
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Robust mesoporous bimetallic yolk–shell catalysts for chemical CO2 upgrading via dry reforming of methane†
Cameron-Alexander Hurd
Price
 a,
Laura
Pastor-Pérez
ab,
Tomas
Ramirez Reina
*a and
Jian
Liu
*ac
a,
Laura
Pastor-Pérez
ab,
Tomas
Ramirez Reina
*a and
Jian
Liu
*ac
aDepartment of Chemical Engineering and Process Engineering, University of Surrey, Guildford, GU2 7XH, UK. E-mail: t.ramirezreina@surrey.ac.uk; jian.liu@surrey.ac.uk
bLaboratorio de Materiales Avanzados, Departamento de Química Inorgánica – Instituto Universitario de Materiales de Alicante Universidad de Alicante, Apartado 99, E-03080 Alicante, Spain
cState Key Laboratory of Catalysis, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, 457 Zhongshan Road, Dalian 116023, China
First published on 19th June 2018
Abstract
Here, we report the synthesis of mesoporous ZnO/Ni@m-SiO2 yolk–shell particles. The unique ZnO/Ni@m-SiO2 catalysts demonstrate impressive resistance to sintering and coking for dry reforming of methane (DRM). They also display long term stability with high levels of conversion and selectivity, making this catalyst promising for chemical CO2 upgrading.
The use of supported nanoparticulate materials as catalysts is a long-standing practice as they demonstrate impressive catalytic properties when applied to numerous reactions.1–5 However, these materials are plagued by numerous drawbacks, some of which are associated with the harsh environments that reactions often demand, causing them to lose activity over time. This poses a significant cost to all industries that use supported catalysts in the form of unplanned shutdowns and material replacement every year. Although the time period over which a catalytic material remains active is highly variable; iron catalysts for ammonia synthesis lasting several years and cracking catalysts sometimes lasting only seconds,6 deactivation is unavoidable.
Although deactivation is certain for all catalysts, it can be postponed. Through the careful design and development of resistant materials, the active lifetime of these catalysts can be extended. This area of research has attracted significant interest for a number of years, being the topic of a considerable amount of research7–13 and review.14–17 The goal of this research being to develop an economically viable material, ideally not containing noble metals, that are resistant to these forms of deactivation that continue to produce high levels of conversion and reaction performance.
The yolk@shell materials detailed in this paper were designed to tackle two well-known areas of catalytic deactivation: coking and sintering. Where supported metal catalysts are concerned, coking can occur in a number of ways: adsorption to the surface through a “monolayer-multilayer” mechanism,13,18,19 total encapsulation of the material that completely deactivates the catalyst19,20 or fill micro- and meso-pores to prevent or at least limit access of the reactants to the active sites.19,20
Where sintering is concerned, it occurs in several ways: reduction in active surface area due to the growth of the catalytic phase, reduction in the amount of support area due to the collapse of the support phase in addition to the collapse of the porous structure. To prevent these problems, the shell within which our active Ni/ZnO core is held, provides a physical shield to mitigate sintering and also acts as a barrier to prevent the blocking of the active sites through coke formation.
The resultant catalysts were used in the dry reforming of methane (DRM) reaction due to renewed interest in the “green” chemistry approach to produce added value products through chemical recycling CO2, building on the research of other groups21–25 as well as our own.26–28 The chemical recycling of CO2 through the DRM reaction, represented by eqn (1) is a highly endothermic reaction that requires temperatures high enough to favour the process. The later also triggers sintering of active phases and the formation of coke. In fact, given the relatively hard conditions of DRM, it constitutes an excellent “proof-of concept” reaction to validate the yolk–shell approach.
| CH4 + CO2 → 2H2 + 2CO | (1) |
In this scenario we aim to demonstrate the effectiveness of m-SiO2 as a shell material for ZnO–Ni cores within the DRM reaction in the unique “yolk@shell” morphology as a method for preventing the thermal degradation of the active nickel species that does not prevent high levels of conversion.
The schematic preparation method of our encapsulated catalysts along with representative scanning electron microscopy (SEM) image is depicted in Fig. 1 and further explained in the ESI.† The SEM image illustrates a broken yolk shell particle with the core clearly visible through the hole in the shell. This provides not only visual confirmation of the successful encapsulation process but also shows the size of these particles to be approximately 400 nm in diameter with a 200 nm core.
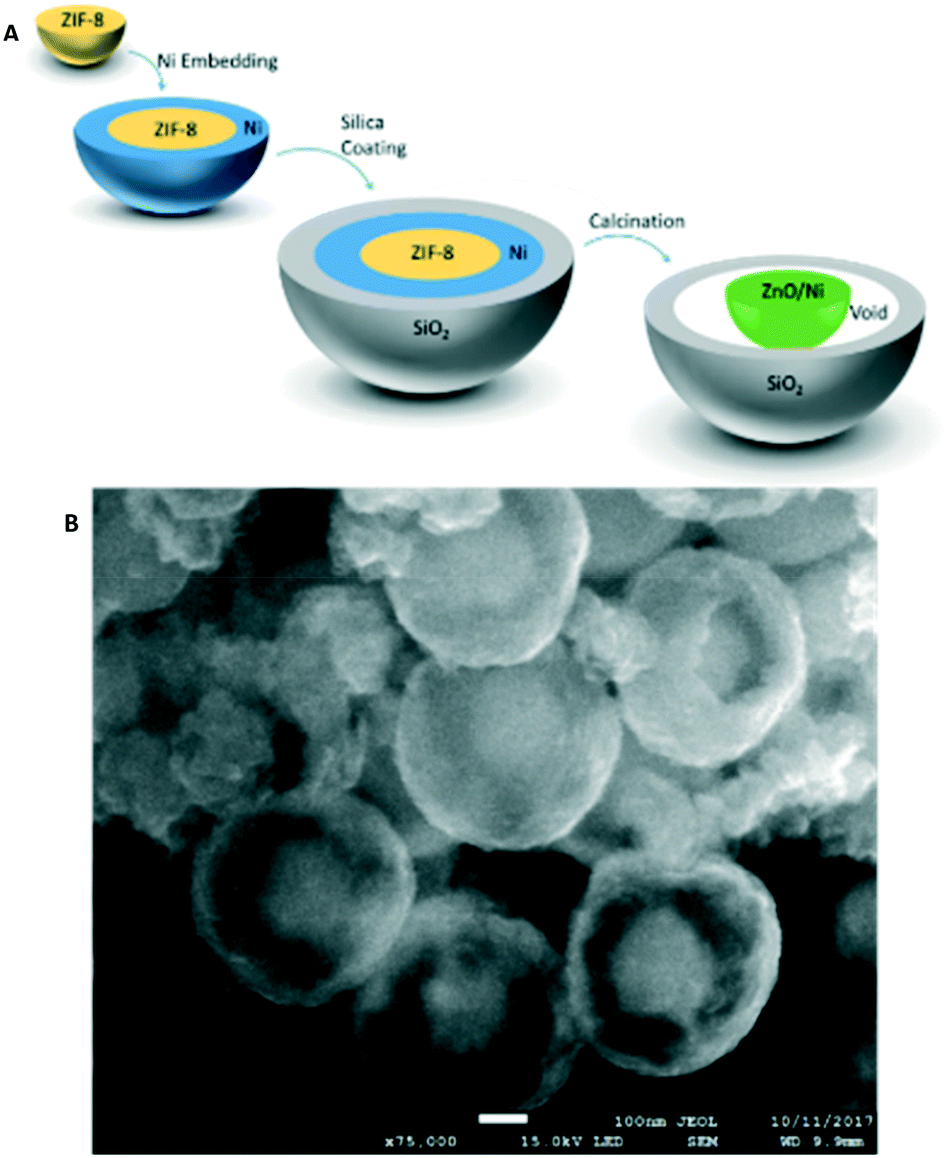 | ||
| Fig. 1 A) A schematic illustration of the synthetic procedure of yolk–shell catalysts. B) SEM image of the resultant ZnO/Ni@m-SiO2 yolk–shell particles, confirming successful encapsulation. | ||
The yolk@shell material as well as reference Ni based catalysts were tested in the DRM reaction using a continuous flow reactor at different temperatures (Fig. 2A), details of the procedure for catalytic testing can be found in the ESI.† As seen in Fig. 2, the increasing reactant conversion and product ratio can be tracked against increasing temperature for all the studied samples. As expected within an endothermic reaction, it can be seen that the material performs best at higher temperatures, with conversion of CO2 reaching 98% at 850 °C. In a similar way, the H2/CO ratio also increases for all the studied catalysts and in the case of the yolk–shell materials it goes from 0.6 at 550 °C to 0.97 at 850 °C. From Fig. 2 it can be inferred that our advanced yolk–shell catalysts present a comparable performance than that exhibited by the standard DRM catalysts. Ni/SiO2–Al2O3 seems to be slightly more active in the low temperature range however the stability of this solid is very poor in comparison to that displayed by the encapsulated catalyst as shown in Fig. S1 (ESI†). Indeed, this stability test reveals the superiority of the yolk–shell catalyst in comparison to benchmark materials which show a rapid deactivation.
 | ||
| Fig. 2 Shows the comparative temperature screening results of our reported material against Ni/Al2O3 and Ni/SiO2–Al2O3 concerning A) CH4 conversion B) CO2 conversion and C) H2/CO ratio. | ||
Fig. 3 represents a longer term stability test and shows that our ZnO/Ni@m-SiO2 particles can be applied for continuous operations displaying good levels of CO2 and CH4 conversion. The long term catalytic performance of this material in the DRM reaction, was examined at a temperature of 850 °C, due to the extraordinary performance achieved under these conditions. The material demonstrated considerable long-term stability and high levels of CO2/CH4 conversion when compared to recent DRM studies (Table S1, ESI†), albeit with some deactivation (96% to 78% for CO2, 89% to 61% for CH4) after 143 hours of continuous operation. Additionally the catalyst produces an excellent syngas stream with a H2/CO ratio close to 1 (the maximum allowed by the nature of the reaction). Such syngas is a highly valuable product for chemical synthesis and can be used to produce fuels (via Fischer–Tropsch), methanol or could be directly utilised to power a solid oxide fuel cell (SOFC).
 | ||
| Fig. 3 Shows the long term stability reactant conversion and H2/CO ratio data of our reported material at 850 °C. | ||
It can be seen however that carbon peaks appear on the post-reaction XRD pattern in Fig. 4A. This coking can be observed on the TEM images (Fig. S2, ESI†) to be taking place on the small amount of unencapsulated nickel/zinc oxide cores that are present throughout the sample. In the same images, it is apparent that the encapsulated cores remain un-coked and thus can be assumed to be successfully resistant.
 | ||
| Fig. 4 A) XRD patterns of the calcined material, a, the activated material, b, and the post reaction sample, c. B) TEM image showing the successful encapsulation of the ZnO/Ni core within the m-SiO2 shell. | ||
Furthermore, when the post-reaction sample underwent Raman Spectroscopy (Fig. S3, ESI†), the carbon deposits were found to be very graphitic in nature, adding to the notion that the formation of such unreactive graphitic carbon filaments are responsible for the deactivation of this material. Carbon deposition above 650 °C is caused by the decomposition of methane and is a common occurrence within the DRM, especially with bulk nickel catalysts that have minimal support interaction.29–31 Similar phenomena have been noted in several papers involving the use of Ni catalysts in the DRM reaction,29,32–34 demonstrating that this method of deactivation is highly probable for our material in particular for the fraction of unencapsulated Ni particles.
The XRD patterns b and c in Fig. 4A, represent the activated and post-reaction catalyst samples, respectively. There is little difference between the two, with the exception of the appearance of carbon peaks in pattern c, the formation of which is in good agreement with Raman spectra.
Ni crystallite size was estimated using the Scherrer equation on the most intense reflections in Fig. 4A. It was found that the Ni crystallite size increased by 2 nm, from 21.5 to 23.5 nm respectively. It can be assumed then that the nickel crystallite size did not increase significantly after the reaction and that the m-SiO2 shell successfully prevented the cores (active phase) from sintering.
This is of particular interest as the operating temperature of the DRM reaction undertaken in this study is significantly above the Tamman and Hüttig temperatures for nickel as shown in the work by Moulijn et al.,35 meaning that sintering should have been observed in the post-reaction sample. The fact that it has not been observed, shows the m-SiO2 shell is providing effective protection to the active particles in the core despite the harsh reaction conditions (high reaction temperature and relatively high space velocities).
All the characterisation data obtained from these samples, including the visual confirmation via TEM (Fig. 4B) and SEM (Fig. 1B), supports the successful synthesis of the yolk–shell particles and the ability of nano-encapsulated catalysts to tolerate metallic sintering.
However, we are aware of the presence of a small amount of unencapsulated nickel/ZnO cores after detailed analysis of the SEM and transition electron microscope (TEM) images (Fig. S2 and S4, ESI†) that are seen as black spots contained in the carbon nanotubes. Here we observe carbon filaments barring access to reactants for both ZnO/Ni cores without shells and our encapsulated material (Fig. S4, ESI†). The presence of unencapsulated cores that are prone to sintering and coking, explains the activity drop observed in Fig. 3.
In any case, this work documents the successful synthesis and effective application of ZnO/Ni@m-SiO2 yolk–shell particles towards the DRM reaction, showing also the benefits of the mesoporous silica shell to prevent the sintering of the active metal cores. Also this work demonstrates the superiority of Ni based yolk–shell catalysts over standard Ni catalysts which are traditionally used in reforming units. Despite still suffering from coking, we postulate that a variation to the shell composition would solve this and allow to, at the very least, maintain the significant performance this material displayed within the DRM, perhaps even improving it.
Efforts are currently ongoing in our lab to improve the synthetic method to ensure fully encapsulated yolk@shell materials as well as fine tune physicochemical properties to improve catalytic performance.
Nevertheless, this seminal work demonstrates the robustness of Ni based yolk@shell catalysts towards sintering and their potential to combat coking. Overall, this new family of encapsulated catalysts display excellent skills for environmental catalysis applications as for example CO2 upgrading via dry reforming.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support for this work was provided by the EPSRC grants EP/J020184/2 and EP/R512904/1, the Royal Society Research Grant RSGR1180353 and partially supported by the Australian Research Council (ARC) through Linkage Project program (LP150101158).References
- A. Y. Khodakov, W. Chu and P. Fongarland, Chem. Rev., 2007, 107, 1692–1744 CrossRef PubMed.
- R. J. White, R. Luque, V. L. Budarin, J. H. Clark and D. J. Macquarrie, Chem. Soc. Rev., 2009, 38, 481–494 RSC.
- G. Prieto, J. Zečević, H. Friedrich, K. P. De Jong and P. E. De Jongh, Nat. Mater., 2013, 12, 34–39 CrossRef PubMed.
- G. A. Somorjai and Y. G. Borodko, Catal. Lett., 2001, 76, 1–5 CrossRef.
- A. Alshammari, V. N. Kalevaru and A. Martin, in Green Nanotechnology - Overview and Further Prospects, InTech, 2016 Search PubMed.
- G. Boskovic and M. Baerns, in Basic Principles Applied Catalysis, Springer, Berlin, Heidelberg, 2004, pp. 477–503 Search PubMed.
- J. C. Marek and L. F. Albright, ACS Symp. Ser., 1982, 202, 123–149 CrossRef.
- P. G. Menon, J. Mol. Catal., 1990, 59, 207–220 CrossRef.
- J. Rostrup-Nielsen and D. L. Trimm, J. Catal., 1977, 48, 155–165 CrossRef.
- C. Bartholomew and R. J. Farrauto, Fundamentals of Industrial Ventilation, Wiley-Interscience, NJ, USA, 2nd edn, 2006 Search PubMed.
- J. B. Butt, Catal. Sci. Technol., 1984, 6, 1–63 Search PubMed.
- P. J. Denny and M. V. Twigg, in Catalyst Deactivation, ed. B. Delmon and G. F. Froment, Elsevier, 1980, vol. 6, pp. 577–599 Search PubMed.
- J. B. Butt and E. E. Petersen, Activation, Deactivation, and Poisoning of Catalysts, Elsevier, 1st edn, 1988, pp. 64–119 Search PubMed.
- M. Argyle and C. Bartholomew, Catalysts, 2015, 5, 145–269 CrossRef.
- D. L. Trimm, Appl. Catal., 1983, 5, 263–290 CrossRef.
- Z. Bian, S. Das, M. H. Wai, P. Hongmanorom and S. Kawi, ChemPhysChem, 2017, 18, 3117–3134 CrossRef PubMed.
- P. Boldrin, E. Ruiz-Trejo, J. Mermelstein, J. M. Bermúdez Menéndez, T. Ramĺrez Reina and N. P. Brandon, Chem. Rev., 2016, 116, 13633–13684 CrossRef PubMed.
- S. Niknaddaf, M. Soltani, A. Farjoo and F. Khorasheh, Pet. Sci. Technol., 2013, 31, 2451–2462 CrossRef.
- C. H. Bartholomew, Appl. Catal., A, 2001, 212, 17–60 CrossRef.
- C. Montero, B. Valle, J. Bilbao and A. G. Gayubo, Chem. Eng. Trans., 2014, 37, 481–486 Search PubMed.
- K. Mette, S. Kühl, H. Düdder, K. Kähler, A. Tarasov, M. Muhler and M. Behrens, ChemCatChem, 2014, 6, 100–104 CrossRef.
- G. Moradi, F. Khezeli and H. Hemmati, J. Nat. Gas Sci. Eng., 2016, 33, 657–665 CrossRef.
- H. Ay and D. Üner, Appl. Catal., B, 2015, 179, 128–138 CrossRef.
- A. Wolfbeisser, O. Sophiphun, J. Bernardi, J. Wittayakun, K. Föttinger and G. Rupprechter, Catal. Today, 2016, 277, 234–245 CrossRef.
- Y. Lou, M. Steib, Q. Zhang, K. Tiefenbacher, A. Horváth, A. Jentys, Y. Liu and J. A. Lercher, J. Catal., 2017, 356, 147–156 CrossRef.
- T. Stroud, T. J. Smith, E. Le Saché, J. L. Santos, M. A. Centeno, H. Arellano-Garcia, J. A. Odriozola and T. R. Reina, Appl. Catal., B, 2018, 224, 125–135 CrossRef.
- M. Wang, Y. Boyjoo, J. Pan, S. Wang and J. Liu, Chin. J. Catal., 2017, 38, 970–990 CrossRef.
- T. Yang, R. Zhou, D.-W. Wang, S. P. Jiang, Y. Yamauchi, S. Z. Qiao, M. J. Monteiro and J. Liu, Chem. Commun., 2015, 51, 2518–2521 RSC.
- E. Lovell, J. Scott and R. Amal, Molecules, 2015, 20, 4594–4609 CrossRef PubMed.
- E. Lovell, Y. Jiang, J. Scott, F. Wang, Y. Suhardja, M. Chen, J. Huang and R. Amal, Appl. Catal., A, 2014, 473, 51–58 CrossRef.
- S. Wang and G. Q. M. Lu, Appl. Catal., B, 1998, 16, 269–277 CrossRef.
- J. R. Rostrup-Nielsen and J. Sehested, in Catalyst Deactivation 2001, Proceedings, Elsevier, 2001, vol. 139, pp. 1–12 Search PubMed.
- T. P. Braga, R. C. R. Santos, B. M. C. Sales, B. R. da Silva, A. N. Pinheiro, E. R. Leite and A. Valentini, Chin. J. Catal., 2014, 35, 514–523 CrossRef.
- J. Zhang, H. Wang and A. K. Dalai, Appl. Catal., A, 2008, 339, 121–129 CrossRef.
- J. A. Moulijn, A. E. van Diepen and F. Kapteijn, Appl. Catal., A, 2001, 212, 3–16 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8re00058a |
| This journal is © The Royal Society of Chemistry 2018 |