
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Small-scale screening of novel biobased monomers: the curious case of 1,3-cyclopentanediol†
G. J. Noordzijab,
C. H. J. T. Dietzc,
N. Leonéb,
C. H. R. M. Wilsens *b and
S. Rastogib
*b and
S. Rastogib
aChemelot InSciTe, Urmonderbaan 20F, NL-6167 RD Geleen, The Netherlands
bAachen-Maastricht Institute of Biobased Materials (AMIBM), Faculty of Science and Engineering, Maastricht University, Brightlands Chemelot Campus, 6167 RD Geleen, The Netherlands. E-mail: karel.wilsens@maastrichtuniversity.nl
cInorganic Membranes and Membrane Reactors, Dept. Chemical Engineering and Chemistry, Eindhoven University of Technology, PO Box 513, Eindhoven, The Netherlands
First published on 29th November 2018
Abstract
In this work, we report on the small scale polycondensation and consecutive analysis of novel polyesters based on the potentially renewable 1,3-cyclopentanediol (CPdiol). To avoid evaporation of monomers during thin-film polymerization reactions, trimer pre-polyesters have been synthesized from the corresponding acid-chlorides with diol monomers. Polymerization of these trimers was explored by thermogravimetric analysis to identify potential side reactions, and to assess the ideal polymerization temperature. In general we observe that trans-1,3-cyclopentanediol exhibits good thermal stability up to 200 °C, whereas thermal dehydration of the alcohol end-groups occurs upon further heating. In contrast, for cis-1,3-cyclopentanediol, the ester bonds of the cyclopentane end-groups become labile, thereby generating carboxylic acid end-groups, and 3-cyclopentenol already at 180 °C. The thermal dehydration reactions yield double bond end-groups, which in turn facilitate cross-linking through cross-coupling and Diels–Alder reactions, leading to an increase in molecular weight. Despite the limited thermal stability of CPdiol, here we demonstrate that polymerization of CPdiol can successfully be achieved in thin-film polycondensation conditions at 180 °C, yielding molecular weights well above 10 kg mol−1.
Introduction
Worldwide, strenuous efforts are taking place to replace oil-based monomers with renewable biomass-based monomers, for both “drop-in” purposes and as new monomers.1–5 However, in particular for new building blocks, it is not assured that these monomers – or the resulting polymeric materials – will function as desired. In order to fully assess such monomers and polymers, generally large amounts (e.g. several kilos) are required to conduct repeatable processing and characterization tests. Unfortunately, academic research often leads to new monomers obtained via e.g. new (bio)catalytic processes having yields in gram scale, or lower. Another major limitation for translations of lab scale synthesis to large scale are the required revenues: significant investment in both time and money is needed to develop new monomers and polymers on a larger scale. To overcome this mismatch in scale, a viable assessment method is desired to polymerize and qualify monomers on a small scale.High-throughput screening equipment is a well-known tool in the fast screening of molecules in e.g. organic synthesis, drug-discovery, and cell-experiments. Also in the field of polymer synthesis, semi-automated high-throughput techniques have been used in solution polymerization,6,7 olefin polymerization,8 and melt-polycondensation.9,10 For example, a small-scale high-throughput melt-polycondensation experiment has been described by Gruter et al.11 for the evaluation of catalyst effectivity in the synthesis of poly(ethylene 2,5-furandicarboxylate) (PEF), commonly considered to be the renewable replacement of poly(ethylene terephthalate) (PET). In general, such screening techniques can lead to faster catalyst development, but can also provide systematic identification of structure–property relation in polymers by varying monomer compositions. An added benefit of small-scale screening experiments is a minimal requirement of monomer availability, making this an attractive technique for assessing the potential of new building blocks. Despite the advantages of high-throughput experimentation, full automation for polymerization reactions can still be challenging due to the time restrictions for polymer characterization. For example, important polymer characterization techniques such as gel-permeating chromatography (GPC) or differential scanning calorimetry (DSC) require relatively long analysis times, potentially becoming the bottleneck in high-throughput screening processes.
The group of Zhang et al.12 has developed a new industrially scalable and cost-effective synthesis route to 1,3-cyclopentanediol (CPdiol) from biomass. These authors reported that CPdiol, usually obtained from non-renewable cyclopentadiene, could be obtained from the aqueous phase rearrangement of furfuryl alcohol, followed by hydrogenation over RANEY® (Scheme 1). CPdiol was obtained as a mixture of cis and trans isomers, which could be further purified via fractional distillation under vacuum.
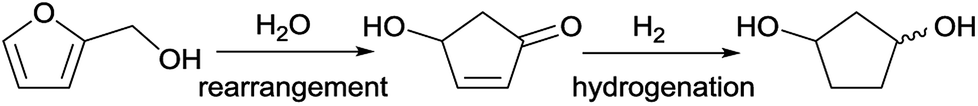 | ||
| Scheme 1 Reaction pathway for the synthesis of 1,3-cyclopentanediol from renewable furfuryl alcohol.12 | ||
Typically in racemic cycloaliphatic monomers the trans isomer results in higher rigidity in the polymer, resulting in an increase in glass transition temperature (Tg). Higher symmetry of the trans isomer also results into more perfect crystals, favoring higher crystallinity, as opposed to the cis isomer. Indeed, this effect is well described for polyesters having cycloaliphatic rings, for example 1,4-cyclohexanediol (CHdiol),13,14 1,4-cyclohexanedimethanol (CHdm),15 and others.14,16–19 In most cases the polyesters with a high trans ratio are semi-crystalline, whereas crystallinity is often lost when reaching, or exceeding, a 50/50 cis/trans mixture. With this literature precedent we expect that the properties of polymers with CPdiol will vary with the cis/trans ratio. Additionally, compared to six-membered rings, the five membered cyclo-aliphatic ring in CPdiol is expected to suppress rigidity and symmetry.20 This is due to the steric configuration of the cyclopentane ring, which cannot adopt the well-known ‘boat’-conformation found in cyclohexane.21 These characteristic differences make CPdiol an interesting candidate for polymerization in order to provide a chemical and physical comparison with CHdiol and CHdm.
Zhang et al.12 polymerized 1,3-CPdiol into polyurethanes, and several (enzymatic) transesterification reactions involving various 1,2- and 1,3-CPdiol structures are known.22–24 However, to the best of our knowledge, polyesters having 1,3-CPdiol have not been reported in literature. This might be attributed to the fact that CPdiol is only available on small scale.
To overcome the challenges of the limited availability of CPdiol, and to investigate the physical properties of polyesters with CPdiol compared to polymers having cyclohexane counterparts, a small-scale polymerization screening method is reported in this publication.
Experimental section
Materials
1,3-Cyclopentanediol (15/85 cis/trans), 1,4-cyclohexanedimethanol (38/62 cis/trans), adipoyl chloride, sebacoyl chloride, terephthaloyl chloride, tin(II) 2-ethyl-hexanoate, and anhydrous chloroform, DMF, THF, and toluene were obtained from Sigma Aldrich. Magnesium(IV) sulfate, 4-dimethylaminopyridine and pyridine were obtained from ACROS. 2,5-Furan-dicarboxylic acid was obtained from ABCR. 1,4-Cyclohexanediol (46/54 cis/trans) was obtained from Alfa Aesar. Ethylene glycol was obtained from TCI. Standard laboratory solvents were obtained from Biosolve. Deuterated solvents were obtained from Buchem BV (Netherlands). The purchased compounds were used directly without further purification, unless otherwise specified.Characterization methods
1H-NMR and 13C-NMR spectra were recorded with a Bruker Ultrashield 300 spectrometer (300 MHz magnetic field). NMR-samples were prepared by dissolving ca. 10 mg of sample in 0.5 mL deuterated solvent, including dimethyl sulfoxide (DMSO-d6), deuterated chloroform (CDCl3), and deuterated trifluoroacetic acid (d-TFA). All spectra were referenced against tetramethylsilane (TMS), or residual solvent peak from the deuterated solvent.Molecular weight (Mn, Mw) and dispersity (Ð) of the polymers were calculated after gel permeation chromatography (GPC) on a PSS SECcurity GPC system using Agilent 1260 Infinity instrument technology. The GPC system was equipped with two PFG combination medium micro-columns with 7 μm particle size (4.6 × 250 mm, separation range 100–1.000.000 Da), a PFG combination medium pre-column with 7 μm particle size (4.6 × 30 mm), and a Refractive Index detector (RI). Distilled 1,1,1,3,3,3-hexafluoroisopropanol (HFIP) containing 0.019% sodium trifluoroacetate was used as mobile phase at 40 °C, with a 0.3 mL min−1 flow rate. The GPC apparatus was calibrated with poly(methyl methacrylate) standards obtained from PSS. GPC samples were prepared by dissolving 5 mg of polymer in 1.5 mL HFIP overnight under constant shaking, the samples were filtered over a 0.2 μm PTFE syringe filter prior to injection.
Thermal stability of compounds and screening of polymerization conditions were performed via thermogravimetric analysis (TGA) using a TA Instruments Q500. Experiments were performed under a nitrogen atmosphere with a heating rate of 10 °C min−1, from room temperature (RT) up to 700 °C. For polymerization screening experiments the sample was heated to the desired reaction temperature (generally 180, 200, or 220 °C) at a rate of 10 °C min−1 and kept isothermal for the desired reaction time. Thermal transition temperatures of the polymers were analyzed via differential scanning calorimetry (DSC) using a TA Instruments DSC Q2000. Typically, two heating and cooling runs were performed at 10 °C min−1, where the first heating was used to erase any thermal history in the samples. The glass-transition (Tg), melt-transition (Tm), crystallization (Tc), and cold-crystallization (Tcc) temperatures were obtained from the second heating and cooling run. DSC samples were prepared by loading 3–5 mg oven-dried samples in Tzero Hermetic Aluminium pans.
Polarized optical microscopy (POM) images were recorded on an Olympus BX53DP 26, equipped with a Linkam HFSX350 Hotstage. POM was used to determine melt-temperatures of samples which could not be measured via DSC (e.g. because of degradation upon melting). POM samples were loaded on a microscopy slide and heated at a rate of 10 °C min−1. The melt-temperature or range was determined by visual aid of the samples becoming isotropic in nature.
Matrix assisted laser desorption/ionization-time of flight-mass spectroscopy (MALDI-ToF-MS) analysis was recorded on a Voyager DESTR from Applied Biosystems (laser frequency 20 Hz, 337 nm and a voltage of 25 kV). The matrix material was DCTB (40 mg mL−1 in HFIP). Potassium trifluoroacetate was added as cationic ionization agent (5 mg mL−1 in HFIP). The polymer sample was dissolved in HFIP (1 mg mL−1), to which the matrix material and the ionization agent were added (5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:5). If the samples were not fully dissolved, the mixture was filtered over a 0.2 μm PTFE syringe filter prior to placing on the target plate.
:1:5). If the samples were not fully dissolved, the mixture was filtered over a 0.2 μm PTFE syringe filter prior to placing on the target plate.
Synthesis methods
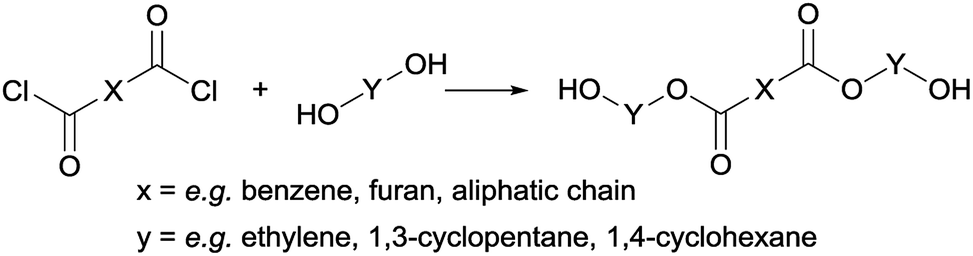 | ||
| Scheme 2 General synthesis route to obtain trimer pre-polyesters. | ||
Polymerization screening methods
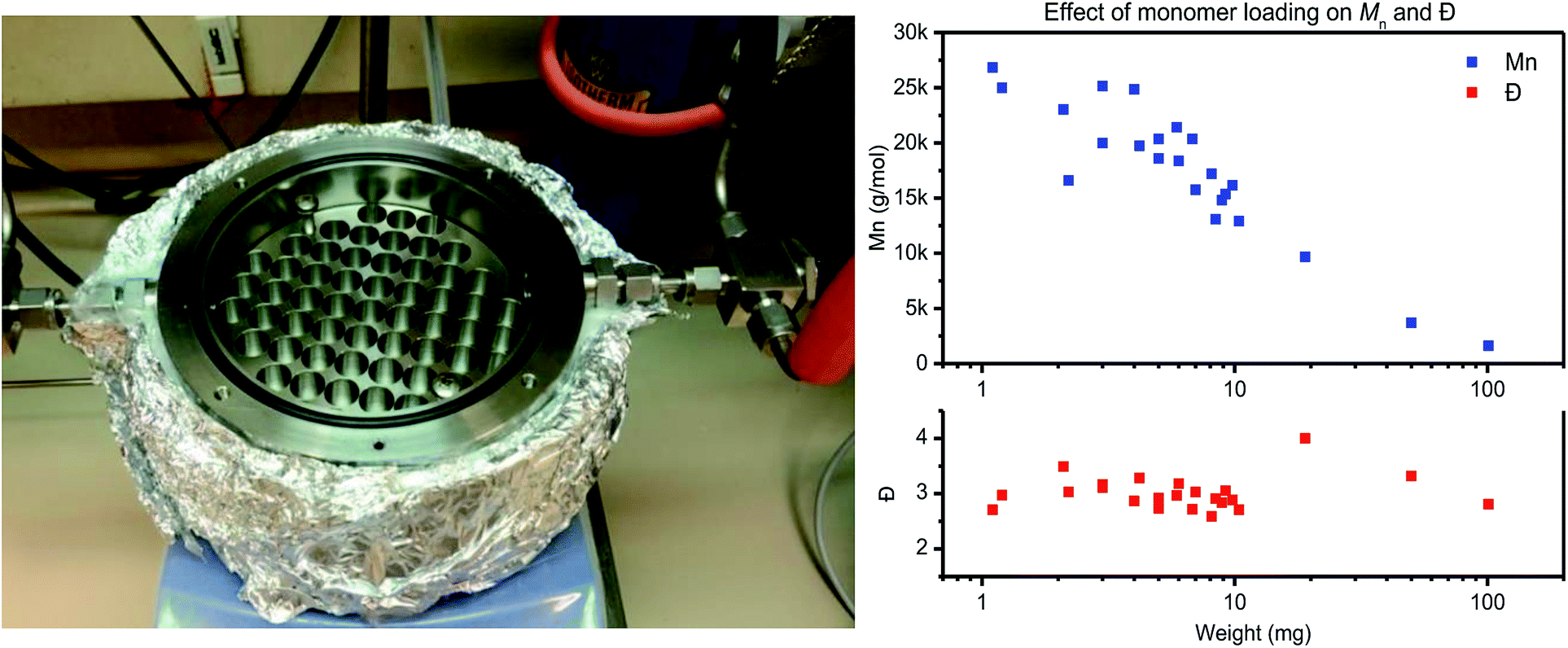 | ||
| Fig. 1 The left image depicts the used stainless-steel reactor with a block for 57 HPLC vials for polycondensation screening reactions. The image on the right depicts the effect of monomer loading (1–100 mg) on Mn (top) and Ð (bottom) for PET polymerized in the same reactor. | ||
The reactor is designed for thin-film polymerization reactions, as thin-film polycondensation reactions do not require stirring for effective condensate removal. Therefore the film thickness should be low enough so that diffusion and mass transfer do not limit the rate of polymerization. For the materials used in this study, placement of 4 mg of polymer material in standard HPLC vial results in a thin-film with a thickness of roughly 200 μm (taking into account an inner vial radius of r = 2.5 mm and assuming a polymer density of ρ = 1.0 g mm−3 (V = (π × r2 × h)/ρ)).
To test the actual effect of monomer loading on the polymerization results, a PET trimer – 1,4-bis(ethyleneglycol-terephthalate) – was polymerized with a loading varying from 1 mg to 100 mg (Fig. 1, right). With increasing monomer loading from 1 mg to 100 mg, the Mn values drops from >25 kg mol−1 to <5 kg mol−1, respectively. For a monomer loading around 10 mg, Mn values around 15 kg mol−1 were consistently obtained on all runs. Therefore, to ensure repeatability of the polymerization experiments, in combination with ease in weighing, further work with this reactor is based on a monomer loading of 10 ± 1 mg. A detailed overview of the polymerization reactions performed for assessment of the repeatability of the thin-film polymerization reactions is provided in the ESI.†
Results and discussion
Trimer synthesis
Following previous work of Sweileh et al.,25 a general synthesis method for the preparation of trimer pre-polyesters, shown in Scheme 2, was developed. Although the use of acid chlorides is not sustainable, it allows for synthesis of trimer pre-polyesters, which do not suffer from evaporation and sublimation during thin-film polycondensation conditions. In other words, these trimer pre-polyesters are excellent candidates for rapid screening and optimization of polymerization conditions. Please note, for bulk polymerizations the use of acid chlorides is unnecessary as dimethyl esters can readily be used.To evaluate the effect of cis/trans ratio in CPdiol, 1,3-cyclopentanediol-furanoate (CP-F) trimers with varying cis content (8%, 20%, 30%, and 40%) were prepared. In spite of the addition of a 3-fold excess of free diol in the reaction mixture, some traces of pentamer and heptamer oligomers were found in addition to the expected trimer oligomers. The presence of trimers, observed from 1H-NMR, LC-MS, and GPC analysis, is exemplified in Fig. 2 for CP-F trimer having a 8/92 cis/trans ratio. A detailed overview of the analysis of the other trimers used in this work is provided in the ESI.† In general, with the used synthesis method, the ratio of trimer/pentamer/heptamer oligomers was 1/0.15/0.01. Nevertheless, all oligomers could be synthesized successfully using the reported synthesis method.
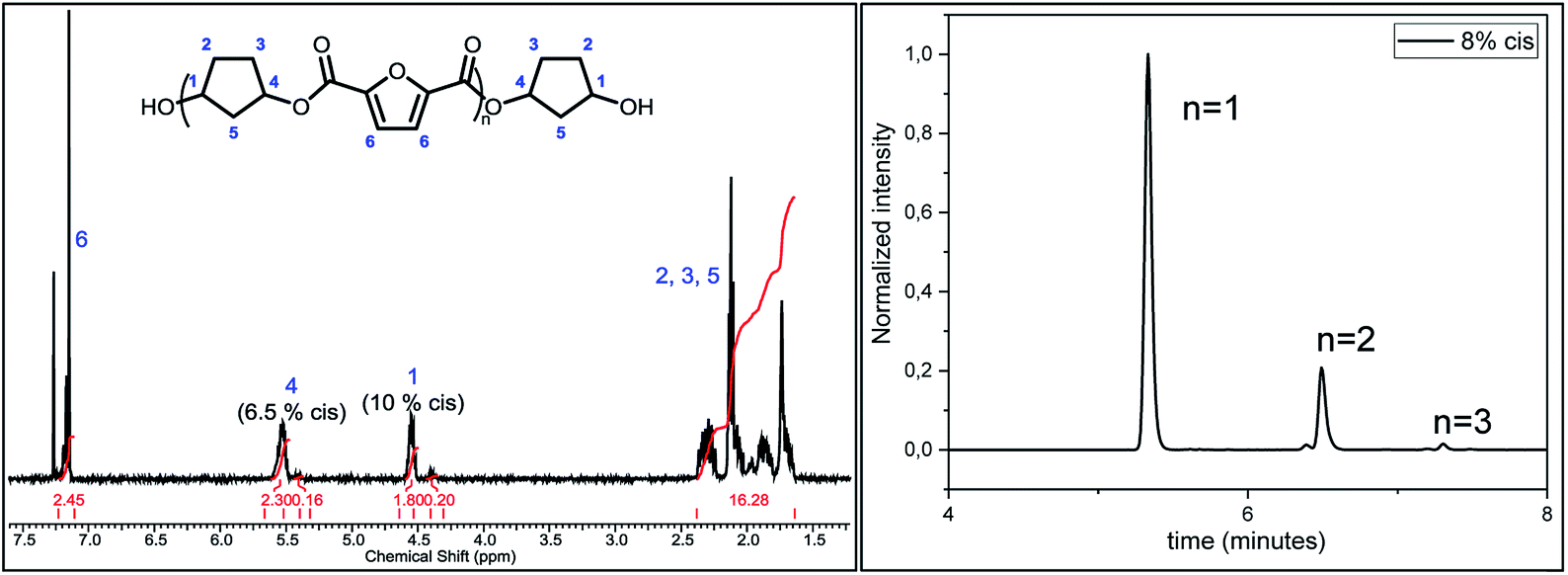 | ||
| Fig. 2 The image on the left depicts the 1H-NMR spectrum of the CP-F trimer with 8/92 cis/trans ratio. The image on the right displays the elution of the same trimer in LC-MS, highlighting the presence of small amounts of pentamer and heptamer. | ||
Interestingly, we observed that the CP-F trimers with the trimers with increasing cis content displayed increased discoloration after drying at 110 °C under vacuum. To investigate the cause of the increased discoloration the oven-dried samples were analyzed with LC-MS. In addition to the trimer/pentamer/heptamer distribution, new peaks with a mass difference of −18, and −84 g mol−1 were identified. The weight loss of 18 g mol−1 likely corresponds to the occurrence of a dehydration reaction, whereas the weight loss of 84 g mol−1 suggests the liberation of a 3-cyclopentenol (CPol) group.
To obtain the products in a pure form, the samples were dissolved in chloroform, resulting in a clear solution in combination with black insoluble particles, which were filtered out. Precipitation in heptane yielded the desired products, ranging from off-white to light brown in color. Interestingly, with increasing cis content, the yield dropped from ∼80% to ∼50%. Overall, these observations suggest that CPdiol exhibits limited thermal stability, where the cis isomer seems less thermally stable than the trans isomer. An image of the discolored products and the analysis of the trimers before and after purification are provided in the ESI.†
Evaluation and optimization of polymerization conditions
TGA studies were performed to ascertain the right polymerization conditions for each trimer and preventing potential degradation resulting from the thermal instability of the CP-F trimers. From the left image in Fig. 3, it can be observed that the CP-F trimers are all thermally stable up to 200 °C, irrespective of the cis/trans ratio. Considering that no evaporation, sublimation, or degradation occurs below 200 °C, polymerization can be performed in TGA. In this case, all observed mass-loss should result from the generation of the condensate CPdiol: the mass loss at the theoretical maximum conversion equals the mass-loss of one CPdiol group of the trimer, which contributes to 31% of the weight.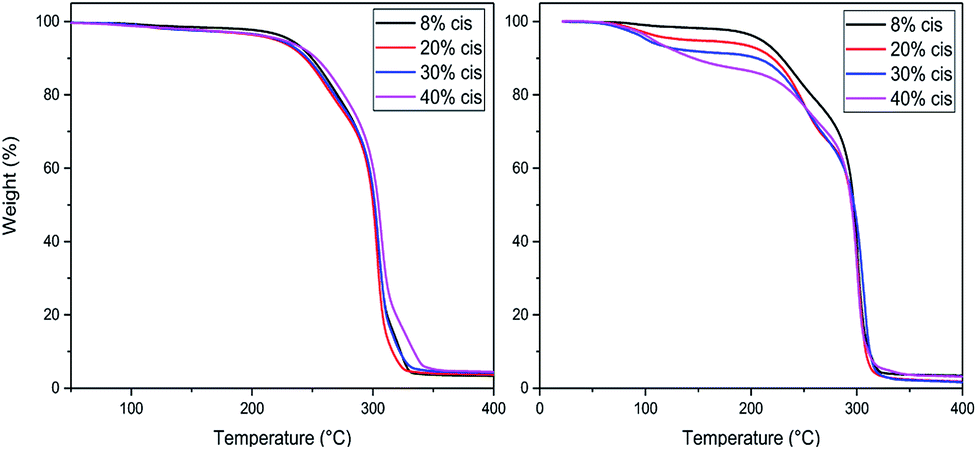 | ||
| Fig. 3 TGA experiments for CP-F trimer, with varying cis content, with a heating ramp of 10 °C min−1, non-catalyzed (left) and catalyzed (right). | ||
To this end, all trimers were mixed with 1 mol% of Sn(II)octanoate catalyst and the same TGA experiments were performed after extensive drying (Fig. 3, right). Indeed, upon the addition of the tin catalyst weight loss is found to start around 100 °C, followed by a stable plateau. Further heating results in degradation, starting above 220 °C. The amount of weight-loss between 100 °C and 200 °C increases with increasing cis-CPdiol content (respectively 3.9, 6.8, 9.1, and 13.5%, Fig. 3, right). These findings indicate that trimers having a higher cis-CPdiol content generate volatiles more rapidly during polymerization, potentially indicating an increased rate of polycondensation.
To identify the optimal polymerization temperature, all trimers were polymerized in the TGA for one hour at 180, 200, and 220 °C (Table 1). As observed previously, an increased weight-loss is observed with increasing cis-fraction, combined with an increased discoloration of the final product. Due to the absence of a vacuum-step in the polymerization in the TGA, the molecular weight of the resulting oligomers remained low, with Mn varying between 500 and 3000 g mol−1. The melting behavior of the polymers was determined via polarized optical microscopy (POM) equipped with a Linkam hot-stage, rather than in DSC due to the limited thermal stability of the samples. The hot-stage was heated at 10 °C min−1, and the visual off-set of melting was noted.
| Polymer | Cis | Pol. T. | W. loss | Color | Mn (g mol−1) | Mw (g mol−1) | Đ | Tm (°C) | Td (°C) |
|---|---|---|---|---|---|---|---|---|---|
| Poly(CP-F) | 8% | 180 °C | 11.1% | Light yellow | 1224 | 2448 | 2.0 | 260 | 285 |
| Poly(CP-F) | 8% | 200 °C | 19.1% | Light orange | 1846 | 3628 | 1.97 | 300 | 308 |
| Poly(CP-F) | 8% | 220 °C | 26.1% | Light brown | 2769 | 6401 | 2.31 | 285 | 295 |
| Poly(CP-F) | 20% | 180 °C | 9.2% | Light orange | 546 | 1206 | 2.21 | 290 | 296 |
| Poly(CP-F) | 20% | 200 °C | 20.3% | Light orange | 1602 | 3484 | 2.17 | 285 | 296 |
| Poly(CP-F) | 20% | 220 °C | 26.8% | Light brown | 2680 | 6772 | 2.53 | 267 | 281 |
| Poly(CP-F) | 30% | 180 °C | 11.8% | Light brown | 525 | 1161 | 2.21 | 245–280 | 280 |
| Poly(CP-F) | 30% | 200 °C | 23.3% | Light brown | 1660 | 3724 | 2.24 | 245–282 | 280 |
| Poly(C-PF) | 30% | 220 °C | 28.9% | Brown | 2342 | 7169 | 3.06 | 254–274 | 274 |
| Poly(CP-F) | 40% | 180 °C | 14.2% | Transp. brown | 663 | 1414 | 2.13 | — | n.d. |
| Poly(CP-F) | 40% | 200 °C | 23.5% | Transp. brown | 2071 | 4503 | 2.18 | — | n.d. |
| Poly(CP-F) | 40% | 220 °C | 29.1% | Dark brown | 2330 | 8129 | 3.49 | — | n.d. |
As expected, all samples showed rapid signs of (further) discoloration with the evolution of gasses directly after melting, indicating that the degradation temperature (Td) is very close to the melting temperature (Tm). Additionally, increasing of the cis-fraction resulted in an increase in Mw and a decrease in Tm, until a fully amorphous material is obtained for the polymer having 40% cis-CPdiol. These findings are in coherence with literature on the cis/trans effect of other cycloaliphatic diols.
The difference in polymerization behavior of varying cis-CPdiol content becomes apparent by analyzing the GPC traces (Fig. 4). In general, the trimer containing 8% cis-CPdiol polymerized rapidly at 180 °C, indicated by the narrow Ð and the relatively high molecular weight compared to the polymer with higher cis-CPdiol content. When increasing the reaction temperature to 200 °C, the rise of a high molecular weight tail is observed in the molecular weight distribution. In fact, this is true for all samples, and is particularly dominant in the sample having 40% cis-CPdiol. Further heating to 220 °C broadens the molecular weight distribution even further, and this effect again becomes more apparent with increasing amount of cis-CPdiol. It should be noted that the oligomers with increasing cis-CPdiol content became increasingly difficulty to dissolve in the HFIP solvent. In fact, especially for the oligomers containing 30 and 40% cis-CPdiol, black insoluble components remained in the HFIP solvent, which were filtered off prior to GPC analysis. This indicates that the GPC traces shown in Fig. 4 might not reflect the true polymer content as not all material has dissolved.
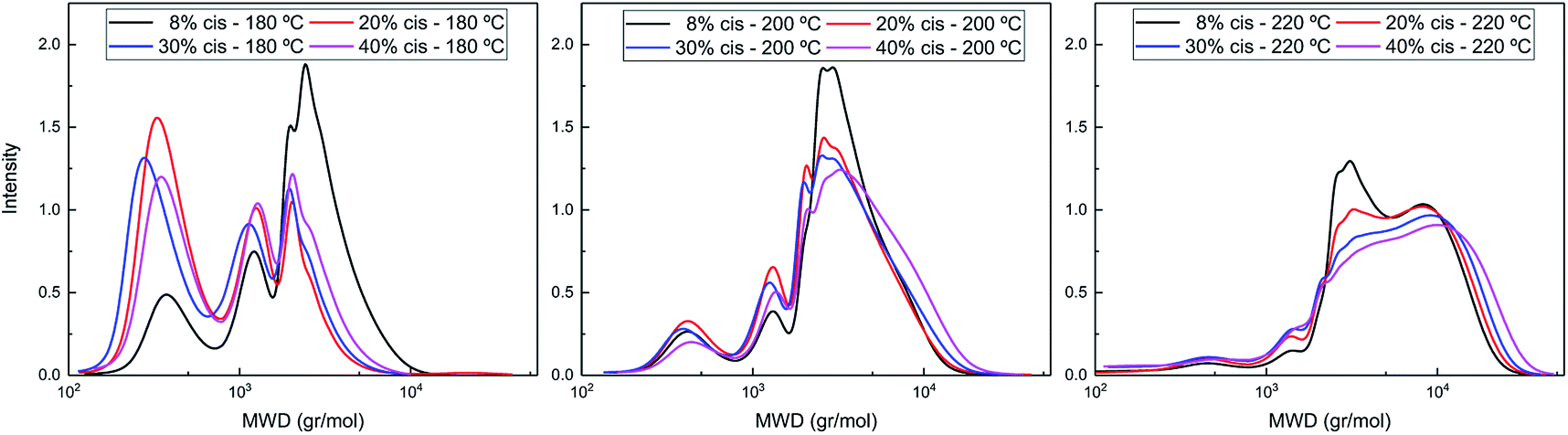 | ||
| Fig. 4 GPC traces of poly(CP-F) polymerized at 180 °C (left), 200 °C (middle), and 220 °C (right), with varying cis-1,3-CPdiol content from 8% (black), 20% (red), 30% (blue) to 40% (purple). | ||
Nevertheless, the formation of insoluble polymer particles suggests the presence of a cross-linking side reaction: the presence of a bimodal distribution is highly unlikely in polycondensation reactions considering the continuously occurring transesterification reaction in combination with Flory's equireactivity theory. Instead, considering the increasing discoloration and decreasing solubility of polymers with increasing cis-CPdiol content, we consider it more likely that cis-CPdiol undergoes a degradative cross-linking reaction in addition to the polycondensation reaction, thereby generating high molecular weight components. To assess whether such a side reaction proceeds during polymerization, the obtained oligomers have been analyzed using MALDI-TOF-MS.
Oligomer analysis using MALDI-ToF-MS
Assuming that the CP-F polycondensation reaction proceeds without side-reactions, only the presence of linear chains with cyclopentanol (CPol) groups on both chain ends is expected (structure 1, Fig. 5). As is observed from LC-MS analysis, thermal dehydration (2, 3), and loss of 1,3-CPol end-group (4, 5) can be expected to occur as a side-reaction during polymerization. The mass distributions of the oligomers after polymerization at 180 °C (blue, top), 200 °C (red, middle), and 220 °C (black, bottom) of 1,3-CP-F trimers having 8% (Fig. 6, left), 20% (Fig. 6, right), 30% (Fig. 7, left), and 40% cis-CPdiol (Fig. 7, right) are reported. The depicted region is selected between 800 and 1050 dalton, in order to show the distribution between repeat units n = 2 (m/z = 806.82) and n = 3 (m/z = 1029.02) of expected main product 1. A more detailed list, the calculated masses of possible chain distributions, and the full MALDI-TOF-MS spectra of the trimers are provided in the ESI section.†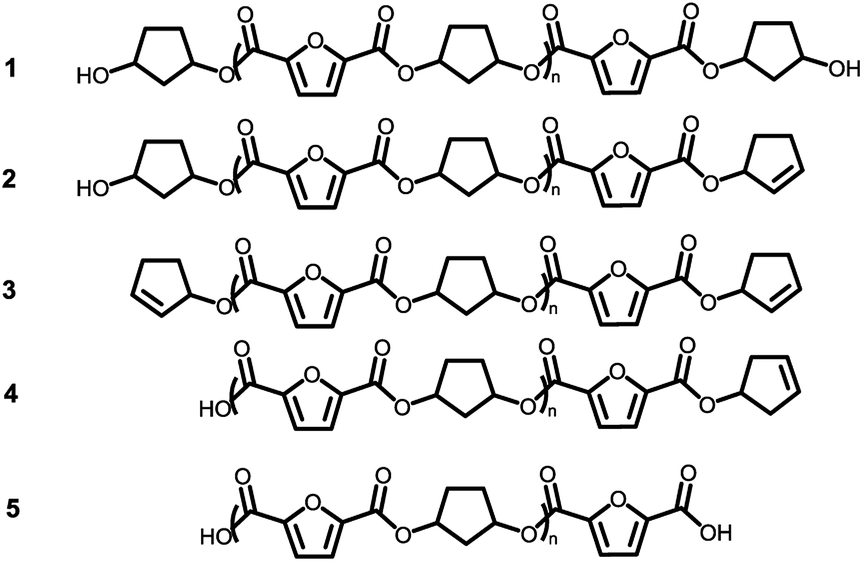 | ||
| Fig. 5 Expected products for poly(CP-F) before and after thermal stability experiments. | ||
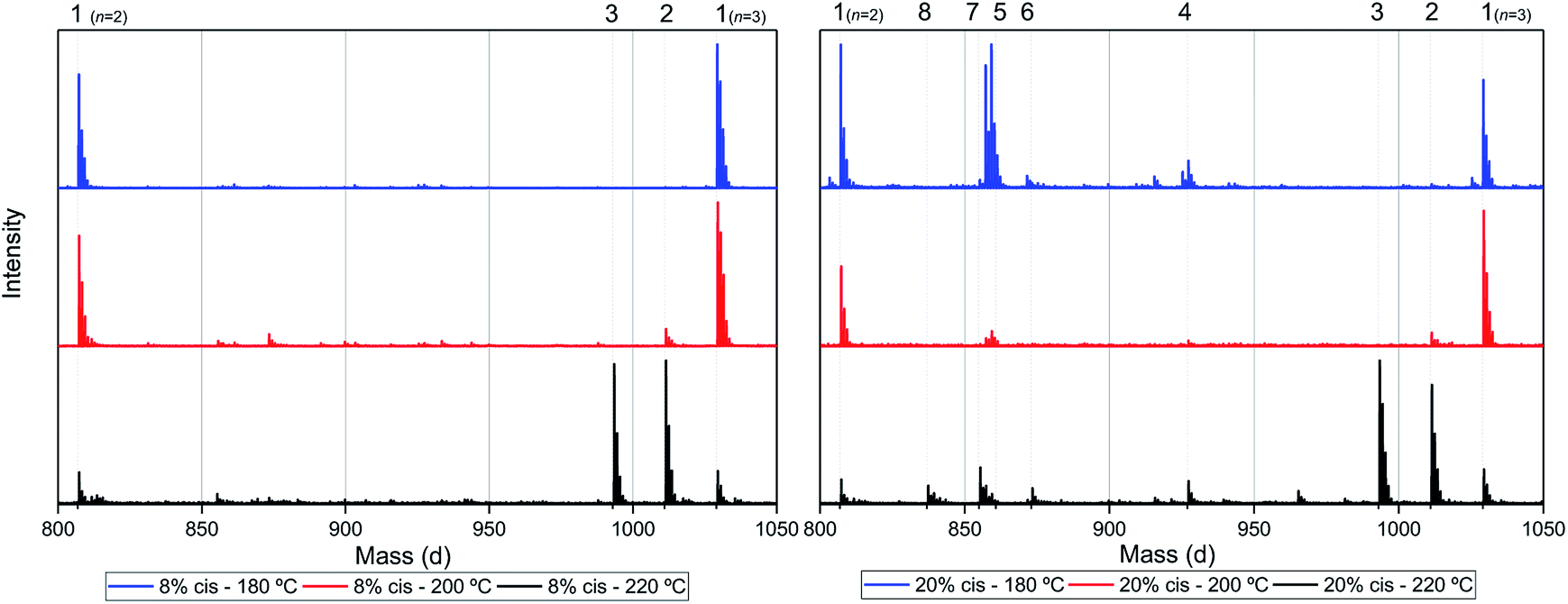 | ||
| Fig. 6 MALDI-ToF-MS spectra of poly(CP-F) polymerized at 180 °C (blue, top), 200 °C (red, middle), and 220 °C (black, bottom), with cis fractions of 8% (left), and 20% (right). Zoomed in on 750–1050 dalton, between repeating units n = 2 (m/z = 806.82) and n = 3 (m/z = 1029.02) of linear diol 1. | ||
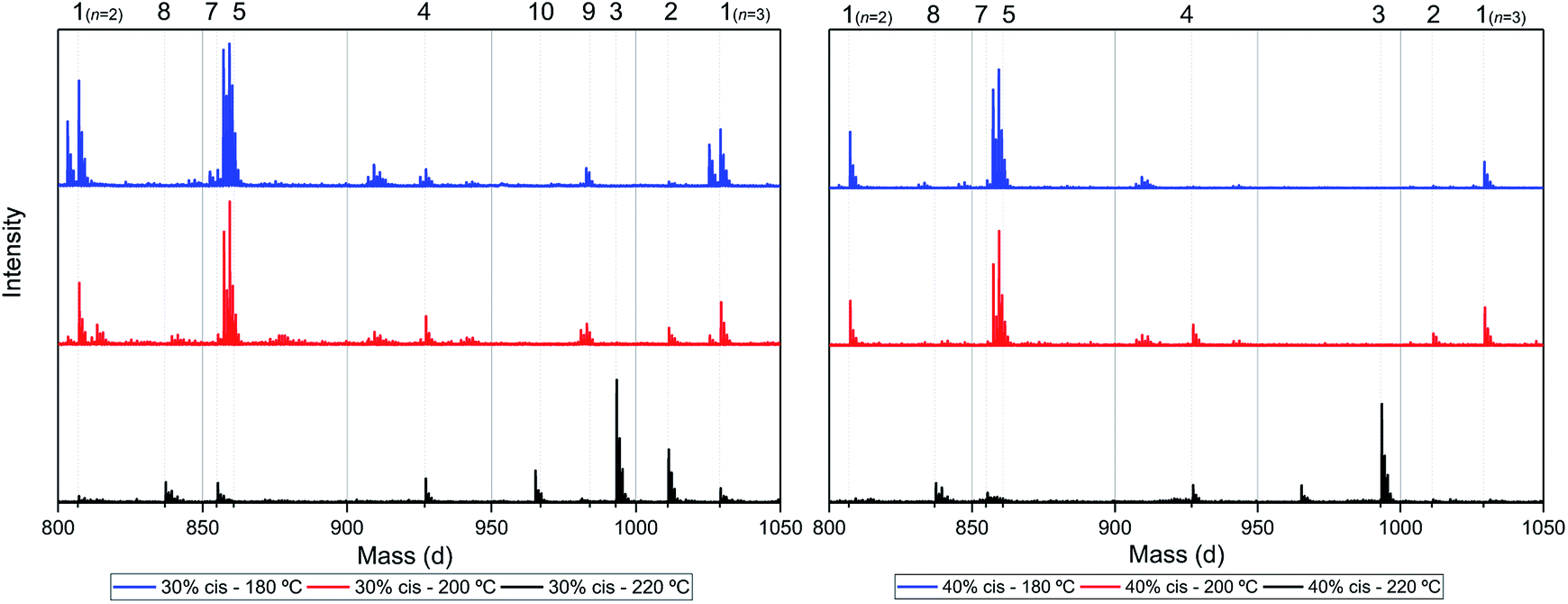 | ||
| Fig. 7 MALDI-ToF-MS spectra of poly(CP-F) polymerized at 180 °C (blue, top), 200 °C (red, middle), and 220 °C (black, bottom), with cis fractions of 30% (left), and 40% (right). Zoomed in on 750–1050 dalton, between repeating units n = 2 (m/z = 806.82) and n = 3 (m/z = 1029.02) of linear diol 1. | ||
As can be seen from Fig. 6, left, the polymer having 8% cis-CPdiol polymerized at 180 °C shows a single distribution of the expected diol product 1 (as is noted on top of the figure). Dehydration of the alcohol end-group takes place when increasing the polymerization temperature, leading to a rise of products 2 and 3 at the expense of product 1, as is clearly visible from the MALDI-ToF-MS spectrum taken at 220 °C. Similar to the samples having 8% cis-CPdiol, an increase in polymerization temperature for the sample having 20% cis-CPdiol results in dehydration of the CPdiol end-groups, as is indicated by the rise in reaction products 2 and 3.
Interestingly, the presence of product 1 becomes less abundant at 180 °C as the cis content increases. Instead, the samples with 20%, 30%, and 40% cis-CPdiol polymerized at 180 °C contain products corresponding to chains with one (structure 4) or two (structure 5) carboxylic acid end-groups. Since the carboxylic acid moiety is not present as starting material, they have to be generated during polymerization. Generally, carboxylic acid groups can be generated through ester hydrolysis by water, (degradative) chain-scission of ester bonds, or complete removal of a 3-cyclopentenol end-group.26,27 Hydrolysis by water would lead to a distribution of chains with a CPol end-group on one side, and a carboxylic acid on the other side. However, such a distribution was not detected in MALDI-TOF-MS analysis (data provided in ESI†). Instead, (degradative) chain-scission of ester bonds would lead to product 4 and 5, however would also inherently lead to a drop in molecular weight, which was not observed in GPC analysis (Fig. 4). Thus it seems likely that only the CPol end-groups have been removed from the product, leading to the observed drop in product 1, and rise in the carboxylic acid terminated products 4 and 5.
An increase in polymerization temperature from 180 °C to 200 °C and 220 °C for CP-F samples having 20%, 30%, and 40% cis-CPdiol results in a decrease in distributions 4 and 5. This could potentially be resulting from thermal decarboxylation of the generated FDCA moieties,26,27 which yields a mono-substituted furan based diene, which can undergo a Diels–Alder reaction with CPene end-groups, also providing a potential route towards branched or cross-linked species. We will elaborate further on this hypothesis later in this work.
To recall, both LC-MS (provided in the ESI†) and the MALDI-TOF-MS analysis depicted in Fig. 6 suggest that thermal dehydration takes place on the external OH group of CPdiol, thereby generating cyclopentene end-groups. These cyclopentene groups are likely to undergo cross-coupling reactions (Scheme 3), leading to products 6, 7, and 8 (Fig. 8), thereby providing a means to build up high molecular weight materials, in addition to the polycondensation reaction. Indeed, a rise in products 6, 7, and 8 can be observed in the samples with increasing cis-CPdiol content, confirming that CPene cross-coupling reactions are occurring. This CPene–CPene coupling reaction also explain the rise in molecular weight observed in GPC analysis of samples having higher cis-CPdiol content (Fig. 4).
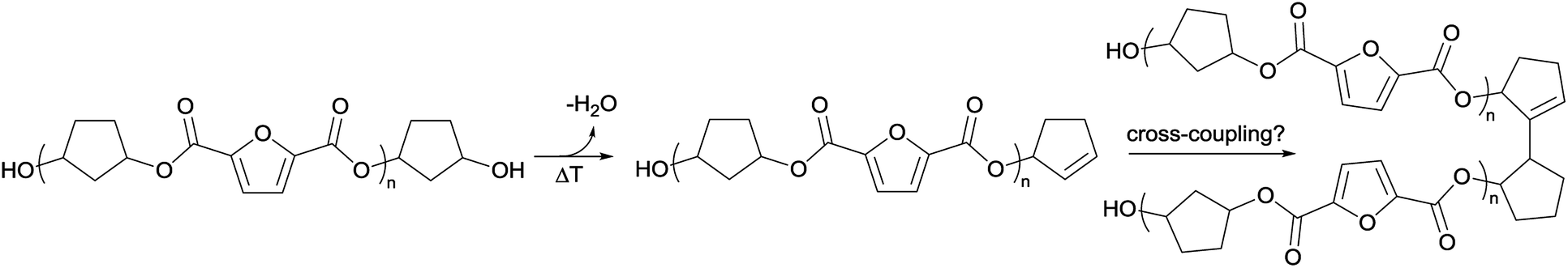 | ||
| Scheme 3 Anticipated degradation process of oligomers. A cyclopentanol end-group can undergo thermal dehydration, generating a cyclopentene end-group, which in turn can undergo a cross-coupling reaction. | ||
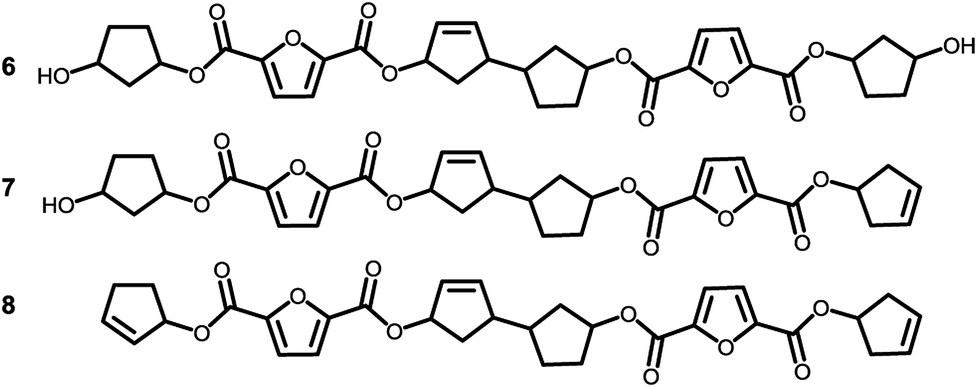 | ||
| Fig. 8 Expected products for poly(CP-F) after CPene cross-coupling reaction and thermal dehydration. | ||
As indicated earlier, an additional chain-extension reaction that can take place is the Diels–Alder addition reaction between decarboxylated furan moieties and CPene groups. This Diels–Alder side reaction would result into the formation of distribution 9, as shown in Scheme 4. Indeed, such a distribution is found in the polymer having 30% cis-CPdiol, (highlighted by dotted line 9 in Fig. 7, left). With increased polymerization temperature to 220 °C, distribution 9 decreases, and a distribution with a mass loss of 18 g mol−1 arises (dotted line 10, Fig. 7). It is likely that distribution 10 corresponds to the dehydrated product of 9. In fact, this distribution is observed in both polymers having 20% and 40% cis-CPdiol, polymerized at 220 °C. These findings provide further proof for the occurrence of a Diels–Alder reaction between the degradation products.
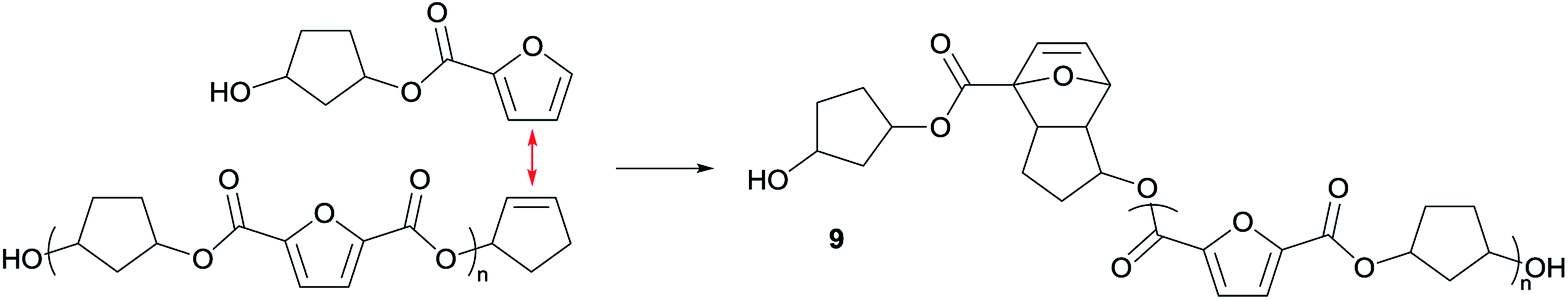 | ||
| Scheme 4 Anticipated Diels–Alder side reaction between a furan group and a CPene group leading to a polymer distribution 9. | ||
It should be noted that the obtained mass-distributions in these MALDI-ToF-MS analysis seem limited to fit only linear chains that are ending with either carboxylic acid, CPol, or CPene end-groups. Although cross-linked or branched mass-distributions are expected given the presence of insoluble components in GPC analysis, such cross-linked or branched fractions are not detected in these MALDI-ToF-MS experiments. Most likely these branched or cross-linked materials are filtered off during sample preparation or are too low in concentration to measure. Nevertheless, the current MALDI-ToF-MS data provides valuable insights in the polymerizability and thermal stability of CPdiol as monomer in polycondensation reactions.
Stability difference cis and trans 1,3-cyclopentanediol
The obtained results in the polymerization stability study suggest that there is an inherent difference in thermal stability between the cis and trans isomer of 1,3-cyclopentanediol: structures with mostly trans-CPdiol end-groups undergo thermal dehydration at temperatures of 200 °C and higher, thereby generating CPene end-groups. Structures with cis-CPdiol end-groups rather undergo degradative ester bond cleavage at 180 °C instead, thereby generating free carboxylic acid groups in addition to CPene end-groups. It is possible that the origin of this stability difference can be found in the different conformations of these two isomers. The conformations of cis- and trans-1,3-cyclopentanediol have been evaluated using theoretical and NMR investigations in the work of Koniotou et al.21 These authors reported that the most stable conformers for cis-CPdiol have the hydroxyl groups in axial positions, allowing for intramolecular hydrogen bonding.28 In contrast, the most stable conformers for trans-CPdiol have the hydroxyl groups in equatorial positions, where intramolecular hydrogen bonding is not observed. When these findings are extrapolated to esters of 1,3-cyclopentanediol, preliminary minimized energy calculations in ChemDraw 3D software show similar axial and equatorial positioning (Fig. 9). These conformations possibly allow cis-CPdiol to undergo intramolecular hydrogen bonding of the external hydroxyl group with the ester bond on the same CP-ring (Fig. 9, left).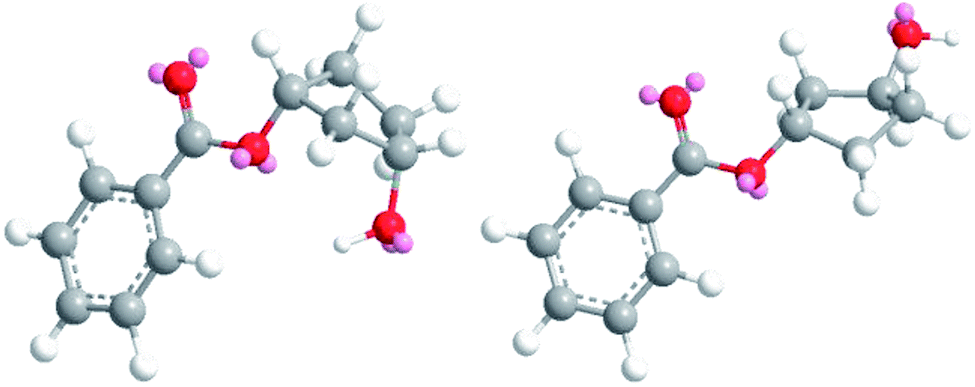 | ||
| Fig. 9 Minimized energy calculations of the mono-benzoate ester of cis-CPdiol (left) and trans-CPdiol (right). Calculated with Chemdraw 3D v17, via MM2 molecular dynamics and minimize energy. | ||
Combining our experimental findings with the preferred conformations of CPdiol esters, suggests that the origin in thermal stability between cis- and trans-CPdiol might be related to the absence or presence of intramolecular hydrogen bonding. Even though in-depth mechanistic studies of the degradation are beyond the scope of this research, a possible degradation mechanism based on the conformations of CPdiol is postulated in Scheme 5. In both cases, β-hydrogen elimination takes place, followed by the breaking of a σ-bond: either C–OH (left), or C–OR (right). Without intramolecular hydrogen bonding it appears that the weakest σ-bond to break is C–OH, leading to dehydration in the trans isomers (Scheme 5, left). However, with intramolecular hydrogen bonding it appears that C–OR becomes the weaker σ-bond, leading to removal of the CPol group, generation an acid-terminated chain (Scheme 5, right).
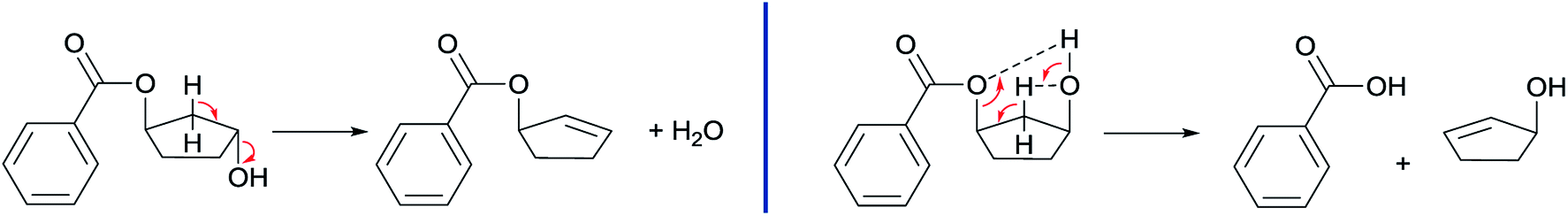 | ||
| Scheme 5 Proposed thermal degradation mechanisms of trans (left) and cis (right) 1,3-CPdiol. | ||
Furthermore, dehydration via β-hydrogen elimination is acid catalyzed, and indeed it is observed that with increasing acid-terminated species 5, dehydrated species (2 and 3) start to form at the expense of diol terminated chains (1) (Fig. 6 and 7). Therefore, it appears that the thermal degradation mechanism of cis-1,3-CPdiol leads to free acid species, which in turn catalyzes the dehydration of the remaining trans 1,3-CPdiol end-groups, leading to an even lower thermal stability of the polymer.
Please note that there is no increase in the low molecular weight fractions observed at the evaluated polymerization temperatures (Fig. 4), indicating that the difference in thermal stability at these temperatures seems to originate solely from the difference in cis or trans end-groups. The thermal stability seems enhanced once the 1,3-CPdiol end-groups are completely polymerized even though ‘normal’ β-hydrogen chain scission still occurs at elevated temperatures.27 Unfortunately, we were unable to study the rate difference in β-hydrogen chain scission between the cis and trans conformers of CPdiol. This is related to the inability of the poly(CP-F) to build up high molecular weight under the explored polymerization conditions.
Assessment of 1,3-cyclopentanediol as building block
In the previous section we reported on the thermal lability of the cis-CPdiol, and the corresponding thermal limitations. Even though presence of 15% cis content in the commercial batch of 1,3-cyclopentanediol limits the polymerization temperature to 180 °C, polyesters could be prepared in small-scale thin-film polycondensation reactions. Apart from poly(CP-F), trimers of CPdiol with adipic acid (A), sebacic acid (S), and terephthalic acid (T) have been synthesized. Furthermore, trimers with structurally related 1,4-cyclohexanediol (CH) (50/50 cis/trans ratio) and 1,4-cyclohexanedimethanol (CHdm) (30/70 cis/trans ratio) have been prepared and polymerized. The polymerizations were performed at 180 °C with 1 mol% of catalyst for 3 hours under vacuum. The resulting polyesters were analyzed for their molecular weight and thermal properties, as is summarized in Table 2. Note, the full details of all polymerization experiments are available in the ESI.† The polymers are abbreviated according to the diol and diacid used, e.g. poly(1,3-cyclopentane-terephthalate) is abbreviated as poly(CP-T).| Mn (g mol−1) | Mw (g mol−1) | Tg (°C) | Tm (°C) | |
|---|---|---|---|---|
| a Degradation upon melt.b Obtained via polarized optical microscopy. | ||||
| CP-T | 5k | 11k | 65 °C | 240–300a °C |
| CP-A | 17k | 57k | −30 °C | Amorphous |
| CP-S | 25k | 92k | −38 °C | 45 °C |
| CH-T | 1.1k | 1.4k | n.o. | 300+b °C |
| CH-A | 13k | 37k | 5 °C | 117 °C |
| CH-S | 16k | 45k | −22 °C | 75 °C |
| CHdm-T | 4k | 7k | n.o. | 240+b °C |
| CHdm-A | 12k | 27k | −30 °C | 92 °C |
| CHdm-S | 18k | 42k | −38 °C | 44 °C |
Poly(CP-T) was obtained as semi-crystalline polymer with an unstable melt above 240 °C, which is likely a result from the previously observed instability of CPdiol based materials. Furthermore, the melting temperature of poly(CP-S) is significantly lower compared to its 1,4-cyclohexanediol based counterpart, i.e. 45 °C versus 75 °C. Instead, the melting behavior of poly(CP-S) is comparable to the polymer containing its 1,4-cyclohexanedimethanol counterpart, as they have similar melting temperatures. Furthermore, poly(CP-A) is fully amorphous, whereas poly(CH-A) and poly(CHdm-A) are both semi-crystalline in nature (Fig. 10, left) unfortunately, the Tg of poly(CH-T) and poly (CHdm-T) could not be observed in DSC analysis, likely due to their high crystallinity. With respect to the glass transition temperature (Tg), polymers containing CPdiol are rather comparable to materials containing CHdmol: both monomers yield polymers with significantly lowered Tg compared to polymers containing CHdiol. These results indicate that polyesters with CPdiol have a reduced rigidity compared to polyesters with CHdiol, and should be compared to the more flexible CHdmol instead. However, although the thermal transitions are comparable, the rate of crystallization of polymers containing CPdiol is significantly faster than for polymers containing CHdmol, as is indicated by the absence of cold-crystallization for poly(CP-S), shown in Fig. 10, right.
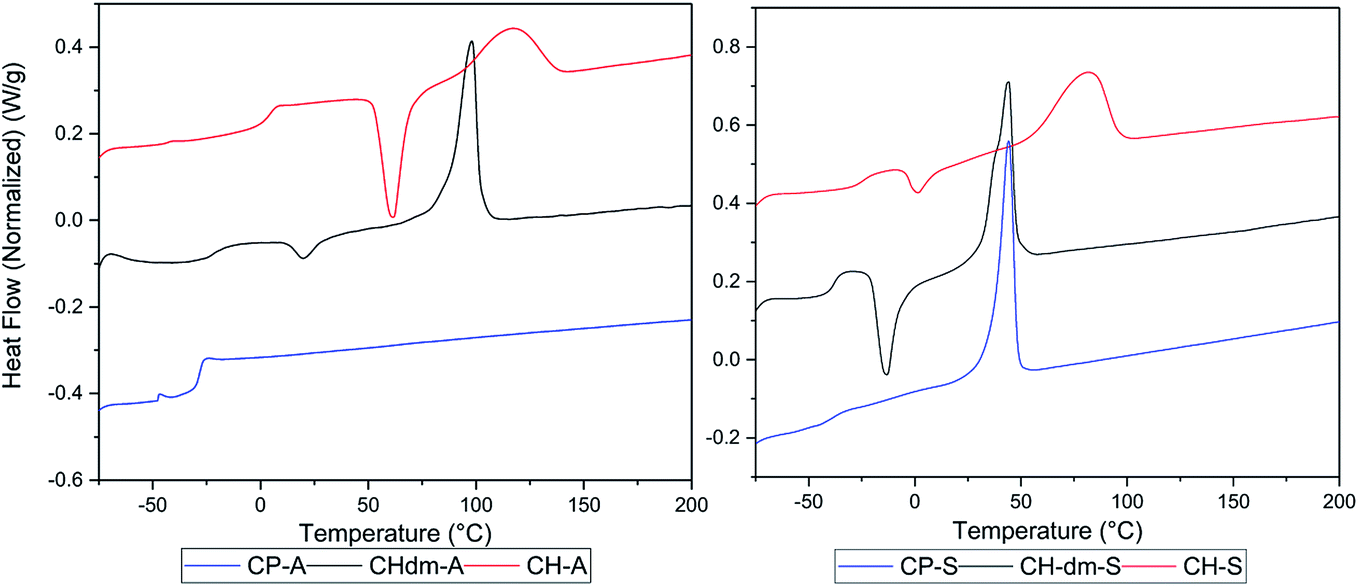 | ||
| Fig. 10 DSC curves of prepared polymers of cycloaliphatic diols 1,3-cyclopentanediol (CP, bottom), 1,4-cyclohexanedimethanol (CHdm, middle), and 1,4-cyclohexanediol (CH, top) with soft blocks adipate (A, left) and sebacate (S, right). Heating and cooling performed with 10 °C min−1, 2nd heating run is shown. | ||
Overall, this preliminary polymerization screening data shows that CPdiol can readily be polymerized under thin-film conditions with various dicarboxylic acids, despite the previously observed degradation reactions. We expect that the application of reduced pressure promotes the polycondensation reaction, and thereby limits the molecular weight build-up by the formed degradation products reported earlier. In fact, under these conditions, Mn values > 15 kg mol−1 are readily obtained for polymers based on CPdiol and sebacic acid or adipic acid. However, the use of high-melting polymers with CPdiol should be avoided due to the inherently unstable melt of CPdiol, preventing melt processing.
Conclusions
A generic synthesis route has been developed to prepare trimer pre-polyesters from various diols, in particular for 1,3-CPdiol, and di-acid-chloride monomers. These trimers were directly used in small-scale thin-film polycondensation reactions as they do not have the drawback of monomer evaporation. Using this approach, TGA was used as a screening technique to identify the thermal stability, and to determine optimal polymerization conditions of the CPdiol based trimers. In general we observed that esters having the trans-CPdiol isomer are found to be stable up to 180 °C, and undergo dehydration at 200 °C and higher. In contrast, esters having the cis-1,3-CPdiol isomer are thermally labile, as they undergo rapid degradation already at 180 °C, which is attributed to the ability to form intramolecular hydrogen bonding. Using the optimal polymerization temperature of 180 °C, high molecular weight polymers (>15 kg mol−1) having 1,3-CPdiol and sebacic acid or adipic acid were successfully made. A comparative study showed that polyesters having CPdiol exhibit lower rigidity and crystallinity than their CHdiol counterpart. Instead, the CPdiol moiety is found to be comparable CHdmol, which can likely be attributed to the less stable half-boat conformation of CPdiol compared to the stable boat-conformation of cyclohexane structures.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was performed under the framework of Chemelot InSciTe and is supported by contributions from the European Regional Development Fund (ERDF) within the framework of OP-Zuid and with contributions from the province of Brabant and Limburg and the Dutch Ministry of Economy.References
- M. Sara, S. K. Brar and J. F. Blais, in Platform Chemical Biorefinery, Elsevier, 2016, pp. 249–283 Search PubMed.
- H. Nakajima, P. Dijkstra and K. Loos, Polymers, 2017, 9, 523 CrossRef.
- M. A. Hillmyer, Science, 2017, 358, 868–870 CrossRef CAS PubMed.
- Y. Zhu, C. Romain and C. K. Williams, Nature, 2016, 540, 354–362 CrossRef CAS PubMed.
- J.-V. Bomtempo, F. Chaves Alves and F. de Almeida Oroski, Faraday Discuss., 2017, 202, 213–225 RSC.
- R. Hoogenboom and U. S. Schubert, Rev. Sci. Instrum., 2005, 76, 062202 CrossRef.
- M. A. R. Meier, R. Hoogenboom and U. S. Schubert, Macromol. Rapid Commun., 2004, 25, 21–33 CrossRef CAS.
- V. Murphy, X. Bei, T. R. Boussie, O. Brümmer, G. M. Diamond, C. Goh, K. A. Hall, A. M. Lapointe, M. Leclerc, J. M. Longmire, J. A. W. Shoemaker, H. Turner and W. H. Weinberg, Chem. Rec., 2002, 2, 278–289 CrossRef CAS PubMed.
- R. A. Potyrailo, J. P. Lemmon and T. K. Leib, Anal. Chem., 2003, 75, 4676–4681 CrossRef CAS PubMed.
- S. Brocchini, K. James, V. Tangpasuthadol and J. Kohn, J. Biomed. Mater. Res., 1998, 42, 66–75 CrossRef CAS PubMed.
- G.-J. M. Gruter, L. Sipos and M. Adrianus Dam, Comb. Chem. High Throughput Screening, 2012, 15, 180–188 CrossRef CAS PubMed.
- G. Li, N. Li, M. Zheng, S. Li, A.-Q. Wang, Y. Cong, X. Wang and T. Zhang, Green Chem., 2016, 3607–3613 RSC.
- J. Wang, X. Liu, Z. Jia, L. Sun, Y. Zhang and J. Zhu, Polymer, 2018, 137, 173–185 CrossRef CAS.
- A. Celli, P. Marchese, L. Sisti, D. Dumand, S. Sullalti and G. Totaro, Polym. Int., 2013, 1210–1217 CrossRef CAS.
- C. J. Kibler, A. Bell and J. G. Smith, J. Polym. Sci., Part A: Gen. Pap., 1964, 2, 2115–2125 CrossRef CAS.
- M. Colonna, C. Berti, E. Binassi, A. Celli, M. Fiorini, P. Marchese, M. Messori and D. J. Brunelle, Polym. Int., 2011, 60, 1607–1613 CrossRef CAS.
- C. Berti, A. Celli, P. Marchese, E. Marianucci, S. Sullalti and G. Barbiroli, Macromol. Chem. Phys., 2010, 211, 1559–1571 CrossRef CAS.
- M. Zhang, R. B. Moore and T. E. Long, J. Polym. Sci., Part A-1: Polym. Chem., 2012, 50, 3710–3718 CrossRef CAS.
- D. R. Kelsey, B. M. Scardino, J. S. Grebowicz and H. H. Chuah, Macromolecules, 2000, 33, 5810–5818 CrossRef CAS.
- T. E. Sandhya, C. Ramesh and S. Sivaram, Macromolecules, 2007, 40, 6906–6915 CrossRef CAS.
- R. J. Abraham and R. Koniotou, Magn. Reson. Chem., 2003, 41, 1000–1008 CrossRef CAS.
- A. Mattson, C. Orrenius, N. Öhrner, C. R. Unelius, K. Hult, T. Norin, M. N. Homsi, F. K. H. Kuske, M. Haugg, N. Trabesinger-Rüf and E. G. Weinhold, Acta Chem. Scand., 1996, 50, 918–921 CrossRef CAS.
- A.-B. L. Fransson, Y. Xu, K. Leijondahl and J.-E. Bäckvall, J. Org. Chem., 2006, 71, 6309–6316 CrossRef CAS PubMed.
- T. Agou, R. Ohata, Y. Mizuhata, N. Tokitoh, H. Fukumoto and T. Kubota, J. Fluorine Chem., 2018, 210, 78–82 CrossRef CAS.
- B. A. Sweileh, H. R. Al-Qalawi and H. A. Mohammad, J. Appl. Polym. Sci., 2014, 131, 39904 CrossRef.
- V. Tsanaktsis, E. Vouvoudi, G. Z. Papageorgiou, D. G. Papageorgiou, K. Chrissafis and D. N. Bikiaris, J. Anal. Appl. Pyrolysis, 2015, 112, 369–378 CrossRef CAS.
- Z. Terzopoulou, V. Tsanaktsis, M. Nerantzaki, D. S. Achilias, T. Vaimakis, G. Z. Papageorgiou and D. N. Bikiaris, J. Anal. Appl. Pyrolysis, 2016, 117, 162–175 CrossRef CAS.
- K. Saegebarth, J. Org. Chem., 1960, 25, 2212–2213 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: ESI contains detailed 2D-NMR analysis of monomers, thin-film polymerization details, and MALDI-ToF-MS data. See DOI: 10.1039/c8ra08811j |
| This journal is © The Royal Society of Chemistry 2018 |