between the concentration of amine and boronic acid catalyst provides some support for mechanisms involving an active species containing both of these components, especially if its formation is dependent on B–N interactions. Crucially, there appears to be a restriction on the quantity of active catalyst that can be generated from free boronic acid (somewhere between 5–10 mol%), which can be lifted at higher amine concentrations.
Summary and conclusions
We have investigated carboxylic acid/amine/boron catalyst systems experimentally and computationally and have shown the potential importance of both the formation of B–X–B species and of B–N interactions in these systems, leading to the proposal of new catalytic cycles for boron-catalysed amidation reactions involving boronic acids, though by extrapolation, boric acid is highly likely to behave similarly. We have demonstrated that whereas boronic acids can catalyse amidation reactions, borinic acids are ineffective as catalysts themselves, and in fact, undergo protodeboronation instead to derive boronic acids in situ. In turn, the boronic acid analogue is catalytically competent and causes downstream amidation catalysis. This work clearly indicates that at least three free coordination sites are needed on the boron atom to enable effective catalysis to take place, and hence, borinic acids are precluded from amidation reactivity. Indeed, our proposed mechanisms can account for this and other observations regarding amidation catalysis. Firstly, the formation of an active dimeric boron catalyst appears to be impossible at the borinic acid oxidation level, as rapid complexation of an acid and amine molecule leads to the formation of a catalytically inactive complex 5. The success of ortho-substituted boronic acids,13 including bifunctional systems,12 as catalysts can also potentially be rationalized by their preference for the formation of dimeric species over trimeric boroxines. Furthermore, this supports our previous observation that cooperative catalysis using two different boronic acids can lead to enhanced yields of amides in challenging cases.15 Our proposed mechanisms are also consistent with a recent report that a complex B/N/O heterocyclic system displays high activity in catalytic amidation reactions.16The findings in this study indicate the clear importance of bringing together multiple techniques to understand the various entities involved in amidation catalysis, i.e. the combined use of NMR (1H, 13C and especially 11B), IR, X-ray crystallography, reactivity studies and the importance of examining multiple potential reaction pathways by theoretical methods, as exemplified by the five related key transition states summarised in Fig. 14. In this case, this comprehensive approach has allowed us to move away from a seemingly ‘accepted’ mechanism17 which arguably has little supporting experimental evidence,11a and to propose instead closely related low energy alternatives. We consider that the mechanism shown in Fig. 13 may be the most likely to be effective under catalytic conditions using boronic acids, due to our experimental observations of the reactivity of dimer 11 with amine, and the direct 11B NMR analysis of catalytic reaction mixtures. The key point though is that this is a guide, and individual systems may change between different modes of action depending upon both catalyst and substrate properties (electronics, substituents, etc.). However, we anticipate that these mechanistic insights will provide valuable assistance for the design of more active boron amidation catalysts in the future.
Experimental
Computational methods
Pathfinder explorations of the potential energy surfaces for putative catalytic cycles all utilised the Def2-TZVPP basis set25 and the B3LYP density functional augmented with the Grimme D3 dispersion energy correction,26 a combination selected after earlier evaluation for catalytic cycles27 and as implemented in the Gaussian 09, versions D01 and E.01 and Gaussian 16 version A.03 programs. A solvent correction using the CPCM method and dichloromethane parameters was employed throughout and all energies are corrected for thermal contributions and entropy by calculation of normal mode frequencies, with transition states exhibiting the required single negative force constant. All free energies correspond to a standard state at 298 K of 0.0445 M (1 atm). Intrinsic reaction coordinates (IRCs) were used to define the start and end species of all transition states. Research data management (RDM) is via deposition into a collection-based data repository28 where access is via the appropriate FAIR data DOI.29General experimental methods
General experimental details and data files are available via a data repository.29 Selected experimental procedures and data are included in the ESI.†Synthetic methods
All synthetic methods and compound characterization data is available via a data repository or in the ESI.†26X-ray crystallography and NMR spectroscopy
Crystallographic and NMR full data files (in Mpublish format) are available via a data repository.29Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Durham University for Doctoral Fellowship funding (SA), GlaxoSmithKline and UCL Chemistry for supporting a PhD studentship (MTS), and Pfizer for providing an EPSRC CASE award (VK). We also thank Dr Dmitry S. Yufit (Chemistry Department, Durham University) for solving X-ray crystal structures of compounds 4a, 5c, 11d and β-7a.Notes and references
- (a) M. F. Lappert, Chem. Rev., 1956, 56, 959–1064 CrossRef CAS; (b) G. Heller, Top. Curr. Chem., 1986, 131, 42–78 CrossRef.
- M. J. D. Bosdet and W. E. Piers, Can. J. Chem., 2009, 87, 8–29 CrossRef.
- E. J. Corey, R. K. Bakshi and S. Shibata, J. Am. Chem. Soc., 1987, 109, 5551–5553 CrossRef CAS.
- T. R. Kelly, A. Whiting and N. S. Chandrakumar, J. Am. Chem. Soc., 1986, 108, 3510–3512 CrossRef CAS.
- (a) B. M. Trost, D. O'Krongly and J. L. Belletire, J. Am. Chem. Soc., 1980, 102, 7595–7596 CrossRef CAS; (b) A. Dal Negro, L. Ungaretti and A. Perotti, J. Chem. Soc., Dalton Trans., 1972, 1639–1643 RSC.
- (a) H. Charville, D. Jackson, G. Hodges and A. Whiting, Chem. Commun., 2010, 46, 1813–1823 RSC; (b) H. Charville, D. A. Jackson, G. Hodges, A. Whiting and M. R. Wilson, Eur. J. Org. Chem., 2011, 5981–5990 CrossRef CAS.
- (a) H. Lundberg, F. Tinnis, N. Selander and H. Adolfsson, Chem. Soc. Rev., 2014, 43, 2714–2742 RSC; (b) R. M. de Figueiredo, J.-S. Suppo and J.-M. Campagne, Chem. Rev., 2016, 116, 12029–12122 CrossRef CAS PubMed.
- (a) R. M. Lanigan and T. D. Sheppard, Eur. J. Org. Chem., 2013, 7453–7465 CrossRef CAS; (b) K. Ishihara, Tetrahedron, 2009, 65, 1085–1109 CrossRef CAS; (c) B. M. Monks and A. Whiting, Sustainable Catalysis: Challenges and Practices for the Pharmaceutical and Fine Chemical Industries, ed. P. J. Dunn, K. K. Hii, M. J. Krische and M. T. Williams, Wiley, Hobokon, NJ, USA, 2013, ch. 5 Search PubMed; (d) F. Ferdousi and A. Whiting, Green Catalytic Direct Amide Bond Formation in Green and Sustainable Medicinal Chemistry: Methods, Tools and Strategies for the 21st Century Pharmaceutical Industry, ed. L. Summerton, H. F. Sneddon, L. C. Jones and J. H. Clark, RSC Green Chemistry series, Cambridge, 2016, ch. 13 Search PubMed.
- P. Tang, Org. Synth., 2005, 81, 262 CrossRef CAS.
- (a) P. Starkov and T. D. Sheppard, Org. Biomol. Chem., 2011, 9, 1320–1323 RSC; (b) R. M. Lanigan, P. Starkov and T. D. Sheppard, J. Org. Chem., 2013, 78, 4512–4523 CrossRef CAS PubMed; (c) V. Karaluka, R. M. Lanigan, P. M. Murray, M. Badland and T. D. Sheppard, Org. Biomol. Chem., 2015, 13, 10888–10894 RSC; (d) R. M. Lanigan, V. Karaluka, M. T. Sabatini, P. Starkov, M. Badland, L. Boulton and T. D. Sheppard, Chem. Commun., 2016, 52, 8846–8849 RSC; (e) T. Maki, K. Ishihara and H. Yamamoto, Org. Lett., 2006, 8, 1431–1434 CrossRef CAS PubMed; (f) M. T. Sabatini, L. T. Boulton and T. D. Sheppard, Sci. Adv., 2017, 3, e1701028 CrossRef PubMed.
- (a) K. Ishihara, S. Ohara and H. Yamamoto, J. Org. Chem., 1996, 61, 4196–4197 CrossRef CAS PubMed; (b) K. Ishihara, S. Ohara and H. Yamamoto, Macromolecules, 2000, 33, 3511–3513 CrossRef CAS; (c) K. Ishihara, S. Ohara and H. Yamamoto, Org. Synth., 2002, 79, 176–185 CrossRef CAS; (d) T. Maki, K. Ishihara and H. Yamamoto, Synlett, 2004, 1355–1358 CAS; (e) A. Sakakura, R. Yamashita, T. Ohkubo, M. Akakura and K. Ishihara, Org. Lett., 2011, 13, 892–895 CrossRef CAS PubMed; (f) A. Sakakura, R. Yamashita, M. Akakura and K. Ishihara, Aust. J. Chem., 2011, 64, 1458–1465 CrossRef CAS; (g) R. Yamashita, A. Sakakura and K. Ishihara, Org. Lett., 2013, 15, 3654–3657 CrossRef CAS PubMed; (h) K. Ishihara and Y. Lu, Chem. Sci., 2016, 7, 1276–1280 RSC.
- (a) K. Arnold, B. Davies, R. L. Giles, C. Grosjean, G. E. Smith and A. Whiting, Adv. Synth. Catal., 2006, 348, 813–820 CrossRef CAS; (b) K. Arnold, A. S. Batsanov, B. Davies and A. Whiting, Green Chem., 2008, 10, 124–134 RSC; (c) K. Arnold, B. Davies, D. Hérault and A. Whiting, Angew. Chem., Int. Ed., 2008, 47, 2673–2676 CrossRef CAS PubMed.
- (a) R. M. Al-Zoubi, O. Marion and D. G. Hall, Angew. Chem., Int. Ed., 2008, 47, 2876–2879 CrossRef CAS PubMed; (b) N. Gernigon, R. M. Al-Zoubi and D. G. Hall, J. Org. Chem., 2012, 77, 8386–8400 CrossRef CAS PubMed.
- T. M. El Dine, J. Rouden and J. Blanchet, Chem. Commun., 2015, 51, 16084–16087 RSC.
- S. Liu, Y. Yang, X. Liu, F. K. Ferdousi, A. S. Batsanov and A. Whiting, Eur. J. Org. Chem., 2013, 5692–5700 CrossRef CAS.
- H. Noda, M. Furutachi, Y. Asada, M. Shibasaki and N. Kumagai, Nat. Chem., 2017, 9, 571–577 CrossRef CAS PubMed.
- (a) T. Marcelli, Angew. Chem., Int. Ed., 2010, 49, 6840–6843 CrossRef CAS PubMed; (b) C. Wang, H.-Z. Yu, Y. Fu and Q.-X. Guo, Org. Biomol. Chem., 2013, 11, 2140–2146 RSC.
- P. Zhang, R. M. Kriegel and J. W. Frost, ACS Sustainable Chem. Eng., 2016, 4, 6991–6995 CrossRef CAS.
- (a) P. Paetzold, P. Bohm, A. Richter and E. Scholl, Z. Naturforsch, 1976, 31b, 754–764 CAS; (b) G. H. L. Nefkens and B. Zwanenburg, Tetrahedron, 1983, 39, 2995–2998 CrossRef CAS.
- B. Garrigues and M. Mulliez, J. Organomet. Chem., 1986, 314, 19–24 CrossRef CAS.
- A. Schäfer, W. Saak, D. Haase and T. Müller, Angew. Chem., Int. Ed., 2012, 51, 2981–2984 CrossRef PubMed.
- (a) B. Wrackmeyer, E. Khan and R. Kempe, Z. Naturforsch., B: J. Chem. Sci., 2008, 63, 275–279 CAS; (b) R. Köster, A. Sporzyński, W. Schüβler, D. Bläser and R. Boese, Chem. Ber., 1994, 127, 1191–1199 CrossRef.
- (a) J. Burés, Angew. Chem., Int. Ed., 2016, 55, 2028–2031 CrossRef PubMed; (b) J. Burés, Angew. Chem., Int. Ed., 2016, 55, 16084–16087 CrossRef PubMed; (c) J. Burés, Top. Catal., 2017, 60, 631–633 CrossRef.
- H. Lundberg, F. Tinnis, J. Zhang, A. G. Algarra, F. Himo and H. Adolfsson, J. Am. Chem. Soc., 2017, 139, 2286–2295 CrossRef CAS PubMed.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104–154118 CrossRef PubMed.
- A. Armstrong, R. A. Boto, P. Dingwall, J. Contreras-García, M. J. Harvey, N. J. Mason and H. S. Rzepa, Chem. Sci., 2014, 5, 2057–2071 RSC.
- M. J. Harvey, A. McLean and H. S. Rzepa, J. Cheminf., 2017, 9, 4 Search PubMed.
- S. Arkhipenko, A. S. Batsanov, M. T. Sabatini, V. Karaluka, T. D. Sheppard, H. S. Rzepa and A. Whiting, Imperial College Research Computing Services data repository, 2016, DOI:10.14469/hpc/1620 and sub-collections therein.
Footnote |
| † Electronic supplementary information (ESI) available: FAIR data for NMR spectra, computational data and ESI for synthetic procedures. CCDC 1551614–1551624 and 1563948–1563950. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7sc03595k and ref. 26. |
| This journal is © The Royal Society of Chemistry 2018 |