
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Continuous low temperature synthesis of MAPbX3 perovskite nanocrystals in a flow reactor†
Xinxing
Liang
 a,
Robert W.
Baker
a,
Kejun
Wu
b,
Wentao
Deng
a,
Dominic
Ferdani
a,
Peter S.
Kubiak
a,
Frank
Marken
a,
Laura
Torrente-Murciano
b and
Petra J.
Cameron
*a
a,
Robert W.
Baker
a,
Kejun
Wu
b,
Wentao
Deng
a,
Dominic
Ferdani
a,
Peter S.
Kubiak
a,
Frank
Marken
a,
Laura
Torrente-Murciano
b and
Petra J.
Cameron
*a
aDepartment of Chemistry, University of Bath, Claverton Down, Bath, BA2 7AY, UK. E-mail: P.J.Cameron@bath.ac.uk
bDepartment of Chemical Engineering and Biotechnology, University of Cambridge, Cambridge, CB3 0AS, UK
First published on 7th August 2018
Abstract
Continuous room temperature synthesis of MAPbX3 perovskite nanocrystals was conducted using a facile flow reactor. The process exhibited outstanding reproducibility and the resulting nanocrystals showed a narrow size distribution, high stability and excellent emissive properties. Their photoluminescence can be tuned by changing the halide composition to cover the visible and near-infrared region.
Organo lead halide perovskites are a class of materials that show high absorption coefficients,1 long carrier diffusion lengths,2 high carrier mobilities3 and low trap densities.4 Due to these unique properties, as well as their easy solution processability, they have been widely investigated as absorbers for solar cells.5,6 Perovskite solar cells show highly competitive efficiencies, increasing from 3.81% in 20097 to 22.1% in less than 10 years.8
Along with the development of bulk perovskite materials, perovskite nanocrystals (NCs) have also attracted significant interest, as they display size-dependent absorption and emission, narrow emission width and high quantum efficiency.9–11 MAPb(Br0.3I0.7)3 (MA = methylammonium) NCs have been used as interlayers9 for perovskite solar cells, placed between a MAPbI3 layer and hole-transporting layer (HTL). The NCs improved the device performance and the increase in efficiency was attributed to improved hole transfer at the interface. CsPbI3 NCs have been used as the absorber layer for solar cells10 with an efficiency of 10.77%, high stability and an open-circuit voltage as high as 1.23 V. Moreover, both CsPbX3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 11 and MAPbX312 NCs have been investigated for applications in LEDs showing narrow bandwidth and easy tunability across visible and near infrared range.
11 and MAPbX312 NCs have been investigated for applications in LEDs showing narrow bandwidth and easy tunability across visible and near infrared range.
Currently, the synthesis of APbX3 perovskite NCs is mainly based on two methods: ligand-assisted re-precipitation (LARP)13 and hot injection (HI).14 In LARP the perovskite precursors are dissolved in a solvent (typically DMF or DMSO) and the nanocrystals precipitate out when the mixture is injected into an anti-solvent (e.g. toluene, hexane). In LARP, bulk material is formed as a by-product together with the nanoparticles, which limits the synthetic yield.13 In HI, a Cs-oleate solution is injected into a PbX2 solution at elevated temperature. This method normally requires temperatures of over 100 °C, and is most commonly used for the synthesis of Cs-based perovskites and rarely for MAPbX3 nanocrystals.15 Although these two methods result in the formation of highly emissive perovskite NCs, the synthesis is typically carried out in batch systems which are characterized by poor heat and mass transport, making the large-scale production difficult due to the limitations on their scale-up.
A promising alternative is the use of flow reactors for the continuous production of perovskite NCs. Flow reactors are characterized by enhanced mass and heat transfer, higher reproducibility and better yield compared with batch synthesis,16,17 and are particularly suitable for large scale production. Flow systems have been successfully used to synthesize a variety of nanomaterials, including metal nanoparticles,18 metal oxides19 and colloidal semiconductors.20,21 Wu et al. demonstrated the synthesis of silver nanoparticles with narrow size distributions in helical microreactors in the absence of capping ligands.18
To date, very little work has been carried out on the flow synthesis of perovskite NCs. Lignos et al.22 reported an elegant flow synthesis of CsPbX3 nanocrystals using a method developed from the HI technique; however the process required not only high temperatures of 120–180 °C, but also the use of an oil carrier fluid which increased the system complexity. Formamadinium lead halide nanocrystals have also been prepared in flow – using microdroplets suspended in a carrier fluid.23,24 Online photoluminescence measurements were used to probe nanocrystal structures as a function of precursor concentration and synthesis temperatures from 40 to 100 °C.
In this communication, we report a facile and highly reproducible continuous flow method for the synthesis of MAPbX3 perovskite NCs using a single-phase system, demonstrating the lack of diffusion limitations during such synthesis, and thus, simplifying the process and its scale-up. In the flow preparation of high quality formamadinium lead halide nanocrystals described by Lignos et al. and Maceiczyk et al.,23,24 the FA containing solution was degassed at RT for 30 min under vacuum and heated to 120 °C under a nitrogen atmosphere for another 30 min. Similarly, the halide precursor solution was degassed for 1 hour at 130 °C under vacuum. Our method gives particles with a slightly wider size distribution, but was carried out at room temperature using all chemicals as received.
The setup developed for the flow synthesis of the perovskite NCs is depicted in Fig. 1. The full experimental procedure is given in the ESI.† Briefly, PbX2 was dissolved in 1-octadecene (ODE) (solution 1) and methylammonium iodide or bromide (MAX) was dissolved in a mixture of ODE and n-butanol (BuOH) (solution 2). Oleic acid (OA) and/or oleylamine (OLA) were added as ligands. Neither solution was degassed before use. Syringe pumps were used to inject the solutions into a 3 meter PTFE reactor immersed in a water bath at 30 °C. The synthesis worked well at room temperature, however a slightly elevated temperature of 30 °C was chosen to avoid any effect of fluctuating lab temperature on the experiment. The perovskite NCs were observed to nucleate quickly, forming small crystals that created a visible colour change in the tube a few seconds after mixing. No fouling of the reactors walls was observed. The product was collected in a container inside an ice bath. The resulting mixture was very concentrated. It was centrifuged to remove most of the suspended material – the resulting supernatant was highly fluorescent. MAPbX3 NCs are known to be unstable, due to the low energy of lead halide perovskite formation and proton exchange reactions between surfactants and passivation ligands which leads to the facile detachment of the ligands.25 Unfortunately the NCs in ODE were only stable for up to an hour before large scale precipitation of solid was observed. To separate and stabilize the NCs, the pellet of solid material obtained after centrifugation was resuspended in toluene. This suspension was then centrifuged again and the toluene supernatant solution was used without any further treatment.
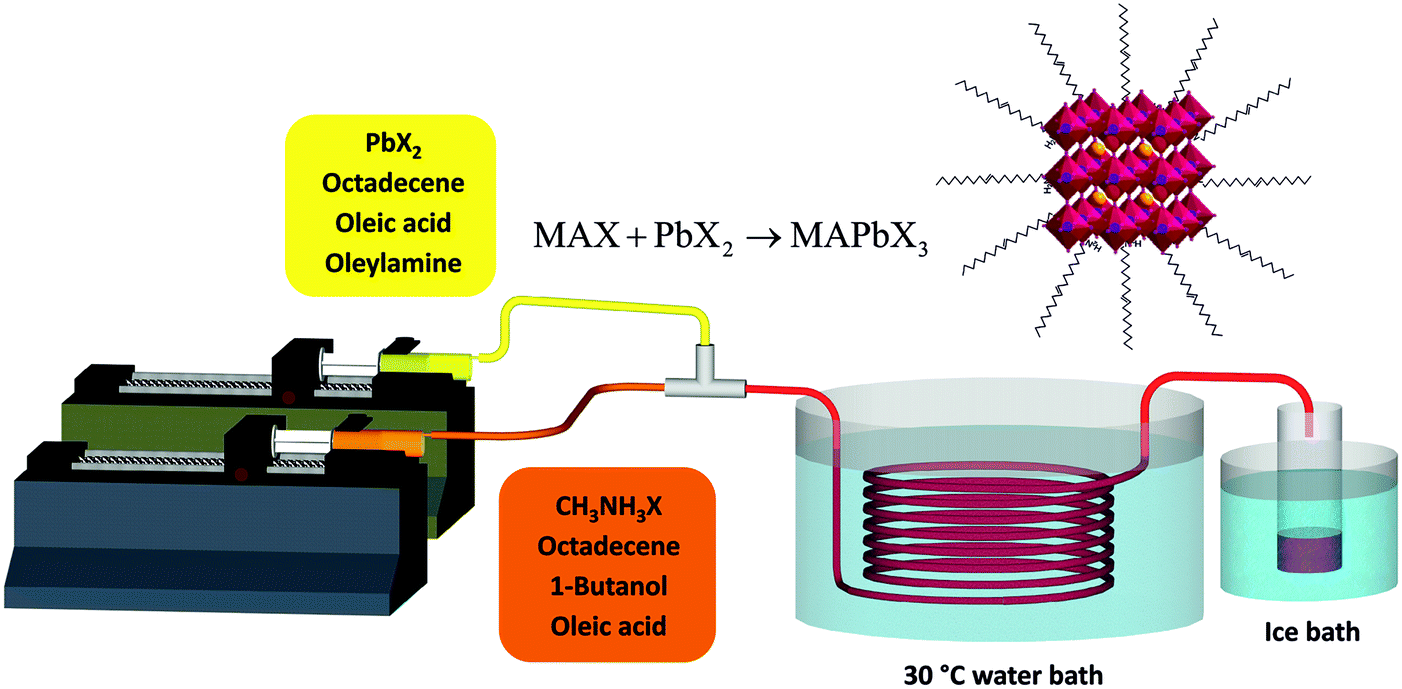 | ||
| Fig. 1 Schematic diagram of the flow reactor system used for the synthesis of MAPbX3 nanocrystals at low temperature. | ||
TEM images of MAPbI3 NCs were taken for freshly prepared nanocrystals (Fig. S6†) and for nanocrystals that had been stored as a suspension in toluene for >8 months. Fig. 2a shows the NCs after 8 months, square particles are clearly visible with a high morphological purity. The NCs were very stable and the size/morphology did not change during storage.
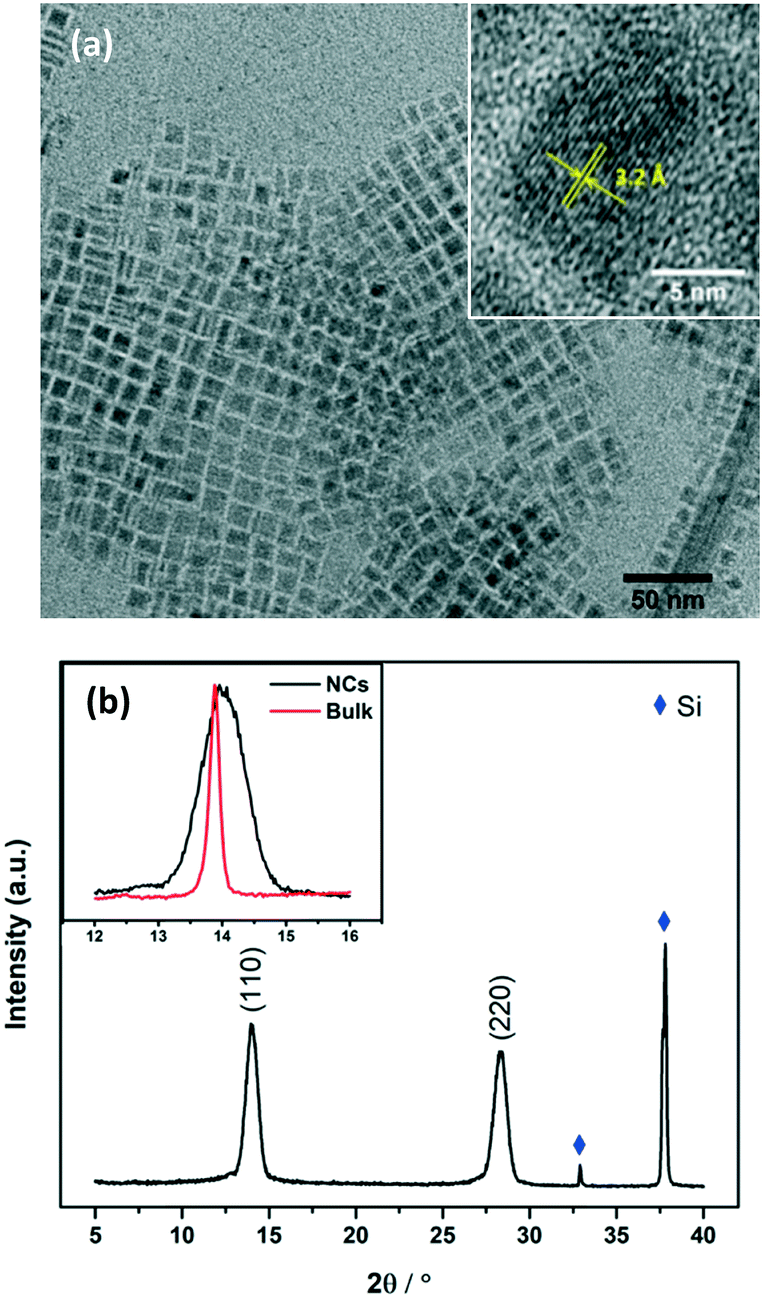 | ||
| Fig. 2 (a) TEM images of MAPbI3 NCs stored for over 8 months. The inset is the high resolution TEM of a typical NC. The interplanar spacing was measured to be 3.2 Å, which is consistent with the (220) plane of tetragonal phase MAPbI3 perovskite.26 (b) XRD patterns of MAPbI3 NCs coated on silicon wafer. The inset is the comparison of the (110) peak of MAPbI3 NCs with that of the bulk film. | ||
The NCs had sizes of 10.5 ± 1.5 nm, as shown in the size distribution histogram in Fig. S1.† The MAPbI3 nanoparticles show a good size distribution, although high temperature hot injection methods have been used to prepare NCs with slightly narrower size distributions. We are currently undertaking a large scale systematic study to test whether similar size distributions can be obtained using our simple room temperature synthesis.
The structure of the crystals was confirmed by X-ray diffraction (XRD) spectra taken for NCs coated onto a piece of silicon wafer (Fig. 2b). Diffraction peaks at 14.0° and 28.3° were observed, corresponding to the (110) and (220) planes of the tetragonal perovskite phase. Notably, the peaks are very broad compared to those observed for the bulk material (see Fig. S2† and the inset of Fig. 2b), confirming the small crystal sizes. Scherrer's equation was used to calculate the average crystal size.
| τ = Kλ/Bcosθ |
The MAPbI3 NCs showed strong absorption and emission in toluene solution (Fig. 3). The absorption onset is at ∼740 nm, and the emission peaks sharply at 745 nm with a narrow FWHM (full width at half maximum) of 39 nm. The absorption and emission are both blue-shifted compared to the bulk material (spectrum shown in Fig. S3†).
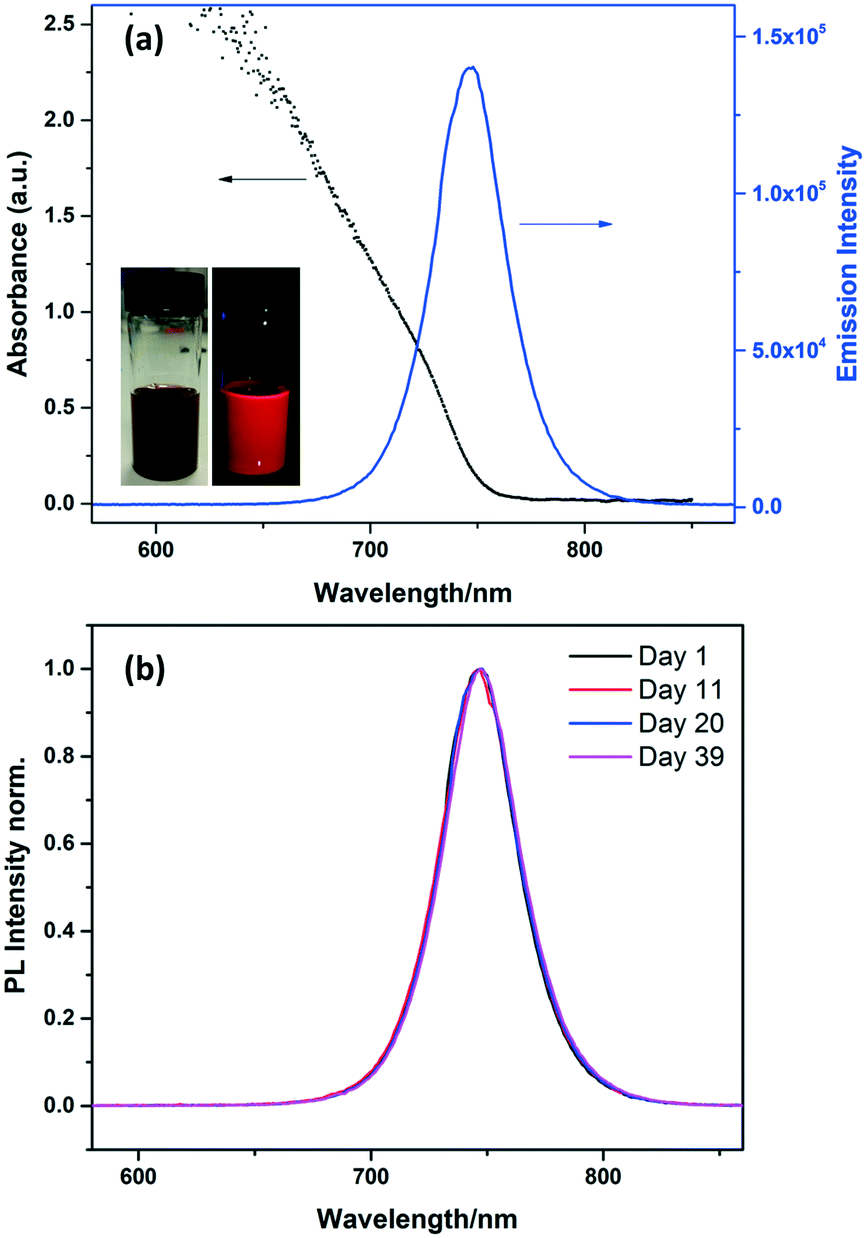 | ||
| Fig. 3 (a) Absorption and photoluminescence of MAPbI3 NCs solution in toluene. The insets show the solution under ambient and UV light. (b) PL spectra taken for MAPbI3 NCs solution over 39 days. The solution was stored in a sealed vial at room temperature in the dark. | ||
In our synthesis, no degassing treatments to the precursor solutions were carried out. For comparison, the precursor solutions were also degassed as they were in Kovalenko's report.23 As shown in Fig. S4,† the PL responses of the samples are very similar. Fig. S5† shows a TEM image of the nanoparticles. The peak positions of the non-degassed sample and the degassed sample showed a small difference of only ∼8 nm, and the peak width of the degassed sample was slightly broader than for our standard procedure. The small difference in the PL spectra indicates a degassing treatment is not required in our synthesis, probably due to the closed system in microreactors which simplifies the synthesis. Notably, the flow synthesis showed outstanding reproducibility, as shown in the PL spectra of eleven independent repeats in Fig. S6.† the emission peaks showed high conformity, centering on 742.4 ± 1.6 nm, with the FWHM of 39.8 ± 0.3 nm. The small deviation indicates the high robustness of the method. While it is extremely difficult to calculate the yield of reactions producing nanoparticles, we are able to make large volumes containing gram quantities of suspended NCs.
Stability is an important problem for perovskite NCs, especially MA-based perovskite NCs which are normally much less stable than Cs-based inorganic perovskite.15,27 As is the case with all NCs, they are prone to aggregation which has to be prevented by the correct choice of stabilising ligands. MAPbX3 perovskites can also be easily decomposed into PbI228 in the presence of polar solvents such as water. To investigate the long term stability of the MAPbI3 NCs in toluene solution, photoluminescence emission spectra were measured at regular intervals for more than one month, as shown in Fig. 3b. The emission peak for the MAPbI3 NC solution did not change over 39 days, with no shifts or broadening observed. This result suggests that the NCs are stable over this time. The stability of the MAPbI3 NCs was also confirmed by the TEM images discussed above which were collected after the NCs had been stored as a suspension in toluene for 8 months.
The facile flow synthesis was adapted for the preparation of MAPbX3 NCs with different halide compositions to tune the luminescence properties. As shown in Fig. 4, the addition of Br shifts the emission peak to lower wavelengths. A wide range of MAPbIxBr3–x compositions was investigated, allowing us to tune the emission between 485 nm and 745 nm. This is a larger range than has previously been reported for CsPbX3,11 and slightly wider than Zhang's report for MAPbX3 NCs13 Zhang et al. reported tuneable emission of MAPbX3 NCs (made by the LARP method) between 515–734 nm when the halide composition was changed from MAPbBr3 to MAPbI3.
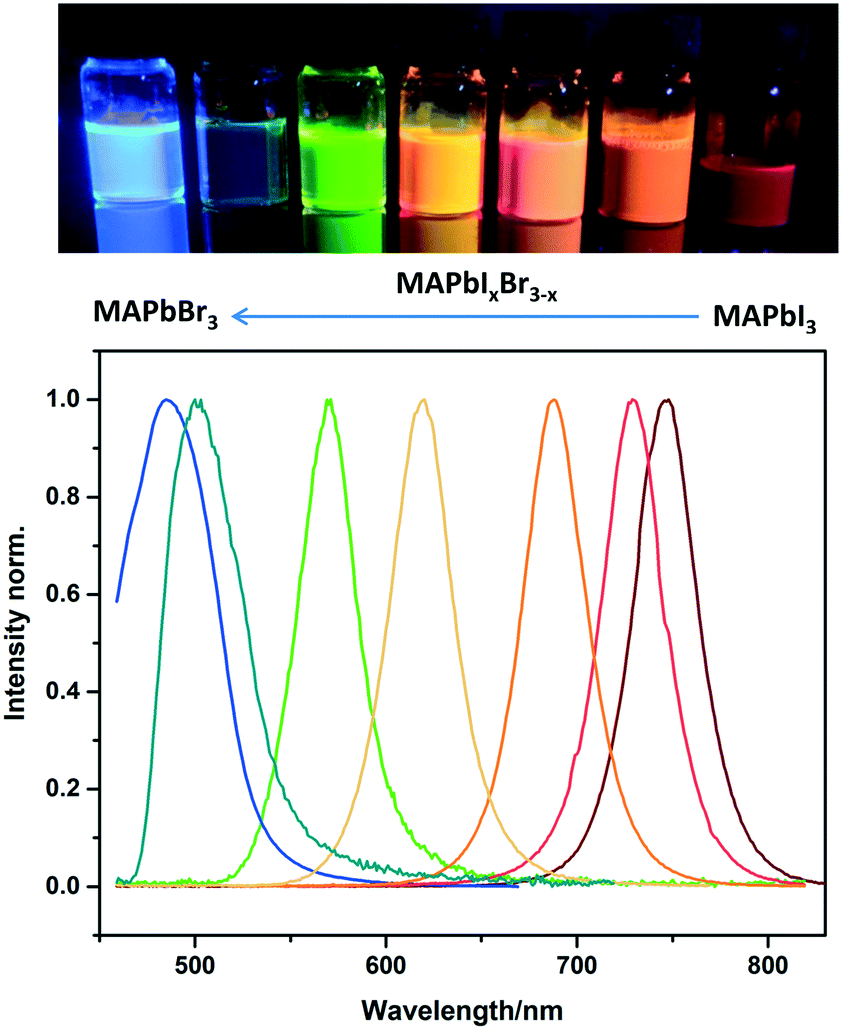 | ||
| Fig. 4 Photo of sample vials under UV illumination and PL spectra of MAPbIxBr3–x perovskite NCs in toluene with different halide compositions. | ||
Our mixed halide nanocrystals also showed high stability, as shown in the long term photoluminescence spectra for MAPbIBr2 and MAPbBr3 NCs displayed in Fig. S8 and S9† respectively. For all of our compositions, excellent colour purity was observed with FWHM below 50 nm which suggests a narrow size distribution.
The photoluminescent quantum yield of the MAPbI3 NCs in toluene was approx. 27%. The QY is not as high as that found for many all-inorganic perovskite NCs, but is comparable with several published reports on organic cation perovskite NCs.12,29–31 Schmidt et al. reported QYs of up to 23% for MAPbBr3 NCs; Hassan et al. reported QYs up to 20% for 2D R2MAn−1PbnI3n+1 perovskite NCs and Weidman et al. reported QYs of up to 22% for perovskite nanoplatelets with various cation, metal and halide compositions. Interestingly, the NCs with different halide compositions showed different morphologies. As shown in Fig. S10,† the MAPbBr3 NCs show a spherical morphology with diameters of 2.8 ± 0.3 nm. The ultra-small size of the MAPbBr3 NCs is believed to be responsible for the emission below 500 nm. The mixed halide NCs showed mixed morphologies between the cubic shape of the MAPbI3 and the spherical ones of the pure MAPbBr3. Additional in-line analysis would be needed to understand the morphology evolution from cubes to dots between the iodide and bromide perovskites and how mixing of the precursors can affect it. Morphology can be improved by controlling the nucleation and growth of the nanoparticles. Previously investigated strategies include modifying the ligand concentration and ratios,32,33 ligand chain length34,35 as well as solvent ratio,36 temperature37 and precursor concentration.38,39 Additional control can be achieved by novel purification strategies such as flow electrophoresis and solvent selection.40,41
In summary, a facile flow process has been developed for the synthesis of organo-lead halide perovskite NCs using a flow reactor. The process was carried out at 30 °C with high reproducibility and, in contrast to existing methods, degassing was not required. The perovskite NCs showed small sizes with narrow size distribution, high stability and excellent emissive properties. The emission peaks for MAPbI3 NCs showed narrow FWHM of below 50 nm and remained stable for at least 39 days. Moreover, the luminescence could be easily tuned by changing the halide composition from blue to near infrared with high colour purity. Further work will focus on the effects of process parameters such as flow rate and temperature on the sizes and luminescent properties of the NCs and their applications in photovoltaic and LED devices.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the financial support from the University of Bath through the 50th Anniversary Excellence Studentship for Overseas Students and the Leche Trust and the Great Britain-China Educational Trust (GBCET) for additional support for XL. LTM acknowledges the EPSRC for a fellowship award (grant number EP/L020432/2). DF and PJC acknowledge funding from the EPSRC Doctoral Training Centre in Sustainable Chemical Technologies: EP/G03768X/1. The authors thank Professor Tony James for providing access to the fluorescence spectrometer.References
- W.-J. Yin, T. Shi and Y. Yan, Adv. Mater., 2014, 26, 4653–4658 CrossRef PubMed.
- S. D. Stranks, G. E. Eperon, G. Grancini, C. Menelaou, M. J. P. Alcocer, T. Leijtens, L. M. Herz, A. Petrozza and H. J. Snaith, Science, 2013, 342, 341–344 CrossRef PubMed.
- H. Oga, A. Saeki, Y. Ogomi, S. Hayase and S. Seki, J. Am. Chem. Soc., 2014, 136, 13818–13825 CrossRef PubMed.
- B. R. Sutherland and E. H. Sargent, Nat. Photonics, 2016, 10, 295–302 CrossRef.
- M. M. Lee, J. Teuscher, T. Miyasaka, T. N. Murakami and H. J. Snaith, Science, 2012, 338, 643–647 CrossRef PubMed.
- W. Nie, H. Tsai, R. Asadpour, J.-C. Blancon, A. J. Neukirch, G. Gupta, J. J. Crochet, M. Chhowalla, S. Tretiak, M. A. Alam, H.-L. Wang and A. D. Mohite, Science, 2015, 347, 522–525 CrossRef PubMed.
- A. Kojima, K. Teshima, Y. Shirai and T. Miyasaka, J. Am. Chem. Soc., 2009, 131, 6050–6051 CrossRef PubMed.
- J.-P. Correa-Baena, A. Abate, M. Saliba, W. Tress, T. Jesper Jacobsson, M. Grätzel and A. Hagfeldt, Energy Environ. Sci., 2017, 10, 710–727 RSC.
- M. Cha, P. Da, J. Wang, W. Wang, Z. Chen, F. Xiu, G. Zheng and Z.-S. Wang, J. Am. Chem. Soc., 2016, 138, 8581–8587 CrossRef PubMed.
- Q. A. Akkerman, M. Gandini, F. Di Stasio, P. Rastogi, F. Palazon, G. Bertoni, J. M. Ball, M. Prato, A. Petrozza and L. Manna, Nat. Energy, 2016, 2, 16194 CrossRef.
- J. Song, J. Li, X. Li, L. Xu, Y. Dong and H. Zeng, Adv. Mater., 2015, 27, 7162–7167 CrossRef PubMed.
- L. C. Schmidt, A. Pertegás, S. González-Carrero, O. Malinkiewicz, S. Agouram, G. Mínguez Espallargas, H. J. Bolink, R. E. Galian and J. Pérez-Prieto, J. Am. Chem. Soc., 2014, 136, 850–853 CrossRef PubMed.
- F. Zhang, H. Zhong, C. Chen, X. Wu, X. Hu, H. Huang, J. Han, B. Zou and Y. Dong, ACS Nano, 2015, 9, 4533–4542 CrossRef PubMed.
- L. Protesescu, S. Yakunin, M. I. Bodnarchuk, F. Krieg, R. Caputo, C. H. Hendon, R. X. Yang, A. Walsh and M. V. Kovalenko, Nano Lett., 2015, 15, 3692–3696 CrossRef PubMed.
- O. Vybornyi, S. Yakunin and M. V. Kovalenko, Nanoscale, 2016, 8, 6278–6283 RSC.
- R. L. Hartman, J. P. McMullen and K. F. Jensen, Angew. Chem., Int. Ed., 2011, 50, 7502–7519 CrossRef PubMed.
- Y. Song, J. Hormes and C. S. S. R. Kumar, Small, 2008, 4, 698–711 CrossRef PubMed.
- K.-J. Wu, G. M. De Varine Bohan and L. Torrente-Murciano, React. Chem. Eng., 2017, 2, 116–128 RSC.
- E. Y. Erdem, J. C. Cheng, F. M. Doyle and A. P. Pisano, Small, 2014, 10, 1076–1080 CrossRef PubMed.
- B. K. H. Yen, A. Günther, M. A. Schmidt, K. F. Jensen and M. G. Bawendi, Angew. Chem., Int. Ed., 2005, 44, 5447–5451 CrossRef PubMed.
- S. Marre, J. Park, J. Rempel, J. Guan, M. G. Bawendi and K. F. Jensen, Adv. Mater., 2008, 20, 4830–4834 CrossRef.
- I. Lignos, S. Stavrakis, G. Nedelcu, L. Protesescu, A. J. DeMello and M. V. Kovalenko, Nano Lett., 2016, 16, 1869–1877 CrossRef PubMed.
- R. M. Maceiczyk, K. Dümbgen, I. Lignos, L. Protesescu, M. V. Kovalenko and A. J. DeMello, Chem. Mater., 2017, 29, 8433–8439 CrossRef.
- I. Lignos, L. Protesescu, D. B. Emiroglu, R. Maceiczyk, S. Schneider, M. V. Kovalenko and A. J. DeMello, Nano Lett., 2018, 18, 1246–1252 CrossRef PubMed.
- H. Sun, Z. Yang, M. Wei, W. Sun, X. Li, S. Ye, Y. Zhao, H. Tan, E. L. Kynaston, T. B. Schon, H. Yan, Z.-H. Lu, G. A. Ozin, E. H. Sargent and D. S. Seferos, Adv. Mater., 2017, 29, 1701153 CrossRef PubMed.
- T. Oku, in Solar Cells - New Approaches and Reviews, ed. L. A. Kosyachenko, InTech, Rijeka, 2015 Search PubMed.
- X. Li, Y. Wu, S. Zhang, B. Cai, Y. Gu, J. Song and H. Zeng, Adv. Funct. Mater., 2016, 26, 2435–2445 CrossRef.
- G. Niu, W. Li, F. Meng, L. Wang, H. Dong and Y. Qiu, J. Mater. Chem. A, 2014, 2, 705–710 RSC.
- Y. Hassan, Y. Song, R. D. Pensack, A. I. Abdelrahman, Y. Kobayashi, M. A. Winnik and G. D. Scholes, Adv. Mater., 2016, 28, 566–573 CrossRef PubMed.
- M. C. Weidman, M. Seitz, S. D. Stranks and W. A. Tisdale, ACS Nano, 2016, 10, 7830–7839 CrossRef PubMed.
- I. Levchuk, A. Osvet, X. Tang, M. Brandl, J. D. Perea, F. Hoegl, G. J. Matt, R. Hock, M. Batentschuk and C. J. Brabec, Nano Lett., 2017, 17, 2765–2770 CrossRef PubMed.
- G. Almeida, L. Goldoni, Q. Akkerman, Z. Dang, A. H. Khan, S. Marras, I. Moreels and L. Manna, ACS Nano, 2018, 12, 1704–1711 CrossRef PubMed.
- Z. Liang, S. Zhao, Z. Xu, B. Qiao, P. Song, D. Gao and X. Xu, ACS Appl. Mater. Interfaces, 2016, 8, 28824–28830 CrossRef PubMed.
- A. Pan, B. He, X. Fan, Z. Liu, J. J. Urban, A. P. Alivisatos, L. He and Y. Liu, ACS Nano, 2016, 10, 7943–7954 CrossRef PubMed.
- S. Sun, D. Yuan, Y. Xu, A. Wang and Z. Deng, ACS Nano, 2016, 10, 3648–3657 CrossRef PubMed.
- M. B. Teunis, M. A. Johnson, B. B. Muhoberac, S. Seifert and R. Sardar, Chem. Mater., 2017, 29, 3526–3537 CrossRef.
- H. Huang, A. S. Susha, S. V. Kershaw, T. F. Hung and A. L. Rogach, Adv. Sci., 2015, 2, 1500194 CrossRef PubMed.
- W. Liu, J. Zheng, S. Cao, L. Wang, F. Gao, K.-C. Chou, X. Hou and W. Yang, Inorg. Chem., 2018, 57, 1598–1603 CrossRef PubMed.
- A. Dutta, S. K. Dutta, S. Das Adhikari and N. Pradhan, ACS Energy Lett., 2018, 3, 329–334 CrossRef.
- H. Lim, J. Y. Woo, D. C. Lee, J. Lee, S. Jeong and D. Kim, Sci. Rep., 2017, 7, 43581 CrossRef PubMed.
- D. Kim, H. K. Park, H. Choi, J. Noh, K. Kim and S. Jeong, Nanoscale, 2014, 6, 14467–14472 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Materials and methods, additional figures. See DOI: 10.1039/c8re00098k |
| This journal is © The Royal Society of Chemistry 2018 |