Catalytic oxidation of aqueous bioethanol: an efficient upgrade from batch to flow†
Received
9th April 2018
, Accepted 14th May 2018
First published on 28th August 2018
Abstract
The heterogeneously catalyzed oxidation of (bio)ethanol to acetic acid is an environmentally friendly alternative to the current industrial Monsanto process. The reaction yields acetic acid from crude bioethanol under mild reaction conditions. The triphasic reaction is technically critical, due to mass and heat transfer limitations and is thus predominantly studied in batch reactor systems. However, in order to advance the industrial implementation of the catalytic route, the operation in flow at the research stage is necessary. It is an efficient, reliable, and safer system for triphasic reactions and allows us to define better performance parameters for a later up-scale of a continuous flow process. Here, we evaluated the aerobic ethanol oxidation in a flow reactor and compared it with a traditional batch system over a gold–titania catalyst under analogous conditions. In both reactors, the reaction mechanism was similar: there was a zero-order dependency in oxygen and a first-order dependency in ethanol. The different reaction orders indicate that oxygen and ethanol interact with different surface sites, possibly ethanol with gold and oxygen with the support. The study showed that the catalytic performance improves in flow by about 30% for conversion and by 10% for acetic acid selectivity. The enhancement is associated mainly with the greater resistance of gold to sintering in the flow reactor. The study underlines the necessity of switching research to flow systems in order to benchmark more efficiently and identify potential catalysts for industrial implementation as well as to enhance our understanding of triphasic reactions.
1. Introduction
The selective aerobic oxidation of alcohols has attracted significant interest of the scientific community, because it leads to a broad range of bulk and speciality chemicals and produces mainly water as a by-product.1–3 The process is fully sustainable when the alcohol originates from biomass, such as bioethanol. Bioethanol is one of the most common bio-fuels, the production of which is increasing rapidly up to 46 × 109 L per year.4 This has led to the study of new pathways to bio-chemicals. The biological partial oxidation of ethanol has been known for centuries as aerobic fermentation to acetic acid (i.e. vinegar).5 However, due to the slow kinetics of fermentation, the current demands for acetic acid (up to 12 × 106 tons per year) is met mainly by processes that utilize fossil resources, such as the carbonylation of methanol.6 The aerobic catalytic oxidation of bioethanol may be a compromise between the requirement for environmentally benign production of chemicals and the fast and efficient processes of the industry.
The most promising heterogeneous catalysts for the partial oxidation of ethanol are gold-based systems, supported on inorganic metal oxides. They are very versatile and have high activity and selectivity in various alcohol oxidation reactions.7–10 The oxidation of gaseous ethanol in a water-free environment produces acetaldehyde as the main product, in considerable excess of oxygen.11 Although acetaldehyde is a valuable market product, about 40% of it is used for the production of acetic acid.12 On the other hand, the liquid phase oxidation of diluted ethanol has high activity at a milder temperature and produces acetic acid as the main product.13,14 The presence of water is important for product selectivity, which switches from acetaldehyde to acetic acid with increasing dilution.14Eqn (1) shows the net oxidation reaction of ethanol to acetic acid. Jørgensen et al. proposed a reaction mechanism via intermediates (eqn (2)–(5)) based on experimental measurements over titania-supported gold: ethanol adsorbs on the catalyst and forms ethoxy species (2), which dehydrogenates to acetaldehyde (3) and further oxidises to acetic acid (4), or combusts to carbon dioxide (5).14 Ethyl acetate is also a by-product, likely from the esterification of acetic acid and ethanol.14 Although adsorbed oxygen is considered as the oxidant, it has not been experimentally defined. The dissociative adsorption of oxygen on the gold surface is highly unlikely.15 Density functional theory calculations of a Au (111) surface demonstrated that adsorbed hydroxyl groups lower the activation energy of all three steps (eqn (2)–(4)) compared to a clean surface.16,17 Experimentally, the oxidation of diluted ethanol in a basic environment resulted in acetic acid that contained oxygen from water (H2O18), indicating that water is more than just a solvent.16 A specific reaction mechanism has yet to be determined and the cause of the selectivity switch is not yet known.
| | | CH3CH2OH + O2 → CH3COOH + H2O | (1) |
Experimental measurements of aqueous ethanol oxidation, especially kinetic and mechanistic tests, are very challenging due to the triphasic reaction, which induces problems such as mass and heat transfer. Hence, most experimental research is done in batch systems at long residence times and high gas pressures to ensure an excess of dissolved oxygen in the liquid phase.13,14,18–21 Those systems not only hinder scientific research but also differ from conditions in industry, where continuous flow systems are preferred because high throughput and long-term steady-state operation are essential. Continuous systems are considered to be the most suitable for gas–liquid–solid reactions and are implemented in the research of the oxidation of liquid alcohols.22–24 Aqueous ethanol oxidation was initially studied in packed-bed flow reactors.25,26 Operation in flow reduces the residence time of the reaction, favours long-term steady-state performance, and eases the transition to a larger scale.23,27 For research, a flow system enables faster screening of catalysts and specialized reaction testing, such as kinetic measurements and in situ characterization.25,28 Flow reactor systems are sometimes associated with an improvement in product selectivity and mass and heat transfer.27 Furthermore, continuous systems are associated with green chemistry as they are cost and energy efficient and intrinsically safe.29,30
The first step towards the implementation of flow in aqueous ethanol oxidation is a systematic evaluation of its reliability and its advantages over a traditional batch system. Here, we evaluated the activity of a gold–titania catalyst in the oxidation of ethanol in both batch and flow reactors and present a direct comparison of the two systems. The reaction mechanisms were found to be the same, but the effects on the stability of the catalyst differed.
2. Experimental
2.1. Catalyst
A 1% Au/TiO2 (AUROlite™, Strem, Lot: 26622800) commercial catalyst was tested; the material that originated from the same lot was used in both reactor systems and stored in an identical manner. Before testing in batch, the catalyst was crushed and pelletized to achieve a 90–120 μm grain size so that filtration of the catalyst after testing was possible, since no diffusion limitations were detected (ESI†). The catalyst bed in the flow reactor consisted of particles smaller than 90 μm.
2.2. Catalytic testing
The batch experiments were conducted in a series of parallel BR-25 autoclaves purchased from Berghof. The catalyst (100 ± 7 mg and 90–125 μm grain size) and 10 ± 0.03 mL of a 5 ± 0.3 vol% ethanol solution (Fluka, >99.8%) were added to a Teflon®-lined reactor, which was sealed. Thereafter, oxygen (PanGas, 99.999%) was introduced. Each reactor was stirred and heated with a Heidolph MR3002 apparatus, and controlled using a BTC-3000 controller (Berghof).
The flow experiments were performed in a stainless-steel tubular reactor (4 mm internal diameter, 250 mm long) which was heated with a Phoenix Flow Reactor™ unit (ThalesNano) (Fig. 1). The liquid phase was pumped with a Knauer Smartline K120 HPLC pump and the gas with a ThalesNano GasModule™. A Teflon disk back-pressure controller regulated the pressure. The catalyst (1000 ± 5 mg and >90 μm grain size) was mixed with 2000 mg silica (Molar Chemicals) and introduced into the reactor. All the experiments were run using the same catalytic bed, with the exception of the stability tests, during which a fresh catalyst was used. The ethanol stream (5 ± 0.3 vol% ethanol solution, Molar Chemicals, >99.99%) was mixed with oxygen (Linde, 99.5%) in a static mixer and pumped through the catalyst bed. See Table 1 for the standard testing conditions.
 |
| | Fig. 1 Schematic representation of the flow reactor. Aqueous ethanol solution and gaseous oxygen are mixed and pumped through the catalyst bed. The mass flows, temperature, and pressure are constantly controlled (FC, TC, and PC, respectively). The liquid products are collected at the end of the reactor. | |
Table 1 Reaction conditions of catalytic testing in batch and flow reactors. Conditions are kept constant unless otherwise specified
| Batch |
Flow |
|
Where, P0 is the pressure introduced into the batch reactor, PR is the reaction pressure, which in the batch reactor changes with the reaction time, T is the set reaction temperature, mcat. is the catalyst mass, ϕi is the reactant concentration, Vl is the initial liquid volume in the batch, Fi is the reactant volumetric flow rate in the flow reactor, tR is the reaction residence time, and ω is the stirring speed in the batch reactor.
|
|
P
0 = 30 bar, PR = f(t) |
P
R = 30 bar |
|
T = 423 K |
T = 423 K |
|
m
cat. = 100 ± 7 mg |
m
cat. = 1000 ± 5 mg + 2000 mg SiO2 |
|
ϕ
l = 5 ± 0.3 vol% EtOHaq. |
ϕ
l = 5 ± 0.3 vol% EtOHaq. |
|
ϕ
g = 100 vol% O2 |
ϕ
g = 100 vol% O2 |
|
V
l = 10 ml |
F
l = 0.3 ml min−1, Fg = 10 ml min−1 |
|
t
R = 420 min |
t
R = 10.5 min |
|
ω = 500 rpm |
— |
The products were analysed with an Agilent 7890A gas chromatographer equipped with a flame ionization detector (FID). During the analysis, 1 μL of sample was injected at 343 K and carried in a 2 mL min−1 helium flow through the column (DB-FFAP).The temperature of the column was constant at 313 K for 2 min and it was then heated at 8 K min−1 up to 409 K. The FID was fed with 30 mL min−1 hydrogen mixed with 400 mL min−1 air at 573 K. The signal of each compound was calibrated and the calibration line used for quantification was determined by linear regression.
The quantification of the compounds was used to determine the ethanol conversion (X, eqn (6)) and the product selectivity (Si, eqn (7)–(9)), where EtOH is ethanol, AcOH is acetic acid, MeCHO is aldehyde, and EtOAc is ethyl acetate. The average reaction rate (![[r with combining macron]](https://www.rsc.org/images/entities/i_char_0072_0304.gif) , eqn (10)) was defined as the change in the molar concentration of ethanol during the total reaction time. The carbon dioxide selectivity was obtained from the carbon balance (eqn (11)), because it is the only expected gaseous product and the only gaseous carbon-containing product confirmed by GC.14 The gas sample was collected in a 0.6 L Tedlar® bag (Supelco, Lot: 61118LC22G) and analysed qualitatively using an Agilent 3000A Micro GC equipped with three parallel columns: 1. a molecular sieve column with an argon carrier gas and a thermal conductivity detector (TCD); 2. a plot-U column with a helium carrier gas (TCD); 3. an alumina column with a helium carrier gas (TCD). The possibility of carbon species adsorbing on the catalyst was investigated by thermogravimetric analysis, where no significant mass change was observed (ESI†).
, eqn (10)) was defined as the change in the molar concentration of ethanol during the total reaction time. The carbon dioxide selectivity was obtained from the carbon balance (eqn (11)), because it is the only expected gaseous product and the only gaseous carbon-containing product confirmed by GC.14 The gas sample was collected in a 0.6 L Tedlar® bag (Supelco, Lot: 61118LC22G) and analysed qualitatively using an Agilent 3000A Micro GC equipped with three parallel columns: 1. a molecular sieve column with an argon carrier gas and a thermal conductivity detector (TCD); 2. a plot-U column with a helium carrier gas (TCD); 3. an alumina column with a helium carrier gas (TCD). The possibility of carbon species adsorbing on the catalyst was investigated by thermogravimetric analysis, where no significant mass change was observed (ESI†).
| | 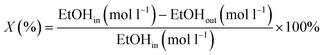 | (6) |
| |  | (7) |
| |  | (8) |
| |  | (9) |
| |  | (10) |
| | | SCO2(%) = 100% − SAcOH − SMeCHO − SEtOAc | (11) |
2.3. Characterization
Scanning transmission electron microscopy (STEM) investigations were performed using an aberration-corrected HD-2700CS STEM microscope (Hitachi).31 The microscope was operated at an acceleration potential of 200 kV (cold field emitter). In the high-angle annular dark-field STEM (HAADF-STEM), the image is generated with incoherently scattered electrons, resulting in an intensity strongly increasing with the atomic number (Z-contrast). The images were recorded with frame times between 20 and 40 s. Analytical investigations were performed with an energy-dispersive X-ray spectrometer (EDXS) attached to the microscope column.
3. Results
3.1. Structure of the catalyst
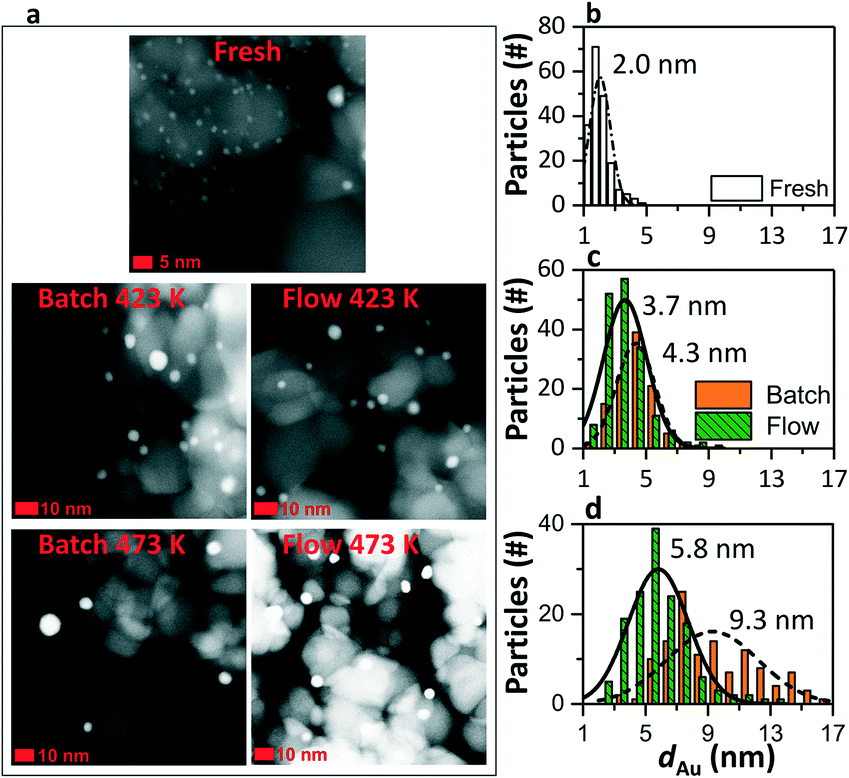
The titania support was defined by XRD as a mixture of 20% rutile and 80% anatase (ESI†), while gold was not detected in any of the patterns (ESI†). Fig. 2a displays the HAADF-STEM images of the fresh and tested catalysts. EDX analysis showed that the bright dots represent gold nanoparticles (ESI†).31Fig. 2b gives the size distribution of the gold particles of the fresh catalyst. The mean particle size was 2.0 nm, determined by size analysis of at least 100 single individual gold particles. Fig. 2c and d show the particle size distribution of the catalysts tested in batch (orange: dashed line) and in flow (green: solid line). The catalysts in both systems have been exposed to reaction conditions for 4 h. After reaction, the catalysts showed an increase in the size of the gold particle. The batch system promoted significant gold sintering, up to 4.3 and 9.3 nm after reaction at 423 and 473 K, respectively. Less sintering was observed after the reaction in flow, with a maximum particle size of 5.8 nm at 473 K.
 |
| | Fig. 2 HAADF-STEM images of fresh and used catalyst (a). The gold particle size was determined using multiple HAADF-STEM images, taking into account at least 100 individual particles: (b) fresh, (c) tested at 423 K, and (d) tested at 473 K (d) in batch (orange – dashed line) and in flow (green – solid line). The mean particle size noted was evaluated based on the normal distribution. | |
3.2. Catalytic activity
Fig. 3 shows the stability test in the flow reactor up to 240 min on-stream under standard conditions, 423 K and 30 bar oxygen (Table 1). The ethanol conversion, which started at 84%, diminished fast to 80% within 60 min and then stabilized at 76% until 240 min. As the conversion decreased within the first 60 min, the selectivity to acetic acid improved from 40 to 46% and remained stable thereafter. The selectivity to acetaldehyde and ethyl acetate also increased by about 2%. Overall, the performance of the catalytic reactor was stable up to 240 min on-stream. The performance was also stable at 473 K (ESI†).
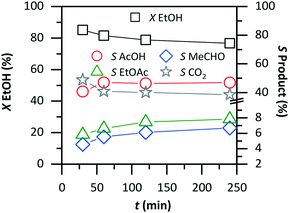 |
| | Fig. 3 Catalyst's stability in the flow reactor at 423 K and 30 bar. The conversion slightly decreases and stabilizes after about 60 min on-stream. Similar evaluation of the catalyst's stability is impossible in batch. | |
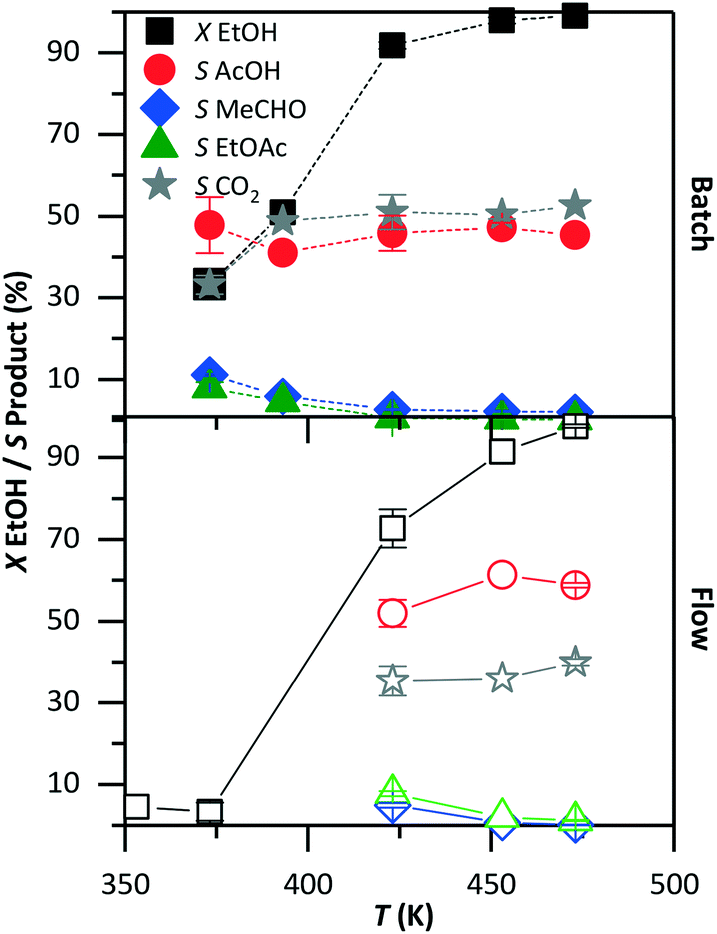
Fig. 4 compares the conversion and product selectivity of the reaction in batch (top graph) and in flow (bottom graph). The conversion increased with temperature in both systems. In the batch system, the ethanol conversion improved from 35% at 373 K to about 100% at and above 453 K. A very low (about 2%) conversion was observed in flow at 373 K. Above 423 K, the conversion started at 73% and reached 98% at 473 K. In the batch reactor, the selectivity to acetic acid was about 45%, independent of temperature. The selectivity to acetaldehyde and ethyl acetate decreased with increasing temperature from 10% to below 5%. The main by-product was carbon dioxide at about 50% selectivity. In flow, the selectivity to acetic acid was 60%, while that for acetaldehyde and ethyl acetate was below 10%. The remaining ethanol was converted to carbon dioxide with 35% selectivity. At 373 K, the product selectivity could not be evaluated with precision due to the low level of conversion.
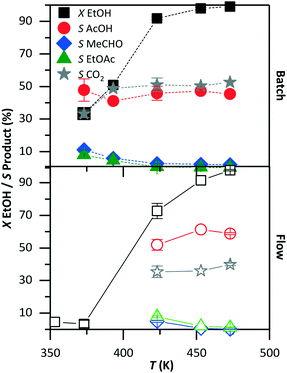 |
| | Fig. 4 Ethanol conversion (X) and product selectivity (S) as a function of temperature in the batch (top) and flow (bottom) systems. The remaining conditions were kept constant as shown in Table 1: 30 bar, 5 vol% ethanol solution, 420 min and 10.5 min in batch and in flow, respectively. | |
Fig. 5a displays the conversion and product selectivity as a function of oxygen partial pressure. The tests at 0.2 bar oxygen partial pressure were carried out in an open-to-the-atmosphere reactor at 358 K to retain the liquid phase. In the batch system (top graph), at a low pressure and temperature, the ethanol conversion was 4% and mainly acetaldehyde was produced with 52% selectivity. A low conversion (10%) was also observed at 0.2 bar oxygen in the flow reactor (lower graph), but the selectivity to acetic acid was 95%.
 |
| | Fig. 5 Ethanol conversion (X) and product selectivity (S) as a function of oxygen partial pressure (a) in the batch (top) and flow (bottom) systems. The remaining conditions were kept constant as shown in Table 1: 423 K, 5 vol% ethanol solution, 420 min and 10.5 min in batch and in flow, respectively. Data at 0.2 bar (yellow-filled symbols) correspond to an open-to-atmosphere reactor at 358 K to sustain the liquid phase. Reaction rate dependency on oxygen partial pressure (b). | |
Between 10 and 50 bar oxygen partial pressure and at 423 K, the conversion in both systems remained constant between 85 and 90%. The product selectivity was also constant, independent of pressure. In the batch reactor, the selectivity to both acetic acid and carbon dioxide stabilized at about 50% while in the flow reactor, acetic acid was the main product with about 60% selectivity; less carbon dioxide was produced than in batch with about 30% selectivity. Small amounts of acetaldehyde and ethyl acetate were observed in both systems. Fig. 5b visualizes the dependency of the reaction rate in oxygen at pressures between 10 and 50 bar. The reaction order in oxygen was zero in both batch (solid symbols) and flow (open symbols).
Fig. 6a shows the effect of ethanol concentration on the catalytic activity. In both systems, the conversion, about 90% in a 5 vol% reactant solution, diminished with increasing ethanol concentration to 50% at 30 vol% of the initial ethanol solution. In the batch reactor at 5 vol% ethanol, the main products were acetic acid and carbon dioxide (each about 50%). With increasing ethanol concentration, the selectivity to acetic acid decreased to 27%; ethyl acetate started to form, resulting in an increase in its selectivity from 1 to 30%. At 30 vol% ethanol solution, acetic acid, carbon dioxide, and ethyl acetate were produced each with about 30% selectivity. In the flow reactor, the selectivity to acetic acid exhibited a maximum of 60% at 10 vol% ethanol solution and decreased to 40% with an increased concentration of the reactant solution. At the same time, more ethyl acetate formed with 23% selectivity. Even at 30 vol% ethanol solution, acetic acid was the main product with 42% selectivity, in contrast to 30% in the batch reactor. Fig. 6b shows the dependency of the reaction rate on ethanol concentration. In both reactors, batch (filled solid symbols) and flow (open symbols), there is approximately a first-order ethanol dependency; the actual slopes were 0.8 and 1.2, respectively.
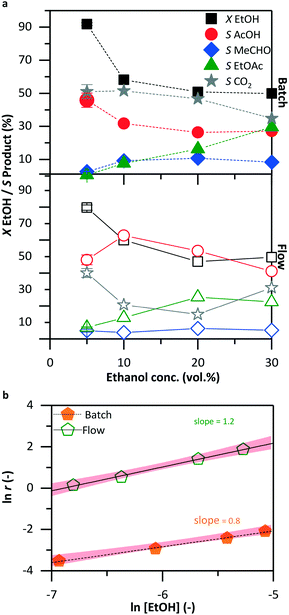 |
| | Fig. 6 Ethanol conversion (X) and product selectivity (S) as a function of ethanol concentration (a) in the batch (top) and flow (bottom) systems. The remaining conditions were kept constant as shown in Table 1: 30 bar, 423 K, 420 min and 10.5 min in batch and in flow, respectively. Reaction rate dependency on ethanol concentration (b). | |
Fig. 7 demonstrates the effect of residence time on the conversion and selectivity in batch (top graph) and in flow (bottom graph). The minimum residence time in batch was 180 min, which corresponded to the time of heating and cooling, during which the reaction proceeded, giving a minimum conversion of about 45%. With a longer residence time, the conversion rose to 90%. The selectivity to acetic acid remained relatively stable between 40 and 47%. The formation of carbon dioxide competed with that of acetic acid, with a selectivity of about 47%. The selectivity to acetaldehyde and ethyl acetate dropped with increasing residence time from 10 to 3 and from 5 to 1%, respectively. The flow system has a maximum residence time of 10.5 min, governed by the limitation of the liquid pump. The conversion improved from 40 to 80% at prolonged reaction times. However, the selectivity to acetic acid decreased by 20%, thus increasing the carbon dioxide selectivity to 40%. At a very short residence time of 2.6 min, there was no carbon dioxide production.
 |
| | Fig. 7 Ethanol conversion (X) and product selectivity (S) as a function of residence time in the batch (top) and flow (bottom) systems. Other conditions were kept constant according to Table 1: 30 bar, 423 K, and 5 vol% ethanol solution. The minimum and maximum residence times are governed by the system's limitations. | |
Fig. 8 shows the conversion level of both systems when both parameters are taken into account. To reach a defined conversion level, the catalyst mass, residence time, or both must be adjusted. It took 14.5 g min−1 at 50% conversion in batch, whereas in flow only 5.5 g min was sufficient. The difference is more apparent at higher conversion, where 90% conversion would require 4.3 times more catalyst or a longer residence time in batch than in flow.
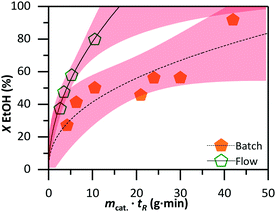 |
| | Fig. 8 Comparison of the required catalyst mass and residence time for achieving the same conversion level in the batch (solid symbols) and flow (open symbols) systems. The lines are obtained by curve fitting (power allometric model) with the red area representing a 97% confidence level. | |
Fig. 9 highlights the difference in selectivity to the liquid products of the two systems. The selectivity to acetic acid was between 55 and 68% in the flow reactor, while in batch it was between 37 and 48%. The smallest difference in selectivity (7%) was found when the conversion approached 100%. At the same time, the selectivity to ethyl acetate was about 10% higher in the flow reactor.
 |
| | Fig. 9 Product selectivity as a function of conversion in batch (solid symbols) and in flow (open symbols). The flow is 7 to 20% more selective towards acetic acid. The lines are obtained by linear fitting with the red area representing a 97% confidence level. | |
4. Discussion
The overall performance of the two systems proves that the triphasic ethanol oxidation is feasible in flow without being detrimental to catalytic performance (Fig. 4 to 9). The stable performance in flow (Fig. 3) indicates that long-term operation is the major advantage of the flow reactor. Long-term stability tests are now feasible with the flow system. Catalyst stability tests in batch, i.e. recycling and reuse of the catalyst, showed a decline in conversion and in acetic acid selectivity.19 Heeskens et al. reasoned that the catalyst deactivation is probably due to the recycling process, filtering and drying, during which a change in the catalyst structure was expected.19 The stable performance in flow justifies the use of one catalytic bed for testing under multiple conditions, while in batch, a fresh catalyst sample is required for each reaction. Hence, the flow operations also have the distinct advantage that the catalyst can be used for longer periods compared to batch cycles, which require new or regenerated catalysts. For example, the ethanol throughput was 18 ml (h−1 g−1) in flow and 14 ml (h−1 g−1) in batch when one batch cycle was taken into account. However, under long-term operation in flow with the same catalyst bed, the throughput can increase indefinitely, for example when 10 batch cycles are taken into account (total 1 g catalyst in both reactors) the batch throughput decreases to 1.5 ml (h−1 g−1).
The overall catalytic performance was better in the flow reactor: when the catalyst mass and residence time were the same; the flow reactor yields a higher ethanol conversion by up to 30% (Fig. 8). This increase in conversion is associated with the lower degree of sintering of the gold particles in flow (Fig. 2). The size of the gold particles is crucial to gold catalysis; the smaller the gold particle, the more active the catalyst.32 The particle size of gold also influences the product selectivity, as exhibited in the epoxidation of propylene over Au/TiO2.32,33 In our case, the selectivity to acetic acid improved in flow by about 10%, independent of the reaction conditions (Fig. 9), while the selectivity to carbon dioxide decreased. While a low and similar selectivity to acetaldehyde was found for both systems, more ethyl acetate formed in flow. The esterification reaction of acetic acid with ethanol (eqn (9)) is an equilibrium reaction that leads to the formation of large amounts of ethyl acetate with increasing ethanol concentration, making it the main product with 50% selectivity when more than 70 wt% ethanol was oxidized.14 The same trend was observed in our batch system with increasing ethanol concentration, but in flow, the ethyl acetate selectivity stabilized at about 30% (Fig. 6). Here, the residence time drives the product selectivity; a short residence time in flow systems may enhance the product selectivity by preventing further chain reactions.27 The residence time also influenced the carbon dioxide selectivity in our flow system (Fig. 7): from 5 to 10 min the selectivity to carbon dioxide increased from 0 to about 50%, at the expense of the acetic acid selectivity. In the batch system, the residence time does not influence the selectivity to acetic acid or carbon dioxide, both of which remain stable at about 50%. This trend is also supported by Aghaei et al. who found 60 and 35%, respectively.20 Hence, in flow, the reaction residence time is a major parameter and fine tuning can lead to optimal selectivity. A highly selective system is more desirable than a system with high conversion, as recycling can improve the overall yield, but low selectivity requires further product separation processes.27
The extensive study under different reaction conditions also provided insight into the reaction kinetics. We found a zero-order dependency in oxygen and first-order in ethanol in both systems (Fig. 5 and 6). This indicates that the reaction mechanism is the same in both systems. Aghaei et al. confirmed those orders by similar studies in batch.20 Their pressure tests at 353, 403, and 433 K showed a stable conversion between 4.5 and 10 bar oxygen partial pressure, indicating a zero-order reaction even below 10 bar. In our study, the only exception was the open-to-the-atmosphere reactor, which led to negligible conversion, both in batch and in flow. At very low pressures, the limiting factor of the reaction was the oxygen mass transfer, as the molal solubility of oxygen in the reactant solution is too low.34 Above 10 bar oxygen partial pressure, oxygen is sufficiently soluble in water, ranging from 0.02 to 0.03 mol O2 per kg H2O, increasing further with oxygen partial pressure. The first-order dependency in ethanol was confirmed by Aghaei et al.20 Although not explicitly defined, a non-zero-dependency in ethanol is observed. Such reaction orders are typically observed for reactions consisting of multiple steps, such as the pathway to acetic acid (eqn (2) to (4)).35 As the surface of the catalyst is fully covered by oxygen (zero-order in oxygen) and adsorbs non-competitively with ethanol, different adsorption sites are expected for the two reactants.
The different adsorption sites of oxygen and ethanol were not identified by our experiments. However, it is argued that oxygen dissociative adsorption on a clean gold surface is highly unlikely.15 Oxygen chemisorption on the titania support is more likely, especially in the presence of oxygen vacancies.36 Similar to carbon monoxide oxidation, oxygen should dissociate on the reducible support rather than on the surface of the noble metal.37,38 With respect to ethanol (eqn (2)), the adsorption and reaction of ethanol on titania would lead to the products acetaldehyde or acetic acid during the reaction over pure titania, which we did not observe. Adsorption on a clean gold surface is also energetically unfavourable at an activation energy of 204 kJ mol−1.16 When the gold surface has some adsorbed hydroxyl groups, the energy decreases by an order of magnitude to 22 kJ mol−1. Our diluted ethanol system probably favours the adsorption of hydroxyl groups on the gold surface, even at neutral pH. Furthermore, the first-order dependency in ethanol indicates that ethanol adsorption is competitive with other species, possibly with hydroxyl groups. The subsequent reaction steps, dehydrogenation to acetaldehyde (eqn (3)) and oxidation to acetic acid (eqn (4)), are also energetically more favourable in the presence of adsorbed hydroxyl groups.16 Here, the presence of molecular oxygen is crucial to regenerate the hydroxyl group to close the catalytic cycle.16 As the oxygen is more likely to dissociatively adsorb on the support, the regeneration of hydroxyl groups must occur on the surface of the support.
5. Conclusions
The aerobic selective oxidation of ethanol was tested over Au/TiO2 in batch and flow systems. Comparison of the catalytic performance in both systems under equivalent reaction conditions confirmed that ethanol oxidation is possible in flow. A main advantage of the flow reactor was the continuous use of one catalyst bed, demonstrating the feasibility of long-term operation when the catalytic performance is stable. The shorter residence time and the use of one catalyst bed in flow can improve the reaction throughput from 1.8 to 18 ml (h−1 g−1). Structural changes on the catalyst were dependent on the reactor system. 2 nm gold particles sintered up to 9 nm in batch, while only 6 nm in flow, which led to improved catalytic performance. The ethanol conversion and selectivity to acetic acid were enhanced by an average of 30% and 10%, respectively. The higher selectivity in flow was also associated with the shorter residence time; in the flow reactor the selectivity was dependent on the residence time. Furthermore, the extensive study revealed a zero-order reaction in oxygen and a first-order reaction in ethanol. The non-competitive adsorption of the two reactants revealed that the reaction mechanism probably proceeds similarly to the oxidation of carbon monoxide: the organic molecule is oxidized through a redox cycle of the reducible support.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We thank Dr. Frank Krumeich and the Electron Microscopy Centre of ETH Zurich for the microscopy analysis. This research was funded by the Swiss National Foundation (SNF 200021E-158188).
References
- T. Mallat and A. Baiker, Chem. Rev., 2004, 104, 3037–3058 CrossRef PubMed.
- C. Parmeggiani, C. Matassini and F. Cardona, Green Chem., 2017, 19, 2030–2050 RSC.
- S. E. Davis, M. S. Ide and R. J. Davis, Green Chem., 2013, 15, 17–45 RSC.
- M. Balat and H. Balat, Appl. Energy, 2009, 86, 2273–2282 CrossRef.
-
C. Le Berre, P. Serp, P. Kalck and G. P. Torrence, Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2014, pp. 1–34 Search PubMed.
- N. Yoneda, S. Kusano, M. Yasui, P. Pujado and S. Wilcher, Appl. Catal., A, 2001, 221, 253–265 CrossRef.
- S. Biella, L. Prati and M. Rossi, J. Catal., 2002, 206, 242–247 CrossRef.
- L. Prati and M. Rossi, J. Catal., 1998, 176, 552–560 CrossRef.
- C. Marsden, E. Taarning, D. Hansen, L. Johansen, S. K. Klitgaard, K. Egeblad and C. H. Christensen, Green Chem., 2008, 10, 168–170 RSC.
- S. Carrettin, Y. Hao, V. Aguilar-Guerrero, B. C. Gates, S. Trasobares, J. J. Calvino and A. Corma, Chem. – Eur. J., 2007, 13, 7771–7779 CrossRef PubMed.
- V. I. Sobolev, K. Y. Koltunov, O. A. Simakova, A.-R. Leino and D. Y. Murzin, Appl. Catal., A, 2012, 433-434, 88–95 CrossRef.
-
M. Eckert, G. Fleischmann, R. Jira, H. M. Bolt and K. Golka, Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2006 Search PubMed.
- C. H. Christensen, B. Jørgensen, J. Rass-Hansen, K. Egeblad, R. Madsen, S. K. Klitgaard, S. M. Hansen, M. R. Hansen, H. C. Andersen and A. Riisager, Angew. Chem., Int. Ed., 2006, 45, 4648–4651 CrossRef PubMed.
- B. Jørgensen, S. Egholm Christiansen, M. L. Dahl Thomsen and C. H. Christensen, J. Catal., 2007, 251, 332–337 CrossRef.
- X. Deng, B. K. Min, A. Guloy and C. M. Friend, J. Am. Chem. Soc., 2005, 127, 9267–9270 CrossRef PubMed.
- B. N. Zope, D. D. Hibbitts, M. Neurock and R. J. Davis, Science, 2010, 330, 74–78 CrossRef PubMed.
- Q. Meng, Y. Shen, J. Xu, X. Ma and J. Gong, Surf. Sci., 2012, 606, 1608–1617 CrossRef.
- K. Q. Sun, S. W. Luo, N. Xu and B. Q. Xu, Catal. Lett., 2008, 124, 238–242 CrossRef.
- D. Heeskens, P. Aghaei, S. Kaluza, J. Strunk and M. Muhler, Phys. Status Solidi B, 2013, 250, 1107–1118 CrossRef.
- P. Aghaei and R. J. Berger, Appl. Catal., B, 2013, 133, 195–203 CrossRef.
- W. Dong, S. Reichenberger, S. Chu, P. Weide, H. Ruland, S. Barcikowski, P. Wagener and M. Muhler, J. Catal., 2015, 330, 497–506 CrossRef.
- N. Zotova, K. Hellgardt, G. H. Kelsall, A. S. Jessiman and K. K. M. Hii, Green Chem., 2010, 12, 2157 RSC.
- X. Ye, M. D. Johnson, T. Diao, M. H. Yates and S. S. Stahl, Green Chem., 2010, 12, 1180 RSC.
- C. V. Navin, K. S. Krishna, C. S. Theegala and C. S. S. R. Kumar, Nanoscale, 2016, 8, 5546–5551 RSC.
- J. Klassen and R. S. Kirk, AIChE J., 1955, 1, 488–495 CrossRef.
- S. Goto and K. Mabuchi, Can. J. Chem. Eng., 1984, 62, 865–869 CrossRef.
- I. Rossetti and M. Compagnoni, Chem. Eng. J., 2016, 296, 56–70 CrossRef.
- M. A. Newton, J. B. Brazier, E. M. Barreiro, S. Parry, H. Emmerich, L. A. Adrio, C. J. Mulligan, K. Hellgardt and K. K. M. Hii, Green Chem., 2016, 18, 406–411 RSC.
- L. Vaccaro, D. Lanari, A. Marrocchi and G. Strappaveccia, Green Chem., 2014, 16, 3680 RSC.
- C. Wiles and P. Watts, Green Chem., 2012, 14, 38–54 RSC.
- F. Krumeich, E. Műller, R. A. Wepf and R. Nesper, J. Phys. Chem. C, 2011, 115, 1080–1083 CrossRef.
- M. Haruta, Catal. Today, 1997, 36, 153–166 CrossRef.
- T. Hayashi, K. Tanaka and M. Haruta, J. Catal., 1998, 178, 566–575 CrossRef.
- D. Tromans, Hydrometallurgy, 1998, 48, 327–342 CrossRef.
-
M. Boudart and G. Djega-Mariadassou, Kinetics of Heterogeneous Catalytic Reactions, Princeton University Press, New York, 1984 Search PubMed.
- G. Lu, A. Linsebigler and J. T. Yates, J. Chem. Phys., 1995, 102, 4657 CrossRef.
- R. Kopelent, J. A. Van Bokhoven, J. Szlachetko, J. Edebeli, C. Paun, M. Nachtegaal and O. V. Safonova, Angew. Chem., Int. Ed., 2015, 54, 8728–8731 CrossRef PubMed.
- M. Lohrenscheit and C. Hess, ChemCatChem, 2016, 8, 523–526 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8re00054a |
|
| This journal is © The Royal Society of Chemistry 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  a,
Tamás
Sipőcz
bc,
Andreas
Nagl
d,
Balázs
Fődi
b,
Ferenc
Darvas
b,
Karin
Föttinger
a,
Tamás
Sipőcz
bc,
Andreas
Nagl
d,
Balázs
Fődi
b,
Ferenc
Darvas
b,
Karin
Föttinger